

This study describes a novel regulatory pathway for the master antioxidant transcription factor Nrf2. The ubiquitin-conjugating enzyme UBE2E3 and its nuclear transport receptor importin-11 coordinately promote the transcriptional activity of Nrf2 by limiting its mitochondrial localization and increasing its residency on target gene promoters.
Abstract
The transcription factor NF-E2 p45–related factor (Nrf2) induces the expression of cytoprotective proteins that maintain and restore redox homeostasis. Nrf2 levels and activity are tightly regulated, and three subcellular populations of the transcription factor have been identified. During homeostasis, the majority of Nrf2 is degraded in the cytoplasm by ubiquitin (Ub)-mediated degradation. A second population is transcriptionally active in the nucleus, and a third population localizes to the outer mitochondrial membrane. Still unresolved are the mechanisms and factors that govern Nrf2 distribution between its subcellular locales. We show here that the Ub-conjugating enzyme UBE2E3 and its nuclear import receptor importin 11 (Imp-11) regulate Nrf2 distribution and activity. Knockdown of UBE2E3 reduces nuclear Nrf2, decreases Nrf2 target gene expression, and relocalizes the transcription factor to a perinuclear cluster of mitochondria. In a complementary manner, Imp-11 functions to restrict KEAP1, the major suppressor of Nrf2, from prematurely extracting the transcription factor off of a subset of target gene promoters. These findings identify a novel pathway of Nrf2 modulation during homeostasis and support a model in which UBE2E3 and Imp-11 promote Nrf2 transcriptional activity by restricting the transcription factor from partitioning to the mitochondria and limiting the repressive activity of nuclear KEAP1.
INTRODUCTION
Oxidative stress is widely held to be a primary contributor to the etiology and progression of numerous pathological conditions, including certain cancers, neurodegenerative disorders, diabetic retinopathy, and age-related macular degeneration (Kowluru and Chan, 2007; Guglielmotto et al., 2009; Branco et al., 2010; Plafker, 2010). As a consequence, significant efforts have been directed at documenting and understanding how cells respond to oxidative challenge and damage. At the nexus of the intracellular endogenous antioxidant defense system is the cap-N-collar transcription factor, NF-E2 p45–related factor (Nrf2). Nrf2 induces the expression of phase II genes that encode for a host of cytoprotective antioxidant and detoxifying enzymes, chaperones, and proteasomal subunits (reviewed in Hayes and Dinkova-Kostova, 2014). These factors cooperate to maintain and, when necessary, restore cellular redox homeostasis by neutralizing free radical stress and facilitating the elimination of damaged proteins, lipids, DNA, and organelles. Nrf2 is highly expressed in muscle, kidney, and lung, with lower levels present in other organs (Moi et al., 1994).
During redox homeostasis, Nrf2 levels are suppressed by the ubiquitin (Ub) proteolytic system (UPS). The UPS consists of an enzyme cascade that attaches one or more Ub molecules onto substrates. The fate of a Ub-tagged substrate varies, depending on the site of attachment, the number of Ub molecules conjugated, and the particular configuration(s) of Ub polymers attached. The canonical pathway regulating Nrf2 polyubiquitylation and degradation is mediated by the multisubunit, E3 ligase Cul3KEAP1 (Itoh et al., 1999; McMahon et al., 2003; Kobayashi et al., 2004; Zhang et al., 2004; Furukawa and Xiong, 2005). The core unit of Cul3KEAP1 comprises the cullin 3 (Cul3) scaffold, KEAP1, and ring of cullin 1 (ROC1). KEAP1 is a substrate adaptor that binds to an N-terminal domain of Cul3 and functions to recruit Nrf2 to the ligase (Kobayashi et al., 2004; Zhang et al., 2004). In an analogous manner, ROC1 binds to a C-terminal domain of Cul3 and recruits a Ub-conjugating enzyme (E2), charged at its active site with Ub, to the complex (Ohta et al., 1999; Reynolds et al., 2008). Through a series of conformational changes, as well as the dynamic attachment and removal of a Ub-like modifier called Nedd8 (e.g., Saha and Deshaies, 2008; Boh et al., 2011), Cul3KEAP1 decorates Nrf2 with polymers of Ub, and ubiquitylated Nrf2 is then delivered to the 26S proteasome for degradation.
In response to an oxidative stress, Nrf2 and KEAP1 dissociate from the Cul3KEAP1 complex. This dissociation results in the stabilization of Nrf2 and its translocation into the nucleus. Nuclear Nrf2 complexes with multiple different small Maf proteins and the heterodimers bind to the antioxidant response elements (AREs) resident within the promoters of phase II target genes (Itoh et al., 1997; Motohashi et al., 2004; Katsuoka et al., 2005). The expression of these cytoprotective proteins, in turn, restores redox equilibrium (reviewed in Sykiotis and Bohmann, 2010; Hayes and Dinkova-Kostova, 2014). Once homeostasis is restored, KEAP1 removes Nrf2 from the nucleus and returns it to the cytoplasm for degradation (Li et al., 2005; Velichkova and Hasson, 2005; Sun et al., 2007). In addition to shuttling between Cul3KEAP1 in the cytoplasm and phase II gene promoters in the nucleus, a population of Nrf2 associates with mitochondria via phosphoglycerate mutase/protein phosphatase 5 (PGAM5), a resident mitochondrial outer membrane protein (Lo and Hannink, 2008). The function of this mitochondrial population is unknown, as are the factor(s) that regulate partitioning of Nrf2 between the nucleus and mitochondria.
In a previous study (Plafker et al., 2010), we discovered that the conserved metazoan E2, UBE2E3 (also known as UbcM2, UbcH9), regulates Nrf2 stability and activity during oxidative challenge. These overexpression studies identified a critical cysteine of UBE2E3, Cys-136, that, when modified with N-ethylmaleimide or mutated to a hydrophobic amino acid (e.g., phenylalanine, tryptophan), promoted binding of the enzyme to Nrf2 in the nucleus. The functional consequence of this binding was an increase in the half-life and activation of the transcription factor. These findings imply that Cys136 of UBE2E3 is oxidatively modified in stressed cells and that endogenous UBE2E3 contributes to the cytoprotective capacity of Nrf2.
In the work presented here, we address the role of endogenous UBE2E3 in Nrf2 distribution and function. Using small interfering RNA (siRNA), immunofluorescence, Nrf2 activity assays, and coprecipitation assays, we discovered that endogenous UBE2E3 is required for the nuclear accumulation and transcriptional activity of Nrf2. Knockdown of the enzyme leads to a reduction in both the basal and inducible levels of nuclear Nrf2 and a concomitant reduction in Nrf2 target gene expression. The decrease in nuclear Nrf2 is accompanied by a redistribution of the transcription factor to a perinuclear “hot spot,” as well as to collapse of the mitochondrial network to the same perinuclear location. We further demonstrate that importin-11 (Imp-11), the nuclear import receptor for UBE2E3, restricts KEAP1 from prematurely extracting Nrf2 off of target gene promoters. Collectively these findings identify a novel mechanism by which the Nrf2 antioxidant defense system is fine-tuned and support a model in which UBE2E3 and Imp-11 promote the nuclear accumulation and activity of Nrf2 by restricting the transcription factor from partitioning to the mitochondria and limiting the repressive activity of nuclear KEAP1.
RESULTS
The UPS plays a central role in countering oxidative stress. It does so by degrading oxidatively damaged proteins (e.g., Dudek et al., 2005) by regulating the stability of cytoprotective proteins (e.g., HIF1α; Salceda and Caro, 1997; Huang et al., 1998; Ohh et al., 2000). Chief among these protective proteins is Nrf2, the master antioxidant transcription factor. We previously reported that the human Ub-conjugating enzyme UBE2E3 contains a putative redox-sensing cysteine that enables the enzyme to directly bind, stabilize, and activate Nrf2 (Plafker et al., 2010). These studies used overexpressed wild-type (wt) and mutant forms of UBE2E3. The present study was undertaken to determine the mechanisms by which endogenous UBE2E3 modulates Nrf2.
Nrf2 drives both the constitutive and stress-induced expression of a battery of antioxidant and detoxification enzymes, chaperones, and proteasome subunits (Kensler et al., 2007; Li and Kong, 2009; Hayes and Dinkova-Kostova, 2014). We therefore first tested whether endogenous UBE2E3 affects Nrf2 target gene expression in either homeostatic or oxidatively challenged cells. Human retinal pigment epithelial cells immortalized with telomerase (RPE-1 cells) were transfected with either a control siRNA (siCON) or siUBE2E3, an siRNA that suppresses UBE2E3 expression (Plafker et al., 2008). The efficiency of siUBE2E3 in reducing expression of the enzyme is shown at the mRNA level (Figure 1), as well as by Western blotting (Figure 2B). At 3 d post–siRNA treatment, cells were exposed to vehicle or tert-butylhydroquinone (tBHQ), a prooxidant that stabilizes and activates Nrf2 (Lee et al., 2001). UBE2E3 knockdown reduced both the basal and oxidant-induced transcriptional activity of Nrf2 (Figure 1, A and B, respectively). mRNA levels of three canonical Nrf2 target genes—NAD(P)H:quinone oxidoreductase 1 (NQO1), glutamylcysteine ligase catalytic subunit (GCLC), and glutamylcysteine ligase modifier subunit (GCLM)—were suppressed by 20–60% in homeostatic cells (Figure 1A) and similarly in tBHQ-treated cells (Figure 1B). The amplitudes of these changes in target gene expression are on scale with perturbations in other Nrf2 regulators (e.g., Ma et al., 2012; Villeneuve et al., 2013). Of interest, induction of a fourth target gene, heme oxygenase-1 (HO-1), was negatively affected after tBHQ treatment, but no statistically significant reduction occurred during homeostasis. Such differential effects on HO-1 induction and other target genes have been reported (e.g., Komatsu et al., 2010) and highlight the diversified nature of ARE promoters and regulatory mechanisms (Malhotra et al., 2010).
FIGURE 1:
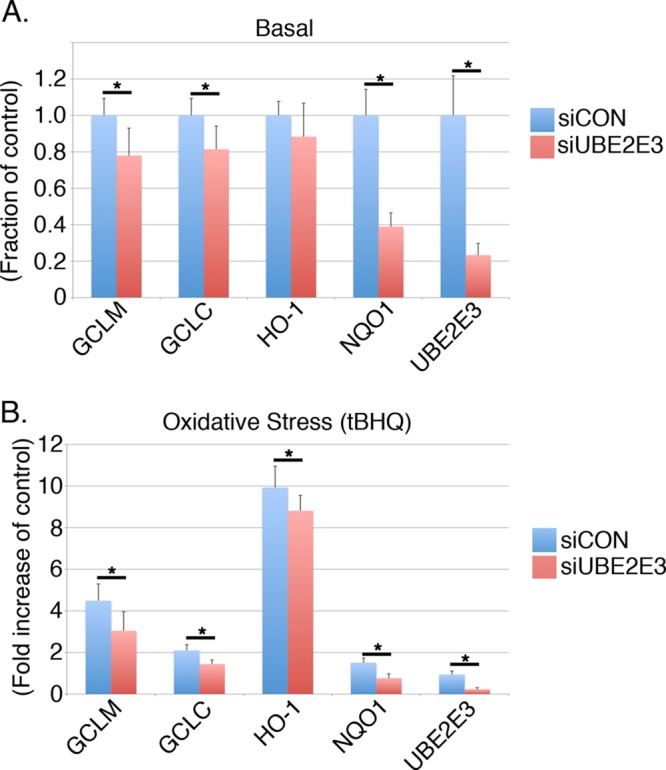
Depletion of UBE2E3 reduces Nrf2 target gene expression. (A) Quantitative PCR data generated from RPE-1 cells treated with siCON (blue bars) or siUBE2E3 (red bars). Specific primers were used to amplify the Nrf2 target genes NQO1, GCLC, GCLR, and HO-1, as well as UBE2E3. (B) Same as A, except that cells were treated for 4 h with tBHQ (oxidant) before harvesting. For A and B, all samples were done in triplicate and results pooled from three independent experiments. Error bars represent ± SD, and asterisks denote statistical significance by ANOVA (p < 0.05).
FIGURE 2:

UBE2E3 or Imp-11 depletion reduces Nrf2 anchoring at ARE. (A) Nrf2 DNA affinity precipitation assay done by combining tBHQ-treated RPE-1 lysates with streptavidin (STV) beads coated with biotinylated oligonucleotides corresponding to either wt or mutant (mut) ARE of NQO1. Input is shown in lane 1, Nrf2 precipitated by wt NQO1 ARE in lane 2, and Nrf2 precipitated by mut NQO1 ARE in lane 3. Nrf2 was detected by Western blotting with an α-Nrf2 antibody. (B) Same assay as A using bead-immobilized, wt ARE NQO1 oligonucleotides and lysates from RPE-1 cells treated with siCON (lanes 1 and 2), siUBE2E3 (lanes 3 and 4), or siImp-11 (lanes 5 and 6). Cells were treated with ethanol (lanes 1, 3, and 5) or tBHQ (lanes 2, 4, and 6). Top and middle panels, α-Nrf2 Western blots; bottom, α-UBE2E3 Western blot demonstrating knockdown of the enzyme. (C) Same assay as B using a single, tBHQ-treated, RPE-1 lysate that was divided into equal aliquots and either mock or UBE2E3 immunodepleted before being combined with bead-immobilized, wt ARE NQO1 oligonucleotides. Top, α-Nrf2 blot shows ARE bound Nrf2; middle, α-Nrf2 blot shows input and Nrf2 remaining postdepletion; bottom, α-UBE2E3 blot shows input and amount of enzyme remaining postdepletion. Graph on the right shows quantitation of ARE-bound Nrf2 from three independent experiments. Asterisk denotes statistical significance by Student's t test. All experiments reported in this figure were done a minimum of three independent times.
After ruling out that the decrease in Nrf2 target gene expression after UBE2E3 depletion was due to a reduction in total Nrf2 levels (Supplemental Figure S1), we performed Nrf2-specific DNA affinity precipitation assays to determine whether UBE2E3 contributes to the anchoring of Nrf2 at the AREs resident in the promoters of target genes. Lysates from siCON- or siUBE2E3-treated cells exposed to tBHQ (to stabilize Nrf2) were combined with a 41–base pair, double-stranded DNA probe representing the ARE sequence from the NQO1 promoter. The probe was biotinylated and immobilized on a streptavidin affinity matrix. The stringency of the assay was established by demonstrating that the wt probe precipitated endogenous Nrf2, whereas a mutant probe did not (Figure 2A). We observed a small but reproducible decrease in Nrf2 recovery from siUBE2E3-treated lysates compared with control lysates (Figure 2B, compare lanes 2 and 4). In contrast, supplementing untreated lysates with recombinant UBE2E3 did not affect the amount of Nrf2 precipitated by the probe (unpublished data). These data indicate that although UBE2E3 is not absolutely required for anchoring Nrf2 at the AREs of target genes, the enzyme does contribute to the amount of Nrf2 that stably associates with target promoter sequences. Given that we previously demonstrated that UBE2E3 is imported into the nucleus by the transport receptor Imp-11 (Plafker and Macara, 2000), we also examined the consequences of Imp-11 knockdown in this assay. Of interest, knockdown of Imp-11 reduced the amount of Nrf2 precipitated by the ARE probe (Figure 2B, lane 6). Imp-11 knockdown efficiency was demonstrated by real-time PCR analysis (see later discussion of Figure 6C), as we could not detect endogenous Imp-11 in RPE-1 lysates with multiple antibodies.
FIGURE 6:

UBE2E3 and importin-11 depletion reduce Keap1 nuclear accumulation. (A) Representative photomicrographs of RPE-1 cells that were treated with the indicated siRNAs and either vehicle (a–f) or LMB (g–l). At 3 d posttransfection, the cells were fixed and immunostained with anti-KEAP1. DNA was counterstained with DAPI. Scale bar, 10 μm. (B) Graph of fold change in nuclear KEAP1 signal (y-axis) as a function of siRNA treatment (x-axis). Nuclear KEAP1 signal was quantified using ImageJ from >250 cells pooled from three independent experiments. Asterisks denote statistically significant differences with p < 0.005, and ns indicates not significant. Error bars represent SD. (C) Quantitative PCR data from RPE-1 cells treated with siCON, siImp-11, or siKEAP1. Specific primers were used to amplify the Nrf2 target genes NQO1, GCLC, GCLR, and HO-1, as well as Imp-11 and KEAP1. Of note, cells were not treated with prooxidant, and these results reflect basal levels of Nrf2 target gene expression. All samples were done in triplicate, and results are pooled from three independent experiments. Error bars represent ± SD, and asterisks denote statistical significance by ANOVA (p < 0.05).
To corroborate the modest effect of UBE2E3 loss on Nrf2 precipitation by the DNA probe, we performed a similar assay in which a tBHQ-treated cell lysate was divided in half. One aliquot was incubated with α-UBE2E3 to immunodeplete the enzyme, and a second aliquot was mock immunodepleted. These lysates were then used in the foregoing DNA affinity precipitation assay. Consistent with the siRNA experiments, removal of the enzyme from the lysate modestly but reproducibly reduced the amount of Nrf2 precipitated by the probe (Figure 2C, top blot and graph). The efficiency of the immunodepletion was demonstrated by Western blotting (Figure 2C, bottom blot).
We next tested whether UBE2E3 affects the nuclear accumulation of Nrf2 in homeostatic or oxidatively stressed cells. RPE-1 cells were treated with siCON or siUBE2E3 and subsequently exposed to vehicle (ethanol) or tBHQ. After fixation and permeabilization, the cells were immunostained to quantify levels of Nrf2 in the nucleus. Nuclear Nrf2 accumulation was largely suppressed in both vehicle-treated siCON and siUBE2E3 cells (Figure 3A, c and i) consistent with constitutive degradation of the transcription factor during homeostasis (Itoh et al., 1999; McMahon et al., 2003; Zhang et al., 2004). Of note, a low level of Nrf2 was detectable in the nuclei of control cells (Figure 3A, b and c) where it induces the basal expression of cytoprotective proteins. UBE2E3-depleted cells had less basal nuclear Nrf2 (Figure 3, A, h and i, and B), consistent with the decreased basal expression of Nrf2 target genes observed in Figure 1A. tBHQ treatment of siCON cells induced the stabilization and nuclear accumulation of Nrf2 (Figure 3A, e and f) whereas cells depleted of UBE2E3 showed reduced nuclear Nrf2 accumulation (Figure 3A, k and l). Quantification of Nrf2 nuclear accumulation (Figure 3B), as well as the ratio of nuclear:cytoplasmic Nrf2 (Supplemental Figure S1B), confirmed the observations from the photomicrographs. Strikingly, the reduced nuclear Nrf2 in siUBE2E3 cells was typically accompanied by an accumulation of the transcription factor at a perinuclear “hot spot” (Figure 3A, i and l, white arrows). These data highlight that although depletion of UBE2E3 does not decrease total Nrf2 levels (Supplemental Figure S1), loss of the enzyme lowers the amount of nuclear Nrf2 in homeostatic and oxidatively stressed cells due to a redistribution of the transcription factor to a perinuclear locale.
FIGURE 3:

UBE2E3 depletion limits basal and induced nuclear accumulation of Nrf2. (A) Representative photomicrographs of RPE-1 cells treated with the indicated siRNAs and either ethanol (EtOH) or oxidant (tBHQ). Cells were fixed, permeabilized, and processed for α-Nrf2 immunostaining (b, e, h, and k). DNA was counterstained with DAPI (a, d, g, and j), and merged images of α-Nrf2 and DAPI are shown (c, f, i, and l). White arrowheads mark examples of perinuclear Nrf2 accumulation. Scale bar, 10 μm. (B) Graph of nuclear Nrf2 immunostaining as a function of treatment. Nuclear Nrf2 in >250 cells/condition was pooled from three independent experiments and quantified using Image J software. The amount of Nrf2 in the nuclei of siCON-treated cells exposed to EtOH was set at a value of 1. Asterisks denote statistically significant differences with p < 0.005. Error bars represent SD.
Because Nrf2 can associate with PGAM5, an atypical protein phosphatase embedded in the outer mitochondrial membrane (Lo and Hannink, 2006, 2008; Takeda et al., 2009), we next assessed whether the cytoplasmic Nrf2 redistribution induced by UBE2E3 knockdown was associated with the mitochondrial network. Strikingly, knockdown of UBE2E3 in unstressed cells induced the normally reticular mitochondrial network to dramatically reorganize and collapse into a juxtanuclear cluster (Figure 4A, b). Costaining for mitochondria and Nrf2 revealed that both occupied the same perinuclear region (Figure 4A, b–d). Furthermore, codepletion of UBE2E3 and Nrf2 blocked the mitochondrial redistribution, indicating that Nrf2 is necessary for the mitochondrial collapse induced by UBE2E3 depletion (Figure 4, B, h, and C). This effect was observed in RPE-1 cells, as well as in HeLa cells (Figure 4C), and similarly occurred in response to tBHQ-induced oxidative stress (unpublished data). Together these findings show that 1) UBE2E3 promotes the nuclear accumulation of Nrf2 during both homeostasis and oxidative stress, 2) depletion of UBE2E3 induces the mitochondrial network and Nrf2 to co-populate a juxtanuclear area and alters the ratio of nuclear to mitochondrial Nrf2, and 3) redistribution of the mitochondrial network requires Nrf2 and/or expression of its target genes.
FIGURE 4:

UBE2E3 depletion induces Nrf2-dependent reorganization of the mitochondrial network. (A) Representative photomicrographs of RPE-1 cells that were treated with siUBE2E3 and 2 d later processed for labeling with DAPI (a) and antibodies against the mitochondrial protein Tom20 (b) and Nrf2 (c). A merge of a–c is shown (d). White arrows mark examples of perinuclear colocalization of mitochondria and Nrf2 (d). (B) Representative photomicrographs of RPE-1 cells treated with the indicated siRNAs. At 2 d post–siRNA transfection, live cells were incubated with MitoTracker to label mitochondria (b, d, f, and h), and 30 min later, the cells were fixed and permeabilized, and the DNA was counterstained with DAPI (a, c, e, and g). White arrows mark examples of clustered mitochondria (d). Scale bars, 10 μm (A, B). (C) Graphs charting the fraction of cells with clustered mitochondria (i.e., clustered/total number of cells) as a function of siRNA treatment in HeLa cells (left) and RPE-1 cells (right). More than 250 cells pooled from three independent experiments were analyzed for each condition. Asterisks denote statistically significant differences with p < 0.005, and error bars represent SD.
Our previous work established that UBE2E3 depletion causes cell cycle arrest characterized by an increase in the cell cycle inhibitor p27Kip1 (Plafker et al., 2008). We therefore tested whether the mitochondrial redistribution induced in siUBE2E3-treated cells was related to this arrest. RPE-1 cells were treated with siCON or siUBE2E3 and 2–3 d later were incubated with the mitochondrial dye MitoTracker before fixation. In parallel, cells were serum starved overnight to induce a cell cycle arrest characterized by p27Kip1 accumulation. The results of these experiments showed that UBE2E3 depletion and serum starvation each led to increased p27Kip1, consistent with cell cycle arrest and exit (Figure 5A, e and h), but yielded distinct changes in the distribution of the mitochondrial network. UBE2E3 suppression caused perinuclear accumulation of the mitochondria (Figure 5A, f, white arrows), whereas serum starvation caused mitochondrial swelling and a vesicular appearance (Figure 5A, i). These data reveal that different inducers of p27Kip1 alter mitochondrial distribution in unique ways, consistent with overlapping but distinct mechanisms underlying the altered mitochondrial phenotypes.
FIGURE 5:

Mitochondrial redistribution induced by UBE2E3 knockdown is dependent on p27Kip1. (A) Representative photomicrographs of RPE-1 cells that were treated with siCON (a–c) or siUBE2E3 (d–f) or were serum starved (g–i). Live cells were labeled with MitoTracker for 30 min and then fixed and immunostained with anti-p27Kip1 (b, e, and h). DNA was counterstained with DAPI (a, d, and g). Experiments were done a minimum of three independent times. (B) Representative photomicrographs of cells treated with the indicated siRNAs and 2 d later labeled with MitoTracker to demonstrate that co-knockdown of p27Kip1 suppresses mitochondrial clustering induced by UBE2E3 depletion. Images captured with a 20× objective. (C) Quantitation of mitochondrial clustering from three independent repeats of experiment shown in B. More than 250 cells were analyzed for each condition. Asterisk denotes statistically significant differences with p < 0.013, and error bars represent SD. (D) Representative photomicrographs of cells treated with the indicated siRNAs and 2 d later labeled with MitoTracker to demonstrate that co-knockdown of Nrf2 suppresses the p27Kip1 elevation induced by UBE2E3 depletion. (E) Quantitation of p27Kip1 expression levels from three independent repeats of experiment shown in D. More than 250 cells were analyzed for each condition. Asterisk denotes statistically significant differences with p < 0.002, and error bars represent SD. Scale bars, 10 μm (A, B, D).
To determine whether the mitochondrial redistribution induced by UBE2E3 depletion requires p27Kip1 elevation, we performed rescue experiments in which cells were co–knocked down for both proteins. Effective p27Kip1 knockdown was confirmed by Western blotting (unpublished data). Quantification of mitochondrial perinuclear accumulation showed that the alteration of mitochondrial distribution induced by loss of UBE2E3 was reversed by codepleting p27Kip1 (Figure 5, B, d, and C). Using a similar co-knockdown rescue strategy, we found that codepletion of Nrf2 reversed the p27Kip1 elevation induced by siUBE2E3 (Figure 5, D, h, and E). This rescue is consistent with the observation that suppression of Nrf2 is required for the proliferation of intestinal stem cells in Drosophila melanogaster (Hochmuth et al., 2011) and can reverse the p27Kip1-dependent arrest of vascular smooth muscle cells by nitro-linoleic acid (Villacorta et al., 2007). Collectively these results demonstrate that the affect of UBE2E3 loss on mitochondrial redistribution is mediated through a p27Kip1-dependent mechanism and that Nrf2 depletion counters this pathway by suppressing the elevation of p27Kip1.
To further investigate the regulation of Nrf2 by UBE2E3, we analyzed whether the enzyme influenced the distribution of KEAP1, the primary regulator of Nrf2 stability, localization, and function. KEAP1 recruits most nascently synthesized Nrf2 for ubiquitylation and subsequent degradation by the proteasome (Itoh et al., 1999; McMahon et al., 2003; Zhang et al., 2004) and additionally represses Nrf2 transcriptional activity by removing it from target gene promoters and returning the transcription factor to the cytoplasm for degradation (Sun et al., 2007). Thus KEAP1 must shuttle between the cytoplasm and nucleus to restrict both the basal and inducible activity of Nrf2. UBE2E3 also constitutively shuttles in and out of the nucleus (Plafker and Macara, 2000), and we therefore examined the effect of UBE2E3 on the nucleocytoplasmic trafficking of KEAP1. To do this, we took advantage of the fungal metabolite leptomycin B (LMB), an inhibitor of the Crm1 pathway that mediates KEAP1 nuclear export (Velichkova and Hasson, 2005). As expected, treatment of cells with LMB trapped KEAP1 inside the nucleus (Figure 6A, h). We quantified the nuclear accumulation of KEAP1 in both control and UBE2E3-depleted cells and found that knockdown of the enzyme reduced nuclear KEAP1 accumulation (Figure 6, A, h vs. j, and B).
Because LMB blocks KEAP1 export, the reduction of nuclear KEAP1 in UBE2E3-depleted cells links the enzyme to KEAP1 import. Sun et al. (2011) found that two transport receptors, KPNA6 and Imp-11, interacted with KEAP1 in a yeast two-hybrid screen and that KPNA6 mediates KEAP1 nuclear import after oxidative stress. The interaction between Imp-11 and KEAP1 was of particular interest to our work, as we had previously demonstrated that Imp-11 mediates UBE2E3 nuclear import (Plafker and Macara, 2000; Plafker et al., 2004). Similar to UBE2E3 knockdown, siRNA-mediated knockdown of Imp-11 modestly reduced the nuclear accumulation of endogenous KEAP1 in unstressed cells treated with LMB (Figure 6, A, l, and B). However, siImp-11 treatment did not negatively affect Nrf2 nuclear accumulation (Supplemental Figure S2). These results predicted that decreased nuclear KEAP1 would increase basal expression of Nrf2 target genes by reducing extraction of Nrf2 from ARE promoters. Consistent with this prediction, we observed an increase in the basal expression levels of a subset of Nrf2 target genes after Imp-11 knockdown (Figure 6C, red bars). As a positive control for increasing the residency of Nrf2 on target gene promoters, we knocked down KEAP1 and observed an increase in the expression of multiple target genes (Figure 6C, green bars). The finding that Imp-11 depletion affected the expression of only two of four target genes and affected HO-1 expression to a greater extent than even KEAP1 knockdown indicates that the affinity of Nrf2 for different AREs varies, as reported previously (Sun et al., 2009), and implies that Nrf2-occupied promoters are differentially sensitive to the influence of Imp-11 and/or KEAP1 nuclear activity.
To further investigate the relationship between UBE2E3, Imp-11, and KEAP1, binding studies were done using Flag-agarose beads and lysates from HEK293T cells overexpressing combinations of Flag-KEAP1, HA3-CUL3, myc-UBE2E3, and HA3-Imp-11. Flag-KEAP1 coprecipitated HA3-CUL3 and myc-UBE2E3 (Figure 7A, lane 7), as well as HA3-Imp-11 (Figure 7A, lane 6), but of interest, pull-down of Imp-11 decreased HA3-CUL3 (Figure 7A, lanes 6 and 8) and effectively abrogated myc-UBE2E3 binding (Figure 7A, lane 8). This competitive binding indicates that Imp-11 interacts with KEAP1 independently of CUL3 and UBE2E3. Using this same transfection and coprecipitation strategy, we observed that Flag-KEAP1 coprecipitated wt UBE2E3 but not the active-site mutant, (C145A) UBE2E3 (Figure 7B, lane 5 vs. 6). These data indicate that KEAP1 selectively engages catalytically activated UBE2E3. We previously found that only catalytically activated UBE2E3, but not the C145A mutant (which cannot be loaded with Ub), interacts with Imp-11 (Plafker et al., 2004). Together these binding data indicate that KEAP1 and Ub-charged UBE2E3 bind mutually exclusively to Imp-11.
FIGURE 7:

UBE2E3 and importin-11 conversely affect KEAP1-CUL3 complex formation. (A) HEK293T cells were transiently transfected with combinations of plasmids encoding Flag-KEAP1, myc-UBE2E3, myc-Imp-11, and HA3-tagged Cul3. Lysates from the transfected cells were then combined with M2 Flag-agarose beads, and the bead-associated proteins were solubilized, resolved by SDS–PAGE, and analyzed by Western blotting with antibodies against the epitope tags (Flag IP). Aliquots of each starting lysate were similarly analyzed (Inputs). Nonspecific binding of proteins to M2 Flag-agarose beads is shown in lanes 1–4 of Flag IP samples. (B) Similar experimental design to A, comparing the binding of wt and catalytically inactive (C145A) myc-UBE2E3 to Flag-KEAP1. Experiments were done a minimum of three independent times. Smeared bands migrating above Flag-KEAP1 likely correspond to oxidized KEAP1 (Hong et al., 2005). (C) Elution assay demonstrating that the binding of UBE2E3, but not KEAP1, to Imp-11 is sensitive to Ran:GTP. Transfected-cell lysates were combined with Flag-agarose, and precipitated complexes were challenged with GTP-containing buffer or recombinant Ran:GTP. Eluted proteins were captured and analyzed alongside proteins that remained bead-associated after the elution challenge. Top, probed with anti-HA to detect HA-tagged Imp-11; bottom, left, probed with anti-myc to detect myc-tagged UBE2E3; bottom, right, probed with anti-Flag to detect Flag-tagged KEAP1. The migration of molecular weight markers is indicated.
To determine whether complex formation between Imp-11 and KEAP1 is consistent with an importin:cargo relationship (i.e., can be dissociated by Ran:GTP; e.g., Plafker and Macara, 2000), we performed binding studies in which anti-hemagglutinin (HA) beads were combined with transfected cell lysates containing HA3-Imp-11 and either Flag-KEAP1 or myc-UBE2E3. Washed complexes were then challenged with either purified Ran:GTP or buffer containing GTP. Eluted Flag-KEAP1 and myc-UBE2E3 were captured, and Western blotting of bound and eluted fractions was done to determine the extent to which Ran:GTP disrupts Imp-11 binding to KEAP1 and UBE2E3. As predicted, UBE2E3 was eluted by Ran:GTP (Figure 7C, lane 3) and resulted in decreased complex formation between the enzyme and Imp-11 (Figure 7C, compare lanes 4 and 5). This result is consistent with Imp-11 transporting and releasing UBE2E3 into the nucleus, a characteristic of canonical nuclear importin:cargo complexes (Plafker and Macara, 2000). In contrast, the Imp-11:KEAP1 complex was resistant to Ran:GTP (Figure 7C, compare lanes 7 and 8 and lanes 9 and 10). These data indicate that KEAP1 is not a canonical import substrate of Imp-11 and, moreover, that the proteins can stably interact in the nucleus.
DISCUSSION
Nrf2 expression is controlled at the transcriptional, translational, and posttranslational levels (e.g., Kwak et al., 2002; Purdom-Dickinson et al., 2007; Sun et al., 2009). Posttranslational regulation is primarily via CUL3KEAP1 targeting of Nrf2 for degradation (Itoh et al., 1999; McMahon et al., 2003; Zhang et al., 2004). KEAP1 also removes Nrf2 from target gene promoters after restoration of homeostasis (Sun et al., 2007). Numerous secondary Nrf2 regulators have been identified, including p62/Sqstm1 (Komatsu et al., 2010), BRCA1 (Gorrini et al., 2013), PALB2/FANCN (Ma et al., 2012), PGAM5 (Lo and Hannink, 2008), and GSK-3β (Salazar et al., 2006), underscoring the need to balance Nrf2 activity with cellular redox status. We identified a novel mechanism of Nrf2-KEAP1 regulation involving UBE2E3 and Imp-11. UBE2E3 is required for partitioning Nrf2 between the nucleus and mitochondria, and complementary to this, Imp-11 enhances basal Nrf2 transcriptional activity by restricting the nuclear function of KEAP1.
There are three subcellular populations of Nrf2 (Jain et al., 2005; Lo and Hannink, 2008). The major share is cytoplasmic and engaged by CUL3KEAP1. A second population is nuclear and resides on target gene promoters. After oxidative challenge, CUL3KEAP1 is inhibited, and the distributions of the cytoplasmic and nuclear populations reverse, resulting in the expression of cytoprotective factors. The third population is associated with mitochondria, but its function and the factors that control it are largely unknown. It is clear, however, that Nrf2 and KEAP1 bind to the outer mitochondrial membrane protein PGAM5 (Lo and Hannink, 2008). We found that UBE2E3 plays an essential role in restricting Nrf2 partitioning to the mitochondria under both basal and oxidative stress conditions (Figures 3 and 4). Relieving this restriction via UBE2E3 knockdown increases Nrf2 localization to mitochondria (Figures 3 and 4) and concomitantly reduces Nrf2 target gene expression (Figure 1). UBE2E3 accomplishes this at least in part by promoting the nuclear accumulation (Figure 3) and ARE residency of Nrf2 (Figure 2, B and C). These data advance our previous studies showing that UBE2E3 interacts with Nrf2 in the nucleus to increase the half-life and activity of the transcription factor (Plafker et al., 2010).
Strikingly, UBE2E3 knockdown induced mitochondrial perinuclear redistribution accompanied by relocalization of Nrf2 to this mitochondrial “hot spot” (Figure 4A). Stress-induced, perinuclear accumulation of mitochondria has been observed after prolonged proteasome inhibition (Muqit et al., 2006; Radke et al., 2008) and hypoxia (Al-Mehdi et al., 2012). UBE2E3 depletion does not induce an overt stress response (e.g., no increase in Hsp60 or Hsp70; unpublished data), yet it does cause a p27Kip1-dependent cell cycle exit (Figure 5A; Plafker et al., 2008). Although this exit per se does not underlie the specific change in mitochondrial distribution induced by UBE2E3 loss (Figure 5A), the elevation of p27Kip1 was necessary, as revealed by rescue experiments in which UBE2E3 and p27Kip1 were codepleted (Figure 5, B and C). Moreover, co-knockdown of Nrf2 also rescues the clustering phenotype by suppressing p27Kip1 elevation (Figures 4, B and C, and 5, D and E). Together these data establish new links between UBE2E3, Nrf2, p27Kip1, and mitochondrial trafficking.
We propose a model for enhanced nuclear Nrf2 residency and activity by UBE2E3 and Imp-11 (Figure 8). Imp-11 binds Ub-charged UBE2E3 in the cytoplasm (Plafker et al., 2004), and the complex translocates into the nucleus, where it is dissociated by Ran:GTP. Released UBE2E3 then engages and promotes the activity of nuclear Nrf2, leaving Imp-11 to bind KEAP1. Once sufficient target gene expression is achieved, KEAP1 dissociates from Imp-11, perhaps via binding to the export receptor Crm1 (Velichkova and Hasson, 2005), and elutes Nrf2 from AREs in preparation for export to the cytoplasm (Sun et al., 2007). In the cytoplasm, the KEAP1-Nrf2 complex associates with CUL3, and Nrf2 is polyubiquitylated and targeted for degradation. If, however, UBE2E3 levels are low, a population of Nrf2 is spared from turnover and is instead distributed to the mitochondria. This model is supported by data showing that UBE2E3 stabilizes and activates Nrf2 in the nucleus (Plafker et al., 2010). In addition, UBE2E3 knockdown decreases Nrf2 target gene expression under both basal and oxidative stress conditions (Figure 1). This dampened transcriptional activity results from a decrease in nuclear Nrf2 (Figure 3) and a concomitant relocalization of Nrf2 to the mitochondria (Figure 4A). Within the nucleus, Imp-11 promotes Nrf2 transcriptional activity by restricting KEAP1 from prematurely extracting Nrf2 off target gene promoters. As a result, in lysates derived from Imp-11–depleted cells, more KEAP1 is available to limit Nrf2 coprecipitation by an ARE probe (Figure 2B). This nuclear function for Imp-11 requires binding KEAP1 in the presence of Ran:GTP, and indeed complex formation between the proteins is resistant to Ran:GTP dissociation (Figure 7C). As predicted by this model, complex formation between Imp-11 and KEAP1 is mutually exclusive, with Imp-11 binding to UBE2E3 (Figure 7A). A seemingly paradoxical result was that Imp-11 knockdown increased the induction of a subset of Nrf2 target genes (Figure 6C), whereas the model predicts decreased transcription based on unrestricted KEAP1 access to nuclear Nrf2. However, the increased target gene expression may be explained by the modest decrease in KEAP1 import after Imp-11 knockdown (Figure 6, A and B). Reduced nuclear KEAP1 would increase the residency time of Nrf2 on certain AREs. This effect of Imp-11 depletion on nuclear KEAP1 accumulation is likely due to disrupted import of UBE2E3 (Plafker et al., 2004). Increased cytoplasmic UBE2E3 binds to and stabilizes CUL3KEAP1 in the cytoplasm (Figure 7B; Plafker et al., 2009), thereby reducing KEAP1 dissociation from CUL3, a necessary step for bulk KEAP1 import (Sun et al., 2007).
FIGURE 8:

Proposed model of UBE2E3:Imp-11 regulation of Nrf2 activity and distribution. Step 1, Ub-charged UBE2E3 binds to Imp-11 and is transported into the nucleus. Step 2, in the nucleus, Ran:GTP binds to Imp-11 and dissociates the UBE2E3:Imp-11 complex. Step 3, the Imp-11-Ran:GTP complex binds KEAP1 and restricts it from prematurely retrieving Nrf2 from target gene promoters. Step 4, activated UBE2E3 engages Nrf2 on the AREs present in the promoters of target genes. Step 5, the export receptor Crm1 exports a KEAP1:Nrf2 complex to the cytoplasm. Step 6, in the presence of UBE2E3, exported KEAP1:Nrf2 associates with the CUL3 ligase to mediate Nrf2 ubiquitylation and degradation. Step 7, inefficient CUL3KEAP1 activity (e.g., due to decreased UBE2E3 levels) promotes Nrf2 enrichment at the mitochondria. Of note, KEAP1 is shown as a single rectangle but is actually a dimer. Unresolved aspects of this model include the mechanisms by which 1) KEAP1 is released from Imp-11, 2) UBE2E3 enhances Nrf2 residency on target gene promoters, and 3) mitochondrial Nrf2 is shielded from CUL3KEAP1. Experimental evidence for steps 1, 2, 4, and 6 comes from our previous studies (Plafker and Macara, 2000; Plafker et al., 2004, 2009, 2010), step 5 comes from the work of others (Velichkova and Hasson, 2005; Sun et al., 2007), and steps 3, 4, and 7 come from the present report.
These studies additionally identify a new function for an import receptor. Imp-11 binds KEAP1 in the nucleus, and complex formation is resistant to Ran:GTP. Only one other mammalian import receptor is known to bind a nuclear cargo in the presence of Ran:GTP, and in this case, Imp-13 was determined to be a bidirectional transporter that delivers multiple cargoes to the nucleus but also exports translation initiation factor eIF1A from the nucleus (Mingot et al., 2001). Imp-11, however, is not exporting KEAP1, but instead regulating the extent with which KEAP1 engages nuclear Nrf2. This discovery expands the repertoire of functions for a nuclear import receptor and highlights a new mechanism for fine-tuning Nrf2 transcriptional activity.
Nrf2 regulation by UBE2E3 opens up many questions, perhaps the foremost being, How does UBE2E3 limit the mitochondrial targeting of Nrf2 while also promoting Nrf2 nuclear residency and transcriptional activity? One explanation is that two populations of UBE2E3 are involved; one, in the cytoplasm, enhances the CUL3KEAP1-mediated turnover of de novo synthesized Nrf2, and the other, in the nucleus, augments Nrf2 residency on AREs. Depletion of UBE2E3 reduces Nrf2 degradation in the cytoplasm, and the excess transcription factor associates with PGAM5 on mitochondria. A second possibility, based on the necessity for an intact UBE2E3 active site, is that the enzyme ubiquitylates factor(s) that promote Nrf2 nuclear entry and/or retention. Thus, in the absence of UBE2E3, less Nrf2 is in the nucleus and more is in the cytoplasm available for capture by PGAM5 at the mitochondria. The mitochondrial “hot spot” induced by UBE2E3 knockdown would create a high local concentration of PGAM5 that could compete with CUL3KEAP1 for cytoplasmic Nrf2. Future experiments will discriminate between these and other models.
MATERIALS AND METHODS
Western blotting
Proteins were boiled in Laemmli buffer, resolved by SDS–PAGE, and transferred to nitrocellulose. Blots were blocked in 5% nonfat milk/TBST (0.1% Tween-20 in Tris-buffered saline) before incubation with primary antibodies. Anti-Flag M2-HRP was diluted 1:5000 in TBST, anti-Myc 9E10 was used at 1:500, and anti-HA 12CA5 was used at 1:1000. Anti-Nrf2 and anti-UBE2E3 antibodies were used as previously described (Plafker et al., 2008; Ziady et al., 2012).
Transfections, immunoprecipitations, and Ran:GTP elutions
HEK293T cells were transfected by the calcium phosphate method (Chen and Okayama, 1987). For immunoprecipitations, cells were lysed in 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, and 1 mM phenylmethylsulfonyl fluoride (PMSF) and centrifuged at 16,000 × g, and the resulting supernatant was mixed with anti-Flag M2 agarose (#A2220; Sigma-Aldrich, St. Louis, MO). After binding at 4°C for 3 h, the bead-associated proteins were washed with ice-cold lysis buffer before being solubilized in 2× concentrated Laemmli buffer and processed for western blotting. The Ran:GTP elution experiments were performed similarly to previous studies (Plafker and Macara, 2000). Briefly, bead-bound complexes were immunoprecipitated from transfected HEK293T cells and combined with either buffer containing GTP or buffer containing 100 μM GTP-loaded Ran. Reactions were incubated for 90 min at 4°C, and eluted proteins were captured and solubilized with 2× Laemmli buffer. Bead-bound proteins were washed with ice-cold phosphate-buffered saline (PBS) and similarly solubilized in preparation for Western blot analysis.
siRNA, immunofluorescence, mitochondrial cluster quantitation, and staining
Knockdown of proteins was achieved by siRNA transfection of RPE-1 cells as previously described (Plafker et al., 2008). At 2–3 d post–siRNA transfection, cells were fixed with 3.7% formaldehyde in PBS for 20 min at room temperature, permeabilized with either −20°C methanol for 2 min or 0.2% Triton X-100 in PBS for 10 min on ice. Cells were then blocked in 3% bovine serum albumin (BSA) in PBS before being incubated with primary antibodies at 4°C overnight. Alexa488 nm, Alex546 nm, and Alex647 nm-conjugated secondary antibodies were used according to the manufacturer's instructions (Life Technologies, Grand Island, NY). DNA was counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Cells were incubated with MitoTracker Red CMXROS (200 nM final concentration in media) for 30 min at 37°C to label mitochondria in live cells. Epifluorescence images were captured using an inverted microscope (Eclipse TE2000; Nikon, Tokyo, Japan) as previously described (Plafker et al., 2004) and processed with Photoshop CS, version 8 (Adobe, San Jose, CA).
Mitochondria were scored as clustered if the typical reticular network was collapsed into an obvious perinuclear hot spot. If such a hot spot was not evident, a cell was scored as not clustered. Four or five fields of cells were randomly chosen for each experimental condition, and graphs were compiled from ≥250 scored cells for each condition pooled from three independent experiments.
ARE binding
The ARE binding assay was performed as described (Sun et al., 2009). For the combined immunodepletion/ARE binding experiment shown in Figure 2C, RPE1 cells were plated in a 10- cm dish, grown to confluency, and treated with 50 μM tBHQ for 4 h. Cells were rinsed with PBS and lysed in 1 ml of RIPA1 buffer (50 mM Tris, pH 7.4, 1% NP40, 0.25% sodium deoxycholate, 150 mM sodium chloride, 1 mM EDTA, 1 mM PMSF), and insoluble debris was pelleted at 16,000 × g for 10 min at 4°C. A 500-μg amount of the cleared lysate was added to two tubes containing either 30 μl of anti-UBE2E3 antibody or PBS, rocked at 4°C for 1 h, and added to 100 μl of protein A–Sepharose for 90 min. The resulting supernatants were used in the ARE binding assay.
Quantitative real-time PCR
siRNA-transfected RPE-1 cells were treated with either 50 μM tBHQ or ethanol for 4 h at 37°C. RNA was extracted from the cells using RNAqueous according to the manufacturer's protocol (Life Technologies). One microgram of RNA was converted to cDNA using qScript cDNA Supermix (Quanta Biosciences, Gaithersburg, MD). Quantitative PCR was performed on a CFX96 thermocycler (Bio-Rad, Hercules, CA) with 25 ng of cDNA, 150 nM final concentration of each primer, and iQ SYBR Green Supermix in a total volume on 10 μl. The primers used were as follows: GCLM forward, 5′-AGT TCC CAA ATC AAC CCA GA-3′, and reverse, 5′-CCA TGT CAA CTG CAC TTC TAG T-3′; GCLC forward, 5′-CAG ACT TTG AGA ACT CTG CCT ATG-3′, and reverse, 5′-AAC ATT CCC TGC AAG ACA GC-3′; HO1 forward, 5′-CCA CCA AGT TCA AGC AGC TC-3′, and reverse, 5′-GCT CTG GTC CTT GGT GTC AT-3′; NQO1 forward, 5′-GAG TCC CTG CCA TTC TGA AA-3′, and reverse, 5′-GTG GAT CCC TTG CAG AGA GT-3′; glyceraldehyde-3-phosphate dehydrogenase forward, 5′-GAA GGT GAA GGT CGG AGT CA-3′, and reverse, 5′-AAT GAA GGG GTC ATT GAT GG-3′; UBE2E3 forward, 5′-GGG CCT AAA GGA GAT AAC A-3′, and reverse, 5′-AGA TTC TGG TAC GGA AAG TAA C-3′; Imp-11 forward, 5′-CAA GAC ACT GGC ATC TAA ACG ACT-3′, and reverse, 5′-CTT ACG CAG CAC TTT CAA TGA TAG-3′; and KEAP1 forward, 5′-CGC TAC GAT GTG GAA ACA GA-3′, and reverse, 5′-CTG GGT CGT AAC ACT CCA CA-3′. The Ct values were averaged from technical triplicates. Transcript levels were normalized to GAPDH, and relative changes in Ct were converted to fold change. All experiments were done three times in triplicate. Statistical significance was calculated using Student's t test and one-way analysis of variance (ANOVA).
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grant R01 GM092900 (to S.M.P.), Grant HR13-182 from the Oklahoma Center for the Advancement of Science and Technology (to S.M.P.), and Grant 1307 from the Beckman Initiative for Macular Research. We thank members of the Plafker laboratory for helpful discussions.
Abbreviations used:
- GCLC
glutamylcysteine ligase catalytic subunit
- GCLM
glutamylcysteine ligase modifier subunit
- HO-1
heme oxygenase-1
- Imp-11
importin-11
- LMB
leptomycin B
- NQO1
NAD(P)H:quinone oxidoreductase 1
- Nrf2
NF-E2 p45–related factor
- PGAM5
phosphoglycerate mutase
- siRNA
small interfering RNA
- tBHQ
tert-butylhydroquinone
- Ub
ubiquitin
- UPS
ubiquitin proteolytic system.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-06-1057) on November 5, 2014.
REFERENCES
- Al-Mehdi AB, Pastukh VM, Swiger BM, Reed DJ, Patel MR, Bardwell GC, Pastukh VV, Alexeyev MF, Gillespie MN. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci Signal. 2012;5:ra47. doi: 10.1126/scisignal.2002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boh BK, Smith PG, Hagen T. Neddylation-induced conformational control regulates cullin RING ligase activity in vivo. J Mol Biol. 2011;409:136–145. doi: 10.1016/j.jmb.2011.03.023. [DOI] [PubMed] [Google Scholar]
- Branco DM, Arduino DM, Esteves AR, Silva DF, Cardoso SM, Oliveira CR. Cross-talk between mitochondria and proteasome in Parkinson's disease pathogenesis. Front Aging Neurosci. 2010;2:17. doi: 10.3389/fnagi.2010.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek EJ, Shang F, Valverde P, Liu Q, Hobbs M, Taylor A. Selectivity of the ubiquitin pathway for oxidatively modified proteins: relevance to protein precipitation diseases. FASEB J. 2005;19:1707–1709. doi: 10.1096/fj.05-4049fje. [DOI] [PubMed] [Google Scholar]
- Furukawa M, Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol. 2005;25:162–171. doi: 10.1128/MCB.25.1.162-171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrini C, Baniasadi PS, Harris IS, Silvester J, Inoue S, Snow B, Joshi PA, Wakeham A, Molyneux SD, Martin B, et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J Exp Med. 2013;210:1529–1544. doi: 10.1084/jem.20121337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guglielmotto M, Tamagno E, Danni O. Oxidative stress and hypoxia contribute to Alzheimer's disease pathogenesis: two sides of the same coin. ScientificWorldJournal. 2009;9:781–791. doi: 10.1100/tsw.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. 2014;39:199–218. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- Hochmuth CE, Biteau B, Bohmann D, Jasper H. Redox regulation by Keap1 and Nrf2 controls intestinal stem cell proliferation in Drosophila. Cell Stem Cell. 2011;8:188–199. doi: 10.1016/j.stem.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong F, Sekhar KR, Freeman ML, Liebler DC. Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. J Biol Chem. 2005;280:31768–31775. doi: 10.1074/jbc.M503346200. [DOI] [PubMed] [Google Scholar]
- Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci USA. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain AK, Bloom DA, Jaiswal AK. Nuclear import and export signals in control of Nrf2. J Biol Chem. 2005;280:29158–29168. doi: 10.1074/jbc.M502083200. [DOI] [PubMed] [Google Scholar]
- Katsuoka F, Motohashi H, Ishii T, Aburatani H, Engel JD, Yamamoto M. Genetic evidence that small maf proteins are essential for the activation of antioxidant response element-dependent genes. Mol Cell Biol. 2005;25:8044–8051. doi: 10.1128/MCB.25.18.8044-8051.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- Kowluru RA, Chan PS. Oxidative stress and diabetic retinopathy. Exp Diabetes Res 2007. 2007:43603. doi: 10.1155/2007/43603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak MK, Itoh K, Yamamoto M, Kensler TW. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: role of antioxidant response element-like sequences in the nrf2 promoter. Mol Cell Biol. 2002;22:2883–2892. doi: 10.1128/MCB.22.9.2883-2892.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Moehlenkamp JD, Hanson JM, Johnson JA. Nrf2-dependent activation of the antioxidant responsive element by tert-butylhydroquinone is independent of oxidative stress in IMR-32 human neuroblastoma cells. Biochem Biophys Res Commun. 2001;280:286–292. doi: 10.1006/bbrc.2000.4106. [DOI] [PubMed] [Google Scholar]
- Li W, Jain MR, Chen C, Yue X, Hebbar V, Zhou R, Kong AN. Nrf2 Possesses a redox-insensitive nuclear export signal overlapping with the leucine zipper motif. J Biol Chem. 2005;280:28430–28438. doi: 10.1074/jbc.M410601200. [DOI] [PubMed] [Google Scholar]
- Li W, Kong AN. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol Carcinog. 2009;48:91–104. doi: 10.1002/mc.20465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo SC, Hannink M. PGAM5, a Bcl-XL-interacting protein, is a novel substrate for the redox-regulated Keap1-dependent ubiquitin ligase complex. J Biol Chem. 2006;281:37893–37903. doi: 10.1074/jbc.M606539200. [DOI] [PubMed] [Google Scholar]
- Lo SC, Hannink M. PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Exp Cell Res. 2008;314:1789–1803. doi: 10.1016/j.yexcr.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Cai H, Wu T, Sobhian B, Huo Y, Alcivar A, Mehta M, Cheung KL, Ganesan S, Kong AN, et al. PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Mol Cell Biol. 2012;32:1506–1517. doi: 10.1128/MCB.06271-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra D, Portales-Casamar E, Singh A, Srivastava S, Arenillas D, Happel C, Shyr C, Wakabayashi N, Kensler TW, Wasserman WW, Biswal S. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010;38:5718–5734. doi: 10.1093/nar/gkq212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon M, Itoh K, Yamamoto M, Hayes JD. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J Biol Chem. 2003;278:21592–21600. doi: 10.1074/jbc.M300931200. [DOI] [PubMed] [Google Scholar]
- Mingot JM, Kostka S, Kraft R, Hartmann E, Gorlich D. Importin 13: a novel mediator of nuclear import and export. EMBO J. 2001;20:3685–3694. doi: 10.1093/emboj/20.14.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci USA. 1994;91:9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motohashi H, Katsuoka F, Engel JD, Yamamoto M. Small Maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the Keap1-Nrf2 regulatory pathway. Proc Natl Acad Sci USA. 2004;101:6379–6384. doi: 10.1073/pnas.0305902101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muqit MM, Abou-Sleiman PM, Saurin AT, Harvey K, Gandhi S, Deas E, Eaton S, Payne Smith MD, Venner K, Matilla A, et al. Altered cleavage and localization of PINK1 to aggresomes in the presence of proteasomal stress. J Neurochem. 2006;98:156–169. doi: 10.1111/j.1471-4159.2006.03845.x. [DOI] [PubMed] [Google Scholar]
- Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2:423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- Ohta T, Michel JJ, Schottelius AJ, Xiong Y. ROC1, a homolog of APC11, represents a family of cullin partners with an associated ubiquitin ligase activity. Mol Cell. 1999;3:535–541. doi: 10.1016/s1097-2765(00)80482-7. [DOI] [PubMed] [Google Scholar]
- Plafker KS, Farjo KM, Wiechmann AF, Plafker SM. The human ubiquitin conjugating enzyme, UBE2E3, is required for proliferation of retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2008;49:5611–5618. doi: 10.1167/iovs.08-1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plafker KS, Nguyen L, Barneche M, Mirza S, Crawford D, Plafker SM. The ubiquitin-conjugating enzyme UbcM2 can regulate the stability and activity of the antioxidant transcription factor Nrf2. J Biol Chem. 2010;285:23064–23074. doi: 10.1074/jbc.M110.121913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plafker KS, Singer JD, Plafker SM. The ubiquitin conjugating enzyme, UbcM2, engages in novel interactions with components of cullin-3 based E3 ligases. Biochemistry. 2009;48:3527–3537. doi: 10.1021/bi801971m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plafker SM. Oxidative stress and the ubiquitin proteolytic system in age-related macular degeneration. Adv Exp Med Biol. 2010;664:447–456. doi: 10.1007/978-1-4419-1399-9_51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plafker SM, Macara IG. Importin-11, a nuclear import receptor for the ubiquitin-conjugating enzyme, UbcM2. EMBO J. 2000;19:5502–5513. doi: 10.1093/emboj/19.20.5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plafker SM, Plafker KS, Weissman AM, Macara IG. Ubiquitin charging of human class III ubiquitin-conjugating enzymes triggers their nuclear import. J Cell Biol. 2004;167:649–659. doi: 10.1083/jcb.200406001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purdom-Dickinson SE, Sheveleva EV, Sun H, Chen QM. Translational control of nrf2 protein in activation of antioxidant response by oxidants. Mol Pharmacol. 2007;72:1074–1081. doi: 10.1124/mol.107.035360. [DOI] [PubMed] [Google Scholar]
- Radke S, Chander H, Schafer P, Meiss G, Kruger R, Schulz JB, Germain D. Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J Biol Chem. 2008;283:12681–12685. doi: 10.1074/jbc.C800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds PJ, Simms JR, Duronio RJ. Identifying determinants of cullin binding specificity among the three functionally different Drosophila melanogaster Roc proteins via domain swapping. PLoS One. 2008:3, e2918. doi: 10.1371/journal.pone.0002918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha A, Deshaies RJ. Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Mol Cell. 2008;32:21–31. doi: 10.1016/j.molcel.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar M, Rojo AI, Velasco D, de Sagarra RM, Cuadrado A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J Biol Chem. 2006;281:14841–14851. doi: 10.1074/jbc.M513737200. [DOI] [PubMed] [Google Scholar]
- Salceda S, Caro J. Hypoxia-inducible factor 1alpha. (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem. 1997;272:22642–22647. doi: 10.1074/jbc.272.36.22642. [DOI] [PubMed] [Google Scholar]
- Sun Z, Chin YE, Zhang DD. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol Cell Biol. 2009;29:2658–2672. doi: 10.1128/MCB.01639-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Wu T, Zhao F, Lau A, Birch CM, Zhang DD. KPNA6 (Importin {alpha}7)-mediated nuclear import of Keap1 represses the Nrf2-dependent antioxidant response. Mol Cell Biol. 2011;31:1800–1811. doi: 10.1128/MCB.05036-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Zhang S, Chan JY, Zhang DD. Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol Cell Biol. 2007;27:6334–6349. doi: 10.1128/MCB.00630-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykiotis GP, Bohmann D. Stress-activated cap’n’collar transcription factors in aging and human disease. Sci Signal. 3:re3. doi: 10.1126/scisignal.3112re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Komuro Y, Hayakawa T, Oguchi H, Ishida Y, Murakami S, Noguchi T, Kinoshita H, Sekine Y, Iemura S, et al. Mitochondrial phosphoglycerate mutase 5 uses alternate catalytic activity as a protein serine/threonine phosphatase to activate ASK1. Proc Natl Acad Sci USA. 2009;106:12301–12305. doi: 10.1073/pnas.0901823106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velichkova M, Hasson T. Keap1 regulates the oxidation-sensitive shuttling of Nrf2 into and out of the nucleus via a Crm1-dependent nuclear export mechanism. Mol Cell Biol. 2005;25:4501–4513. doi: 10.1128/MCB.25.11.4501-4513.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villacorta L, Zhang J, Garcia-Barrio MT, Chen XL, Freeman BA, Chen YE, Cui T. Nitro-linoleic acid inhibits vascular smooth muscle cell proliferation via the Keap1/Nrf2 signaling pathway. Am J Physiol Heart Circ Physiol. 2007;293:H770–776. doi: 10.1152/ajpheart.00261.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve NF, Tian W, Wu T, Sun Z, Lau A, Chapman E, Fang D, Zhang DD. USP15 negatively regulates Nrf2 through deubiquitination of Keap1. Mol Cell. 2013;51:68–79. doi: 10.1016/j.molcel.2013.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24:10941–10953. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziady AG, Sokolow A, Shank S, Corey D, Myers R, Plafker S, Kelley TJ. Interaction with CREB binding protein modulates the activities of Nrf2 and NF-kappaB in cystic fibrosis airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2012;302:L1221–L1231. doi: 10.1152/ajplung.00156.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.