

Proteins of the mitochondrial intermembrane space are oxidatively folded by the incorporation of structural disulfide bonds. Efficient protein oxidation in this highly reducing compartment is possible only because glutaredoxins, which could translate the glutathione redox potential into that of protein thiols, are present at limiting levels.
Abstract
The mitochondrial intermembrane space (IMS) harbors an oxidizing machinery that drives import and folding of small cysteine-containing proteins without targeting signals. The main component of this pathway is the oxidoreductase Mia40, which introduces disulfides into its substrates. We recently showed that the IMS glutathione pool is maintained as reducing as that of the cytosol. It thus remained unclear how equilibration of protein disulfides with the IMS glutathione pool is prevented in order to allow oxidation-driven protein import. Here we demonstrate the presence of glutaredoxins in the IMS and show that limiting amounts of these glutaredoxins provide a kinetic barrier to prevent the thermodynamically feasible reduction of Mia40 substrates by the IMS glutathione pool. Moreover, they allow Mia40 to exist in a predominantly oxidized state. Consequently, overexpression of glutaredoxin 2 in the IMS results in a more reduced Mia40 redox state and a delay in oxidative folding and mitochondrial import of different Mia40 substrates. Our findings thus indicate that carefully balanced glutaredoxin amounts in the IMS ensure efficient oxidative folding in the reducing environment of this compartment.
INTRODUCTION
Oxidative protein folding leads to the acquisition of stable protein folds, mediates disulfide-dependent multimerization, and facilitates compartmental retention. This process is catalyzed in three compartments—the bacterial periplasm, the endoplasmic reticulum (ER), and the mitochondrial intermembrane space (IMS; Riemer et al., 2009; Bulleid and Ellgaard, 2011; Depuydt et al., 2011; Stojanovski et al., 2012). Disulfide formation follows the same general principles: oxidoreductases that contain an oxidized redox-active dicysteine motif interact with and oxidize substrates. They are kept oxidized by sulfhydryl oxidases that couple de novo disulfide formation to electron transfer onto oxygen.
In the cytosol and the mitochondrial matrix, disulfide formation is prevented by a highly reduced glutathione pool and by a set of reducing enzymes of the thioredoxin and glutaredoxin (Grx) families. In addition, in compartments that facilitate thiol oxidation, reducing pathways are active, for example, in counterbalancing nonnative or unwanted disulfides (Chakravarthi et al., 2006; Gleiter and Bardwell, 2008; Appenzeller-Herzog, 2011; Banhegyi et al., 2012; Hagiwara and Nagata, 2012). In the bacterial periplasm, dedicated machinery keeps specialized oxidoreductases reduced by shuffling electrons from cytosolic NADPH over the plasma membrane (Gleiter and Bardwell, 2008). To prevent a potentially futile cross-talk between oxidizing and reducing processes in the periplasm, the processes are separated by large kinetic barriers, that is, the apparent rate constants for unwanted thiol–disulfide reactions are 103- to 107-fold slower than the ones for their favored counterparts (Rozhkova et al., 2004).
Similarly, oxidation pathways in the ER are exposed to reducing processes. The cytosolic glutathione reductase system constantly reduces glutathione disulfide (GSSG) to glutathione (GSH; Chakravarthi et al., 2006), which can then traverse the ER membrane (Banhegyi et al., 1999) and in the ER lumen directly reduce oxidoreductases (Jessop and Bulleid, 2004). In the ER, the very same oxidoreductases participate in both oxidizing and reducing pathways. They are thus maintained in semioxidized redox states (i.e., comprise pools of reduced and oxidized oxidoreductases; Appenzeller-Herzog and Ellgaard, 2008; Appenzeller-Herzog et al., 2010; Kim et al., 2012). This is achieved by carefully balancing oxidizing and reducing pathways. First, the activity of the sulfhydryl oxidase Ero1 is attenuated upon hyperoxidizing ER conditions (Sevier et al., 2007; Baker et al., 2008; Appenzeller-Herzog et al., 2010). This prevents full oxidation of oxidoreductases. Second, the limited influx of GSH from the cytosol impedes full equilibration of the ER glutathione pool with its cytosolic counterpart, resulting in a more-oxidized glutathione pool in the ER compared with the cytosol (i.e., a less-negative glutathione redox potential EGSH:EGSH[ER] in the range −208 to −237 mV (Merksamer et al., 2008; van Lith et al., 2011; Birk et al., 2013), and EGSH[cytosol] in the range −286 to −320 mV (Ostergaard et al., 2004; Hu et al., 2008; Braun et al., 2010; Morgan et al., 2011; Kojer et al., 2012)). This EGSH[ER] prevents the full reduction of ER oxidoreductases and ensures efficient formation of native disulfides (Appenzeller-Herzog et al., 2010; Kim et al., 2012).
Our knowledge of redox homeostasis in the IMS is still very limited. In the IMS, oxidative protein folding is performed by the oxidoreductase Mia40, which folds and oxidizes substrate proteins and is reoxidized by the sulfhydryl oxidase Erv1 (Deponte and Hell, 2009; Riemer et al., 2009; Chatzi and Tokatlidis, 2012; Stojanovski et al., 2012). In vivo, Mia40 is present in a partially reduced state (∼70–80% oxidized, 20–30% reduced; Kojer et al., 2012). The Mia40 redox state is mainly controlled by Erv1 and the IMS glutathione pool (Kojer et al., 2012). The glutathione pools of the cytosol and the IMS are connected by porins in the outer membrane of mitochondria that ensure rapid equilibration. Thus, in contrast to EGSH[ER], EGSH[IMS] and EGSH[cytosol] are almost identical (−301 vs. −306 mV; Figure 1A; Kojer et al., 2012). This constitutes an important challenge: if IMS proteins were to fully equilibrate with the highly reducing IMS glutathione pool, a significant portion of these proteins would exist in a reduced state (up to 33% reduced for proteins like Tim9 and Tim13; Supplemental Table S1), which would particularly lead to considerable problems during oxidation-dependent protein import. However, a number of experimental studies indicated that many IMS proteins are completely oxidized in vivo, and even disulfide-linked Mia40–substrate complexes are surprisingly stable (Brandes et al., 2011; Bottinger et al., 2012; Kojer et al., 2012; Weckbecker et al., 2012; Bode et al., 2013).
FIGURE 1:

The mitochondrial intermembrane space harbors glutaredoxins. (A) Scheme of the IMS redox environment. EGSH[IMS] is in the same range as EGSH[cytosol] (Kojer et al., 2012). Thus it remains unclear how reduction of IMS proteins by GSH is prevented to ensure efficient oxidative folding. (B) Strategy to identify potential IMS-localized Grxs. Redox-sensitive green fluorescent protein (roGFP2) depends on nearby dithiol Grx for rapid equilibration with the local glutathione pool. (C) Scheme of the oxidant recovery assay. Yeast cells containing different roGFP2 sensors were grown to mid log phase in galactose medium and incubated with 20 mM diamide for 5 min. Then the oxidant was removed, and the recovery of the respective roGFP2 sensors was monitored by ratiometric fluorescence measurements (λem = 511 nm, λex = 405 and 488 nm). The graphs depict the resulting degree of sensor oxidation, OxD (for calculation see Supplemental Methods), with OxD = 1 for the fully oxidized probe and OxD = 0 for the fully reduced probe. (D) Targeting of roGFP2 sensors to the cytosol, matrix (Su9 presequence), and IMS (b2 presequence), respectively. For confirmation of localization, see Supplemental Figure S1. (E–G) Oxidant recovery assays using cytosolic (E), matrix (F), and IMS-localized (G) roGFP2 sensors in wild-type, GRX1 and GRX2 deletion, and GRX1, GRX2 double-deletion cells. Reported values are the mean of three independent experiments. Error bars are the means ± SD. (H) Steady states of roGFP2 sensors in cells described in E–G. Reported values are the mean of three independent experiments. Error bars are the means ± SD.
We therefore wondered how oxidative protein folding in the IMS is balanced with GSH-dependent protein reduction. A potential control point is to prevent the direct interaction of GSH and the respective IMS proteins. Indeed, in vitro reconstitution experiments demonstrated that Mia40 is only slowly reduced by GSH (Bien et al., 2010). However, they also showed that GSH in principle is capable of reducing disulfide-linked substrate–Mia40 intermediates (Bien et al., 2010). Possible candidates that could translate the redox state of the local glutathione pool to the redox state of Mia40 and its substrates are members of the dithiol Grx family. Saccharomyces cerevisiae hosts three dithiol Grxs—Grx1, Grx2, and Grx8 (Holmgren et al., 2005; Herrero et al., 2006). Grx1 and Grx2 have a high affinity to glutathione and react rapidly and specifically with it, whereas Grx8 has only little Grx activity, and no physiological function has been found for Grx8 (Eckers et al., 2009). All three dithiol Grxs localize to the cytosol. In addition to its cytosolic isoform, a second Grx2 variant exists, which is translated from an alternative upstream start codon. This second Grx2 species contains a mitochondrial targeting sequence and is thereby directed into the mitochondrial matrix (Pedrajas et al., 2002; Porras et al., 2010). Of importance, no Grx has been localized to the IMS in S. cerevisiae (Kojer et al., 2012; Toledano et al., 2013), although a recent study found hints of an IMS localization of human Grx1 (Pai et al., 2007).
In this study, we demonstrate that the IMS harbors glutaredoxin activity. This activity is mainly exerted by Grx2. A fraction of the cytosolic form of Grx2 that lacks a mitochondrial targeting sequence is imported into the IMS. In the IMS, Grx activity mediates the influence of the IMS glutathione pool on IMS proteins. This includes, but certainly is not restricted to, the regulation of the Mia40 redox state. Of importance, Grx activity in the IMS is limiting because overexpression of an IMS-targeted Grx2 variant shifts Mia40 to a more-reduced redox state. Conversely, GRX deletion hampers reduction of Mia40 by GSH. Furthermore, upon overexpression of Grx2, oxidative folding of the IMS protein and Mia40 substrate Atp23 is strongly delayed. Similarly, under these conditions, the copper chaperone for Sod1 (Ccs1), which is a dually localized protein whose IMS import depends on oxidation by Mia40, does not accumulate in the IMS. This indicates that the levels of Grx have to be carefully balanced so as to enable efficient oxidative folding and oxidation-dependent import into the IMS.
RESULTS
The mitochondrial IMS harbors glutaredoxin activity
We previously showed that EGSH[IMS] is in the range of EGSH[cytosol] (Kojer et al., 2012). We therefore asked how the IMS can favor disulfide bond formation despite this very reducing glutathione pool (Figure 1A). We hypothesized that one explanation could be the slow direct reaction of most proteins with glutathione. In the cytosol and the matrix, dithiol Grx(s) rapidly translate EGSH to surrounding protein thiols. However, in the yeast IMS, no such Grx has been identified. Because the absence of Grxs would explain the existence of an oxidizing pathway in a reducing environment, we first tested for the existence of dithiol Grxs in the IMS.
The biochemical detection of potential IMS-localized Grxs by fractionation approaches is complicated by the presence of large amounts of Grx1 and 2 in the cytosol and of Grx2 in the matrix. We thus first used a functional approach based on the redox-sensitive green fluorescent protein roGFP2. This protein efficiently and specifically equilibrates with the surrounding glutathione pool, but only if a dithiol Grx is present (Gutscher et al., 2008). Differences in the redox state of roGFP2 can be read out as changes in its fluorescence excitation spectrum (Dooley et al., 2004). Fusion proteins consisting of human Grx1 and roGFP2 (Grx1-roGFP2) proved to be elegant tools to dynamically measure physiological EGSH (Morgan et al., 2011; Kojer et al., 2012). These sensor proteins showed similar EGSH values in the cytosol, the matrix, and the IMS under physiological conditions (Kojer et al., 2012). We varied this assay and expressed roGFP2 on its own, reasoning that endogenous Grx activities could thereby be measured in vivo (Figure 1B; Ostergaard et al., 2004; Gutscher et al., 2008).
We first used a dynamic and highly sensitive approach and analyzed the recovery of roGFP2 after oxidative shock (Figure 1C; Kojer et al., 2012). To this end, we placed the roGFP2 probe in the cytosol, where Grx1 and Grx2 serve as redundant glutaredoxin pair (for sensor localization see Figure 1D and Supplemental Figure S1). When wild-type cells were challenged with a diamide pulse, the roGFP2 sensor became oxidized (represented as degree of oxidation, OxD), but it completely recovered within <10 min (Figure 1E). In contrast, roGFP2 remained oxidized in a GRX1, GRX2 double-deletion mutant. Additional GRX8 deletion did not exert any additional effect (Supplemental Figure S2). GRX1 and GRX2 single mutants recovered rapidly, confirming their overlapping activities. The situation looked similar in the matrix (Figure 1F), with the difference that the deletion of GRX2 was sufficient to eliminate Grx activity. Next we placed roGFP2 into the IMS (Figure 1G). In the wild type, IMS-targeted roGFP2 recovered only slowly, indicating that the IMS does contain Grx activity but only little. Analyses of the single mutants indicated that this low activity is mainly exhibited by Grx2 and to only a small degree by Grx1. Thus some Grx2 (and presumably even lower amounts of Grx1) are present in the IMS. However, the catalytic Grx capacity is by far smaller than in the cytosol or the matrix.
Next we analyzed the redox states of roGFP2 in the different compartments at steady state (Figure 1H). In the cytosol, roGFP2 was reduced even in GRX1, GRX2 double-deletion mutants. This is consistent with the overlap of the cytosolic Grx and thioredoxin activities (Draculic et al., 2000; Kumar et al., 2011). In the matrix, the redox sensor remained reduced as long as Grx2 was present. In the IMS, the sensor was significantly more oxidized, indicating that even at steady state, roGFP2 was not efficiently reduced by the glutathione pool of this compartment. In summary, these measurements indicate that there is (low) Grx activity in the IMS mainly due to the presence of a fraction of Grx2 in this compartment. However, the activity is so low that even at steady state the glutathione pool does not fully equilibrate with the redox sensor. Because the main Grx activity in the IMS can be attributed to Grx2, we focus in the following on assessing the role of Grx2.
A part of the cytosolic form of glutaredoxin 2 resides in the IMS
Grx2 is a dually localized protein that is found in the cytosol and the mitochondrial matrix. In addition, Grx2 has been reported to be present at the outer membrane of mitochondria (Porras et al., 2010). The cytosolic and mitochondrial forms of the protein are both encoded from the same gene and translated from different start codons in the mRNA (Figure 2A). Translation from the first start codon results in a protein containing a mitochondrial targeting sequence, whereas translation from the second start codon leads to a shorter cytosolic version of Grx2. To determine which of these forms affects IMS-localized roGFP2, we analyzed strains devoid of GRX1, GRX2, and GRX8 that additionally harbored plasmids encoding either the short cytosolic version of Grx2 (lacking the first 34 amino acids of Grx2, cyto-Grx2) or the matrix form of Grx2 (expressing the M35V mutant of Grx2, mito-Grx2; Figure 2B). When we analyzed the redox state of roGFP2 in these strains, we found cytosolic roGFP2 always to be reduced, whereas the redox state of matrix-localized roGFP2 could be complemented only by the expression of mito-Grx2 but not by cyto-Grx2 (Figure 2C). Conversely, in the IMS, roGFP2 exhibited the redox state it had in wild-type cells only when cyto-Grx2 was expressed in the GRX1, GRX2, GRX8 triple-deletion background (Figure 2C). We confirmed this influence of cyto-Grx2 in the oxidant recovery assay. Only the expression of cyto-Grx2 enabled roGFP2 recovery in the IMS after oxidative shock in a ΔΔΔgrx1,2,8 strain (Figure 2, D and E).
FIGURE 2:

The cytosolic form of Grx2 becomes imported into the IMS. (A) Scheme depicting the GRX2 gene and the two different protein variants resulting from it. Grx2 is a dually localized protein that is found in the cytosol and the mitochondrial matrix. The cytosolic and mitochondrial forms of the protein are both encoded from the same gene and translated from different start codons in the mRNA. Translation from the first start codon results in a protein containing a mitochondrial targeting sequence (mito-Grx2), whereas translation from the second start codon leads to a shorter cytosolic version of Grx2 (cyto-Grx2). For experimental analyses, we designed two plasmids that exclusively express either cyto-Grx2 (Δ1-34-Grx2) or mito-Grx2 (M35V-Grx2.). (B) Expression test of the two Grx2 variants in whole cells. Cells devoid of GRX1, GRX2, and GRX8 additionally harboring cyto- or mito-Grx2 were grown to mid log phase in SD media, lysed, and analyzed by SDS–PAGE and immunoblotting using antibodies directed against Grx2 and Mrpl40 as loading control. (C) Steady states of the cytosolic and mitochondrial roGFP2 sensors (roGFP2, b2- roGFP2, Su9- roGFP2) in GRX1, GRX2, and GRX8 deletion cells harboring cyto- or mito-Grx2. Experiments were performed as described in Figure 1. Reported values are the mean of three independent experiments. Error bars are the means ± SD. (D, E) Recovery kinetics after diamide shock on GRX1, GRX2, and GRX8 deletion cells harboring cyto-Grx2 (D) or mito-Grx2 (E). Cells expressing roGFP2, Su9-roGFP2, and b2-roGFP2 were analyzed as described in Figure 1. Reported values are the mean of three independent experiments. Error bars are the means ± SD. (F) Mitochondria from GRX1, GRX2, GRX8 deletion cells harboring b2-roGFP2 were isolated and analyzed in a fluorescence spectrometer upon incubation with glutathione reductase (GR), NADPH, GSH, and Triton X-100 (TX-100) as indicated. After isolation of mitochondria, the respective probes are fully oxidized (=100% oxidized roGFP2). The incubation with DTT at the end of the kinetics served as control for fully reduced proteins (=0% oxidized roGFP2). (G) Mitochondrial fractionation. Mitochondria were isolated from GRX1, GRX2, GRX8 deletion cells that additionally harbored a plasmid expressing cyto-Grx2. After isolation, mitochondria were digitonin and protease treated as indicated and analyzed by SDS–PAGE and Western blot against Grx2, Pgk1 (cytosol), Mia40 (IMS), and Mrpl40 (matrix). To detect Grx2, it was precipitated from the mitochondrial fractions by immunoprecipitation.
The influence of cyto-Grx2 on IMS-localized roGFP2 indicated that cytosolic Grx2 is imported into the mitochondrial IMS. To test whether it was cytosol-localized cyto-Grx2 that indirectly influenced roGFP2 in the IMS, we performed in vitro assays with purified mitochondria (Figure 2F). To this end, we isolated mitochondria from ΔΔΔgrx1,2,8 cells harboring IMS-localized roGFP2. We incubated these mitochondria with a GSH-regenerating system (glutathione reductase, NADPH, GSH) and either purified Grx2 or buffer in the presence and absence of a detergent to solubilize mitochondrial membranes. In isolated mitochondria, IMS-localized roGFP2 was in the oxidized state. When we treated intact mitochondria with Grx2 and the GSH-regenerating system, roGFP2 remained oxidized during the time of the experiment. Only upon lysis of mitochondrial membranes with Triton X-100 did addition of Grx2 and the GSH-regenerating system result in IMS-roGFP2 reduction. These data indicate that cyto-Grx2 cannot influence IMS-localized roGFP2 indirectly but has to reside in the IMS to do so.
We therefore next verified that cyto-Grx2 can indeed be found in mitochondria at steady state (Figure 2G). To this end, we expressed cyto-Grx2 in a ΔΔΔgrx1,2,8 strain to exclude that matrix-localized Grx2 interferes with subsequent fractionation approaches. We isolated mitochondria from these cells and treated them with increasing amounts of digitonin to dissolve mitochondrial membranes in a stepwise manner. This treatment was followed by protease treatment to remove exposed proteins. Using this approach, we found that a fraction of cyto-Grx2 was protected from protease treatment in intact mitochondria. On digitonin treatment, this cyto-Grx2 fraction behaved like the IMS protein Mia40, indicating its IMS localization (Figure 2G). Taken together, we find that a portion of the cytosolic form of Grx2 is present in the mitochondrial IMS. In the IMS, cyto-Grx2 allows the equilibration of roGFP2 (and likely other IMS proteins) with the local glutathione pool.
Glutaredoxin 2 translates the redox potential of the IMS glutathione pool into the redox state of the oxidoreductase Mia40
We previously described an influence of the IMS glutathione pool on Mia40, which leads to a Mia40 redox state that in wild-type cells is in part reduced (Kojer et al., 2012). Because Mia40 reacts only slowly directly with glutathione (Bien et al., 2010), we asked whether IMS-localized Grx2 translates dynamics of the IMS glutathione pool to Mia40 (Figure 3A). We therefore analyzed the Mia40 redox state in strains lacking GRX2 (Figure 3B). In wild-type cells, Mia40 was oxidized to 65 ± 8%, whereas in cells lacking GRX2, a larger fraction of Mia40 was oxidized (86 ± 7%). We also analyzed the Mia40 redox state in cells with decreased levels of Erv1, the enzyme that reoxidizes Mia40. In these strains, Mia40 was almost completely reduced in the presence and absence of Grx2 (Figure 3B). To detail our findings, we analyzed the recovery of the Mia40 redox state after oxidative shock in cells with low Erv1 levels (Figure 3C). We found a recovery of Mia40 from its fully oxidized form with a half-time of 4 min in Erv1-depleted cells, whereas in cells additionally lacking GRX2, this recovery time was extended to >10 min (Figure 3D). In line with our previous findings, we also confirmed that it is the cytosolic form of Grx2 that influences the Mia40 redox state (Supplemental Figure S3).
FIGURE 3:
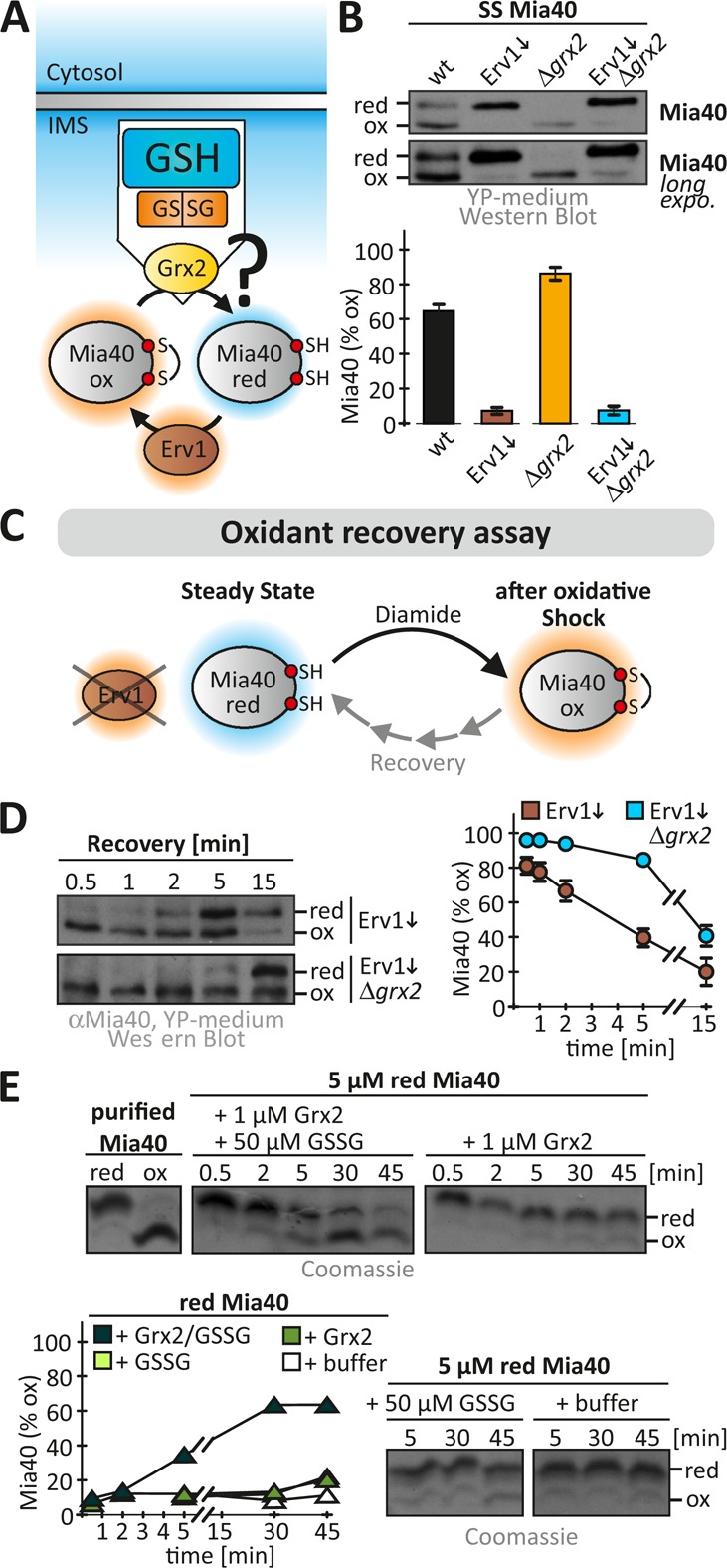
Grx2 directly influences the Mia40 redox state. (A) Scheme depicting redox influences on the IMS oxidoreductase Mia40. Mia40 is oxidized by the sulfhydryl oxidase Erv1 and is reduced by the local glutathione pool potentially mediated by IMS-localized Grxs. (B) Redox state of Mia40 in the indicated yeast cells. Cells grown to mid log phase in YP medium containing glucose to lower Erv1 levels were precipitated with TCA. The resulting pellet was resuspended in a buffer containing 20 mM N-ethylmaleimide (NEM). The samples were analyzed on nonreducing SDS–PAGE, followed by immunoblotting against Mia40. SS, steady state. Reported values are the mean of three independent experiments. Error bars are the means ± SD. (C) Scheme of oxidant recovery assay. Yeast cells were incubated with 20 mM diamide for 5 min, the oxidant was removed, and the recovery of the Mia40 redox state was assessed at different times using an NEM-based alkylation gel shift assay. (D) Oxidant recovery assays in wild-type and Δgrx2 cells, both depleted of Erv1. Reported values are the mean of three independent experiments. Error bars are the means ± SD. (E) In vitro reconstitution of the Grx2-Mia40 disulfide exchange. Reoxidation of purified reduced Mia40 was followed in the presence of GSSG, Grx2, or buffer or a combination of all three. The redox state of Mia40 was assessed at the indicated times using AMS.
Although we could demonstrate that Grx2 mediates the equilibration of Mia40 with the local glutathione pool, we did not know whether Grx2 acts directly on Mia40. We therefore reconstituted the Mia40-Grx2 system in vitro. Starting with 5 μM reduced purified Mia40, we followed its oxidation in a buffer composed of 50 μM GSSG in the presence and absence of 1 μM purified Grx2 (Figure 3E). In the absence of either Grx2 or GSSG, Mia40 did not become oxidized. Only when Grx2 and GSSG were both added was Mia40 efficiently oxidized, indicating that Grx2 is sufficient to catalyze the electron transfer between glutathione and Mia40. Taking the results together, we find Grx2 to control the reducing influences on the redox state of Mia40 in vivo. To this end, Grx2 equilibrates with the local glutathione pool and directly interacts with Mia40, which parallels the interaction of Grx2 and roGFP2.
Glutaredoxin 2 amounts in the IMS are limiting for equilibration of the local glutathione pool with Mia40 and roGFP2
In wild-type cells, recovery of cytosolic and matrix roGFP2 took place with almost similar kinetics as for Grx1-roGFP2 (Figure 4A). In contrast, IMS-localized roGFP2 recovered much more slowly than Grx1-roGFP2 after oxidative shock (Figures 1G and 4A). We thus wanted to investigate whether Grx2 amounts in the IMS are limiting for fast probe recovery. To this end, we used a plasmid encoding the cytosolic form of Grx2 fused to the mitochondrial targeting signal of cytochrome b2 (b2-Grx2). On expression of b2-Grx2, the recovery kinetics of roGFP2 in the IMS paralleled that of Grx1-roGFP2 (Figure 4B). This finding was also reflected in a more-reduced steady-state OxD of IMS-localized roGFP2 upon b2-Grx2 expression (Figure 4C).
FIGURE 4:

The levels of Grx2 in the IMS are limiting for the efficient equilibration of roGFP2 and Mia40 with the local glutathione pool. (A) Oxidant recovery assay using cytosolic, IMS, and matrix-localized roGFP2 and Grx1-roGFP2 sensors in wild-type cells. The experiment was performed as described in Figure 1H. Reported values are the mean of three independent experiments. Error bars are the means ± SD. (B) Oxidant recovery assay using cytosolic, IMS, and matrix-localized roGFP2 sensors in wild-type cells or cells expressing IMS-targeted Grx2 (b2-Grx2). The experiment was performed as described in Figure 1C. Reported values are the mean of three independent experiments. Error bars are the means ± SD. (C) Steady states of cytosolic, IMS. and matrix-localized roGFP2 and Grx1-roGFP2 in wild-type cells and cells expressing b2-Grx2. The experiment was performed as described in Figure 1G. Reported values are the mean of three independent experiments. Error bars are the means ± SD. (D–F) Redox states of Mia40. The experiment was performed as described in Figure 3B. Redox states were analyzed in wild-type cells and cells additionally containing either b2-Grx2 (D, E) or a catalytically inactive variant of b2-Grx2 (C27S/C30S) (E). In E, cells were grown in the presence or absence of BSO. (F) Mia40 redox states were also assessed in a GRX1, GRX2, GRX8 deletion strain in the presence or absence of b2-Grx2 or b2-Grx2(C27S/C30S). Reported values are the mean of three independent experiments. Error bars are the means ± SD. (G) Scheme detailing the role of limiting amounts of IMS-localized Grx2 in preventing the full equilibration of Mia40 with the surrounding glutathione pool.
If Grx2 levels are limiting for roGFP2 recovery in the IMS, it appeared likely that they also prevent Mia40 from fast equilibration with the local glutathione pool. We therefore analyzed the Mia40 redox state in strains expressing b2-Grx2 and found Mia40 to be more reduced at steady state compared with the wild-type situation (Figure 4D). This reducing influence of b2-Grx2 on Mia40 relied on the redox-active cysteines in b2-Grx2 since the expression of the corresponding redox-inactive C27S/C30S mutant did not result in a more reduced Mia40 compared with the control situation (Figure 4E). Moreover, the effect of b2-Grx2 overexpression could be reversed by incubation with buthionine sulfoximine (BSO; Figure 4E). This chemical blocks glutathione biosynthesis, thereby decreasing cellular glutathione levels, which results in a more-oxidizing glutathione pool in the IMS, the cytosol, and the matrix (Supplemental Figure S4). We could confirm the effects of b2-Grx2 variants when we analyzed the Mia40 redox state in the ΔΔΔgrx1,2,8 deletion strain (Figure 4F). In summary, this implies that endogenous amounts of Grx2 are limiting for equilibration of Mia40 with the local glutathione pool (Figure 4G).
Limiting amounts of Grx2 are critical for efficient oxidative folding of Mia40 substrates
Next we wanted to investigate whether changes in the Mia40 redox state and a better coupling of the glutathione and the IMS protein thiol pools by b2-GRX2 overexpression would affect oxidative folding of Mia40 substrates. To this end, we followed the oxidation of three different IMS proteins: Cox19, Atp23, and Ccs1 (Figure 5A). Cox19 is a twin-CX9C protein that contains four cysteines and forms two disulfides (Bien et al., 2010). Atp23 contains 10 cysteines and in its mature form is mainly oxidized (four or five disulfides; Weckbecker et al., 2012). Ccs1 is dually localized between cytosol and IMS. The fast formation of one disulfide by Mia40 drives accumulation of Ccs1 in the IMS and competes with cytosolic folding of Ccs1, which prevents mitochondrial import (Kloppel et al., 2011).
FIGURE 5:

Unbalanced levels of Grx2 delay oxidative protein folding in the IMS. (A) Scheme depicting the three Mia40 substrates analyzed in this study. Atp23 and Cox19 are fully imported into the IMS, whereas only a minor portion of Ccs1 localizes to the IMS. During import, disulfide bonds are formed in these substrates: one in Ccs1, two in Cox19, and four or five in Atp23. These substrates cover the complete range of known Mia40 substrates in S. cerevisiae. (B) Scheme of the oxidation assay to follow oxidative folding in intact yeast cells. (C) mmPEG24 alkylation of Atp23-HA. Cells were radioactively labeled and precipitated with TCA, and samples were treated with buffer or tris(2-carboxyethyl)phosphine (TCEP) and mmPEG24. Samples were analyzed by SDS–PAGE and autoradiography. (D) Oxidation kinetics of Atp23-HA in wild- type cells expressing or not expressing b2-Grx2. Experiments were performed as described in B. (E) Oxidation kinetics of Atp23-HA in wild-type cells expressing either b2-Grx2 or b2-Grx2(C27S/C30S). Experiments were performed as described in B. (F) Mitochondrial accumulation of Ccs1 in wild-type mitochondria containing or lacking b2-Grx2. Mitochondria isolated from the indicated strains were analyzed by SDS–PAGE and immunoblotting against Ccs1 and Mrpl40.
To analyze the oxidative folding of Atp23 and Cox19 in vivo, we used a dynamic pulse-chase approach (Figure 5B; Fischer et al., 2013). Yeast cells were incubated for a short time with [35S]methionine, which was subsequently competed away by addition of an excess of cold methionine. After different incubation periods (chase) in which protein import and oxidation could proceed, oxidation/thiol-disulfide exchange was stopped by precipitation with trichloroacetic acid (TCA). The redox state was read out by alkylation with mmPEG24, and then proteins of interest were enriched by immunoprecipitation. When we analyzed Cox19, we found that it became oxidized with a half-time of ∼1 min (Supplemental Figure S5) and that modulation of the levels of IMS-localized glutaredoxins (deletion or specific overexpression) exerted only a mild effect on the oxidation kinetics of this, by comparison relatively small and simple substrate.
We thus turned to Atp23, which presumably possesses a stronger challenge to the IMS oxidation machinery owing to its size and complex disulfide pattern. We verified that Atp23-hemagglutinin (HA) could be specifically enriched, and the modified and unmodified versions of the protein could be clearly distinguished on SDS–PAGE (Figures 5C). When we analyzed the oxidative folding of Atp23-HA in wild-type cells, we found that it became oxidized with a half-time of ∼4 min (Figure 5D). However, in wild-type cells expressing b2-GRX2, oxidation of Atp23-HA was strongly affected and proceeded with half-time of >8 min (Figure 5D; for analogous experiments in a GRX1, GRX2, GRX8 deletion background, see Supplemental Figure S6). Thus, upon deviation of IMS Grx amounts from wild-type levels, oxidative folding of Atp23-HA was hampered. To confirm that the observed effects of b2-GRX2 expression were not artifacts of overexpressing an additional protein in the IMS, we tested the oxidative folding of Atp23-HA in a strain that overexpressed catalytically inactive Grx2(C27S/C30S). Indeed, under these conditions, we did not observe strong deviations of the kinetics of Atp23-HA oxidation from the respective wild-type kinetics (Figure 5E), supporting that it is the redox activity of Grx2 that leads to the slowdown of oxidation of the complex substrate Atp23-HA. In line with our kinetic data, we also found small corresponding changes in the steady-state levels of Atp23-HA (Supplemental Figure S7).
Next we tested whether Grx2 levels also influenced the distribution of Ccs1 between cytosol and mitochondria. Unfortunately, we could not employ our oxidation assay for this protein because only very small amounts of the total protein are oxidized and imported into the IMS. Instead, we analyzed the steady-state levels of Ccs1 in isolated mitochondria. We hypothesized that a failure to become oxidized with fast kinetics would prevent IMS accumulation of Ccs1 because the protein would in the meantime fold in the cytosol. We indeed found the mitochondrial levels of Ccs1 to be diminished upon overexpression of b2-GRX2 (Figure 5F). Taking our results together, we conclude that excess amounts of Grx2 in the IMS impair the oxidative folding of substrates like Atp23 and Ccs1.
DISCUSSION
In this study, we identify Grx2 as the predominant glutaredoxin in the yeast IMS and find that a small portion of the cytosolic form of Grx2 becomes imported into the IMS. We provide evidence that the very low amounts of Grx2 in the IMS exert a kinetic control that enables disulfide bond formation in a highly reducing environment (Figure 6). This gatekeeper function of Grx2 is critical, because upon full equilibration with the glutathione pool, a significant portion of disulfide-containing IMS proteins would be present in a reduced state even if most of these proteins do have very negative reduction potentials (Supplemental Table S1). Because the import of many if not most IMS proteins depends on the formation of disulfides, impaired oxidative folding would consequently deplete those proteins. Indeed, we find that overexpression of Grx2 in the IMS results in the delay of oxidative protein folding and protein import. It remains unclear at which step excess amounts of Grx influence protein oxidation. Grx2 might directly reduce already formed disulfides in Mia40 substrates, or it might reduce the critical mixed disulfide between Mia40 and its substrates that precedes disulfide formation and serves in translocating substrates over the outer membrane. Finally, in the presence of increased amounts of Grx2 in the IMS, a larger fraction of Mia40 is reduced, which might delay substrate oxidation. This would be in line with findings that depletion of Erv1, which results in increased levels of reduced Mia40, hampers substrate oxidation (Kojer et al., 2012; Fischer et al., 2013).
FIGURE 6:
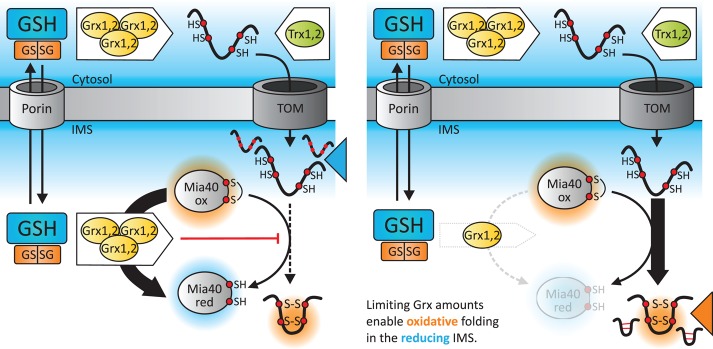
Model for the role of glutaredoxins in controlling the biogenesis of Mia40 substrates. Mia40 substrates are synthesized in the cytosol and maintained there in a reduced state by a highly reducing glutathione pool and high activities of the Grx (mammalian cells; Banci et al., 2013) and the Trx (yeast; Durigon et al., 2012) systems. In the IMS, the glutathione pool is as reducing as its cytosolic counterpart due to rapid equilibration over porins (Kojer et al., 2012). Nonetheless, due to limited glutaredoxin activity in the IMS, fast reduction of proteins is prevented, and disulfide bond formation by Mia40 thus can proceed.
Of note, deletion of GRXs also hampers oxidative folding in the IMS (Supplemental Figure S6), although to a lesser extent than IMS-Grx overexpression, indicating a beneficial function of low amounts of Grx for oxidative protein folding. Under those conditions, a larger fraction of Mia40 is oxidized, and the reduction of IMS-localized roGFP2 after oxidative shock is strongly impaired. The beneficial function of Grx for protein folding might be, for example, deglutathionylation of Mia40 substrates before their oxidation, or it could serve in an isomerization pathway. In vitro data support the existence of such a pathway in Mia40-dependent disulfide formation because in a reconstituted system, GSH participates in proofreading folding intermediates, thereby accelerating oxidative folding (Bien et al., 2010).
Thus, during biogenesis of Mia40 substrates, reducing systems exert influences at different steps (Figure 6). In the cytosol, excess amounts of thioredoxins and Grxs in combination with a very reducing glutathione pool ensure the maintenance of reduced precursors, which is essential for import (Durigon et al., 2012; Banci et al., 2013). Conversely, the limiting amounts of Grxs in the IMS then allow oxidative folding despite a similarly reducing glutathione pool of IMS and cytosol. In addition, the IMS-localized Grxs might partake in proofreading reactions, although this remains to be shown.
Our data also provide an explanation for previously reported differences in EGSH[IMS]. Using the unfused sensor rxYFP, Hu et al. (2008) observed a more-oxidizing EGSH[IMS] (−255 mV) than we did using the Grx1-roGFP2 probe (−301 mV; Kojer et al., 2012). With roGFP2, we also observed such a more-oxidized OxD in the IMS compared with Grx1-roGFP2. This OxD[roGFP2] became more reduced upon coexpression of Grx2 in the IMS (Figure 4C). Of importance, as for Grx1-roGFP2, recovery after oxidative shock of IMS-roGFP2 was dependent on the cytosolic glutathione reductase system (Supplemental Figure S8), confirming that the IMS glutathione pool is in contact with the cytosol. We conclude that Grx1-roGFP2 truly measures EGSH in a given compartment, including the IMS, whereas unfused probes (roGFP2, rxYFP) sense the redox environment in the same way proteins thiols do. Thus unfused probes and Grx1-roGFP2 report similar E values for the cytosol where high Grx activities are present but result in different electrochemical potentials in the IMS, where glutaredoxin amounts are limiting.
The gatekeeper function of Grx2 is dependent on its concentration in the IMS. It is thus interesting that cyto-Grx2 (and presumably also Grx1) distributes between the cytosol and the IMS. Because the cytosolic form of Grx2 lacks a mitochondrial targeting signal, it therefore likely follows, like other IMS proteins, a folding trap pathway for IMS import. In the future it will be exciting to define precisely the import pathway of Grx2 and understand how it is regulated. In addition to Grxs, the IMS also contains a thioredoxin and thioredoxin reductase (Vogtle et al., 2012), and it will be of interest to address their role in IMS redox homeostasis and their cross-talk with the glutathione-Grx system. Taking our results together, our study provides an excellent example of how during evolution different strategies have evolved to enable oxidative protein folding and avoid futile cross-talk between reducing and oxidizing cellular pathways. In the bacterial periplasm, GSH is absent, and the protein-dependent reducing pathway is kinetically separated from the oxidizing machinery; in the ER, the influx of GSH over the ER membrane appears to be limited; and in the IMS, the exchange of reduction equivalents between the glutathione pool and protein thiols/disulfides is kinetically controlled by limiting the amounts of glutaredoxins.
MATERIALS AND METHODS
Plasmids, strains, and antibodies
For primers, plasmids, and yeast strains, refer to Supplemental Tables S2 and S3. The following antibodies were used: anti-roGFP2 (Kojer et al., 2012), anti-Pgk1 (Invitrogen, Carlsbad, CA), anti-Erv1, anti-Mia40, anti-Cox19 (Bien et al., 2010), anti-Mrpl40 (Gruschke et al., 2010), anti-HA (Sigma-Aldrich, St. Louis, MO), and anti-Ccs1 (Kloppel et al., 2011). The antibody against Grx2 was raised in rabbits immunized with heterologously expressed and purified yeast Grx2.
Isolation of mitochondria, analyses of protein amounts, and digitonin-based subfractionation of mitochondria
These were performed as described previously (Kloppel et al., 2011; Longen et al., 2014).
In vivo measurements of fluorescence sensors and redox-state analyses of Mia40
Measurements were performed as detailed previously (Kojer et al., 2012). For a description of OxD and EGSH calculations, refer to the Supplemental Methods.
Pulse-chase assay to assess the oxidation kinetics of Mia40 substrates
Yeast cells were grown in galactose-containing medium. Cells at 14 OD600 (∼1.5 × 108 cells) were radioactively labeled with [35S]methionine for 3 min. Radioactive methionine was competed away by addition of excess cold methionine. After different incubation periods, oxidation was stopped by TCA precipitation. Redox states were analyzed as described previously (Fischer et al., 2013). First, the TCA pellet was resuspended in a buffer containing mmPEG24. Then specific proteins were immunoprecipitated and analyzed by nonreducing SDS–PAGE and autoradiography.
Protein purification and in vitro assay
Mia40 was expressed from pGEX-6P-1 and purified as described (Bien et al., 2010). Grx2 was expressed from pRSETA with a hexahistidine tag. Affinity purification with nickel-nitriloacetic acid beads was performed as recommended by the manufacturer. Grx2 and Mia40 were dialyzed in buffer A (10 mM NaPi buffer, pH 7.4, 10 mM NaCl). The experiment was performed in the presence of 0.5% O2. Purified Mia40 was reduced by adding 5 mM dithiothreitol (DTT) for 10 min at 25°C. DTT was removed by gel filtration using a PD Mini Trap G-10 column (GE Healthcare, Freiburg, Germany). Then 5 μM Mia40 was incubated with both 1 μM Grx2 and 50 μM GSSG (Sigma-Aldrich) or with 1 μM Grx2 or 50 μM GSSG. After incubation at 30°C, the reaction was stopped by adding 12% TCA. The resulting pellet was resuspended in sample buffer (60 mM Tris HCl, pH 6.8, 2% [wt/vol] SDS, 10% glycerol, 0.05% [wt/vol] bromphenol blue) containing 1 mM 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS) and incubated 1 h at 25°C. Samples were analyzed by nonreducing SDS–PAGE and Coomassie staining.
Supplementary Material
Acknowledgments
We thank Vera Nehr and Andrea Trinkaus for excellent technical help and members of the Riemer lab for critical discussions. We thank Bruce Morgan for discussions and critical reading of the manuscript. Templates for Grx2 plasmids were kindly provided by J. A. Bárcena (University of Córdoba, Córdoba, Spain). This work was supported in part by funding from the Deutsche Forschungsgemeinschaft and the Landesschwerpunkt Membrantransport (RIMB) to J.R.
Abbreviations used:
- AMS
4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid
- BSO
buthionine sulfoximine
- EGSH
glutathione redox potential
- ER
endoplasmic reticulum
- GR
glutathione reductase
- Grx
glutaredoxin
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- HA
hemagglutinin
- IMS
intermembrane space of mitochondria
- SS
steady state
- TCA
trichloroacetic acid
- TCEP
tris(2-carboxyethyl)phosphine.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-10-1422) on November 12, 2014.
J.R. and K.K. designed the study and the experiments. K.K., V.P., and G.C. performed the experiments. All authors analyzed the experiments. J.R. wrote the manuscript with input from K.K. and J.H.
The authors declare no conflict of interest.
REFERENCES
- Appenzeller-Herzog C. Glutathione- and non-glutathione-based oxidant control in the endoplasmic reticulum. J Cell Sci. 2011;124:847–855. doi: 10.1242/jcs.080895. [DOI] [PubMed] [Google Scholar]
- Appenzeller-Herzog C, Ellgaard L. In vivo reduction-oxidation state of protein disulfide isomerase: the two active sites independently occur in the reduced and oxidized forms. Antioxid Redox Signal. 2008;10:55–64. doi: 10.1089/ars.2007.1837. [DOI] [PubMed] [Google Scholar]
- Appenzeller-Herzog C, Riemer J, Zito E, Chin KT, Ron D, Spiess M, Ellgaard L. Disulphide production by Ero1alpha-PDI relay is rapid and effectively regulated. EMBO J. 2010;29:3318–3329. doi: 10.1038/emboj.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker KM, Chakravarthi S, Langton KP, Sheppard AM, Lu H, Bulleid NJ. Low reduction potential of Ero1alpha regulatory disulphides ensures tight control of substrate oxidation. EMBO J. 2008;27:2988–2997. doi: 10.1038/emboj.2008.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banci L, Barbieri L, Luchinat E, Secci E. Visualization of redox-controlled protein fold in living cells. Chem Biol. 2013;20:747–752. doi: 10.1016/j.chembiol.2013.05.007. [DOI] [PubMed] [Google Scholar]
- Banhegyi G, Lusini L, Puskas F, Rossi R, Fulceri R, Braun L, Mile V, di Simplicio P, Mandl J, Benedetti A. Preferential transport of glutathione versus glutathione disulfide in rat liver microsomal vesicles. J Biol Chem. 1999;274:12213–12216. doi: 10.1074/jbc.274.18.12213. [DOI] [PubMed] [Google Scholar]
- Banhegyi G, Margittai E, Szarka A, Mandl J, Csala M. Crosstalk and barriers between the electron carriers of the endoplasmic reticulum. Antioxid Redox Signal. 2012;16:772–780. doi: 10.1089/ars.2011.4437. [DOI] [PubMed] [Google Scholar]
- Bien M, Longen S, Wagener N, Chwalla I, Herrmann JM, Riemer J. Mitochondrial disulfide bond formation is driven by intersubunit electron transfer in Erv1 and proofread by glutathione. Mol Cell. 2010;37:516–528. doi: 10.1016/j.molcel.2010.01.017. [DOI] [PubMed] [Google Scholar]
- Birk J, Meyer M, Aller I, Hansen HG, Odermatt A, Dick TP, Meyer AJ, Appenzeller-Herzog C. Endoplasmic reticulum: reduced and oxidized glutathione revisited. J Cell Sci. 2013;126:1604–1617. doi: 10.1242/jcs.117218. [DOI] [PubMed] [Google Scholar]
- Bode M, Longen S, Morgan B, Peleh V, Dick TP, Bihlmaier K, Herrmann JM. Inaccurately assembled cytochrome c oxidase can lead to oxidative stress-induced growth arrest. Antioxid Redox Signal. 2013;18:1597–1612. doi: 10.1089/ars.2012.4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottinger L, Gornicka A, Czerwik T, Bragoszewski P, Loniewska-Lwowska A, Schulze-Specking A, Truscott KN, Guiard B, Milenkovic D, Chacinska A. In vivo evidence for cooperation of Mia40 and Erv1 in the oxidation of mitochondrial proteins. Mol Biol Cell. 2012;23:3957–3969. doi: 10.1091/mbc.E12-05-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes N, Reichmann D, Tienson H, Leichert LI, Jakob U. Using quantitative redox proteomics to dissect the yeast redoxome. J Biol Chem. 2011;286:41893–41903. doi: 10.1074/jbc.M111.296236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun NA, Morgan B, Dick TP, Schwappach B. The yeast CLC protein counteracts vesicular acidification during iron starvation. J Cell Sci. 2010;123:2342–2350. doi: 10.1242/jcs.068403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulleid NJ, Ellgaard L. Multiple ways to make disulfides. Trends Biochem Sci. 2011;36:485–492. doi: 10.1016/j.tibs.2011.05.004. [DOI] [PubMed] [Google Scholar]
- Chakravarthi S, Jessop CE, Bulleid NJ. The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep. 2006;7:271–275. doi: 10.1038/sj.embor.7400645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzi A, Tokatlidis K. The mitochondrial intermembrane space: a hub for oxidative folding linked to protein biogenesis. Antioxid Redox Signal. 2012;19:54–62. doi: 10.1089/ars.2012.4855. [DOI] [PubMed] [Google Scholar]
- Deponte M, Hell K. Disulphide bond formation in the intermembrane space of mitochondria. J Biochem. 2009;146:599–608. doi: 10.1093/jb/mvp133. [DOI] [PubMed] [Google Scholar]
- Depuydt M, Messens J, Collet JF. How proteins form disulfide bonds. Antioxid Redox Signal. 2011;15:49–66. doi: 10.1089/ars.2010.3575. [DOI] [PubMed] [Google Scholar]
- Dooley CT, Dore TM, Hanson GT, Jackson WC, Remington SJ, Tsien RY. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J Biol Chem. 2004;279:22284–22293. doi: 10.1074/jbc.M312847200. [DOI] [PubMed] [Google Scholar]
- Draculic T, Dawes IW, Grant CM. A single glutaredoxin or thioredoxin gene is essential for viability in the yeast Saccharomyces cerevisiae. Mol Microbiol. 2000;36:1167–1174. doi: 10.1046/j.1365-2958.2000.01948.x. [DOI] [PubMed] [Google Scholar]
- Durigon R, Wang Q, Ceh Pavia E, Grant CM, Lu H. Cytosolic thioredoxin system facilitates the import of mitochondrial small Tim proteins. EMBO Rep. 2012;13:916–922. doi: 10.1038/embor.2012.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckers E, Bien M, Stroobant V, Herrmann JM, Deponte M. Biochemical characterization of dithiol glutaredoxin 8 from Saccharomyces cerevisiae: the catalytic redox mechanism redux. Biochemistry. 2009;48:1410–1423. doi: 10.1021/bi801859b. [DOI] [PubMed] [Google Scholar]
- Fischer M, Horn S, Belkacemi A, Kojer K, Petrungaro C, Habich M, Ali M, Kuttner V, Bien M, Kauff F, et al. Protein import and oxidative folding in the mitochondrial intermembrane space of intact mammalian cells. Mol Biol Cell. 2013;24:2160–21670. doi: 10.1091/mbc.E12-12-0862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleiter S, Bardwell JC. Disulfide bond isomerization in prokaryotes. Biochim Biophys Acta. 2008;1783:530–534. doi: 10.1016/j.bbamcr.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruschke S, Grone K, Heublein M, Holz S, Israel L, Imhof A, Herrmann JM, Ott M. Proteins at the polypeptide tunnel exit of the yeast mitochondrial ribosome. J Biol Chem. 2010;285:19022–19028. doi: 10.1074/jbc.M110.113837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutscher M, Pauleau AL, Marty L, Brach T, Wabnitz GH, Samstag Y, Meyer AJ, Dick TP. Real-time imaging of the intracellular glutathione redox potential. Nat Methods. 2008;5:553–559. doi: 10.1038/nmeth.1212. [DOI] [PubMed] [Google Scholar]
- Hagiwara M, Nagata K. Redox-dependent protein quality control in the endoplasmic reticulum: folding to degradation. Antioxid Redox Signal. 2012;16:1119–1128. doi: 10.1089/ars.2011.4495. [DOI] [PubMed] [Google Scholar]
- Herrero E, Ros J, Tamarit J, Belli G. Glutaredoxins in fungi. Photosynth Res. 2006;89:127–140. doi: 10.1007/s11120-006-9079-3. [DOI] [PubMed] [Google Scholar]
- Holmgren A, Johansson C, Berndt C, Lonn ME, Hudemann C, Lillig CH. Thiol redox control via thioredoxin and glutaredoxin systems. Biochem Soc Trans. 2005;33:1375–1377. doi: 10.1042/BST0331375. [DOI] [PubMed] [Google Scholar]
- Hu J, Dong L, Outten CE. The redox environment in the mitochondrial intermembrane space is maintained separately from the cytosol and matrix. J Biol Chem. 2008;283:29126–29134. doi: 10.1074/jbc.M803028200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessop CE, Bulleid NJ. Glutathione directly reduces an oxidoreductase in the endoplasmic reticulum of mammalian cells. J Biol Chem. 2004;279:55341–55347. doi: 10.1074/jbc.M411409200. [DOI] [PubMed] [Google Scholar]
- Kim S, Sideris DP, Sevier CS, Kaiser CA. Balanced Ero1 activation and inactivation establishes ER redox homeostasis. J Cell Biol. 2012;196:713–725. doi: 10.1083/jcb.201110090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloppel C, Suzuki Y, Kojer K, Petrungaro C, Longen S, Fiedler S, Keller S, Riemer J. Mia40-dependent oxidation of cysteines in domain I of Ccs1 controls its distribution between mitochondria and the cytosol. Mol Biol Cell. 2011;22:3749–3757. doi: 10.1091/mbc.E11-04-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojer K, Bien M, Gangel H, Morgan B, Dick TP, Riemer J. Glutathione redox potential in the mitochondrial intermembrane space is linked to the cytosol and impacts the Mia40 redox state. EMBO J. 2012;31:3169–3182. doi: 10.1038/emboj.2012.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar C, Igbaria A, D’Autreaux B, Planson AG, Junot C, Godat E, Bachhawat AK, Delaunay-Moisan A, Toledano MB. Glutathione revisited: a vital function in iron metabolism and ancillary role in thiol-redox control. EMBO J. 2011;30:2044–2056. doi: 10.1038/emboj.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longen S, Woellhaf MW, Petrungaro C, Riemer J, Herrmann JM. The disulfide relay of the intermembrane space oxidizes the ribosomal subunit mrp10 on its transit into the mitochondrial matrix. Dev Cell. 2014;28:30–42. doi: 10.1016/j.devcel.2013.11.007. [DOI] [PubMed] [Google Scholar]
- Merksamer PI, Trusina A, Papa FR. Real-time redox measurements during endoplasmic reticulum stress reveal interlinked protein folding functions. Cell. 2008;135:933–947. doi: 10.1016/j.cell.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan B, Sobotta MC, Dick TP. Measuring E(GSH) and H(2)O(2) with roGFP2-based redox probes. Free Radic Biol Med. 2011;51:1943–1951. doi: 10.1016/j.freeradbiomed.2011.08.035. [DOI] [PubMed] [Google Scholar]
- Ostergaard H, Tachibana C, Winther JR. Monitoring disulfide bond formation in the eukaryotic cytosol. J Cell Biol. 2004;166:337–345. doi: 10.1083/jcb.200402120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai HV, Starke DW, Lesnefsky EJ, Hoppel CL, Mieyal JJ. What is the functional significance of the unique location of glutaredoxin 1 (GRx1) in the intermembrane space of mitochondria. Antioxid Redox Signal. 2007;9:2027–2033. doi: 10.1089/ars.2007.1642. [DOI] [PubMed] [Google Scholar]
- Pedrajas JR, Porras P, Martinez-Galisteo E, Padilla CA, Miranda-Vizuete A, Barcena JA. Two isoforms of Saccharomyces cerevisiae glutaredoxin 2 are expressed in vivo and localize to different subcellular compartments. Biochem J. 2002;364:617–623. doi: 10.1042/BJ20020570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porras P, McDonagh B, Pedrajas JR, Barcena JA, Padilla CA. Structure and function of yeast glutaredoxin 2 depend on postranslational processing and are related to subcellular distribution. Biochim Biophys Acta. 2010;1804:839–845. doi: 10.1016/j.bbapap.2009.12.012. [DOI] [PubMed] [Google Scholar]
- Riemer J, Bulleid N, Herrmann JM. Disulfide formation in the ER and mitochondria: two solutions to a common process. Science. 2009;324:1284–1287. doi: 10.1126/science.1170653. [DOI] [PubMed] [Google Scholar]
- Rozhkova A, Stirnimann CU, Frei P, Grauschopf U, Brunisholz R, Grutter MG, Capitani G, Glockshuber R. Structural basis and kinetics of inter- and intramolecular disulfide exchange in the redox catalyst DsbD. EMBO J. 2004;23:1709–1719. doi: 10.1038/sj.emboj.7600178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevier CS, Qu H, Heldman N, Gross E, Fass D, Kaiser CA. Modulation of cellular disulfide-bond formation and the ER redox environment by feedback regulation of Ero1. Cell. 2007;129:333–344. doi: 10.1016/j.cell.2007.02.039. [DOI] [PubMed] [Google Scholar]
- Stojanovski D, Bragoszewski P, Chacinska A. The MIA pathway: a tight bond between protein transport and oxidative folding in mitochondria. Biochim Biophys Acta. 2012;1823:1142–1150. doi: 10.1016/j.bbamcr.2012.04.014. [DOI] [PubMed] [Google Scholar]
- Toledano MB, Delaunay-Moisan A, Outten CE, Igbaria A. Functions and cellular compartmentation of the thioredoxin and glutathione pathways in yeast. Antioxid Redox Signal. 2013;18:1699–1711. doi: 10.1089/ars.2012.5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Lith M, Tiwari S, Pediani J, Milligan G, Bulleid NJ. Real-time monitoring of redox changes in the mammalian endoplasmic reticulum. J Cell Sci. 2011;124:2349–2356. doi: 10.1242/jcs.085530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogtle FN, Burkhart JM, Rao S, Gerbeth C, Hinrichs J, Martinou JC, Chacinska A, Sickmann A, Zahedi RP, Meisinger C. Intermembrane space proteome of yeast mitochondria. Mol Cell Proteomics. 2012;11:1840–1852. doi: 10.1074/mcp.M112.021105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weckbecker D, Longen S, Riemer J, Herrmann JM. Atp23 biogenesis reveals a chaperone-like folding activity of Mia40 in the IMS of mitochondria. EMBO J. 2012;31:4348–4358. doi: 10.1038/emboj.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.