

A central ubiquitin ligase involved in endoplasmic reticulum (ER)–associated protein degradation is the HRD-ligase. The ER-luminal subunit Hrd3 cooperates with the cochaperone Scj1 in clearing misfolded proteins from the ER.
Abstract
Misfolded proteins of the secretory pathway are extracted from the endoplasmic reticulum (ER), polyubiquitylated by a protein complex termed the Hmg-CoA reductase degradation ligase (HRD-ligase), and degraded by cytosolic 26S proteasomes. This process is termed ER-associated protein degradation (ERAD). We previously showed that the membrane protein Der1, which is a subunit of the HRD-ligase, is involved in the export of aberrant polypeptides from the ER. Unexpectedly, we also uncovered a close spatial proximity of Der1 and the substrate receptor Hrd3 in the ER lumen. We report here on a mutant Hrd3KR that is selectively defective for ERAD of soluble proteins. Hrd3KR displays subtle structural changes that affect its positioning toward Der1. Furthermore, increased quantities of the ER-resident Hsp70-type chaperone Kar2 and the Hsp40-type cochaperone Scj1 bind to Hrd3KR. Of note, deletion of SCJ1 impairs ERAD of model substrates and causes the accumulation of client proteins at Hrd3. Our data imply a function of Scj1 in the removal of malfolded proteins from the receptor Hrd3, which facilitates their delivery to downstream-acting components like Der1.
INTRODUCTION
Protein folding in the endoplasmic reticulum (ER) is error-prone, and even in unstressed cells, a considerable fraction of the secretory proteome will not attain its native conformation. Such defective conformers are selected by a protein quality control (PQC) system in the ER and dislocated into the cytoplasm, where they are decomposed by the ubiquitin proteasome system (Hirsch et al., 2009; Brodsky, 2012; Ruggiano et al., 2014). This process is termed ER-associated protein degradation (ERAD). A key component of ERAD is the HMG-CoA reductase degradation ligase (HRD-ligase). This multimeric ER membrane–embedded ubiquitin ligase was first identified in the yeast Saccharomyces cerevisiae but is conserved in all eukaryotic organisms. Hrd1, the central component of this assembly, is anchored in the ER membrane by six transmembrane segments and exposes a RING finger domain into the cytoplasm (Bays et al., 2001). Ubiquitylation of Hrd1 substrates is catalyzed by the ubiquitin-conjugating enzyme Ubc7, which is recruited to the ligase by its activating factor, Cue1 (Biederer et al., 1997; Bagola et al., 2013). An adaptor protein, Ubx2, brings the cytoplasmic Cdc48/Npl4/Ufd1 complex to the ligase, which targets ubiquitylated substrates from the ER to the proteasome (Neuber et al., 2005; Schuberth and Buchberger, 2005). Hrd1 forms oligomers upon binding to the scaffolding protein Usa1 (Horn et al., 2009). In addition, Usa1 recruits the small membrane protein Der1, which participates in the dislocation of client proteins from the ER lumen into the cytoplasm (Carvalho et al., 2006; Horn et al., 2009, Mehnert et al., 2014). In the ER lumen, Hrd1 is associated with Hrd3, which in turn binds to the lectin Yos9 (Gardner et al., 2000; Carvalho et al., 2006; Denic et al., 2006; Gauss et al., 2006a).
The HRD-ligase targets structurally corrupted soluble proteins from the lumen as well as aberrant integral ER membrane proteins for proteolysis. However, it remains to be determined how this protein complex specifically identifies these highly diverse types of client molecules and how it discriminates terminally misfolded polypeptides from species that are in the process of productive maturation. Genetic data imply that the Hrd3/Yos9 heterodimer is involved in this process (Denic et al., 2006; Gauss et al., 2006a; Izawa et al., 2012). The lectin Yos9 preferentially binds N-linked glycan structures on aberrant glycoproteins, which are most likely generated by the sequential action of the mannosidases Mns1 and Htm1 (Quan et al., 2008; Gauss et al., 2011). The activity of these enzymes is believed to destine unfolded proteins for ERAD and thereby delimitates the time a newly imported glycoprotein is given to attain its native conformation in the ER. Hrd3 exposes a large domain into the ER lumen containing numerous SEL1-like repeats (SLRs), which share a high degree of structural similarity with tetratricopeptide repeats (TPRs). TPRs and SLRs are believed to mediate protein–protein interactions (D’Andrea and Regan, 2003). Indeed, Hrd3 binds misfolded proteins in vivo (Gauss et al., 2006b). Moreover, the Hrd3/Yos9 unit was previously shown to interact with the ER-resident Hsp70-type chaperone Kar2—in mammals termed BiP—which proposes a close functional relation of these proteins (Denic et al., 2006). These findings support the idea that Hrd3 constitutes the main substrate receptor of the HRD-ligase and that this protein selects client molecules by binding to malfolded regions. Yos9 refines the specificity of the receptor by identifying glycan moieties, which are primarily found on glycoproteins that have already undergone futile folding attempts. Although this model sounds attractive, the precise role of Hrd3 in substrate detection is unclear. In cells depleted of Hrd3, Hrd1 is rapidly degraded, thereby destructing the overall activity of the ligase (Gardner et al., 2000). This phenomenon hampered a detailed analysis of Hrd3 function in vivo. Moreover, recent work identified regions within the transmembrane segments of Hrd1 that selectively contribute to the degradation of individual membrane-bound substrates, suggesting that they directly participate in the recognition of misfolded proteins (Sato et al., 2009). Therefore selection of some Hrd1 client molecules may not rely on Hrd3 function.
In this study, we report on a mutant variant Hrd3KR, which is specifically defective for the turnover of soluble ERAD substrates. Hrd3KR displays slight structural aberrations that affect its alignment to the downstream-acting protein Der1. We also find that Hrd3 associates with the ER-resident Hsp40-type cochaperone Scj1 and that the Hrd3KR variant binds larger quantities of this protein. Intriguingly, deletion of SCJ1 blocks the degradation of ERAD substrates and causes strong accumulation of such polypeptides at Hrd3. These observations imply that binding to Hrd3 initiates ERAD of soluble ER proteins and that Scj1 contributes to this process by promoting the release of polypeptides from Hrd3 and thereby enables their delivery to downstream-acting factors like the dislocation apparatus.
RESULTS
Mutations in Hrd3 affect turnover of soluble clients
Yeast Hrd3 contains a large ER luminal domain predicted to contain six to nine SLRs. The SLR motif shares a highly similar α-helical architecture with tetratricopeptide repeats and is commonly believed to arrange protein–protein interactions (Ponting et al., 1999; D’Andrea and Regan, 2003). Owing to low sequence similarity, the accurate detection of individual SLRs in a protein sequence is difficult. We propose a positioning of the SLRs in Hrd3 according to a calculation by the program TPRpred, but other alignments have been discussed as well (Karpenahalli et al., 2007; Mittl and Schneider-Brachert, 2007). Of note, the location of the most carboxy-terminal SLR in Hrd3 is essentially the same in all models. Sequential alignment of Hrd3 to homologues from other species identified a patch of several amino acid residues within the most carboxy-terminal SLR that are highly conserved (Figure 1A). To assess the importance of this region for Hrd3 function, we changed some of these residues to alanine, leucine, and isoleucine, respectively, and monitored the effect of these mutants on ERAD. As determined in cycloheximide decay assays, the generated Hrd3 variants, with the exception of Hrd3 E639A, are stable proteins (Figure 1B). Although Hrd1 becomes short-lived in the absence of its binding partner Hrd3, we did not observe Hrd1 degradation in any strain expressing the stable Hrd3 variants, indicating that these proteins were correctly assembled with Hrd1. Pulse-chase analysis revealed that Hrd3 K652I/R653L (Hrd3KR) and Hrd3 N645I/R646I (Hrd3NR) displayed a significant defect in the turnover of the prevalent soluble ERAD substrate CPY* (Figure 1C). The other Hrd3 variants had only marginal effects on ERAD (Supplemental Figure S1A). Replacement of K652 and R653 with alanine (Hrd3 K652A R653A) also substantially affected CPY* processing, which further underscores the relevance of these residues for Hrd3 function (Supplemental Figure S1B). A three-dimensional structure prediction of Hrd3 generated by the Phyre2 homology program (Kelley et al., 2009) with a confidence level of 100% revealed that SLRs in Hrd3 build a superhelix with the A-helices forming the inner concave surface (Supplemental Figure S1C). K652 and R653 in the B-helix of the most carboxy-terminal SLR appear to be located at the surface, supporting a function of these residues in establishing protein–protein interactions. Intriguingly, the Hrd3KR mutant also blocked the turnover of another soluble ERAD substrate, PrA*, whereas proteolysis of membrane-bound targets of the HRD-ligase, such as 6xMyc-Hmg2, and ectopically expressed CD4 was not affected (Figure 1, D and E, and Supplemental Figure S1D). To investigate whether the degradation deficiency of Hrd3KR is specific for a certain type of misfolding or for the topology of a polypeptide, we analyzed the turnover of CT*, which is also a substrate of the HRD-ligase. CT* encompasses full-length CPY* fused to a transmembrane segment that anchors it in the ER membrane (Taxis et al., 2003). Unlike the degradation of CPY*, processing of CT* was not affected in Hrd3KR cells, which supports the notion that Hrd3KR is selectively defective for the turnover of soluble ERAD substrates and that the type of lesion on a target protein does not contribute to the mutant phenotype (Supplemental Figure S1E). Concordantly processing of KWW, a membrane-bound Hrd1 substrate that is degraded in a Der1-dependent manner (Vashist and Ng, 2004), was also not impaired in Hrd3KR cells (Supplemental Figure S1F). In summary, we created a Hrd3 mutant that allows the discrimination of Hrd3 function in the processing of soluble and membrane-bound client molecules. Furthermore, these results imply that particular parts in the luminal domain of Hrd3 are exclusively required for the targeting of soluble polypeptides to the HRD-ligase.
FIGURE 1:

Conserved residues are important for Hrd3 function. (A) Schematic drawing of the Hrd3 protein (top). Positions of the signal sequence (green), the transmembrane segment (blue), and the predicted nine SEL1-like repeats (orange) according to a calculation by TPRpred (Karpenahalli et al., 2007). Bottom, ClustalW2 (McWilliam et al., 2013) alignment of the most carboxy-terminal SLR of Hrd3 from S. cerevisiae (Q05787), and its homologues from Candida glabrata (Q6FNV5), Caenorhabditis elegans (CAB01505), Mus musculus (NP_001034178), and Homo sapiens (NP_005056). Conserved residues are shaded black. Predicted secondary structure elements are shown at the top of the sequences (helices A and B). (B) Stability of Hrd3 variants mutated for the given residues and Hrd1 in these strains as determined in a cycloheximide decay assay. The mutated Hrd3 versions were expressed from a plasmid in a yeast strain deleted for endogenous HRD3. Sec61 served as a loading control. (C–E) Pulse-chase analysis to monitor the turnover of CPY*, PrA*, and 6xMyc-Hmg2. The given Hrd3 variants were expressed from either the corresponding genomic locus (C, D) or from low-copy-number plasmids in cells disrupted for endogenous HRD3 (∆hrd3) (E). Quantification of three independent experiments with SD of the mean.
Hrd3KR binds increased amounts of chaperones
We next investigated the assembly of Hrd3KR into the ligase complex. To this end, we immunoprecipitated hemagglutinin (HA) epitope–tagged variants of Hrd3 or Hrd3KR from cell lysates prepared under nondenaturing conditions and analyzed the samples by immunoblotting. Of note, both HA-Hrd3KR and the wild-type protein precipitated equal amounts of the partner proteins Yos9 and Hrd1, as well as other of components of the HRD-ligase, like Usa1 and Ubx2 (Figure 2A). Hrd3 forms oligomers in the ER (Horn et al., 2009), and we wondered whether the KR mutation affects this feature. Therefore we expressed combinations of HA-tagged variants of Hrd3 or Hrd3KR along with untagged versions in yeast cells and precipitated the HA-Hrd3 proteins. We were able to detect equal amounts of untagged Hrd3 in all precipitates, indicating that the KR mutation did not affect oligomerization of this protein (Figure 2B). Intriguingly, we copurified increasing amounts of the ER-resident Hsp70-type chaperone Kar2 and the Hsp40- type cochaperone Scj1 with HA-Hrd3KR (Figure 2A). Even more striking, we also detected significantly more of the substrate CPY* in the HA-Hrd3KR precipitate. To figure out whether the KR mutation leads to a general enrichment of CPY* at the HRD-ligase, we monitored the association of this substrate with the downstream-acting components Der1 and Hrd1 in Hrd3KR- expressing cells by immunoprecipitation. We expected to preferentially detect direct binding of these proteins to client molecules due to the relatively weak and transient nature of such interactions. Indeed, in precipitates of Der1-Myc or Hrd1-HA from extracts of cells expressing either Hrd3 or Hrd3KR, the amount of copurified CPY* was virtually the same (Figure 2C). Thus it appears that the KR mutation causes an accumulation of CPY* directly at Hrd3. In addition, these results demonstrate that the enhanced substrate association does not simply result from increased steady-state levels of CPY* in Hrd3KR cells. In vitro experiments using purified Hrd3 expressed in Sf9 insect cells provided further evidence for direct binding of this receptor to aberrantly folded protein structures. Chemically denatured luciferase aggregates upon dilution into aqueous buffer due to nonnative hydrophobic interactions. Progressive clustering of luciferase increases the light scattering, which can be measured in a photometer. The addition of luminal domain of Hrd3 (Hrd3lum) to such an assay suppressed the aggregation of luciferase in a concentration-dependent manner to a similar extend as Escherichia coli–expressed DnaJ (Supplemental Figure S2A). Bovine serum albumin (BSA) did not impede luciferase agglomeration. In contrast to a combination of DnaK, DnaJ, and GrpE, however, the addition of Hrd3lum failed to promote refolding of chemically denatured luciferase (Supplemental Figure S2B). These results indicated that Hrd3 prevented the aggregation of a denatured protein by preserving it in an unfolded soluble state.
FIGURE 2:
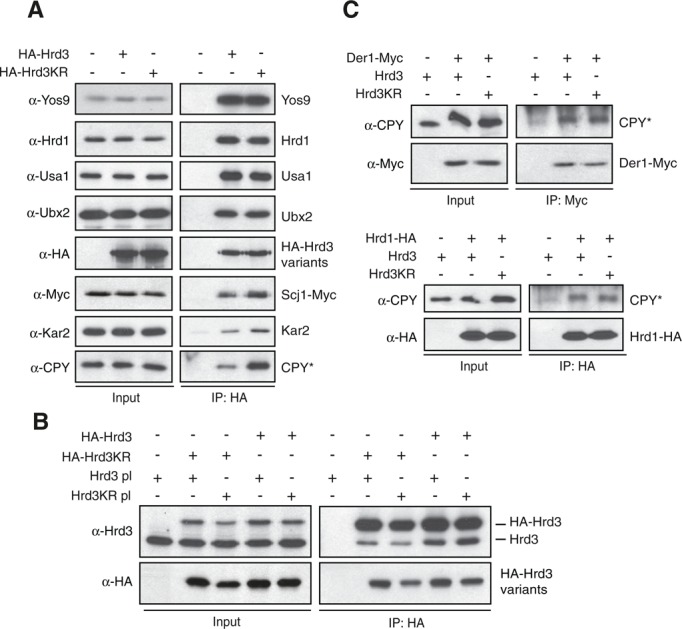
Hrd3KR is fully integrated into the HRD-ligase. (A) Microsomes prepared from ∆hrd3 cells expressing plasmid-encoded HA-Hrd3 or HA-Hrd3KR were solubilized with NP40, and the Hrd3 variants were precipitated with anti-HA antibodies under nondenaturing conditions. The bound proteins were analyzed by SDS–PAGE and immunoblotting using the indicated antibodies. (B) HA-tagged Hrd3 variants were precipitated from lysates of yeast cells expressing HA-Hrd3 or HA-Hrd3KR and containing plasmids encoding Hrd3 or Hrd3KR under nondenaturing conditions. The precipitates were analyzed by immunoblotting with the given antibodies. (C) Plasmid-borne Hrd3 or Hrd3KR were expressed in ∆hrd3 Der1-Myc (left) or ∆hrd3 Hrd1-HA (right) cells. Der1-Myc and Hrd1-HA were then purified from solubilized microsomal preparations and the precipitates analyzed by immunoblotting with the indicated antibodies.
The KR mutation causes moderate structural changes in Hrd3
By employing site-specific in vivo photo-cross-linking, we recently detected close spatial proximity of the membrane protein Der1 and the ER luminal exposed parts of Hrd3 (Mehnert et al., 2014). We used this approach to screen for alterations in Hrd3KR that would affect its activity in substrate processing. In short, plasmids encoding selected Myc epitope–tagged Der1 variants for the labeling with the photoactivatable cross-linker p-benzoyl-l-phenylalanine (pBpa) at a defined position were transformed into yeast cells expressing Hrd3 or Hrd3KR (Chen et al., 2007). After activation of the cross-linker by exposure to ultraviolet (UV) light, the cells were lysed under denaturing conditions, pBpa-Der1-Myc was immunoprecipitated, and the samples were analyzed by immunoblotting. Of note, Hrd3 and Hrd3KR displayed subtle differences in the cross-linking pattern to pBpa-labeled Der1. Most prominently, the intensity of the Hrd3KR cross-link to position G38 and to a lesser extent also to position R33 in the ER-luminal loop 1 of Der1 was significantly reduced (Figure 3, A and B). Conversely, the amount of the cross-linking product with position Q52 of Der1 appeared to be slightly increased in the Hrd3KR mutant (Figure 3B). These observations imply that the KR mutation causes moderate structural changes in Hrd3, which affect the spatial orientation of this protein toward Der1. Such small alterations may disturb the correct placement of target molecules within the ligase complex and prevent efficient handover to downstream-acting components. Indeed, we were unable to detect significant deviations in the cross-linking pattern of pBpa-Der1 to CPY* compared with the wild-type strain, indicating that this substrate does not accumulate at the Hrd3-Der1 interface in the Hrd3KR mutant (Figure 3C).
FIGURE 3:
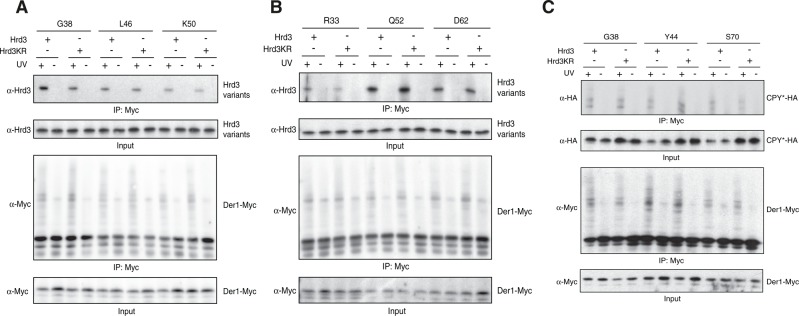
Hrd3KR displays subtle structural alterations. (A) Yeast cells expressing endogenous Hrd3 or Hrd3KR were transformed with high-copy plasmids derived from pMM075 encoding Der1-Myc variants controlled by the CUP1 promoter that each contained an amber stop codon at the indicated positions (Mehnert et al., 2014). The expression of a suppressor tRNA and a corresponding tRNA synthetase allows the incorporation of the photoreactive amino acid analogue pBpa at these sites (Chen et al., 2007). Where indicated, cells were irradiated with UV light, followed by cell lysis and immunoprecipitation of Der1-Myc under denaturing conditions. The precipitates were analyzed by SDS–PAGE and immunoblotting. Labeling with pBpa was carried out for positions in the first luminal loop of Der1. (B) Same as in A, but Der1-Myc was labeled with pBpa at positions in the first (R33) or the second transmembrane domain (D62) or the first luminal loop (Q52). (C) Cells expressing HA-tagged CPY* and Hrd3 or Hrd3KR were transformed with Der1-Myc construct for the incorporation of pBpa at positions in the first luminal loop (G38 and Y44) or in the second transmembrane domain (S70). The experiment was analyzed as in A.
The Hsp40-type cochaperone Scj1 is required for ERAD
Previous studies based on genetic experiments suggested a function of the ER-resident Hsp70-type chaperone Kar2 in conjunction with the Hsp40 type cochaperones Scj1 and Jem1 in the disposal of ERAD substrates (Nishikawa et al., 2001; Buck et al., 2010). Hsp40 type proteins contain a so-called DnaJ domain and control substrate processing of Hsp70 chaperones by regulating their ATP hydrolysis (Kampinga and Craig, 2010). In addition to Scj1 and Jem1, Erj5 and Sec63 also belong to the group of Hsp40-type proteins that reside in the yeast ER. Sec63 is an essential component of the translocation machinery that promotes the import of proteins from the cytoplasm into the ER (Park and Rapoport, 2012). Mutants in SEC63 display pleiotropic phenotypes caused by a general impairment to maintain ER homeostasis. To avoid indirect effects generated by the ambiguous nature of these phenotypes, we did not further investigate a role of Sec63 in ERAD. Cells lacking Scj1, Jem1, or Erj5 are viable, and the detailed molecular function of these proteins in ER protein maturation is still elusive (Nishikawa et al., 2001; Carla Fama et al., 2007; Buck et al., 2010). In immunoprecipitation experiments, we found a previously unnoticed interaction of Hrd3 with the Hsp40-type cochaperone Scj1 (Figure 2A). Intriguingly, the amount of bound Scj1, as well as of Kar2, was considerably increased in the Hrd3KR mutant. Therefore we set out to investigate any involvement of Scj1 in ERAD. As determined by pulse-chase analysis, the deletion of SCJ1 but not of JEM1 or ERJ5 strongly affected CPY* turnover (Figure 4A). Similarly, cells lacking Scj1 but not Jem1 were impaired for PrA* degradation (Figure 4B). In contrast, processing of the membrane-bound substrates 6xMyc-Hmg2 and KWW was barely delayed in the absence of Scj1 (Figure 4C and Supplemental Figure 1F). Deletion of SCJ1 may affect the assembly of the HRD-ligase and thereby interfere with its activity toward selected ERAD substrates. Therefore we precipitated HA-Hrd3 or HA-Hrd3KR from extracts of either wild-type or ∆scj1 cells under nondenaturing conditions. We detected equal amounts of Yos9, Hrd1, and Ubx2 in the precipitates, suggesting a correct integration of Hrd3 into the HRD-ligase in both strains (Figure 4D). However, the amount of Kar2 was considerably increased in the HA-Hrd3 precipitates of SCJ1-deleted cells.
FIGURE 4:

Scj1 is required for ERAD. (A–C) Quantification of radioactive pulse-chase analysis to monitor the degradation of CPY* (A), PrA* (B), or 6xMyc-Hmg2 (C) in strains of the given genotype. Mean values of three (A) or two (B, C) experiments. (D) HA-Hrd3 or HA-Hrd3KR expressed from the chromosomal locus were immunoprecipitated from lysates of yeast strains of the indicated genotypes. The precipitates were analyzed by immunoblotting using the given antibodies.
Substrate accumulates at Hrd3 in the absence of Scj1
Scj1 could assist the identification of malfolded proteins and thus be involved in the recruitment of client molecules by Hrd3. We therefore investigated the effect of Scj1 on CPY* processing by precipitating HA-Hrd3 from lysates of cells lacking either Jem1 or Scj1 and recorded the amount of bound CPY* by immunoblotting. Strikingly, deletion of SCJ1 but not of JEM1 resulted in extensive accumulation of the ERAD substrate CPY* at Hrd3 (Figure 5A). This effect was not enhanced in Hrd3KR cells, indicating that Scj1 and Hrd3 function in the same pathway for CPY* processing (Figure 5B). In line with this, we observed that CPY* turnover was impaired to a similar extent in cells expressing Hrd3KR, deleted for SCJ1, or harboring both the Hrd3KR and Δscj1 mutations (Figure 5C). Strong overexpression of CPY* caused only a very moderate increase in the amount of Scj1 that was copurified with HA-Hrd3, although substantially more substrate was detected in the precipitate (Figure 5D). This finding strongly suggests that the association of Scj1 (and possibly also of Kar2) with Hrd3 is not indirectly mediated via the binding of either factor to client molecules. Instead, Scj1 and Hrd3 interact in a substrate-independent manner.
FIGURE 5:
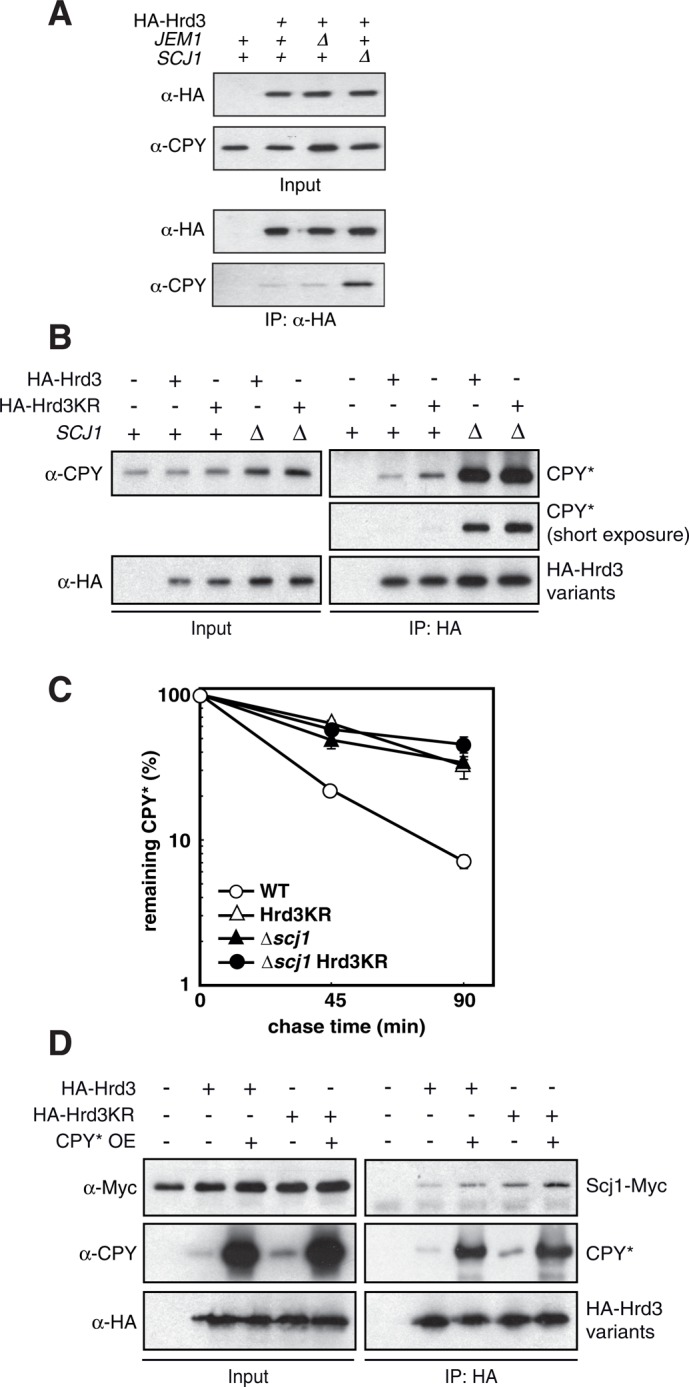
Accumulation of substrate at Hrd3 in cells lacking Scj1. (A) HA-Hrd3 expressed from the chromosomal locus was purified from lysates of yeast cells of the given genotype and the precipitates analyzed by immunoblotting. (B) HA-Hrd3 or HA-Hrd3KR expressed from plasmids in cells deleted for hrd3 was purified from lysates prepared under nondenaturing conditions and the precipitates analyzed by immunoblotting with the given antibodies. (C) Pulse- chase experiment to determine the degradation of CPY* in yeast cells of the indicated genotype. Mean values of three independent experiments with SD of the mean. (D) Lysates were prepared from cells lacking Hrd3 and containing plasmids for the expression of Hrd3 or Hrd3KR. The HA-tagged Hrd3 variants were then immunoprecipitated and the precipitates analyzed by immunoblotting. Where indicated, CPY* was overexpressed using a multi-copy-number plasmid.
We also wanted to determine the site of substrate accumulation on Hrd3 in the absence of Scj1 in more detail. To this end, we performed in vivo cross-linking with pBpa-labeled Der1 variants in either wild-type or Δscj1 cells and analyzed these reactions for the amount of the cross-linked substrate CPY*-HA. Of note, we detected significantly more pBpa-Der1/CPY* cross-linking products for positions in the Der1 luminal loop 1 than for membrane-embedded parts in strains lacking Scj1 when compared with wild-type cells (Figure 6A). This observation demonstrates that in the absence of Scj1, ERAD substrates arrest at Hrd3 in close proximity to the ER luminally exposed parts of Der1. Of importance, this result also implies that substrate accumulation in Hrd3KR and Δscj1 strains occurs at different sites at the HRD-ligase (compare cross-linking patterns in Figures 3C and 6A). Consequently the overexpression of Scj1 in Hrd3KR cells was unable to trigger the release of client molecules from the mutated receptor as determined in immunoprecipitation experiments (Figure 6B) and failed to rescue the ERAD defect observed in such yeast strains (Figure 6C).
FIGURE 6:
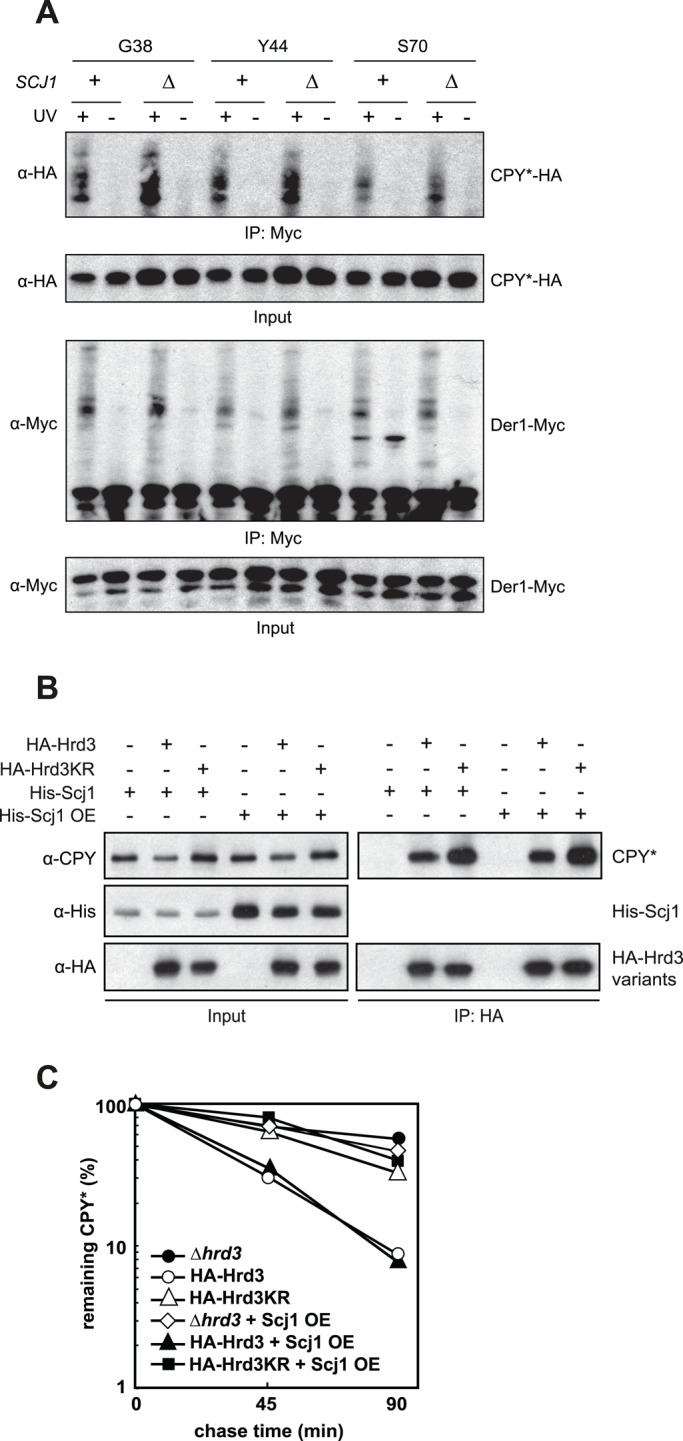
The deletion of Scj1 causes substrate accumulation at the Hrd3-Der1 interface. (A) Site-specific in vivo photo-cross-linking of Der1-Myc to CPY*-HA in cells of the given genotype. Plasmid-encoded Der1-Myc labeled with pBpa at positions in the first luminal loop (G38 and Y44) or in the second transmembrane domain (S70) was immunoprecipitated from cell lysates after UV irradiation using anti-Myc antibodies. The precipitates were analyzed by immunoblotting with the indicated antibodies. (B) ∆hrd3 ∆scj1 cells were transformed with plasmids encoding HA-Hrd3, HA-Hrd3KR, and constructs that express 1xHis-Scj1 from a low- copy-number vector (His-Scj1, pMM100) or a high-copy-number vector (His-Scj1 OE, pMM101). HA-tagged Hrd3 was immunoprecipitated, and the samples were analyzed by immunoblotting. His-Scj1 could not be detected in the precipitate due to strong cross-reactions with the precipitating antibodies. (C) Scj1 was overexpressed in yeast cells of the given genotype, and the degradation of CPY* was monitored in radioactive pulse chase experiments. Mean values of three independent experiments.
DISCUSSION
Terminally misfolded proteins of the secretory pathway are efficiently cleared from the ER by the membrane-bound HRD-ligase complex. In this study, we provide evidence for a function of Hrd3 as the major substrate receptor for soluble ER luminal client proteins of this ligase. A mutation in the most carboxy-terminal SLR of Hrd3 termed Hrd3KR causes a specific defect in the ligase complex to process soluble client molecules. Of interest, the KR mutation is located in a region of Hrd3 that was implicated to mediate the interaction to its partner Hrd1 (Gauss et al., 2006b). Moreover, this part of the mammalian Hrd3 homologue Sel1L is required to suppress proliferation in tumor growth (Cattaneo et al., 2004). We were not able to detect major rearrangements within the HRD-ligase in cells expressing Hrd3KR. However, we observed slight structural changes that seem to affect proper alignment of Hrd3KR to other subunits of this complex and thereby most likely impede the transfer of bound client molecules to downstream-acting factors. We also noticed a significant accumulation of substrate at the Hrd3KR protein, indicating that the efficient detachment of clients may also depend on the correct orientation of Hrd3 to other factors of the HRD-ligase. Indeed, substrate molecules do not accumulate at the Hrd3-Der1 interface in this mutant. Strikingly, we also detected an association of Hrd3 with the Hsp40-type cochaperone Scj1. This observation implied a previously unnoticed functional interplay of Hrd3 and the molecular protein-folding machinery of the ER. Hsp40-type proteins regulate ATP hydrolysis of Hsp70-type chaperones via their DnaJ domain and thereby modulate substrate processing by these enzymes (Kampinga and Craig, 2010). Deletion of SCJ1 impaired turnover of soluble ERAD substrates but had little effect on the degradation of a membrane-bound one. Along with the finding that the combination of the Hrd3KR mutation with a deletion of SCJ1 had no additive effect on CPY* turnover, this observation implies that both proteins act in the same pathway of ERAD. Because we observed substantially increased amounts of client molecules and Kar2 associated with Hrd3 in cells depleted for Scj1, we do not assume a function of this cochaperone in substrate recruitment. Instead, Scj1 appears to participate in the release of polypeptides from the Hrd3 receptor, which is a prerequisite for their proper processing by the HRD-ligase. In this way, Scj1 probably regulates the association of substrate–chaperone complexes with Hrd3.
Although speculative at the moment, we propose the following model for the functional interplay of Hrd3 and Scj1 in ERAD (Figure 7). The association of a misfolded luminal polypeptide with Hrd3 initiates its degradation. It remains to be determined whether aberrant proteins directly bind to this receptor. Such a scenario is supported by our in vitro assays. Alternatively, Hrd3 may recognize specific assemblies containing malfolded polypeptides in complex with molecular chaperones. Next Scj1, probably in conjunction with the Hsp70-type chaperone Kar2, will promote the release of bound substrates from the Hrd3 receptor. The accumulation of Kar2, Scj1, and substrate at the Hrd3KR mutant suggests that the activity of Scj1 may be enhanced toward Hsp70–substrate complexes when they are presented by Hrd3 in a defined manner. Detachment of a client molecule from Hrd3 most likely does not immediately direct it to ERAD but allows its escape into the lumen for repeated folding attempts as well. However, the engagement of a polypeptide with Hrd3 will bring it into close spatial proximity to the membrane protein Der1. This small subunit of the HRD-ligase was recently shown to facilitate membrane insertion of ERAD substrates, which triggers their dislocation into the cytoplasm (Mehnert et al., 2014). As such, Der1 displays a weak affinity for malfolded proteins, which might suffice to take over substrates detached from Hrd3 (Gauss et al., 2006b; Mehnert et al., 2014). We would expect that the efficient transfer of cargo from Hrd3 to Der1 relies on a tight spatial organization of both proteins within the HRD-ligase. Indeed, we previously observed such a close arrangement in in vivo cross-linking experiments (Mehnert et al., 2014). Concordantly our data imply that the correct alignment of Hrd3 and Der1 is critical for substrate processing because the Hrd3KR mutant, which displays moderate aberrations in its orientation toward Der1, is impaired in ERAD. In summary, we suggest that the selection of ERAD substrates involves the reiterated interaction with and Scj1-stimulated release from Hrd3, which will increase the probability of a polypeptide to interact with Der1 and subsequently to be routed for degradation by the ERAD machinery.
FIGURE 7:
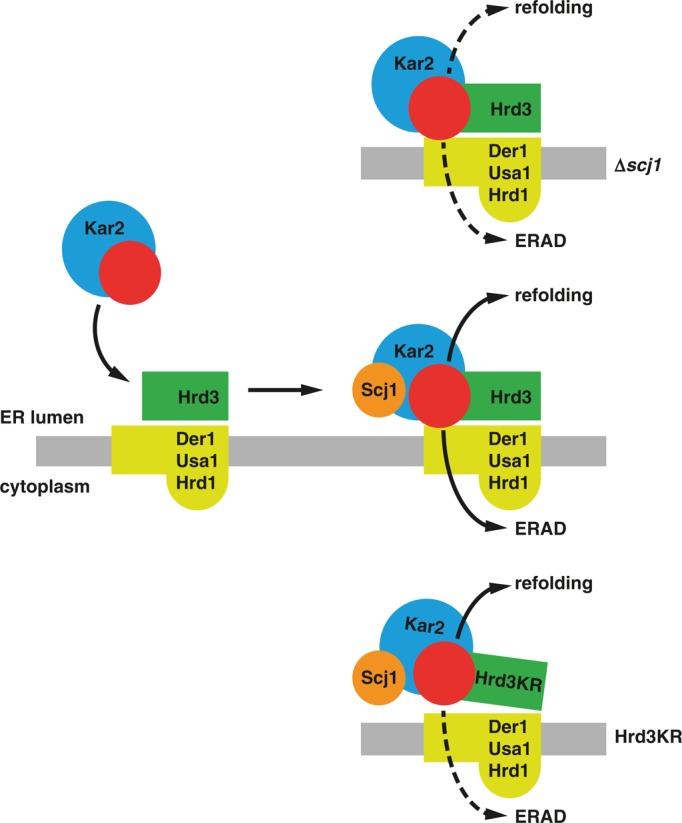
Proposed model for the orchestrated function of Scj1 and Hrd3 in ERAD. Unfolded proteins (red), possibly in complex with molecular chaperones like Kar2, bind to Hrd3. The DnaJ-like protein Scj1 stimulates the dissociation of this transient assembly, which causes the detachment of the client protein and Kar2 from Hrd3. The unfolded polypeptide may then be released into the ER lumen, where it will again engage components of the folding machinery (refolding). Alternatively, it may get in contact with Der1, which resides in close spatial proximity to Hrd3. Der1 displays a weak affinity for aberrantly folded proteins, which might suffice to take over cargo released from Hrd3. At the same time, Der1 facilitates insertion of malfolded proteins into a dislocation apparatus, which will direct these polypeptides for ubiquitylation by Hrd1 and degradation by cytoplasmic proteasomes (ERAD). In the absence of Scj1 (∆scj1), the detachment of substrate from Hrd3 is impaired, which results in the accumulation of malfolded substrates at the Hrd3/Der1 interface. Conversely, the KR mutation causes a structural rearrangement in Hrd3 that blocks the efficient transfer of substrate molecules to Der1 and their dislocation into the cytoplasm.
This scenario shows functional parallels to the posttranslational import of proteins into mitochondria. Here substrate molecules containing internal targeting sequences associate with cytoplasmic Hsp70s. This complex specifically binds to the TPR motifs present in the outer membrane receptors Tom70 and 71 (Young et al., 2003). Remarkably, Tom70 and 71 share a similar structural organization with Hrd3: they are anchored to the membrane by a single transmembrane segment and expose a large domain containing TPRs or the highly related SLRs toward the site of substrate recruitment. Client molecules bound to Tom70/71 are then handed over to the general substrate receptors of the outer membrane Tom20/Tom22 and subsequently inserted into the mitochondrial membranes (Baker et al., 2007; Kutik et al., 2007). Not much is known on how substrate proteins are detached from Tom70/71 and how they are transferred to downstream-acting components. The regulated release of cargo by the activity of Hsp40 cofactors may trigger this process. Intriguingly, the yeast DnaJ-like protein Djp1 was shown to be required for Tom70-dependent mitochondrial protein import (Papic et al., 2013). In addition, in mammals, various cytoplasmic Hsp40-type proteins participate in mitochondrial protein import; however, their contribution to this process remains unclear (Bhangoo et al., 2007).
In mammalian cells, the protein disulfide isomerase ERdj5 is required for the turnover of aberrant secretory polypeptides (Dong et al., 2008; Ushioda et al., 2008). Of interest, this protein contains a DnaJ domain and binds to the Hsp70-type chaperone BiP as well as to the Hrd3 homologue Sel1L (Ushioda et al., 2013; Williams et al., 2013). Similar to yeast Scj1, ERdj5 could therefore promote dissociation of cargo from BiP and Sel1L and thereby facilitate its processing by downstream-acting factors of the ERAD pathway. Thus the folding machinery of the ER may contribute to the degradation of aberrant proteins in several ways. On one hand, components of the ER-PQC participate in the identification of malfolded polypeptides. Selection of defective proteins possibly involves the prolonged association of such conformers with folding enzymes and their subsequent delivery to the receptors of the ERAD ubiquitin ligases. Quite unexpectedly, particular elements of the ER-PQC also facilitate the release of malfolded polypeptides from the receptors, which stimulates the routing of such species to the dislocation apparatus and thereby supports the dynamics of substrate processing in the course of ERAD.
MATERIALS AND METHODS
Antibodies
Monoclonal anti-HA (HA-7) and anti-Myc (9E10) antibodies (Sigma-Aldrich, St. Louis, MO), monoclonal and polyclonal anti-CPY antibodies (Abcam, Cambridge, UK), and polyclonal antiserum directed against CD4 (Santa Cruz Biotechnology, Dallas, TX) are commercially available. Polyclonal anti–proteinase A antiserum was kindly provided by Dieter H. Wolf (University of Stuttgart, Stuttgart, Germany). Polyclonal antibodies specific for Hrd3, Hrd1, Sec61, Kar2, Yos9, Ubx2, and Usa1 were raised in rabbits by immunization with affinity-purified proteins expressed in E. coli.
Yeast strains
All S. cerevisiae strains used in this study were haploid descendants of DF5, and their genotypes are given in Supplemental Table S1. Standard protocols were followed for the preparation of media and the transformation of cells by the lithium acetate method. PCR-based techniques were used to construct strains expressing C-terminally or internally epitope-tagged variants of Hrd3, Jem1, and Scj1 from their chromosomal locus under control of their native promoter (Longtine et al., 1998; Knop et al., 1999).
Plasmids
Plasmids used in this study are listed in the Supplemental Table S2. CT* and KWW were expressed from plasmids pWO804 (Taxis et al., 2003) and pSM101 (Vashist and Ng, 2004), respectively. The PrA* expression vector pTX339 was constructed by ligating NotI-XhoI fragments released from plasmid pES163 (Spear and Ng, 2005) into the NotI-XhoI sites of yeast vector pRS415. CD4 was heterologously expressed in yeast from plasmid pBM108 (Meusser and Sommer, 2004). HRD3-expressing plasmids were generated by PCR amplification of the open reading frame from genomic DNA using the primers FZ81 5′ attggtaccTGATGTAGAGCAGTAACTGGAAG and FZ80 5′ tatgagctcGTATCACCTTCGCCAATGC and cloned into vector pRS317, thereby generating plasmid pJU301, or into pRS316, generating plasmid pFZ063. HRD3 mutants were generated using the QuikChange Site-Directed Mutagenesis kit (Stratagene, Santa Clara, CA), generating plasmid pFZ29 when using pJU301 as a template or pFZ064 using pFZ063. Plasmid pRH244 (Hampton et al., 1996) was used to introduce 6xMyc-Hmg2 into yeast strains.
Immunoprecipitations
Logarithmically growing cells were collected and washed with water supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF). We disrupted 100 OD600 of cells with glass beads in 400 μl of ice-cold IP-32 buffer (50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES]–NaOH, pH 7.2, 50 mM NaCl, 125 mM KOAc, 2 mM MgAc2, 1 mM EDTA, 10 μM CaCl2, 3% glycerol, 1 mM PMSF, pH 7.5). We added 1.2 ml of IP-32 buffer and removed cellular debris by centrifugation at 1000 × g for 5 min at 4°C. Microsomes were collected by centrifugation at 20,000 × g for 20 min at 4°C and solubilized in 1.2 ml of IP-32 lysis buffer (IP-32 supplemented with 0.5% NP-40). Lysates were cleared by centrifugation at 20,000 × g for 10 min at 4°C, and epitope-tagged proteins were precipitated with anti-HA or anti-Myc antibodies coupled to protein A Sepharose beads (Amersham, Pittsburgh, PA) at 4°C overnight. Beads were washed three times with 1 ml of IP-32 lysis buffer, and bound proteins were eluted with SDS sample buffer before analysis by SDS–PAGE and immunoblotting.
Pulse-chase analysis
Pulse-chase experiments were performed as described (Gauss et al., 2006b). Cells were pulse labeled with 35S-radiolabeled methionine and cysteine (Perkin Elmer, Waltham, MA) and chased into medium containing unlabeled amino acids. Samples were taken at the indicated time points and lysed, and the protein of interest was precipitated using specific antibodies. CPY*- and PrA*-containing precipitates were treated with endoglycosidase F (Roche, Basel, Switzerland). Samples were then separated by SDS–PAGE. Quantification of the signals was done on a Typhoon LFA9500 laser scanner (GE Healthcare, Buckinghamshire, UK) using the Image Quant software.
Cycloheximide decay assay
A detailed description can be found in Meusser et al. (2004). Logarithmically growing cells were diluted into fresh medium, and protein synthesis was inhibited by addition of cycloheximide to a concentration of 1 mg/ml. Aliquots were taken at indicated time points. Cells were lysed, and the proteins were analyzed by SDS–PAGE and immunoblotting. The signals were quantified using Cy5-labeled secondary antibodies (Life Technologies) and a Typhoon FLA9500 laser scanner employing the Image Quant software.
Site-specific in vivo photo-cross-linking
Yeast cells were transformed with plasmid pGK1-pBpa encoding a suppressor tRNA and the corresponding tRNA synthetase (Chen et al., 2007). In addition, the yeast cells expressed plasmid-encoded Der1-Myc constructs containing an amber stop codon at defined positions (derived from pMM075). For the incorporation of the cross-linker, yeast cells were grown in synthetic medium supplemented with 0.4 mM photoreactive amino acid analogue pBpa overnight. Expression of the pBpa-labeled Der1-Myc constructs was induced by the addition of 1 mM CuSO4 for 2 h. Activation of the cross-linker occurred by UV irradiation at λ = 365 nm for 45 min on ice (B-100AP; UVP, Upland, CA). After cell lysis and removal of the debris by centrifugation, Der1-Myc was immunoprecipitated with anti-Myc antibodies. The cross-linking products were eluted from the beads with dithiothreitol (DTT)-containing sample buffer and analyzed by SDS–PAGE, followed by immunodetection using specific antibodies. A detailed description of the cross-linking procedure can be found elsewhere (Mehnert et al., 2014).
Purification of Hrd3
The coding sequence of the luminal domain of Hrd3 (residues 21–768) was cloned in pFastBac 1 vector (Invitrogen, Carlsbad, CA) under the Polyhedrin promoter (pRG69). Recombinant baculoviral stock was generated from Sf9 insect cells according to the manufacturer's instructions. For protein expression, 30 ml of baculovirus was added to 3 l Sf9 cells in Sf-900 II SFM medium (Invitrogen) at a density of 2 million cells/ml and expression carried out for 48 h. Cells were pelleted, washed once with phosphate-buffered saline (PBS), and suspended in PBS supplemented with 1 mM DTT, 1 mM PMSF, 5 μM E64, and 0.5% Nonidet P40. Cells were lysed in a Dounce homogenizer and incubated for 2 h at 4°C. The lysate was cleared at 100,000 × g for 1 h at 4°C and loaded onto glutathione Sepharose 4 fast flow equilibrated in the same buffer. After overnight binding at 4°C and washing of the beads, Prescission protease was added and the beads incubated for 6 h in the cold room. The cleaved Hrd3lum protein in the supernatant was recovered and concentrated in an Ultracel centrifugal filter (Millipore, Billerica, MA), and the final protein concentration was determined by the Lowry method.
Light scattering experiments
Firefly luciferase was denatured in 6 M GdmHCl for 30 min at 25°C. The denatured protein was diluted into refolding buffer (20 mM HEPES-KOH, pH 7.5, 50 mM KCl, 2 mM MgCl2) to a final concentration of 100 nM in the presence of indicated concentrations of proteins in a fluorescence cuvette. Light scattering at 350 nm was recorded in a Fluoromax 4 fluorimeter (GE Healthcare) at 25°C in a time course.
Luciferase activity measurements
Chemically denatured firefly luciferase (luc) was diluted in refolding buffer (50 mm HEPES, pH 7.65, 120 mm KCl, 10 mm MgCl2,), 3 mM ATP, 10 mg/ml BSA, and 5 mM DTT to a final concentration of 100 nM. Where indicated, 5 μM DnaK, 1 μM DnaJ, 3 μM GrpE (kind gifts from Bernd Bukau, University of Heidelberg, Heidelberg, Germany), or 200 nM Hrd3 was added. After a short incubation, the reactions were stopped in 25 mM Tris-phosphate, 2 mM DTT, 2 mM EDTA, 10% glycerol, 1% Triton X-100, and 1 mg/ml BSA, and the enzymatic activity was determined in a Lumat LB 9507 luminometer (Berthold Technologies, Midland, ON, Canada) in the presence of Luciferase assay buffer (Promega, Madison, WI) according to the manufacturer's protocol.
Supplementary Material
Acknowledgments
We thank Corinna Volkwein, Angelika Wittstruck, and Mareen Kamarys for excellent technical support and all members of the laboratory of Thomas Sommer for fruitful discussions and critical reading of the manuscript. Dieter H. Wolf is acknowledged for providing plasmid pWO804 and antibodies against proteinase A. Purified DnaJ protein was a kind gift of Jeffrey Brodsky (University of Pittsburgh, Pittsburgh, PA). We thank Bernd Bukau for DnaJ, DnaK, and GrpE proteins. Davis Ng (National University of Singapore, Singapore) kindly provided plasmids pES163 and pSM101. The Deutsche Forschungsgemeinschaft generously supports research in the laboratory of T.S. (SFB 740, Priority Program 1365, German-Israel Project Cooperation DIP).
Abbreviations used:
- CPY
carboxypeptidase Y
- ER
endoplasmic reticulum
- ERAD
ER-associated protein degradation
- ER-PQC
ER-protein quality control
- HA
hemagglutinin
- HMG-CoA
3-hydroxy-3-methylglutaryl-coenzyme A
- HRD-ligase
HMG-CoA reductase degradation ligase
- pBpa
p-benzoyl-l-phenylalanine
- PrA
proteinase A
- SLR
SEL1-like repeats
- TPR
tetratricopeptide repeats.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-07-1202) on November 26, 2014.
*These authors contributed equally to this work.
Present addresses: †Institute of Molecular Systems Biology, Swiss Federal Institute of Technology Zurich, 8093 Zurich, Switzerland;
‡Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA 98109.
REFERENCES
- Bagola K, von Delbruck M, Dittmar G, Scheffner M, Ziv I, Glickman MH, Ciechanover A, Sommer T. Ubiquitin binding by a CUE domain regulates ubiquitin chain formation by ERAD E3 ligases. Mol Cell. 2013;50:528–539. doi: 10.1016/j.molcel.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Baker MJ, Frazier AE, Gulbis JM, Ryan MT. Mitochondrial protein-import machinery, correlating structure with function. Trends Cell Biol. 2007;17:456–464. doi: 10.1016/j.tcb.2007.07.010. [DOI] [PubMed] [Google Scholar]
- Bays NW, Gardner RG, Seelig LP, Joazeiro CA, Hampton RY. Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat Cell Biol. 2001;3:24–29. doi: 10.1038/35050524. [DOI] [PubMed] [Google Scholar]
- Bhangoo MK, Tzankov S, Fan AC, Dejgaard K, Thomas DY, Young JC. Multiple 40-kDa heat-shock protein chaperones function in Tom70-dependent mitochondrial import. Mol Biol Cell. 2007;18:3414–3428. doi: 10.1091/mbc.E07-01-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederer T, Volkwein C, Sommer T. Role of Cue1p in ubiquitination and degradation at the ER surface. Science. 1997;278:1806–1809. doi: 10.1126/science.278.5344.1806. [DOI] [PubMed] [Google Scholar]
- Brodsky JL. Cleaning up: ER-associated degradation to the rescue. Cell. 2012;151:1163–1167. doi: 10.1016/j.cell.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck TM, Kolb AR, Boyd CR, Kleyman TR, Brodsky JL. The endoplasmic reticulum-associated degradation of the epithelial sodium channel requires a unique complement of molecular chaperones. Mol Biol Cell. 2010;21:1047–1058. doi: 10.1091/mbc.E09-11-0944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carla Fama M, Raden D, Zacchi N, Lemos DR, Robinson AS, Silberstein S. The Saccharomyces cerevisiae YFR041C/ERJ5 gene encoding a type I membrane protein with a J domain is required to preserve the folding capacity of the endoplasmic reticulum. Biochim Biophys Acta. 2007;1773:232–242. doi: 10.1016/j.bbamcr.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho P, Goder V, Rapoport TA. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–373. doi: 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- Cattaneo M, Canton C, Albertini A, Biunno I. Identification of a region within SEL1L protein required for tumour growth inhibition. Gene. 2004;326:149–156. doi: 10.1016/j.gene.2003.10.021. [DOI] [PubMed] [Google Scholar]
- Chen S, Schultz PG, Brock A. An improved system for the generation and analysis of mutant proteins containing unnatural amino acids in Saccharomyces cerevisiae. J Mol Biol. 2007;371:112–122. doi: 10.1016/j.jmb.2007.05.017. [DOI] [PubMed] [Google Scholar]
- D’Andrea LD, Regan L. TPR proteins: the versatile helix. Trends Biochem Sci. 2003;28:655–662. doi: 10.1016/j.tibs.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Denic V, Quan EM, Weissman JS. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell. 2006;126:349–359. doi: 10.1016/j.cell.2006.05.045. [DOI] [PubMed] [Google Scholar]
- Dong M, Bridges JP, Apsley K, Xu Y, Weaver TE. ERdj4 and ERdj5 are required for endoplasmic reticulum-associated protein degradation of misfolded surfactant protein C. Mol Biol Cell. 2008;19:2620–2630. doi: 10.1091/mbc.E07-07-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RG, Swarbrick GM, Bays NW, Cronin SR, Wilhovsky S, Seelig L, Kim C, Hampton RY. Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J Cell Biol. 2000;151:69–82. doi: 10.1083/jcb.151.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauss R, Jarosch E, Sommer T, Hirsch C. A complex of Yos9p and the HRD ligase integrates endoplasmic reticulum quality control into the degradation machinery. Nat Cell Biol. 2006a;8:849–854. doi: 10.1038/ncb1445. [DOI] [PubMed] [Google Scholar]
- Gauss R, Kanehara K, Carvalho P, Ng DT, Aebi M. A complex of Pdi1p and the mannosidase Htm1p initiates clearance of unfolded glycoproteins from the endoplasmic reticulum. Mol Cell. 2011;42:782–793. doi: 10.1016/j.molcel.2011.04.027. [DOI] [PubMed] [Google Scholar]
- Gauss R, Sommer T, Jarosch E. The Hrd1p ligase complex forms a linchpin between ER-lumenal substrate selection and Cdc48p recruitment. EMBO J. 2006b;25:1827–1835. doi: 10.1038/sj.emboj.7601088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, Gardner RG, Rine J. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol Biol Cell. 1996;7:2029–2044. doi: 10.1091/mbc.7.12.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch C, Gauss R, Horn SC, Neuber O, Sommer T. The ubiquitylation machinery of the endoplasmic reticulum. Nature. 2009;458:453–460. doi: 10.1038/nature07962. [DOI] [PubMed] [Google Scholar]
- Horn SC, Hanna J, Hirsch C, Volkwein C, Schutz A, Heinemann U, Sommer T, Jarosch E. Usa1 functions as a scaffold of the HRD-ubiquitin ligase. Mol Cell. 2009;36:782–793. doi: 10.1016/j.molcel.2009.10.015. [DOI] [PubMed] [Google Scholar]
- Izawa T, Nagai H, Endo T, Nishikawa S. Yos9p and Hrd1p mediate ER retention of misfolded proteins for ER-associated degradation. Mol Biol Cell. 2012;23:1283–1293. doi: 10.1091/mbc.E11-08-0722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpenahalli MR, Lupas AN, Soding J. TPRpred: a tool for prediction of TPR-, PPR- and SEL1-like repeats from protein sequences. BMC Bioinformatics. 2007;8:2. doi: 10.1186/1471-2105-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Knop M, Siegers K, Pereira G, Zachariae W, Winsor B, Nasmyth K, Schiebel E. Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast. 1999;15:963–972. doi: 10.1002/(SICI)1097-0061(199907)15:10B<963::AID-YEA399>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Kutik S, Guiard B, Meyer HE, Wiedemann N, Pfanner N. Cooperation of translocase complexes in mitochondrial protein import. J Cell Biol. 2007;179:585–591. doi: 10.1083/jcb.200708199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013;41:W597–W600. doi: 10.1093/nar/gkt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehnert M, Sommer T, Jarosch E. Der1 promotes movement of misfolded proteins through the endoplasmic reticulum membrane. Nat Cell Biol. 2014;16:77–86. doi: 10.1038/ncb2882. [DOI] [PubMed] [Google Scholar]
- Meusser B, Sommer T. Vpu-mediated degradation of CD4 reconstituted in yeast reveals mechanistic differences to cellular ER-associated protein degradation. Mol Cell. 2004;14:247–258. doi: 10.1016/s1097-2765(04)00212-6. [DOI] [PubMed] [Google Scholar]
- Mittl PR, Schneider-Brachert W. Sel1-like repeat proteins in signal transduction. Cell Signal. 2007;19:20–31. doi: 10.1016/j.cellsig.2006.05.034. [DOI] [PubMed] [Google Scholar]
- Neuber O, Jarosch E, Volkwein C, Walter J, Sommer T. Ubx2 links the Cdc48 complex to ER-associated protein degradation. Nat Cell Biol 7. 2005:993–998. doi: 10.1038/ncb1298. [DOI] [PubMed] [Google Scholar]
- Nishikawa SI, Fewell SW, Kato Y, Brodsky JL, Endo T. Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J Cell Biol. 2001;153:1061–1070. doi: 10.1083/jcb.153.5.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papic D, Elbaz-Alon Y, Koerdt SN, Leopold K, Worm D, Jung M, Schuldiner M, Rapaport D. The role of Djp1 in import of the mitochondrial protein Mim1 demonstrates specificity between a cochaperone and its substrate protein. Mol Cell Biol. 2013;33:4083–4094. doi: 10.1128/MCB.00227-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Rapoport TA. Mechanisms of Sec61/SecY-mediated protein translocation across membranes. Annu Rev Biophys. 2012;41:21–40. doi: 10.1146/annurev-biophys-050511-102312. [DOI] [PubMed] [Google Scholar]
- Ponting CP, Schultz J, Milpetz F, Bork P. SMART: identification and annotation of domains from signalling and extracellular protein sequences. Nucleic Acids Res. 1999;27:229–232. doi: 10.1093/nar/27.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan EM, Kamiya Y, Kamiya D, Denic V, Weibezahn J, Kato K, Weissman JS. Defining the glycan destruction signal for endoplasmic reticulum-associated degradation. Mol Cell. 2008;32:870–877. doi: 10.1016/j.molcel.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiano A, Foresti O, Carvalho P. Quality control: ER-associated degradation: protein quality control and beyond. J Cell Biol. 2014;204:869–879. doi: 10.1083/jcb.201312042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato BK, Schulz D, Do PH, Hampton RY. Misfolded membrane proteins are specifically recognized by the transmembrane domain of the Hrd1p ubiquitin ligase. Mol Cell. 2009;34:212–222. doi: 10.1016/j.molcel.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuberth C, Buchberger A. Membrane-bound Ubx2 recruits Cdc48 to ubiquitin ligases and their substrates to ensure efficient ER-associated protein degradation. Nat Cell Biol. 2005;7:999–1006. doi: 10.1038/ncb1299. [DOI] [PubMed] [Google Scholar]
- Spear ED, Ng DTW. Single, context-specific glycans can target misfolded glycoproteins for ER-associated degradation. J Cell Biol. 2005;169:73–82. doi: 10.1083/jcb.200411136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taxis C, Hitt R, Park SH, Deak PM, Kostova Z, Wolf DH. Use of modular substrates demonstrates mechanistic diversity and reveals differences in chaperone requirement of ERAD. J Biol Chem. 2003;278:35903–35913. doi: 10.1074/jbc.M301080200. [DOI] [PubMed] [Google Scholar]
- Ushioda R, Hoseki J, Araki K, Jansen G, Thomas DY, Nagata K. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science. 2008;321:569–572. doi: 10.1126/science.1159293. [DOI] [PubMed] [Google Scholar]
- Ushioda R, Hoseki J, Nagata K. Glycosylation-independent ERAD pathway serves as a backup system under ER stress. Mol Biol Cell. 2013;24:3155–3163. doi: 10.1091/mbc.E13-03-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashist S, Ng D. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol. 2004;165:41–52. doi: 10.1083/jcb.200309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JM, Inoue T, Banks L, Tsai B. The ERdj5-Sel1L complex facilitates cholera toxin retrotranslocation. Mol Biol Cell. 2013;24:785–795. doi: 10.1091/mbc.E12-07-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JC, Hoogenraad NJ, Hartl FU. Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell. 2003;112:41–50. doi: 10.1016/s0092-8674(02)01250-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.