

Association of the circadian Per2 factor to p53 results in cytosol–nuclear shuttling of the complex and further association to Mdm2. The trimeric complex remains in the nucleus until a genotoxic signal frees p53, allowing for a transcriptional checkpoint response.
Abstract
Circadian period proteins influence cell division and death by associating with checkpoint components, although their mode of regulation has not been firmly established. hPer2 forms a trimeric complex with hp53 and its negative regulator Mdm2. In unstressed cells, this association leads to increased hp53 stability by blocking Mdm2-dependent ubiquitination and transcription of hp53 target genes. Because of the relevance of hp53 in checkpoint signaling, we hypothesize that hPer2 association with hp53 acts as a regulatory module that influences hp53's downstream response to genotoxic stress. Unlike the trimeric complex, whose distribution was confined to the nuclear compartment, hPer2/hp53 was identified in both cytosol and nucleus. At the transcriptional level, a reporter containing the hp21WAF1/CIP1 promoter, a target of hp53, remained inactive in cells expressing a stable form of the hPer2/hp53 complex even when treated with γ-radiation. Finally, we established that hPer2 directly acts on the hp53 node, as checkpoint components upstream of hp53 remained active in response to DNA damage. Quantitative transcriptional analyses of hp53 target genes demonstrated that unbound hp53 was absolutely required for activation of the DNA-damage response. Our results provide evidence of the mode by which the circadian tumor suppressor hPer2 modulates hp53 signaling in response to genotoxic stress.
INTRODUCTION
Transcription of period genes oscillates in a circadian manner and is essential for maintaining a functional clock that is driven by interacting transcription-translation–based autoregulatory feedback loops (for review, see Takahashi et al., 2008). Three homologues (period 1–3) have been identified in mammals, whose levels oscillate in the suprachiasmatic nuclei, where the master clock is located, and in peripheral tissues (Albrecht et al., 2007). Period (Per), cryptochrome (Cry), casein kinase Iε/δ (CKIε/δ), circadian locomoter output cycles kaput (Clock), and brain and muscle Arnt-like protein 1 (Bmal1) are the main players responsible for driving the primary negative-feedback loop of the clock, where transcriptional activation, heterodimerization, and translocation maintain the dynamics of the process (for review, see Takahashi et al., 2008).
In recent years, it has been determined that clock component roles have expanded beyond their strict function as modulators of the organism's adaptive response to environmental cues (i.e., light/dark cycles) to include regulating sleep–wake cycles and release of hormones, maintaining the body's thermoregulation, and having a role in photoperiodism. Extensive work in various organismal systems has identified clock factors as obligatory intermediates that control numerous physiological processes directly relevant to human diseases and behavioral disorders (for review, see Takahashi et al., 2008). For example, a Bmal1/Mop3 (Arnt1)–null mutant mouse exhibits infertility, decreased body weight usually associated with abnormal gluconeogenesis and lipogenesis, premature aging, and sleep fragmentation (Bunger et al., 2000; Rudic et al., 2004; Laposky et al., 2005; Shimba et al., 2005; Kondratov et al., 2006), whereas a ClockΔ19 (antimorph) mouse mutant is hyperphagic and obese, hypersensitive to chemotherapeutic agents, and exhibits a manic phenotype (Naylor et al., 2000; Rudic et al., 2004; Gorbacheva et al., 2005; Turek et al., 2005). Others, such as the Cry1 and Cry2 double- null mutant mouse, exhibit a delay in tissue regeneration as monitored in the liver (Matsuo et al., 2003). Csnk1e (CK1εtau mutant) mutant animals have an enhanced metabolic but reduced growth rate (Oklejewicz et al., 1997; Lucas et al., 2000), whereas both Csnk1d (CK1δ)- and timeless-null mutations result in a lethal phenotype (Gotter et al., 2000; Xu et al., 2005). Additional physiological phenotypes, including diminished pupillary light reflex, impaired temporal regulation of metabolism and feeding, age-related autoimmune diseases, and defects in skeletal muscle regeneration, have also been described in mice exhibiting mutations in other clock-related genes, such as Rora, b, and c, Dec 1 and 2, Opn4, Vip, Vipr2, and Nocturnin (for review, see Takahashi et al., 2008). Moreover, deletion and mutations of mouse Period genes result in numerous changes in an animal's phenotype, including shortening or loss of the circadian period (in the case of Per1 and Per2 double-null mutant mice), sensitization of animals to drugs, improper alcohol intake, altered glucose metabolism, and abnormal cellular proliferation (Zheng et al., 1999, 2001; Shearman et al., 2000; Bae et al., 2001; Cermakian et al., 2001; Fu et al., 2002). Of interest, neither Per1- nor Per3-null mutant mice exhibit a phenotype that is reminiscent of that observed in animals in which the expression of checkpoint proteins is compromised (Fu et al., 2002); however, Per2-null mutant mice do. Accordingly, Per2-null mice show increased hyperplastic growth, tumor development, and severe morbidity, a phenotype that is accompanied by hair graying and hair loss, which is exacerbated in the presence of genotoxic stress (Fu et al., 2002). Although there have been attempts to identify molecular signatures responsible for the Per2-null phenotype (Fu et al., 2002), the mechanistic foundation that further supports the observed phenotype is lacking.
Connections between clock molecules and the cellular DNA damage response have been identified. In Neurospora crassa, the clock gene period 4 was identified as an orthologue of the mammalian checkpoint kinase 2 (Chk2) gene (Pregueiro et al., 2006). In colon cancer cell lines, overexpression of Per1 sensitizes cells to DNA damage–induced apoptosis by a yet-unknown mechanism that involves interaction with Chk2 (Gery et al., 2006). However, unlike Per2, Per1 does not seem to act as a tumor suppressor, since homozygous Per1 mutant mice display only a shorter circadian period with reduced precision and stability, and ablation of the per1 gene does not affect cell proliferation (Zheng et al., 2001). Finally, the human Timeless (hTim) protein seems to be required for the phosphorylation and activation of Chk1 by the ataxia telangiectasia and Rad3-related protein (ATR), whereas Per3 appears to physically bind to both ATM and Chk2. However, the role of hTim in circadian regulation of mammalian cells is controversial (Unsal-Kacmaz et al., 2005), and the per3 gene product is not necessary to sustain circadian rhythmicity in mice (Shearman et al., 2000). Overall these studies suggest a scenario in which multiple circadian players converge on a multilevel cellular clock that links environmental conditions to the biochemical and genetic machinery of the cell to influence cell cycle progression and the response to genotoxic stress.
More recently, we performed interaction studies using human Period 2 (hPer2) as bait to map and identify protein–protein interactions and endogenous protein partner complexes (Gotoh et al., 2014). The transcription factor and checkpoint-component p53 (human p53 transcription factor [hp53]) is among the novel hPer2 interactors. Association of hPer2 to the C-terminus region of hp53 results in formation of a trimeric complex in which the oncogenic protein murine [human] double minute-2 (Mdm2) is bound to the N-terminus of hp53. As a result, hPer2 promotes hp53 stability by a mechanism that involves inhibition of hp53 ubiquitination by Mdm2 (Gotoh et al., 2014). The end result is the intersection of circadian and checkpoint components at the key hp53 node and the modulation of the hp53 transcriptional response. Our findings are in agreement with the observation that endogenous p53 is largely undetectable in thymocytes from Per2-null mice and that de novo accumulation of p53 seems to occur several hours after the insult is applied in Per2-null animals (Fu et al., 2002), an observation that goes along with our findings of hPer2 acting as a transcriptional regulator of the TP53 gene (Gotoh et al., 2014).
A number of additional findings indirectly point toward cross-talk between Per2 function and the p53-mediated DNA damage response; however, it remains unclear how Per2 relates to that process mechanistically. For example, it is known that overexpression of Per2 results in reduced cellular proliferation and increased apoptosis in lung and mammary carcinoma, but not in embryonic fibroblast NIH3T3 cells, by a transcriptional mechanism that involves the up-regulation of proapoptotic components (i.e., TP53 and BAX [encodes the Bcl-2-associated X protein, Bax]) and the simultaneous attenuation of antiapoptotic transcripts, including MYC, BCL2L1, and BCL2 (Hua et al., 2006). Sun et al. (2010) expanded these findings to leukemia cells by showing that Per2 overexpression promotes p53-dependent G2/M arrest by down-regulation of CCNB1 and MYC expression followed by apoptosis. In line with these observations is the finding that overexpression of Per2 in hematopoietic cancer cell lines results in a phenotype that includes growth inhibition, cell cycle arrest, apoptosis, and loss of clonogenic ability (Gery and Koeffler, 2009). More recently, the known Ser662Gly (S662G) mutation in Per2, responsible for familial advanced sleep phase syndrome, has been linked to enhanced resistance to x-ray–induced apoptosis and increased E1A- and RAS-mediated oncogenic transformation (Gu et al., 2012). Accordingly, animals bearing the Per2S662G mutation show accelerated tumorigenesis in a p53R172H/+ background. This effect is independent of the length of the circadian cycle but influences the relative phases of expression of p53-regulated, clock-controlled cell cycle genes (i.e., CDKN1a [encoding cyclin-dependent kinase inhibitor p21, p21CIP1/WAF1] and CCND1; Gu et al., 2012). More recently, we showed that hPer2 acts on hp53 by controlling its stability and activity in unstressed conditions (Gotoh et al., 2014) and hypothesized that exposure to genotoxic stress triggers a rapid, hp53-mediated transcriptional checkpoint response by releasing hp53 from a preformed, nucleus-localized, hPer2/hp53 stable endogenous complex. Our findings establish the spatial distribution of the various hPer2, hp53, and Mdm2 complexes, the need for hPer2 association with hp53 to maintain basal levels of this protein, and the relevance of hPer2/hp53 dissociation for an effective hp53-mediated DNA damage checkpoint response.
RESULTS
Spatial organization and shuttling of circadian molecules are common themes when it comes to understanding how oscillations are generated and sustained in biological clock systems. They are also common subjects when considering how sensor components segregate signals in response to stress conditions at various points in the cell cycle. In light of our previous findings in which the circadian factor hPer2 directly binds the tumor suppressor and checkpoint component hp53 in unstressed cells, we now ask whether the complex's spatial organization, as well as association, is critical for an effective hp53-mediated transcriptional response under stress conditions.
Subcellular distribution of hPer2/hp53 complexes
To assess whether functional compartmentalization of the hp53/hPer2 complex occurs, we performed cell fractionation experiments and monitored the formation of polyubiquitinated hp53 complexes and hPer2 binding in extracts from cells treated with proteasome inhibitors (Figure 1, A and B). Human colon carcinoma HCT-116 (HCT116) cells (p53+/+) were cotransfected with myc-hp53 and FLAG-hPer2 or empty vector (–) and treated with MG132 (+MG132) or vehicle (–MG132). Total, cytosolic, and nuclear fractions were analyzed for hp53/hPer2 complex formation and the presence of polyubiquitinated hp53. Input levels of myc-hp53 and FLAG-hPer2, as well as of endogenous hp53 and hPer2 levels, were monitored in each fraction (Supplemental Figure S1, A and B) and normalized to those of myc-hp53 for the experiment shown in Figure 1A. Unlike nontreated MG132 cells, immunoprecipitation of myc-hp53 showed the presence of stable myc-hp53(Ub)n forms in total (T) and cytosolic (C) fractions and, to a lesser extent, in the nuclear (N) fraction of samples treated with MG132 (Figure 1A, bottom, lanes 1–3 vs. lanes 7–9). These results most likely represent the effect of proteasome inhibitors in preserving the myc-hp53 ubiquitinated complexes and accumulation in the cytosol as the preferred site for their ubiquitin-mediated degradation.
FIGURE 1:

Distribution of the hPer2/hp53 complex among cellular compartments. (A) HCT116 cells were transfected with pCS2+myc-hp53 and either pCS2+FLAG-hPer2 (+) or empty vector (–) and maintained in complete medium for 20 h before adding or not (control; –MG132) MG132 (50 μM) and ubiquitin aldehyde (5 nM). Cells were maintained an additional 4 h before harvesting. Lysates (6.4 × 105 cells) were used to prepare the cytosolic (C) and total (T) fractions, whereas 32 × 105 cells were used for nuclear (N) preparation. Total, cytosolic, and nuclear extracts were incubated with α-myc antibody and protein A beads in NP40 lysis buffer containing MG132 and ubiquitin aldehyde. Washed samples were analyzed by immunoblotting using specific antibodies. Ubiquitinated myc-hp53 complexes (myc-hp53(Ub)n) are indicated between brackets. Immunoblot data from a single experiment repeated three times with similar results. Quantification of the sample's ubiquitinated signal was performed using ImageJ, version 1.45 (National Institutes of Health software package; Schneider et al., 2012; bar graph). (B) HCT116 lysates (2.8 × 105 cells for T/C; 14 × 105 cells for N) from pCS2+FLAG-Mdm2– and pCS2+myc-hPer2– cotransfected cells treated or not (–MG132) with 50 μM MG132 and 5 nM ubiquitin aldehyde were immunoprecipitated using α-FLAG and protein A beads and analyzed by immunoblotting for endogenous hp53 (bottom) and myc- and FLAG-expressed proteins (top and middle, respectively).
Significantly, overexpression of FLAG-hPer2 followed by myc-hp53 binding abrogated the formation of hp53(Ub)n in both nuclear and cytosolic fractions (Figure 1A, bottom, lanes 7–9 vs. lanes 10–12), in agreement with the subcellular distribution of hPer2 in those compartments (Figure 1A, top, lanes 10–12, and Supplemental Figure S1B) and the proposed role of hPer2 in modulating hp53 polyubiquitination. These results establish a physical and functional presence of hp53/hPer2 complex in the cytosol and nucleus. Further support comes from results shown in Figure 1A (lanes 1–6), in which studies similar to the ones described earlier were conducted in the absence of MG132 (–MG132), allowing the proteasomal machinery to be fully functional. As a result, hp53(Ub)n forms were undetected (lanes 1–6), and only trace amounts of hPer2 were associated with myc-hp53 in immunoprecipitated nuclear samples (Figure 1A, top and bottom, lane 6) even when input amounts were comparable to those used in the +MG132 experiment (Supplemental Figure S1B, top, lanes 4–6 vs. lanes 10–12). As previously shown, Per2 is degraded by the proteasomal pathway unless associated (Yagita et al., 2002), and thus the level of FLAG-hPer2 was expected to be low in both the cytosolic and nuclear compartments in the absence of MG132 but detectable if associated to a protein counterpart. In sum, our data establish that 1) the hPer2/hp53 complex can be found in both the nuclear and cytosolic subcellular compartments and 2) physical interaction between hp53 and hPer2 might influence hPer2 degradation as well.
Next we asked whether Mdm2 associates with the hPer2/hp53 once in the nucleus. To answer this question, we cotransfected HCT116 cells with FLAG-Mdm2 and myc-hPer2 and looked for formation of the trimeric complex with endogenous hp53 (Figure 1B). Transfected cells were incubated in the absence (–) or presence (+) of MG132 and subjected to cellular fractionation. Consistent with their endogenous distribution, input myc-hPer2 was found in both cytosolic and nuclear fractions (Supplemental Figure S1C, lanes 2 and 3; Gotoh et al., 2014), and FLAG-Mdm2 preferably distributed in the nuclear fraction (Supplemental Figure S1C, lane 2), although some signal was detected in the cytosol, most likely as a result of MG132 addition and inhibition of self-ubiquitination (Supplemental Figure S1C, lane 5). Consequently, endogenous hp53 levels were also increased in MG132-treated samples (Supplemental Figure S1C, lanes 2 and 3 vs. lanes 5 and 6).
Analysis of α-FLAG–immunoprecipitated samples shows that FLAG-Mdm2 associated with myc-hPer2 and hp53 in the nuclear fraction of cells treated with MG132, whereas the trimeric complex was less conspicuous but still detectable in the absence of the proteasome inhibitor (Figure 1B, lane 3 vs. lane 6). Of interest, myc-hPer2 did not seem to be associated with the Mdm2/hp53 complex in the cytosolic fraction (Figure 1B, lanes 2 and 5) but likely formed a two-component complex with hp53 in the absence of ubiquitination (Figure 1A, lanes 11 and 12). Because the input levels of myc-hPer2 were comparatively similar in –MG132 and +MG132 samples (Supplemental Figure S1C, top), one might speculate that the trace levels of hPer2 associated with the FLAG-Mdm2/hp53 complex within the nuclear fraction in MG132-untreated cells (Figure 1B, lane 3) were the result of either hPer2 being a substrate of Mdm2-mediated ubiquitination once the complex was in place or the action of an additional nuclear E3-ligase for hPer2 (i.e., β-TRCP; Ohsaki et al., 2008). Further experiments need to be done to test both possibilities. As expected, FLAG-Mdm2 was detected in association with hp53 in the cytosolic compartment, in agreement with previous findings (Freedman and Levine, 1998), and relative amounts of FLAG-Mdm2/hp53 complex increased in MG132-treated samples (Figure 1B, lane 2 vs. lane 5). In summary, our data establish that hPer2/hp53/Mdm2 exists only in the nucleus and that the hPer2/hp53 and hp53/Mdm2 complexes can be readily detected in both nuclear and cytosolic fractions.
Functional insights into the hPer2/hp53 complex
We previously identified the hp53/hPer2 complex within the nuclear compartment (Figure 1). Thus we next asked whether hp53 transcriptional activity was compromised when bound to hPer2. To answer this question, we first needed to generate a form of the hp53/hPer2 complex that would be constitutively bound and not dissociate once formed in cells. For this task, we generated a chimera set in which either hPer2 full-length or glutathione S-transferase was cloned downstream of hp53 and immediately after a flexible linker (called hp53(ch)hPer2 and hp53(ch)GST hereafter, respectively; Figure 2A). The rationale behind this design is that hPer2 would interact with hp53 by flipping over and forming a stable, covalently bound complex through their domain interactions, which would be nondissociable as seen using biofluorescence complementation assays (unpublished data). As a result, we expect to find the ubiquitination of hp53 compromised without altering its compartmentalization. Therefore hp53(ch)hPer2 would be an adequate tool to evaluate the effect of a nondissociable hp53/hPer2 complex in hp53 downstream gene activation.
FIGURE 2:

In vitro ubiquitination of hp53 is compromised when stably bound to hPer2. (A) Schematic representation of all chimeras designed for this study. Constructs were 5′ FLAG-tagged, myc-tagged, or untagged upstream of hp53, followed by downstream cloning of GST, hPer2, or hPer2(356-574/683-872) open reading frames. A linker encoding for six Gly was inserted between both the hp53 and hPer2 genes and the two hPer2-coding fragments. (B) In vitro transcribed and translated FLAG-hp53, FLAG-hp53(ch)hPer2(356-574/683-872), FLAG-hp53(ch)GST, myc-Mdm2, and myc-hPer2 proteins were used for ubiquitination experiments. When indicated, myc-hPer2 and either FLAG-hp53 or FLAG-hp53(ch)GST were preincubated; thus the complex was formed before adding myc-Mdm2. For the FLAG-hp53(ch)hPer2(356-574/683-872) chimera, the translated protein and myc-Mdm2 were incubated together before the ubiquitination reaction took place. Ubiquitination was carried out as described in Materials and Methods. FLAG-tagged proteins were immunoprecipitated with α-FLAG/protein A beads and blotted using α-ubiquitin antibody. Membranes were then stripped and reprobed with α-FLAG and -myc antibodies to detect complex bound proteins. Asterisk indicates IgG heavy chain. Immunoblot data from a single experiment repeated three times with similar results. Quantification of the sample's ubiquitinated signal was performed using ImageJ, version 1.45 (bar graph). Statistical comparisons were evaluated by t test. #p ≤ 0.05; ##p < 0.005.
To further functionally validate the hp53(ch)hPer2 chimera, we evaluated the ubiquitination status of hp53 in the complex (Figure 2B). In vitro ubiquitination assays were performed using recombinantly expressed proteins (FLAG-hp53, FLAG-hp53(ch)GST, FLAG-hp53(ch)hPer2(356-574/683-872), and FLAG-hp53(ch)hPer2) preincubated, or not (–), with myc-hPer2, followed by myc-Mdm2 addition. Once the ubiquitination reaction took place, hp53 and its chimera proteins were immunoprecipitated and analyzed for ubiquitin incorporation by immunoblotting (Figure 2B and Supplemental Figure S2). Results show that FLAG-hp53 and FLAG-hp53(ch)GST behave similarly with respect to overall ubiquitination status when prebound to hPer2 and compared with their controls in the absence of hPer2 addition (Figure 2B, bottom, lane 7 vs. lane 8 and lane 2 vs. lane 3). Consistent with its role as a stable complex, ubiquitination of FLAG-hp53(ch)hPer2(356-574/683-872) and FLAG-hp53(ch)hPer2 closely resembled the basal signal obtained with just hp53 (or hp53(ch)GST) when preincubated with hPer2 (Figure 2B, bottom, lane 5 vs. lanes 3 and 8; Supplemental Figure S2, lane 3 vs. lane 5), further validating the chimera as an nondissociable hp53/hPer2 complex mimetic. In all cases, binding components were confirmed by immunoblotting and are shown in Figure 2B (top and middle).
Next we compared the localization of hp53(ch)hPer2 with that of myc-tagged hp53 and its chimera form (hp53(ch)GST) in human non–small cell lung carcinoma-1299 (H1299) cells (p53-null). Accordingly, we transfected H1299 cells with the various constructs and monitored their subcellular localization by fluorescence microscopy. Representative pictures are shown in Figure 3 and Supplemental Figure S3. In most normal cells, p53 is cytoplasmic; however, it is primarily located in the nucleus in rapidly growing normal cells, transformed cells, and various tumor cells, including those from breast and colon (Liang and Clarke, 2001). In agreement, myc-hp53 and myc-hp53(ch)GST were largely detected in the nuclear compartment (Figure 3 and Supplemental Figure S3, i and ii). The hp53(ch)hPer2 chimera was detected in both cytosolic and nuclear compartments (Figure 3 and Supplemental Figure S3, vi), in agreement with hPer2/hp53 distribution as in Figure 1A. Moreover, hp53(ch)hPer2 was recognized by α-p53 and α-Per2 antibodies targeting conformational native epitopes in both proteins, suggesting that the integrity of the folding in the complex was not compromised. Comparable results were also obtained with tagged forms of the chimera complex (unpublished data). Profile plotting of signal intensity along cross sections of cells transfected with the various constructs unambiguously determined their distribution and levels of expression (Supplemental Figure S3).
FIGURE 3:

Relevance of hPer2-interacting domains for hp53 localization. H1299 cells were transfected with myc-tagged forms of hp53, hp53(ch)GST, hp53(ch)hPer2(356-574/683-872), NLS-hp53(ch)GST, and NLS-hp53(ch)hPer2(356-574/683-872) (i–v) or the untagged form of hp53(ch)hPer2 (vi). Proteins were visualized by confocal microscopy using α-myc-Cy3– conjugated primary antibody (i–v) or α-p53 and -Per2 primary antibodies and α-mouse Cy3– and α-rabbit Alexa Fluor 488 (Life Technologies)–conjugated secondary antibodies, respectively (vi). Actin fibers and DNA were stained with phalloidin Alexa Fluor 488 and Syto60 (Life Technologies), respectively. Merge images (right) were from protein staining, phalloidin, and DNA (i–v) and protein and DNA (vi). A Nikon ECLIPSE TE2000-E microscope and NIS-Elements AR 3.0 software were used to record images. Scale bars, 10 μm.
Localization of p53 in the nucleus is the result of the presence of three monopartite nuclear localization signals (NLSs) located within the C-terminus of p53, with NLS1 (316PQPKKKP322) being the most active (Shaulsky et al., 1990). Remarkably, it is the C-terminus of hp53 that interacts with hPer2, and, thus, it seems reasonable that hp53 NLSs would be occluded at the interface of the two interacting molecules. Moreover, the strongest NLS described in hPer2 so far is a bipartite sequence located between residues 778 and 794 (Yagita et al., 2002) that in hPer2 directly interacts with hp53 and most likely would not be exposed. Therefore we ask what element(s) actually drive the hp53(ch)hPer2 complex to the nucleus. To answer this question, we generated additional chimeras of hp53 in which the hPer2-interacting fragments 356–574 and 683–872 (which contain the hPer2 NLS) were cloned downstream of hp53 (contains all NLSs) and were separated by flexible linkers (called hp53(ch)hPer2(356-574/683-872) hereafter; Figure 2A). As shown in Figure 3 and Supplemental Figure S3iii, hp53(ch)hPer2(356-574/683-872) failed to localize in the nucleus of H1299 cells and readily accumulated in the cytoplasmic compartment, suggesting that neither NLS was functional and that hp53(ch)hPer2 translocation had most likely been driven by a yet-to-be identified NLS in hPer2 or that there is an additional cargo protein associated with the complex involved in translocation. To evaluate this possibility, we engineered an NLS sequence, in this case from SV40 (Kalderon et al., 1984), inserted it upstream of hp53(ch)hPer2(356-574/683-872), and monitored its localization in H1299 cells. Of interest, the sole addition of this NLS resulted in hp53(ch)hPer2(356-574/683-872) shuttling to the nucleus, supporting the existence of a yet-to-be identified component related to hPer2 that provides a signal for transportation (Figure 3 and Supplemental Figure S3, iii vs. v). Finally, we showed that hCry1 was able to bind hPer2/hp53 when tested in vitro, a result that points to the existence of multiple regulators of diverse biochemical nature. Binding of hCry1 could be explained as the result of its interaction with the C-terminus of hPer2 (Yagita et al., 2002), a region that does not overlap with the binding site mapped for hp53 (Supplemental Figure S4A; Gotoh et al., 2014). Despite this result, this last finding needs to be taken with caution, as we failed to find compelling evidence to prove the existence of an endogenous hPer2/hCry1/hp53 complex in cells, a result that might reflect its transient nature or simply expose the low efficiency of the immunoprecipitating antibodies that were used.
hp53 transcriptional activity is compromised when bound to hPer2
First, we investigated whether shuttling of the hPer2/hp53 complex from the cytosol to the nucleus was sufficient to trigger hp53-mediated downstream signaling (Figure 4 and Supplemental Figure S4B). Accordingly, we transfected H1299 cells (p53-null) with various chimera constructs, as well as with wild-type hp53, and monitored the expression of the cyclin-dependent inhibitor human p21WAF1/CIP1 (hp21WAF1/CIP1), a transcriptional target of p53, by immunoblotting. Transfection of cells with hp53, FLAG-hp53(ch)GST, and FLAG-NLS-hp53(ch)GST resulted in localization of all three proteins in the nucleus (Figure 3, i, ii, and iv) and hp21WAF1/CIP1 expression (Figure 4, lanes 2, 3, and 5, and Supplemental Figure S4B, lanes 3 and 4). As expected, transfection of H1299 with FLAG-hp53(ch)hPer2(356-574/683-872) did not lead to hp21WAF1/CIP1 expression, as the complex remained sequestered in the cytosol (Figures 3iii and 4, lane 4). Of interest, and despite the addition of an NLS to favor shuttling, transfection of cells with either FLAG-NLS-hp53(ch)hPer2(356-574/683-872) (Figure 4, lane 6) or FLAG-hp53(ch)hPer2 (Supplemental Figure S4B, lane 6) did not result in hp21WAF1/CIP1 expression. These findings show that other events besides translocation are involved in triggering hPer2-mediated hp53 downstream signaling.
FIGURE 4:
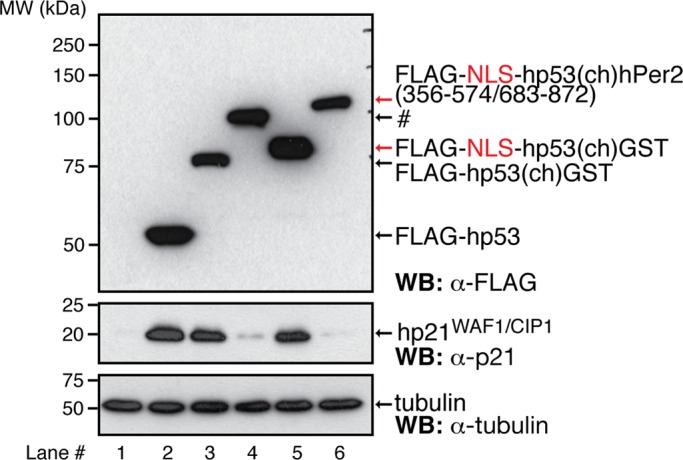
Binding of hPer2 to hp53 prevents hp21 from being expressed. H1299 cells were transfected with pCS2+FLAG-hp53, -hp53(ch)GST, -NLS-hp53(ch)GST, -hp53(ch)hPer2(356-574/683-872) (labeled #), or -NLS-hp53(ch)hPer2(356-574/683-872) and harvested 24 h later. Cell lysates (∼40 μg) were resolved by SDS–PAGE and recombinant (top left and right) and endogenous proteins (middle and bottom left and right) detected by immunoblotting using α-p21, -FLAG, and -tubulin antibodies.
To further investigate the contribution of hPer2 in p53-mediated signaling, we tested the ability of hPer2 to modulate the reporter activity of a p53-responsive promoter CDKN1a (called hp21WAF1/CIP1-luc hereafter) in the context of H1299 cells cotransfected with various hp53 and hPer2 chimeras when exposed, or not, to genotoxic stimuli (Figure 5, A and B). Chimeric transfections did not alter cell viability (Supplemental Figure S5). Unlike cells transfected with FLAG-hp53(ch)hPer2, H1299 cells show a roughly fourfold increase in hp21WAF1/CIP1-luc activity compared with empty vector–transfected cells when expressing comparative levels of either FLAG-hp53 or hp53(ch)GST in the absence of radiation (–γ-IR; Figure 5B and Supplemental Figure S6A). As expected, H1299 cells transfected with either FLAG-hp53 or hp53(ch)GST and exposed to genotoxic stress (+γ-IR) showed a greater enhancement of hp21WAF1/CIP1 activity (∼50% more than similar untreated cells) that is down-regulated to basal levels when cells were transfected with FLAG-hp53(ch)hPer2 instead (Figure 5B and Supplemental Figure S6A). Moreover, this result seems to be independent of the radiation dose, as shown in Supplemental Figure S7. In accordance, hp21WAF1/CIP1 activity remained low in FLAG-hp53(ch)hPer2(356-574/683-872)–transfected cells despite overexpression of the recombinant proteins and relocalization of the FLAG-NLS-hp53(ch)hPer2(356-574/683-872) chimera to the nucleus (Figure 3 and Supplemental Figures S4B, S6B, and S8E). Collectively these results suggest that when bound to hPer2, hp53 is unable to perform its transcriptional function despite the chimera being localized in the same cellular compartment (Figures 3 and 5B).
FIGURE 5:

The hPer2 protein maintains hp53 transcriptionally inactive when complexed. (A) Schematic representation of the approach followed. In all cases, H1299 cells were harvested after transfection and before irradiation (γ-IR, 5 Gy; t = 0). (B, C) Cells harvested 2 h after irradiation. (D) Time points taken at 2-h intervals after irradiation. (B) H1299 cells were cotransfected with the reporter hp21WAF1/CIP1-luc construct, pCS2+FLAG-hp53, pCS2+FLAG-hp53(ch)GST, pCS2+FLAG-hp53(ch)hPer2, or EV, with pCMV-β-gal as internal control. Extracts from cells treated (+γ-IR) or not (–γ-IR) with radiation were assayed for luciferase and β-galactosidase activities. The experiment was replicated thrice; error bars indicate SEM, and data were evaluated by ANOVA using Bonferroni posthoc test (SPSS). ###p ≤ 0.001. (C) H1299 cells were transfected with either pCS2+FLAG-hp53 or pCS2+FLAG-hp53(ch)hPer2 and treated (+γ-IR) or not (–γ-IR) with radiation. The qRT-PCR data were normalized to the levels of expression in untreated FLAG-hp53–transfected cells (–γ-IR). Data are presented as the mean ± SEM from three independent experiments performed in triplicate. Statistical comparisons were evaluated by ANOVA using Bonferroni or Games-Howell posthoc analyses when needed (SPSS). NS, not significant; #p ≤ 0.05; ##p ≤ 0.01; ###p ≤ 0.001. (D) H1299 cells were transfected with pCS2+FLAG-hp53, pCS2+FLAG-hp53(ch)GST, or pCS2+FLAG-hp53(ch)hPer2 and treated (+γ-IR) or not (–γ-IR) with radiation as indicated in A and Materials and Methods. Aliquots of lysates taken at different times (20 μg) were blotted using specific antibodies (α-Chk1 and α-p21 for endogenous Chk1 kinase and hp21WAF1/CIP1, respectively; α-FLAG for FLAG-hp53, FLAG-hp53(ch)GST, and hp53(ch)hPer2; α-Chk1-Ser345 for phosphorylation in Ser345 of endogenous Chk1; and α-p53-Ser15 for phosphorylation in Ser15 in FLAG-hp53, FLAG-hp53(ch)GST, and hp53(ch)hPer2). Tubulin was used as a loading control (bottom). Asterisk indicates a nonspecific signal.
We then expanded our studies to examine the transcriptional effect of hp53(ch)hPer2 chimera on other hp53 target genes (i.e., SFN [encodes 14-3-3σ], BAX, MYC, CDKN1a, GADD45α [encodes growth arrest and DNA damage–inducible protein 45α, Gadd45α]) by measuring mRNA levels using quantitative reverse-transcription PCR (qRT-PCR). The rationale behind this experiment is that hp53(ch)hPer2 would counteract the effect of hp53 (or hp53(ch)GST) in gene expression due to its incapability of dissociating hPer2 from hp53, thus acting as a dominant-negative complex. Moreover, we expect that, whereas genotoxic stress (+γ-IR) would exacerbate specific p53-mediated gene expression, hp53(ch)hPer2 would continue to maintain a negative effect despite the stimuli.
As shown in Figure 5C, untreated H1299 cells (–γ-IR) transfected with hp53(ch)hPer2 showed down-regulation of both SFN and CDKN1a expression relative to the values obtained for hp53 transfection. Conversely, MYC exhibited positive regulation, whereas BAX and GADD45α remained largely unchanged. These results are in agreement with the positive role of hp53 in transcription of SFN and CDKN1a and its repressor role toward MYC. Three independent controls were simultaneously tested for all analyzed genes and incorporated as supplementary material (Supplemental Figure S8, A–C). There were no significant differences in gene expression when 1) empty vector (EV)–transfected cells were exposed, or not, to γ-IR, ruling out off-target effects (Supplemental Figure S8A), 2) cells transfected with either FLAG-hp53(ch)GST or FLAG-hp53 showed comparative levels of expression for all transcripts in the absence of radiation (Supplemental Figure S8B), and 3) under genotoxic stress (Supplemental Figure S8C). In all cases, gene expression levels were relative to those obtained by transfecting FLAG-hp53.
Next H1299 cells were exposed to genotoxic stress (+γ-IR) and transcripts amplified and compared relative to that of FLAG-hp53 (–γ-IR) (Figure 5C). As expected, both SFN and CDKN1a transcripts increased in response to radiation, confirming activation of the hp53 pathway (Figure 5C, white vs. black bars). The relevance of hPer2 dissociation for hp53 activity was evident when similar transcript levels were measured in extracts from hp53(ch)hPer2-transfected cells and were found significantly down-regulated (Figure 5C, black vs. black dashed bars). In other cases, neither hp53 nor hp53(ch)hPer2 transfections affected the expression of hp53 target genes (i.e., GADD45α and BAX) independently of whether cells were exposed or not and collected shortly after genotoxic stress (–/+ γ-IR). Finally, MYC expression was upregulated, approximately threefold, in hp53(ch)hPer2-transfected H1299 cells relative to FLAG-hp53, opposing its repressor role on MYC expression. Similar experiments were performed using FLAG-hp53(ch)hPer2(356-574/683-872), a chimera construct that effectively blocks hp53 ubiquitination (Supplemental Figure S8D) but remains secluded in the cytosolic compartment unless an NLS is added (Figure 3). As expected, transcript analyses of FLAG-hp53(ch)hPer2(356-574/683-872)–H1299 transfected cells showed a pattern of expression that resembles the one observed when cells were transfected with hp53(ch)hPer2 and opposite to that resulting from hp53 transfection (Supplemental Figure S8D). Taken together, our data support a model in which hp53 binding to hPer2 is required for maintaining basal levels of hp53 in the nucleus in an inactive transcriptional state by forming a stable complex. Moreover, our findings imply that unbound hp53 from hPer2/hp53 is an absolute requirement to signaling downstream in response to DNA damage.
The hPer2 protein directly influences checkpoint signaling
In an attempt to gain better understanding of the role of hPer2 in hp53-mediated checkpoint signaling, we investigated the integrity of the checkpoint pathway upstream of hp53 and for hp21WAF1/CIP1 expression in various transfected scenarios in irradiated H1299 cells (Figure 5D and Supplemental Figure S8F). Phosphorylation of downstream ATM/ATR kinase (i.e., Ser345 in Chk1) precedes p53 stabilization and is a marker of checkpoint activation (for review, see Meek, 2009). Therefore H1299 cells were transfected with EV, FLAG-hp53, FLAG-hp53(ch)GST, FLAG-hp53(ch)hPer2, or FLAG-hp53(ch)hPer2(356-574/683-872) and exposed, or not, to genotoxic stress. Extracts were analyzed for endogenous active forms of hChk1 and hp53 and levels of hp21WAF/CIP1 (Figure 5D and Supplemental Figure S8F). First, we monitored hChk1 protein in all samples to find steady levels of expression throughout the time course analyzed in both exposed and transfected samples (Figure 5D and Supplemental Figure S8F). In addition, activation of Chk1 by phosphorylation in Ser345 was detected as early as 2 h postirradiation in exposed samples from transfected cells (+γ-IR), thus confirming both checkpoint activation and the integrity of the p53 upstream signal cascade in all cases (Figure 5D and Supplemental Figure S8F). On the other hand, phosphorylation of Ser15 of hp53 was detected exclusively in extracts from FLAG-hp53 and FLAG-hp53(ch)GST cells and in response to radiation (Figure 5D), suggesting that, if present, hp53 would get activated. Of interest, neither FLAG-hp53(ch)hPer2 nor FLAG-hp53(ch)hPer2(356-574/683-872) was phosphorylated on Ser15 of the hp53 portion of the chimera, implying that this site might not be accessible when the complex with hPer2 is formed (Figure 5D and Supplemental Figure S8F). Together our present data and previous studies support a model in which binding of hPer2 to hp53 modulates its stability and compromises hp53 function (Figure 6; Gotoh et al., 2014).
FIGURE 6:

Proposed model of hPer2 and hp53 interaction and function. Newly synthesized hp53, hPer2, and Cry localize in the cytosolic compartment where the Cry/hPer2/CKIε/δ (pool I) is formed, and hPer2 is incorporated in one or more complexes that constitutes pool II. Pool I: As extensively reviewed, cytosolic hPer2 is phosphorylated by casein kinase I ε/δ (CKIε/δ) and initially degraded by the ubiquitin proteasome pathway (Ko and Takahashi, 2006). Later, Cry accumulates and associates with hPer2/CKIε/δ, and this complex translocates to the nucleus, where Cry disrupts the Clock/Bmal1-associated transcriptional complex, resulting in inhibition of CRY, PER, and REB-ERVα and derepression of BMAL1 transcription and modulation of the expression of other clock-controlled genes (shown as “circadian circuit” for simplicity). Pool II: The hPer2 protein associates with cytosolic hp53, forming a stable complex (Gotoh et al., 2014) that translocates to the nuclear compartment and keeps hp53 in a functionally inactive but stable state, ensuring that basal levels of hp53 exist (“priming state”). This heterodimer eventually incorporates Mdm2, forming a trimeric and stable Mdm2/hp53/hPer2 complex. In response to, for example, a genotoxic stress (labeled as γ-IR in the cartoon), the trimeric complex disassembles by a yet-unknown mechanism, which leads to release of hp53 and downstream activation of genes involved in cell cycle arrest and DNA repair. An amplification loop exists in which hPer2, alone or in association with an unidentified partner, transcriptionally activates TP53, further sustaining the hp53-mediated response (boxed in orange). Alternatively, cytosolic hp53 enters the nucleus, where it is targeted by Mdm2 and either polyubiquitinylated and degraded back into the cytosol or monoubiquitinated and translocated to a different compartment, as has been described (for review, see Kruse and Gu, 2009).
DISCUSSION
The connection of circadian components to various aspects of cell division promises a better understanding of how cells sense and respond to changes in environmental conditions. At the transcriptional level, the integration of clock core components to multiple signaling networks is evident from studies carried out using genome-wide RNA interference screening analysis and high-throughput microarray technologies (Duffield et al., 2002; Panda et al., 2002; Mullenders et al., 2009; Zhang et al., 2009). Indeed, these studies have thoroughly identified genes whose knockdown directly modulates the circadian clock and others whose expression is controlled by clock core components (Duffield et al., 2002; Panda et al., 2002; Mullenders et al., 2009; Zhang et al., 2009). Regardless of the analysis platform, it is clear that cell cycle and circadian components are interlocked through genetic, protein interaction, and physiological mechanisms and that perturbation of the circadian system affects cell growth and proliferation (Fu et al., 2002; Matsuo et al., 2003; Miller et al., 2007). Accordingly, circadian-regulated cell cycle genes such as CCND1, WEE1, MYC, GADD45α, and CDKN1a are known to show periodic patterns of expression in a 24-h cycle, a finding further strengthened by the identification of Per2 and Bmal1 as direct modulators of WEE1, MYC, and CDKN1a expression (Grundschober et al., 2001; Fu et al., 2002; Matsuo et al., 2003; Grechez-Cassiau et al., 2008). Further theoretical analyses of protein interaction networks helped place gene products that are directly or indirectly associated to known clock components, including various cell cycle modulators, in a global interactome map, which has provided clues about the many aspects of cell physiology directly regulated by the clock (for review, see Zhang and Kay, 2010).
An initial question relates to the subcellular localization of hp53, hPer2, and Mdm2 and their respective complexes. Our studies establish the presence of the hp53/hPer2 complex in both cytosolic and nuclear compartments, whereas the presence of the trimeric complex with Mdm2 seems to be restricted to the nucleus (Figure 1 and Supplemental Figure S1). Our chimera experiments provide evidence of distinct roles of hPer2 in both subcellular compartments. We believe that, when in the cytosol, hPer2 provides the signal needed for the hp53/hPer2 complex to translocate to the nucleus, whereas in the nucleus, hPer2 helps to maintain hp53 in a stable, inactive form until a stimulus (either external or internal) is applied to the biological system (Figure 6). This model is largely supported by our previous work (Gotoh et al., 2014) and experiments showing that 1) blockage of the hp53 C-terminus NLSs by binding to hPer2 domains (hp53(ch)hPer2(356-574/683-872)), but not full-length hPer2 (hp53(ch)hPer2), prevents hp53 from nuclear localization (Figure 3, iii and vi, and Supplemental Figure S3), 2) addition of NLS to the same construct relocalizes the chimera protein in the nucleus (Figure 3v and Supplemental Figure S3), 3) only a constitutive hp53/hPer2-bound chimera (hp53(ch)hPer2) localizes in both compartments (Figure 3vi and Supplemental Figure S3), suggesting a role for hPer2 in hp53 translocation, and, finally, 4) constitutive association of hPer2 to hp53 in the form of [hp53(ch)hPer2] prevents hp53 from exerting its transcriptional activity in cells even in the presence of genotoxic stress (Figure 5 and Supplemental Figures S7 and S8). At present, it cannot be formally excluded that hPer2, hCry1, and hp53 or a fraction of these molecules could, a priori, form a trimeric complex in the cell. This is largely due to our finding that in vitro– expressed proteins were able to form multiple stable complexes (Supplemental Figure S4A). In addition, rhythmic hPer2 heterodimerizes with hCry1 through feedback to the nucleus and sustains oscillations. Thus, it is plausible that two pools of hPer2 might exist—one bound to hp53 and a separate pool that associates with its circadian counterpart. Nevertheless, the two pools of hPer2 might not be exclusive and might serve to assign distinct functions to hPer2 in each of these pools.
Once the hp53/hPer2 complex is in the nucleus, Mdm2 binds to the N-terminus domain of hp53, forming a trimeric complex in which hp53 ubiquitination does not take place (Figures 1B and 6; Gotoh et al., 2014). This is in addition to the already established canonical p53 pathway, in which nuclear trafficking of hp53 is followed by Mdm2 binding and either monoubiquitination and nuclear export or p53 polyubiquitination and proteasomal degradation (incorporated in Figure 6; Honda et al., 1997; Li et al., 2003). We speculate that, under physiological conditions, hp53 remains an inactive component of the hp53/hPer2/Mdm2 and hp53/hPer2 complexes and is therefore unable to modulate the expression of downstream target genes (Figures 5 and 6 and Supplemental Figure S4).
Regulation of stress-mediated p53 activation has largely relied on a well-established model in which p53 stabilization precedes activation. Stabilization is primarily accomplished via the release of p53 from its interaction with Mdm2 by a mechanism that involves checkpoint activation, p53 phosphorylation, and inhibition of p53's interaction with Mdm2 (for review, see Meek, 2009), thus preventing p53 ubiquitination and proteasomal degradation (for review, see Kruse and Gu, 2009). Once stabilized, p53 is further posttranslationally modified, and its tetrameric form binds to DNA at specific high- and low-affinity p53 response elements, which regulates the expression of target genes via the recruitment of coactivators and corepressors. Despite being capable of providing an explanation for the many modes by which p53 controls gene expression, the canonical model is not sufficient to explain more recent genetic studies. Therefore further refinement has been introduced. As a result, the most recently updated model takes into consideration the many stress types that converge in p53, the various posttranslational modifications to which p53 is subjected (i.e., acetylation, sumoylation), and the tissue-specific function of p53. The end result is a model in which regulatory redundancy among posttranslational modifications is a common theme and serves to permit p53 to sense different signals and intensities, which allows for a signaling response that is properly modulated and tissue specific (for review, see Vousden, 2009). As expected, there is also a myriad of p53 binding partners, whose interactions exert selective influences on p53 target genes, ranging from Mdm2 and MdmX negative regulators to orphan receptors, and for which a wealth of studies already exists (for review, see Vousden, 2009). Thus some common steps exist in the regulation of p53 activity (stabilization, modification, protein interaction, and promoter-specific activation), but many players differentially intervene in each step to influence the outcome of the response.
Questions arise about how free hp53 accumulates in response to radiation and what the role of hPer2 is in this event. Although we cannot provide a definitive mechanism for how the transition from hPer2 bound to hPer2 free leads to hp53 accumulation and transcriptional response (Figure 6, dashed arrow), it does not seem to be globally associated with hPer2 dissociation and shuttling, as is the case for HCT116 cells (Supplemental Figure S10A), but, more likely, to be specific for certain cell types, as is for HEK293 cells (Supplemental Figure S10B). In addition, it also seems linked to posttranslational events, as we found that hPer2 is associated with checkpoint kinases in response to radiation (unpublished data). Finally, we cannot rule out the existence of additional players in this response, including other circadian factors that may act by sequestering hPer2 away from (or “pulling hPer2 out of”) the complex with hp53 when cells are exposed to a genotoxic stimulus. As a result, our model stresses the need for the existence of an hp53 tetramer that is free of hPer2 in order for its transcriptional activity to take place (Figures 5 and 6 and Supplemental Figure S4); nonetheless, more in-depth studies need to be performed to uncover the exact mechanism.
An additional component in the model refers to the regulation of TP53 (encodes p53) transcription by hPer2, a process we propose that acts as a form of amplification loop that helps sustain the initial hPer2/hp53-mediated response (Supplemental Figure S9; Gotoh et al., 2014). The relevance of hPer2-mediated TP53 transcription is exposed when the expression of hp53 downstream target genes in the context of hPer2 overexpression is analyzed (Supplemental Figure S9A; Gotoh et al., 2014). Based on our proposed model, overexpression of hPer2 should lead to stabilization of hp53 and inactivation of its transcriptional activity as a result of hPer2/hp53 complex formation. However, Gotoh et al. (2014) reported that this is not the case and that whereas hp53 is stabilized by hPer2 overexpression, transcription of the hp53 downstream target genes remains largely unaltered. A way to reconcile these data is to monitor TP53 transcription in the context of hPer2 overexpression (Supplemental Figure S9, A and B; Gotoh et al., 2014). Accordingly, our data show that whereas cells expressing hPer2 (HCT116 cell, p53+/+) up-regulate TP53 (Supplemental Figure S9A), the pool of hp53 complexed to hPer2 is only a fraction of the total level of hp53 in the cell, and thus “free” hp53 still exists and is able to transcriptionally activate downstream target genes (Supplemental Figure S9, A and B). This concept is further validated in H1299 cells (p53-null), in which hPer2 overexpression does not result in CDKN1a expression but FLAG-hp53 and -hPer2 cotransfection does when the level of hp53 is greater than that of hPer2 (Supplemental Figure S9C). Finally, an interesting aspect of our work is the level within the checkpoint hierarchy at which hPer2 intersects this pathway, the hp53 node. Because cancer development largely relies on sustained inactivation of the p53 pathway, the existence of factors that respond to environmental signals and influence hp53 stability encourages the search for unconventional drug targets and would certainly provide a new direction as to when and how to treat various cancers.
Our results, which establish a connection between the circadian regulatory system and the DNA-damage response mechanism at the level of hPer2–hp53 interaction, have important implications for our understanding of the etiology of cancer and the development of novel treatment modalities. Because cancer initiation and progression largely rely on a variety of DNA-damage repair mechanisms going awry in normal cells, thus facilitating tumor growth, the identification of hPer2 as a factor that responds to environmental signals and stabilizes hp53 provides a unique opportunity to evaluate the influence of the environment in the development of, for example, sporadic forms of cancers. Furthermore, our findings encourage a search for unconventional drug targets and treatment regimens based on the dynamics of the circadian control system.
MATERIALS AND METHODS
Plasmid constructs
The FLAG-tagged, myc-tagged, and untagged chimera constructs of hp53 and hPer2 were generated as follows. The hp53 cDNA clone was amplified by PCR and the stop codon removed before cloning into the LIC site of either pCS2+FLAG or myc tag vectors. For the GST chimera construct of hp53, the GST sequence was cloned downstream of hp53 into the SalI/XhoI sites in either vector. A glycine linker of six residues was introduced between hp53 and GST to favor flexibility. These chimera constructs are called FLAG-hp53(ch)GST and myc-hp53(ch)GST throughout the text. For the hPer2 chimera of hp53, the two hPer2-interacting fragments (residues 356–574 and 683–872) were subcloned downstream of hp53 following essentially the approach described except that hPer2(356-574) and hPer2(683-872) fragments were sequentially cloned into the SalI/XhoI and SalI/SalI sites, respectively. As was the case with the GST chimera construct, six glycine residues were added between hp53 and hPer2(356-574) and between the hPer2 fragments. These chimera constructs are referred to as FLAG-hp53(ch)hPer2(356-574/683-872) and myc-hp53(ch)hPer2(356-574/683-872). Chimeras containing an NLS were generated by cloning the NLS sequence encoding the TPPKKKRKVEDP residues from SV40 (Rexach and Blobel, 1995) upstream of hp53 using the appropriate pCS2+ as the template. The constructs are called FLAG-NLS-hp53(ch)GST, myc-NLS-hp53(ch)GST, FLAG-NLS-hp53(ch)hPer2(356-574/683-872), and myc-NLS-hp53(ch)hPer2(356-574/683-872). Full-length Per2-containing chimeras were generated as follows. The hPer2 cDNA was cloned downstream of hp53 into the SalI/XbaI sites. A six-glycine linker was genetically engineered between hp53 and hPer2 sequences to allow for flexible rotations. The full-length chimeras are referred to as FLAG-hp53(ch)hPer2, myc-hp53(ch)hPer2, and hp53(ch)hPer2. The hp21WAF1/CIP1-Luc reporter plasmid was a kind gift from Daiqing Liao (University of Florida, Gainesville, FL) and is described in Zhao et al. (2003).
Cell culture and transfections
The HCT116 and H1299 cell lines were purchased from the American Type Culture Collection and maintained according to the manufacturer's recommendations. For transfection experiments, cells were seeded in six- or 12-well plates until they reached 50–80% confluence. Transfections were optimized using Lipofectamine LTX (Life Technologies, Grand Island, NY) for HCT116 and H1299 following the manufacturer's instructions. Otherwise, transfections in all cell lines were in HyClone HyQ-RS reduced serum medium (Thermo Scientific, Waltham, MA) for 4 h for H1299 and HCT116. Proteins were then allowed to express at 37°C/5% CO2 in the appropriate medium containing 10% fetal bovine serum without antibiotics, after which they were either collected or further synchronized. Extracts for protein analysis were prepared in NP-40 lysis buffer containing 10 mM Tris-HCl (pH 7.5), 137 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 80 mM β-glycerophosphate, 1 mM Na3VO4, 10 mM NaF, and protease inhibitors (10 μM leupeptin, 1 μM aprotinin A, and 0.4 μM pepstatin).
In vitro binding assays
In vitro transcription and translation of pCS2+myc-hPer2, myc-Mdm2, myc-hCry1, FLAG-hPer2, and FLAG-hp53 were carried out using the SP6 high-yield TNT system (Promega, Madison, WI) following the manufacturer's instructions, although, unlike the standard procedure, the reaction was cold. Aliquots (1–4 μl) of indicated recombinant proteins were preincubated for 20 min at room temperature to allow the complex to form before adding NP40 lysis buffer. Immunoprecipitation of the various complexes was carried out essentially as described.
Immunoprecipitation and immunoblot assays
For (co)immunoprecipitation experiments, transfected cells were harvested in lysis buffer, and extracts (∼100 μg) were incubated with either α-FLAG M2 agarose beads (Sigma-Aldrich, St. Louis, MO) or α-myc (9E10) beads (Santa Cruz Biotechnology, Dallas, TX) for either 2 h or overnight at 4°C with rotation before washing. Where indicated, immunoprecipitations were carried out in a two-step procedure, with extracts being incubated with the antibody (α-myc, α-FLAG, or α-p53) overnight at 4°C before the addition of protein A beads (50% slurry; Sigma-Aldrich). Sample beads were then washed with lysis buffer, resolved by SDS–PAGE, and analyzed by immunoblotting using specific primary antibodies (α-FLAG [Sigma-Aldrich], α-myc [Santa Cruz Biotechnology], α-Per2 [Sigma-Aldrich]), α-p53 [Santa Cruz Biotechnology], and α-ubiquitin [Enzo Biomol, Farmingdale, NY]). When indicated, the purity of the various cellular fractions was monitored by immunoblotting using α-tubulin (Sigma-Aldrich) and α-lamin A/C (Santa Cruz Biotechnology) antibodies for the cytosolic and nuclear fractions, respectively. In all cases, horseradish peroxidase–conjugated α-rabbit or α-mouse immunoglobulin G (IgG) secondary antibodies (GE Healthcare Life Sciences, Buckinghamshire, UK; Cell Signaling, Danvers, MA) were used for immunoblotting following standard procedures. Chemiluminescence reactions were performed using the SuperSignal West Pico substrate (Pierce, Rockford, IL).
In other experiments, aliquots of total cell extracts were analyzed for expression of endogenous proteins. Circadian-synchronized HCT116 cells were collected at different times after synchronization and extracts (20–100 μg) analyzed for endogenous expression of hPer2, hp53, Mdm2, hCry1, and tubulin using specific antibodies. In addition, H1299 cells transfected with FLAG-hp53, FLAG-hp53(ch)GST, FLAG-hp53(ch)hPer2(356-574/683-872), FLAG-hp53(ch)hPer2, or EV were treated or not (control; –γ-IR) with ionizing radiation (5 Gy; +γ-IR; Di Leonardo et al., 1994) and collected at different times after irradiation. Extracts (20 μg) were analyzed for expression of endogenous and recombinant proteins as well as posttranslational modifications by immunoblotting using α-FLAG, -Chk1 (Santa Cruz Biotechnology), -Chk1-Ser345 (Cell Signaling), -p53-Ser15 (Cell Signaling), -p21 (Cell Signaling), and -tubulin (Sigma-Aldrich) antibodies.
Finally, endogenous hp21 expression was monitored in extracts (30–40 μg) from H1299 cells transfected with FLAG-hp53, FLAG-hp53(ch)GST, FLAG-NLS-hp53(ch)GST, FLAG-hp53(ch)hPer2(356-574/683-872), NLS-FLAG-hp53(ch)hPer2(356-574/683-872), FLAG-hp53(ch)hPer2, FLAG-hp53(ch)hPer2, or EV using an α-p21 antibody (Cell Signaling). Fusion proteins were detected using α-FLAG antibody as described. In all cases, tubulin levels were monitored by immunoblotting and used as a loading control.
RNA extraction and qRT-PCR
Cell samples were obtained from cultures and treated or not (control; –γ-IR) with ionizing radiation (5 Gy; +γ-IR) 2 h before collection. In other experiments, cells were transfected and/or circadian synchronized before radiation treatment. Total RNA was extracted from cell pellets using the Trizol Reagent (Life Technologies) following the manufacturer's instructions. RNA was quantified by spectrophotometric reading at 260 nm and analyzed for quality assurance using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) at the Virginia Bioinformatics Institute Proteomics Core Facility (Blacksburg, VA). qRT-PCR was conducted essentially as previously described (Yang et al., 2008). Briefly, total RNA was pretreated with DNaseI (Promega) at 37°C for 30 min, and a 1-μg sample was used as a template for first-strand cDNA synthesis using the iScript cDNA Synthesis system (Bio-Rad, Hercules, CA). qRT-PCR assay was performed using IQ SYBR Green Supermix (Bio-Rad) as follows: 10 ng of cDNA (50 ng for the 14-3-3σ gene) was added to a 20-μl reaction volume containing the indicated primers for amplification (see Supplemental Methods and Supplemental Table S1). Real-time assays were performed in triplicate on a MyIQ single-color Real-Time PCR Detection instrument (Bio-Rad). Data were collected and analyzed with Optical System Software, version 1.0. The glyceraldehyde-3-phosphate dehydrogenase and β-actin genes were used as internal controls to compute the relative expression level (ΔCT) for each sample. The fold change of gene expression in each sample was calculated as 2−ΔΔCt.
Immunofluorescence microscopy
H1299 cells were cultured on coverslips for 24 h and then transfected with pCS2+myc-hp53, -hp53(ch)GST, -hp53(ch)hPer2(356-574/683-872), -NLS-hp53(ch)GST, or -NLS-hp53(ch)hPer2(356-574/683-872) or untagged hp53(ch)hPer2 using Lipofectamine LTX (Life Technologies). After transfection, cells were fixed (3.7% formaldehyde/phosphate-buffered saline [PBS]/0.5% Triton X-100), washed with PBS/0.5% Triton X-100 and then 0.1% Triton X-100, and blocked with goat serum at room temperature for 30 min. Subcellular localization of myc fusion proteins was detected using an anti-myc-Cy-3–conjugated antibody (Sigma–Aldrich). Localization of untagged chimera recombinant protein (hp53(ch)hPer2) was visualized using α-p53 (Sigma-Aldrich) and -Per2 antibodies (Santa Cruz Biotechnology). Actin and nuclei were visualized by incubating fixed cells with phalloidin-Alexa 488 (Life Technologies) and SYTO 60 (Life Technologies). Fluorescence was visualized using a Nikon Eclipse TE2000-E microscope equipped with a Cascade II E2V CCD97 camera (Photometrics, Tucson, AZ) at 488, 568, and 647 nm. Images were processed using the NIS-Elements AR 3.0 Nikon software.
Gene reporter activity
H1299 cells were seeded onto 12-well plates and cotransfected with ∼200 ng of hp21WAF1/CIP1-luciferase (Luc) and pCS2+FLAG-hp53, -hp53(ch)GST, -hp53(ch)hPer2, or -hp53(ch)hPer2(356-574/683-872) or empty vector (pCS2+FLAG) each. The pCMV-β-gal (∼200 ng) plasmid was included as an internal control. Cells were treated or not (controls; –γ-IR) with ionizing radiation (5 Gy; +γ-IR) and harvested 2 h later. Reporter activity was measured using the Bright-Glo Luciferase Assay System (Promega) according to the manufacturer's instructions. Readings were recorded from a Glomax Luminometer, and results were normalized for expression of β-gal, which was determined separately using the Galacto-Light Plus System (Bio-Rad). Experiments were done in triplicate and repeated at least twice.
Statistical analyses
Data were processed using either a two-tailed unpaired Student's t test or an analysis of variance (ANOVA) followed by either Bonferroni or, when necessary, Games-Howell post hoc test (SPSS; IBM, Armonk, NY). Levene's test was used to determine homogeneity of variances, whereas data normality was examined using Shapiro–Wilk W test and Box–Cox Y transformation (JMP 9; SAS) and applied when necessary. Values of p ≤ 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
We thank John Tyson, Jill Sible, Daniel Capelluto, and James Maller for critical reading of the manuscript and all members of the Finkielstein laboratory for help and discussions. We also thank J. Webster for comments and editing of the manuscript. We are grateful to Steven L. McKnight (University of Texas Southwestern Medical Center, Dallas, TX) and Daiqing Liao for providing us with the hPer2 cDNA and pWAF1-Luc constructs, respectively. We also acknowledge C. Santos and J. Yang for generating reagents used throughout this research. This work was supported by a National Science Foundation CAREER Award (MCB-0844491), the Avon Foundation (02-2009-033), the Fralin Life Science Institute (F441598), and the Susan G. Komen Foundation (BCTR0706931) to C.V.F.
Abbreviations used:
- BAX
encodes the Bcl-2-associated X protein (Bax)
- CDKN1a
encodes cyclin-dependent kinase inhibitor p21 (p21CIP1/WAF1)
- GADD45α
encodes growth arrest and DNA damage–inducible protein 45α (Gadd45α)
- H1299
human non–small cell lung carcinoma cells
- HCT116
human colorectal carcinoma-116 cells
- hp53
human p53 transcription factor
- hPer2
human Period 2
- Mdm2
murine [human] double minute-2
- SFN
encodes 14-3-3σ.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-05-0994) on November 19, 2014.
*These authors contributed equally.
†Present address: Department of Biology, University of Padova, Via U. Bassi, 58/B, 35121, Padova, Italy
T.G. provided data in Figures 1, 2, 4, and 5D and Supplemental Figures S1, S2, S4, S8F, S9, B and C, and S10 and contributed with reagents in Figures 3 and 5 and Supplemental Figures S3, S5, and S8. M.V.-C. performed the experiments shown in Figures 3 and 5, B and C, and Supplemental Figures S3, S8, A–E, and S9A, contributed in the execution of the experiments shown in Figure 5D and Supplemental Figures S1, S8F, and S9C, and performed the statistical analyses except for those mentioned otherwise. J.L. performed the experiments shown in Supplemental Figures S6 and S7 (top and middle) and S9C. S.S. provided Supplemental Figures S5 and S7 (bottom) and the statistical analyses shown in Supplemental Figures S5, S7, and S9. C.V.F., T.G., and M.V.-C. analyzed the overall data, refined the hypothesis, and proposed the model. C.V.F. supervised and coordinated all investigators for the project and wrote the manuscript.
REFERENCES
- Albrecht U, Bordon A, Schmutz I, Ripperger J. The multiple facets of Per2. Cold Spring Harb Symp Quant Biol. 2007;72:95–104. doi: 10.1101/sqb.2007.72.001. [DOI] [PubMed] [Google Scholar]
- Bae K, Jin X, Maywood ES, Hastings MH, Reppert SM, Weaver DR. Differential functions of mPer1, mPer2, and mPer3 in the SCN circadian clock. Neuron. 2001;30:525–536. doi: 10.1016/s0896-6273(01)00302-6. [DOI] [PubMed] [Google Scholar]
- Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, Simon MC, Takahashi JS, Bradfield CA. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103:1009–1017. doi: 10.1016/s0092-8674(00)00205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cermakian N, Monaco L, Pando MP, Dierich A, Sassone-Corsi P. Altered behavioral rhythms and clock gene expression in mice with a targeted mutation in the Period1 gene. EMBO J. 2001;20:3967–3974. doi: 10.1093/emboj/20.15.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Leonardo A, Linke SP, Clarkin K, Wahl GM. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev. 1994;8:2540–2551. doi: 10.1101/gad.8.21.2540. [DOI] [PubMed] [Google Scholar]
- Duffield GE, Best JD, Meurers BH, Bittner A, Loros JJ, Dunlap JC. Circadian programs of transcriptional activation, signaling, and protein turnover revealed by microarray analysis of mammalian cells. Curr Biol. 2002;12:551–557. doi: 10.1016/s0960-9822(02)00765-0. [DOI] [PubMed] [Google Scholar]
- Freedman DA, Levine AJ. Nuclear export is required for degradation of endogenous p53 by MDM2 and human papillomavirus E6. Mol Cell Biol. 1998;18:7288–7293. doi: 10.1128/mcb.18.12.7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Pelicano H, Liu J, Huang P, Lee C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell. 2002;111:41–50. doi: 10.1016/s0092-8674(02)00961-3. [DOI] [PubMed] [Google Scholar]
- Gery S, Koeffler HP. Per2 is a C/EBP target gene implicated in myeloid leukemia. Integr Cancer Ther. 2009;8:317–320. doi: 10.1177/1534735409352084. [DOI] [PubMed] [Google Scholar]
- Gery S, Komatsu N, Baldjyan L, Yu A, Koo D, Koeffler HP. The circadian gene per1 plays an important role in cell growth and DNA damage control in human cancer cells. Mol Cell. 2006;22:375–382. doi: 10.1016/j.molcel.2006.03.038. [DOI] [PubMed] [Google Scholar]
- Gorbacheva VY, Kondratov RV, Zhang R, Cherukuri S, Gudkov AV, Takahashi JS, Antoch MP. Circadian sensitivity to the chemotherapeutic agent cyclophosphamide depends on the functional status of the CLOCK/BMAL1 transactivation complex. Proc Natl Acad Sci USA. 2005;102:3407–3412. doi: 10.1073/pnas.0409897102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh T, Vila-Caballer M, Santos CS, Liu J, Yang J, Finkielstein CV. The circadian factor Period 2 modulates p53 stability and transcriptional activity in unstressed cells. Mol Biol Cell 25, 3081–3093. 2014 doi: 10.1091/mbc.E14-05-0993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotter AL, Manganaro T, Weaver DR, Kolakowski LF, Jr, Possidente B, Sriram S, MacLaughlin DT, Reppert SM. A time-less function for mouse timeless. Nat Neurosci. 2000;3:755–756. doi: 10.1038/77653. [DOI] [PubMed] [Google Scholar]
- Grechez-Cassiau A, Rayet B, Guillaumond F, Teboul M, Delaunay F. The circadian clock component BMAL1 is a critical regulator of p21WAF1/CIP1 expression and hepatocyte proliferation. J Biol Chem. 2008;283:4535–4542. doi: 10.1074/jbc.M705576200. [DOI] [PubMed] [Google Scholar]
- Grundschober C, Delaunay F, Puhlhofer A, Triqueneaux G, Laudet V, Bartfai T, Nef P. Circadian regulation of diverse gene products revealed by mRNA expression profiling of synchronized fibroblasts. J Biol Chem. 2001;276:46751–46758. doi: 10.1074/jbc.M107499200. [DOI] [PubMed] [Google Scholar]
- Gu X, Xing L, Shi G, Liu Z, Wang X, Qu Z, Wu X, Dong Z, Gao X, Liu G, et al. The circadian mutation PER2(S662G) is linked to cell cycle progression and tumorigenesis. Cell Death Differ. 2012;19:397–405. doi: 10.1038/cdd.2011.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- Hua H, Wang Y, Wan C, Liu Y, Zhu B, Yang C, Wang X, Wang Z, Cornelissen-Guillaume G, Halberg F. Circadian gene mPer2 overexpression induces cancer cell apoptosis. Cancer Sci. 2006;97:589–596. doi: 10.1111/j.1349-7006.2006.00225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalderon D, Richardson WD, Markham AF, Smith AE. Sequence requirements for nuclear location of simian virus 40 large-T antigen. Nature. 1984;311:33–38. doi: 10.1038/311033a0. [DOI] [PubMed] [Google Scholar]
- Ko CH, Takahashi JS. Molecular components of the mammalian circadian clock. Hum Mol Genet. 2006;15((Spec No 2)):R271–R277. doi: 10.1093/hmg/ddl207. [DOI] [PubMed] [Google Scholar]
- Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core component of the circadian clock. Genes Dev. 2006;20:1868–1873. doi: 10.1101/gad.1432206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laposky A, Easton A, Dugovic C, Walisser J, Bradfield C, Turek F. Deletion of the mammalian circadian clock gene BMAL1/Mop3 alters baseline sleep architecture and the response to sleep deprivation. Sleep. 2005;28:395–409. doi: 10.1093/sleep/28.4.395. [DOI] [PubMed] [Google Scholar]
- Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W. Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science. 2003;302:1972–1975. doi: 10.1126/science.1091362. [DOI] [PubMed] [Google Scholar]
- Liang SH, Clarke MF. Regulation of p53 localization. Eur J Biochem. 2001;268:2779–2783. doi: 10.1046/j.1432-1327.2001.02227.x. [DOI] [PubMed] [Google Scholar]
- Lucas RJ, Stirland JA, Mohammad YN, Loudon AS. Postnatal growth rate and gonadal development in circadian tau mutant hamsters reared in constant dim red light. J Reprod Fertil. 2000;118:327–330. doi: 10.1530/jrf.0.1180327. [DOI] [PubMed] [Google Scholar]
- Matsuo T, Yamaguchi S, Mitsui S, Emi A, Shimoda F, Okamura H. Control mechanism of the circadian clock for timing of cell division in vivo. Science. 2003;302:255–259. doi: 10.1126/science.1086271. [DOI] [PubMed] [Google Scholar]
- Meek DW. Tumour suppression by p53: a role for the DNA damage response. Nat Rev Cancer. 2009;9:714–723. doi: 10.1038/nrc2716. [DOI] [PubMed] [Google Scholar]
- Miller BH, McDearmon EL, Panda S, Hayes KR, Zhang J, Andrews JL, Antoch MP, Walker JR, Esser KA, Hogenesch JB, Takahashi JS. Circadian and CLOCK-controlled regulation of the mouse transcriptome and cell proliferation. Proc Natl Acad Sci USA. 2007;104:3342–3347. doi: 10.1073/pnas.0611724104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullenders J, Fabius AW, Madiredjo M, Bernards R, Beijersbergen RL. A large scale shRNA barcode screen identifies the circadian clock component ARNTL as putative regulator of the p53 tumor suppressor pathway. PLoS One. 2009;4:e4798. doi: 10.1371/journal.pone.0004798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor E, Bergmann BM, Krauski K, Zee PC, Takahashi JS, Vitaterna MH, Turek FW. The circadian clock mutation alters sleep homeostasis in the mouse. J Neurosci. 2000;20:8138–8143. doi: 10.1523/JNEUROSCI.20-21-08138.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsaki K, Oishi K, Kozono Y, Nakayama K, Nakayama KI, Ishida N. The role of {beta}-TrCP1 and {beta}-TrCP2 in circadian rhythm generation by mediating degradation of clock protein PER2. J Biochem. 2008;144:609–618. doi: 10.1093/jb/mvn112. [DOI] [PubMed] [Google Scholar]
- Oklejewicz M, Hut RA, Daan S, Loudon AS, Stirland AJ. Metabolic rate changes proportionally to circadian frequency in tau mutant Syrian hamsters. J Biol Rhythms. 1997;12:413–422. doi: 10.1177/074873049701200503. [DOI] [PubMed] [Google Scholar]
- Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, Schultz PG, Kay SA, Takahashi JS, Hogenesch JB. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109:307–320. doi: 10.1016/s0092-8674(02)00722-5. [DOI] [PubMed] [Google Scholar]
- Pregueiro AM, Liu Q, Baker CL, Dunlap JC, Loros JJ. The Neurospora checkpoint kinase 2: a regulatory link between the circadian and cell cycles. Science. 2006;313:644–649. doi: 10.1126/science.1121716. [DOI] [PubMed] [Google Scholar]
- Rexach M, Blobel G. Protein import into nuclei: association and dissociation reactions involving transport substrate, transport factors, and nucleoporins. Cell. 1995;83:683–692. doi: 10.1016/0092-8674(95)90181-7. [DOI] [PubMed] [Google Scholar]
- Rudic RD, McNamara P, Curtis AM, Boston RC, Panda S, Hogenesch JB, Fitzgerald GA. BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol. 2004;2:e377. doi: 10.1371/journal.pbio.0020377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaulsky G, Goldfinger N, Ben-Ze’ev A, Rotter V. Nuclear accumulation of p53 protein is mediated by several nuclear localization signals and plays a role in tumorigenesis. Mol Cell Biol. 1990;10:6565–6577. doi: 10.1128/mcb.10.12.6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearman LP, Jin X, Lee C, Reppert SM, Weaver DR. Targeted disruption of the mPer3 gene: subtle effects on circadian clock function. Mol Cell Biol. 2000;20:6269–6275. doi: 10.1128/mcb.20.17.6269-6275.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimba S, Ishii N, Ohta Y, Ohno T, Watabe Y, Hayashi M, Wada T, Aoyagi T, Tezuka M. Brain and muscle Arnt-like protein-1 (BMAL1), a component of the molecular clock, regulates adipogenesis. Proc Natl Acad Sci USA. 2005;102:12071–12076. doi: 10.1073/pnas.0502383102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun CM, Huang SF, Zeng JM, Liu DB, Xiao Q, Tian WJ, Zhu XD, Huang ZG, Feng WL. Per2 inhibits k562 leukemia cell growth in vitro and in vivo through cell cycle arrest and apoptosis induction. Pathol Oncol Res. 2010;16:403–411. doi: 10.1007/s12253-009-9227-0. [DOI] [PubMed] [Google Scholar]
- Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet. 2008;9:764–775. doi: 10.1038/nrg2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, Laposky A, Losee-Olson S, Easton A, Jensen DR, et al. Obesity and metabolic syndrome in circadian clock mutant mice. Science. 2005;308:1043–1045. doi: 10.1126/science.1108750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unsal-Kacmaz K, Mullen TE, Kaufmann WK, Sancar A. Coupling of human circadian and cell cycles by the timeless protein. Mol Cell Biol. 2005;25:3109–3116. doi: 10.1128/MCB.25.8.3109-3116.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Xu Y, Padiath QS, Shapiro RE, Jones CR, Wu SC, Saigoh N, Saigoh K, Ptacek LJ, Fu YH. Functional consequences of a CKIdelta mutation causing familial advanced sleep phase syndrome. Nature. 2005;434:640–644. doi: 10.1038/nature03453. [DOI] [PubMed] [Google Scholar]
- Yagita K, Tamanini F, Yasuda M, Hoeijmakers JH, van der Horst GT, Okamura H. Nucleocytoplasmic shuttling and mCRY-dependent inhibition of ubiquitylation of the mPER2 clock protein. EMBO J. 2002;21:1301–1314. doi: 10.1093/emboj/21.6.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Kim KD, Lucas A, Drahos KE, Santos CS, Mury SP, Capelluto DG, Finkielstein CV. A novel heme-regulatory motif mediates heme-dependent degradation of the circadian factor period 2. Mol Cell Biol. 2008;28:4697–4711. doi: 10.1128/MCB.00236-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang EE, Kay SA. Clocks not winding down: unravelling circadian networks. Nat Rev Mol Cell Biol. 2010;11:764–776. doi: 10.1038/nrm2995. [DOI] [PubMed] [Google Scholar]
- Zhang EE, Liu AC, Hirota T, Miraglia LJ, Welch G, Pongsawakul PY, Liu X, Atwood A, Huss JW, 3rd, Janes J, et al. A genome-wide RNAi screen for modifiers of the circadian clock in human cells. Cell. 2009;139:199–210. doi: 10.1016/j.cell.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao LY, Liu Y, Bertos NR, Yang XJ, Liao D. PCAF is a coactivator for p73-mediated transactivation. Oncogene. 2003;22:8316–8329. doi: 10.1038/sj.onc.1206916. [DOI] [PubMed] [Google Scholar]
- Zheng B, Albrecht U, Kaasik K, Sage M, Lu W, Vaishnav S, Li Q, Sun ZS, Eichele G, Bradley A, Lee CC. Nonredundant roles of the mPer1 and mPer2 genes in the mammalian circadian clock. Cell. 2001;105:683–694. doi: 10.1016/s0092-8674(01)00380-4. [DOI] [PubMed] [Google Scholar]
- Zheng B, Larkin DW, Albrecht U, Sun ZS, Sage M, Eichele G, Lee CC, Bradley A. The mPer2 gene encodes a functional component of the mammalian circadian clock. Nature. 1999;400:169–173. doi: 10.1038/22118. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.