

Abstract
We studied promoter methylation (PM) of 11 genes in Peripheral Blood Lymphocytes (PBLs) and tissues of hepatitis C virus (HCV) associated hepatocellular carcinoma (HCC) and chronic hepatitis (CH) Egyptian patients. The present study included 31 HCC with their ANT, 38 CH and 13 normal hepatic tissue (NHT) samples. In all groups, PM of APC, FHIT, p15, p73, p14, p16, DAPK1, CDH1, RARβ, RASSF1A, O6MGMT was assessed by methylation-specific PCR (MSP). APC and O6-MGMT protein expression was assessed by immunohistochemistry (IHC) in the studied HCC and CH (20 samples each) as well as in a different HCC and CH set for confirmation of MSP results. PM was associated with progression from CH to HCC. Most genes showed high methylation frequency (MF) and the methylation index (MI) increased with disease progression. MF of p14, p73, RASSF1A, CDH1 and O6MGMT was significantly higher in HCC and their ANT. MF of APC was higher in CH. We reported high concordance between MF in HCC and their ANT, MF in PBL and CH tissues as well as between PM and protein expression of APC and O6MGMT. A panel of 4 genes (APC, p73, p14, O6MGMT) classifies the cases independently into HCC and CH with high accuracy (89.9%), sensitivity (83.9%) and specificity (94.7%). HCV infection may contribute to hepatocarcinogenesis through enhancing PM of multiple genes. PM of APC occurs early in the cascade while PM of p14, p73, RASSF1A, RARB, CDH1 and O6MGMT are late changes. A panel of APC, p73, p14, O6-MGMT could be used in monitoring CH patients for early detection of HCC. Also, we found that, the methylation status is not significantly affected by whether the tissue was from the liver or PBL, indicating the possibility of use PBL as indicator to genetic profile instead of liver tissue regardless the stage of disease.
Keywords: Hepatitis C virus-genotype 4, Chronic hepatitis, Hepatocellular carcinoma, Promoter methylation
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common solid tumor worldwide and the fourth leading cause of cancer-related death [1]. It accounts for approximately 600,000 deaths per year [2] and it shows a wide geographical variation with low incidence areas in North America and Europe, and high incidence areas in Africa and Asia. In Egypt the incidence of HCC has doubled in the past 10 years, thus it is now the second most incident and lethal cancer in men after lung cancer [3]. The heavy burden of HCC parallels the high rates of HCV infection while hepatitis B virus (HBV) rates have declined after the introduction of the vaccine in 1992 [4,5]. Although it has been estimated that 80% of HCC occurs in cirrhotic livers, the exact molecular mechanisms underlying virus-associated hepatocarcinogenesis are still unclear.
Multiple genetic aberrations of oncogenes and tumor suppressor genes have been identified, which control hepatocytes proliferation, differentiation, maintenance of genomic integrity and death [6,7]. In addition, recent studies suggest aberrant DNA (PM) as an alternative mechanism of tumor pathogenesis because the hypermethylated promoters often lack transcriptional activity, which could result in gene inactivation [8]. DNA methylation refers to the addition of a methyl group to the cytosine residue in CpG dinucleotides. Normally, clustered CpG dinucleotides (CpG islands) are not methylated regardless of their transcriptional status, whereas in tumor cells, methylation of CpG islands in the promoter regions of many tumor suppressor genes (TSGs) and growth regulatory genes effectively silences those genes. Since different types of cancer show distinct DNA methylation profiles, it is possible to develop cancer- type specific methylation signatures [9]. The power of PM as a marker derives not only from its ability to be detected in a wide variety of samples, from fresh specimens to body fluids and archival paraffin-embedded tissues, but also from the defined localization of the lesion in promoter CpG islands of the genes. This could be an early important event in carcinogenesis and could also be of importance for treatment or prognostication [10]. DNA methylation profiles in Egypt has not been well studied, though it has the highest prevalence of HCV infection in the world with approximately 14% of the population infected, and seven million have chronic HCV induced liver disease [11].
We sought to assess DNA methylation patterns in Egyptian patients with HCV associated chronic hepatitis and HCC using a panel of genes that are commonly hypermethylated in other solid tumors (p14, p15, p16, p73, APC, FHIT, DAPK1, CDH1, RARβ, RASSF1A, and O6MGMT) in order to understand the role of epigenetic silencing in this patient population. The studied groups included 38 HCV/genotype-4-associated CH patients with matched PBL in 20 of them and 31 HCC cases with their ANT. Thirteen NHT obtained from healthy individuals, were used as a control group. The prognostic impact of aberrant PM was also assessed through correlations between methylation patterns and the clinic-pathological features of the studied patients.
Methodology
Study design
This prospective study encompassed three groups. The first group included 31 HCC cases, of which, 23 cases had enough adjacent normal tissue (ANT) samples to be assessed. The second group included: A) 20 cases of chronic CH patients with cirrhosis from which tissue samples and Peripheral Blood Lymphocytes (PBLs) were collected and 18 cases of asymptomatic carriers (ASC), from which tissue samples only were collected. The third group was a control group in which normal hepatic tissue (NHT) samples were obtained from 13 liver transplantation donors matched for age (±5 years) and sex.
HCC samples were obtained from patients who underwent surgical resection of their tumors at the National Cancer Institute (NCI), Cairo, Egypt. Whereas CH samples were obtained from the Endemic medicine department, Kasr Al-Aini School of Medicine, Cairo University. All cases were assessed for viral profile as a part of the routine clinical workup. All HCC and CH cases were positive for HCV/genotype-4 and negative for HBV by serological tests and/or HBV-DNA by real time PCR (qRT PCR). Histopathological diagnosis and grading of the HCC cases were done according to the World Health Organization (WHO) classification criteria [12] and staging was performed according to the American Joint Committee on Cancer [13]. Grading and staging of CH patients were performed according to the pathology activity index [14]. A written informed consent was obtained from each patient and the Institutional Review Boards of the National Cancer Institute and Kasr Al-Aini School of Medicine, Cairo University, reviewed the study protocol which was in accordance with the 2007 Declaration of Helsinki. All patients’ characteristics were collected from the patients’ records and illustrated in Table 1.
Table 1.
Clinical features of the studied groups.
| Variables | CAH cases (38) |
HCC cases (31) |
p Value | ||
|---|---|---|---|---|---|
| Mean | Range | Mean | Range | ||
| Age (years) | 40.0 | (1–61) | 57 | (38–78) | <0.0001 |
| WBCs | 5.9 | (3.2–108) | 5.4 | (2.5–26.6) | <0.0001 |
| RBCs | 14 | (4.17–15.5) | 4.2 | (3.6–9.3) | |
| HG | 14.7 | (11.3–17) | 13.1 | (9.3–16.9) | <0.0023 |
| Platelets | 202.5 | (98–377) | 195 | (33–356) | <0.0001 |
| AST | 48.5 | (14–297) | 67 | (7–432) | |
| ALT | 52 | (4–209) | 58 | (10–480) | |
| Alk | 90 | (34–282) | 96 | (35–387) | |
| Albumin | 4.3 | (2.6–40) | 3.3 | (2.5–6.0) | |
| Total bilirubin | 0.99 | (0.28–23.4) | 1.00 | (0.20–4.1) | |
| Direct bilirubin | 0.55 | (0.1–12.1) | 0.4 | (0.2–2.6) | |
Only significant p values are illustrated.
DNA extraction
DNA was extracted from PBL according to standard protocols (6). Briefly, equal volume of equilibrated phenol (pH 7.0–7.5) was added to samples and vortexed. The upper aqueous layer was removed and an equal volume of phenol/chloroform (1:1) was added and vortexed. The upper aqueous layer was removed again and an equal volume of chloroform/isoamyl alcohol (24:1) was added and vortexed. This was followed by the addition of 3 M Sodium acetate (pH 4.7–5.2), DNA precipitation by ice-cold ethanol and overnight incubation at −80 °C. The fluid was decanted and the DNA pellet was dissolved in sterile water. DNA was extracted from fresh tissue samples as previously described [15].
Bisulphate conversion and methylation-specific polymerase chain reaction (MSP)
The extracted DNA was subjected to bisulfate treatment followed by MSP using the primer sequences and the methylation-specific PCR conditions illustrated in Table 2. DNA methylation of CpG islands for p14, p15, p16, p73, APC, FHIT, DAPK1, CDH1, RARβ, RASSF1A and O6MGMT genes was determined using specific primers for methylated (M) and unmethylated (UM) DNA [16]. Negative control samples without DNA were included in each set of PCR. PCR products were analyzed on 4% ethidium bromide-stained agarose gels and visualized under ultraviolet illumination (Fig. 1).
Table 2.
Primers sequences and conditions of the methylation specific PCR (MSP).
| Gene | Primers | Annealing temperature (1C) | MgCl2 | Cycles |
|---|---|---|---|---|
| CDH1 (M) | TAATTAGCGGTACGGGGGGC CGAAAACAAACGCCGAATACG | 59 | 4.5 | 32 |
| CDH1 (U) | TTAGTTAATTAGTGGTATGGGGGGTGG ACCAAACAAAAACAAACACCAAATACA | 59 | 4.5 | 32 |
| DAPK (M) | GGATAGTCGGATCGAGTTAACGTC CCCTCCCAAACGCCGA | 59 | 4.5 | 35 |
| DAPK (U) | GGAGGATAGTTGGATTGAGTTAATGTT CAAATCCCTCCCAAACACCAA | 59 | 4.5 | 35 |
| p73 (M) | GGACGTAGCGAAATCGGGGTTC ACCCCGAACATCGACGTCCG | 64 | 4.5 | 35 |
| p73 (U) | AGGGGATGTAGTGAAATTGGGGTTT ATCACAACCCCAAACATCAACATCCA | 60 | 4.5 | 35 |
| O6O6-MGMT (M) | TTTCGACGTTCGTAGGTTTTCGC GCACTCTTCCGAAAACGAAACG | 56 | 3.5 | 35 |
| O6O6-MGMT (U) | TTTGTGTTTTGATGTTTGTAGGTTTTTGT AACTCCACACTCTTCCAAAAACAAAACA | 57 | 4.5 | 35 |
| p14 (M) | GTGTTAAAGGGCGGCGTAGC AAAACCCTCACTCGCGACGA | 54 | 4.5 | 35 |
| p14 (U) | TTTTTGGTGTTAAAGGGTGGTGTAGT CACAAAAACCCTCACTCACAACAA | 56 | 4.5 | 35 |
| p15 (M) | GCGTTCGTATTTTGCGGTT CGTACAATAACCGAACGACCGA | 57 | 3.5 | 35 |
| p15 (U) | TGTGATGTGTTTGTATTTTGTGGTT CCATACAATAACCAAACAACCAA | 59 | 4.5 | 35 |
| p16 (M) | TTATTAGAGGGTGGGGCGGATCGC CCACCTAAATCGACCTCCGACCG | 68 | 1.5 | 33 |
| p16 (U) | TTATTAGAGGGTGGGGTGGATTGT CCACCTAAATCAACCTCCAACCA | 58 | 4.5 | 33 |
| FHIT (M) | TTGGGGCGCGGGTTTGGGTTTTTACGC CGTAAACGACGCCGACCCCACTA | 71–63 | 1.5 | 32 |
| FHIT (U) | TTGGGGTGTGGGTTTGGGTTTTTATG CATAAACAACACCAACCCCACTA | 64 | 1.5 | 33 |
| APC (M) | TATTGCGGAGTGCGGGTC TCAACGAACTCCCGACGA | 62 | 3.5 | 35 |
| APC (U) | GTGTTTTATTGTGGAGTGTGGGTT CCAATCAACAAACTCCCAACAA | 62 | 1.5 | 35 |
| RASSF1A (M) | TTCGTCGTTTAGTTTGGATTTTG CCGATTAAACCCGTACTTCG | 56 | 1.5 | 35 |
| RASSF1A (U) | TGTTGTTTAGTTTGGATTTTGG TACAACCCTTCCCAACACAC | 59 | 3.5 | 35 |
| RARβ (M) | TCGAGAACGCGAGCGATTCG GACCAATCCAACCGAAACGA | 62 | 1.5 | 35 |
| RARβ (U) | TTGAGAATGTGAGTGAATTGA AACCAATCCAACCAAAACAA | 59 | 1.5 | 35 |
Fig. 1.
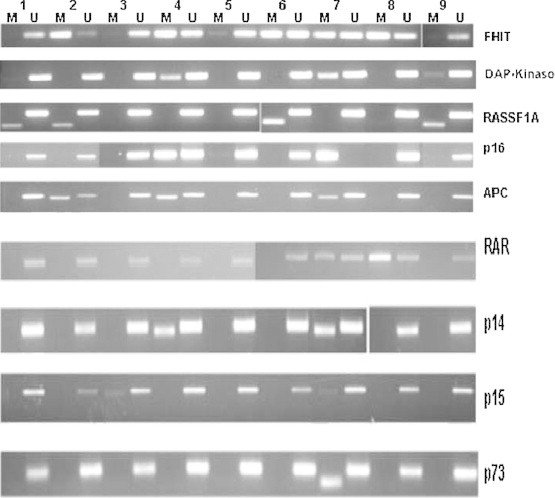
Methylation-specific PCR analyses of nine representative HCC samples (labeled 1–9 on the top). Each gene is indicated on the right. Both methylated (M) and unmethylated (U) reactions were amplified for each bisulfite-treated DNA and run in a 4% agarose gel.
Immunohistochemistry
Protein expression of APC and O6MGMT was assessed in 20 cases of HCC and 20 cases of CH which were assessed for PM by MSP as well as in a confirmatory set of 107 HCC, 52 CH cases and 40 NHT samples to confirm the results of the MSP using the tissue microarray (TMA) technique. Two (5 μm) thick sections were obtained from each TMA block on positive charged slides to be used for immunohistochemistry. Sections were deparaffinized, rehydratedin graded alcohols and the standard streptavidirin–biotin–peroxidase technique was performed [17] using the following antibodies: rabbit anti-human O6MGMT (EPR-4397, Epitomics, USA 1:100) and the rabbit anti-human APC (EP-701Y, Epitomics, USA 1:50). Antigen retrieval was performed by microwave pretreatment in 0.01 M citrate buffer (pH 7.4) and then the primary antibody was applied and incubated overnight at 4 °C in a humidified chamber. After three washes in PBS, the secondary antibody and the avidin–biotin complex (ABC) were applied to slides with diaminobenzidine (DAB) as a chromogen and Mayer’s hematoxylin as a counterstain. To evaluate the specificity of the antibodies, known positive and negative tissues were used as controls. Assessment was based on a cytoplasmic staining pattern for APC and on nuclear expression for O6MGMT.
Statistical methods: The data comprised of information about the presence or absence of PM of the 11 genes in four distinct groups, HCC (31) T, CH with cirrhosis (20) C, ASC (18) A, and healthy controls (NHT, n = 13) B. In the HCC group, 23 HCC cases had data for the tumor and the ANT whereas the remaining eight cases, had data for the tumor and the corresponding PBL but not on ANT. In the CH group, data for tissues was paired with the corresponding PBL. We used one-way ANOVA test to detect differences in the available clinicopathological variables between disease states. For each of these variables, the corrected p-values were reported. Logistic regression with a random effect analysis (non-linear mixed model) was used to determine differences across categories within a group and methylation status controlling for the subject effect for CH and HCC groups. Logistic regression analysis (Proc Logistic) was used to determine differences in methylation status and we reported the interaction and the main effects. The interaction effects measure any synergistic or antagonistic effect of the methylation status (methylated versus unmethylated) and the disease site (normal or tumor and liver or PBL); disease state (healthy controls, ASC, CH with cirrhosis or HCC). All statistical tests were performed using the SAS software package (version 9.2, SAS, Cary, NC).
Results
Clinical findings
There was a significant difference (corrected for multiplicity) between the studied groups regarding age (p-value <0.0001), HG (p value <0.0023), platelets (p value <0.0001) and WBCs (p value <0.0001). In all cases, the HCC group was significantly different from CH patients with cirrhosis and the asymptomatic carrier groups as in Table 1.
Methylation index (MI)
Calculation of the MI (defined as the ratio between the number of methylated genes and the number of total genes analyzed for each sample) was done for all cases. The MI ranged from 0 to 0.55 in CH cases (average: 0.27), and from 0.27 to 0.90 in HCC (average: 0.36). The difference between both groups was statistically insignificant (p > 0.05).
DNA methylation in normal hepatic tissues
PM of the 11 tested genes was assessed in 13 NHT samples. None of the samples showed PM of p15, p73, RARβ, RASSF1A or O6MGMT. PM of the p14 was detected in 46.2% of the cases followed by APC, which was methylated in 30.8% of the cases. A significant difference in methylation frequency (MF) between NHT and CH groups was reported for APC, FHIT, DAPK and RASSF genes as shown in Fig. 2 and Table 3.
Fig. 2a.

Methylation of 11 genes in hepatocellular carcinoma patients. * Dark squares depict methylation and blank squares depict unmethylation.
Fig. 2b.

Methylation of 11 genes in patients with chronic liver diseases. * Dark squares depict methylation and blank squares depict unmethylation.
Fig. 2c.
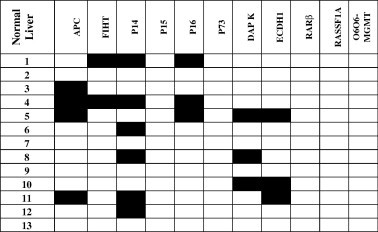
Methylation of 11 genes in normal liver individuals. * Dark squares depict methylation and blank squares depict unmethylation.
Table 3.
Methylation profile of the 11 genes in CAH, HCC and normal liver tissues.
| Genes | Normal liver N = 13 (%) | Chronic hepatitis (CH) |
Hepatocellular carcinoma HCC |
p-Value* |
|||
|---|---|---|---|---|---|---|---|
| (Tissue) (38) (%) | (PBL) (20) (%) | (HCC) (31) (%) | (ANT) (31) (%) | (CH and HCC) | (CH and ANT) | ||
| APC | 4 (30.8) | 33 (86.8) | 16 (80) | 13 (41.9) | 14 (45.2) | <0.001 | <0.001 |
| FHIT | 2 (15.4) | 20 (52.6) | 6 (30) | 21 (67.7) | 20 (64.5) | 0.204 | 0.005 |
| P15 | 0 (0) | 0 (0) | 0 (0) | 5 (16.1) | 5 (16.1) | 0.010 | –# |
| P73 | 0 (0) | 8 (21.1) | 1 (5.0) | 26 (83.9) | 23 (74.2) | <0.001 | <0.001 |
| P14 | 6 (46.2) | 17 (44.7) | 10 (50) | 28 (90.3) | 28 (90.3) | <0.001 | <0.001 |
| P16 | 3 (23.1) | 15 (39.5) | 9 (45) | 14 (45.2) | 19 (61.3) | 0.634 | 0.390 |
| DAPK | 3 (23.1) | 22 (57.9) | 12 (60) | 21 (67.7) | 22 (71) | 0.401 | 0.023 |
| RARβ | 0 (0) | 0 (00) | 0 (0) | 5 (16.1) | 3 (9.7) | 0.015 | –# |
| RASSF | 0 (0) | 26 (68.4) | 20 (100) | 31 (100) | 31 (100) | 0.001 | <0.001 |
| O6O6-MGMT | 0 (0) | 10 (26.3) | 10 (50.0) | 21 (67.7) | 20 (64.5) | <0.001 | <0.001 |
| CDH1 | 3 (23.1) | 7 (18.4) | 8 (40.0) | 17 (54.8) | 14 (45.2) | 0.002 | 0.004 |
p-Values ⩽ 0.05 are considered significant.
Numbers are too small for a valid statistical analysis.
Analysis of significant difference of DNA methylation within the CAH group
To understand aberrant DNA methylation of the selected 11 genes in CAH group (n = 38) to determine whether there are differences within CAH-tissues and CAH-PBL groups across the methylation profiles, data were analyzed according to b analysis approach (Generalized Linear Mixed Models) correcting for multiplicity using a Bonferroni adjustment. Our results as shown in Fig. 3A–K indicate that there are no interaction effects between methylation status and disease site among groups. This means that methylation status is not significantly affected by whether the tissue was from the liver or from the PBL. However, statistical values for APC (Fig. 3A; p-value = 0.03) and p16 (Fig. 3F; p-value = 0.04) would be considered significant for the un-adjusted criteria. There are significant differences between methylated and unmethylated states for APC, p14, p73, p16, DAPK1, and RASSF1A. None of the genes were different across tissue and PBL groups, albeit APC had a p-value of 0.04. The interaction in APC (Fig. 3A) is evidenced by the change from 0.95 to 0.10 from methylated to unmethylated state for the chronic liver tissue and a smaller change of 0.80–0.40 from methylation to unmethylation for the PBL group.
Fig. 3.
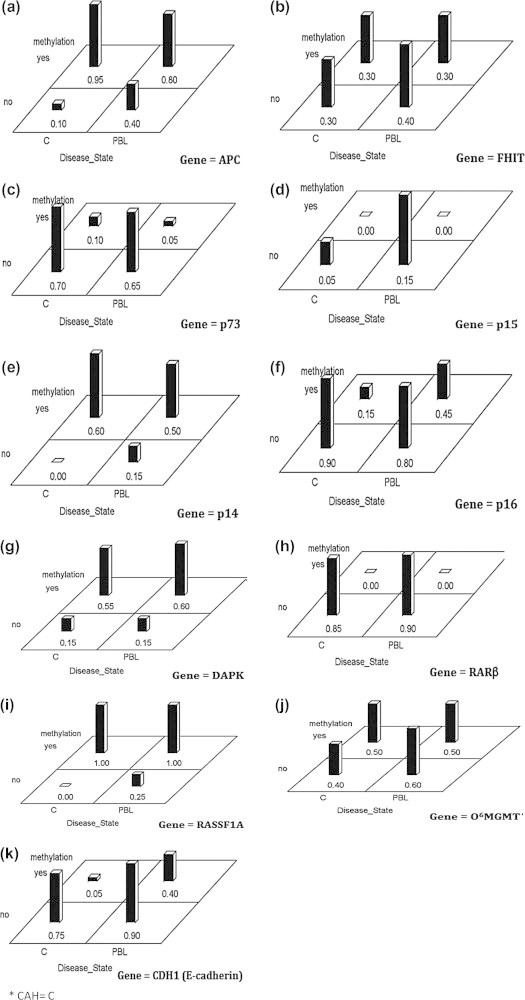
Differences across methylation profiles within CAH* cases between tissues and PBL.
Analysis of Significant difference of DNA methylation with HCC groups
To understand aberrant DNA methylation of the selected 11 genes in HCC, we followed the same technique of data analysis as with CAH group to determine DNA methylation status of genes in 31 HCCs and their adjacent non-cancerous tissues. We used a Bonferroni correction with 0.0045 (.05/11) as our cut-off for significance. Our results indicate that there are no interactions among the tissue sites and methylation status for any of the genes. As shown in Fig. 4A–K, there were differences in the methylation status for the genes RASSF1A (Fig. 4I), FHIT (Fig. 4B), APC (Fig. 4A), p14 (Fig. 4E), p73 (Fig. 4C), RARβ (Fig. 4H), O6MGMT (Fig. 4J), and DAPK1 (Fig. 4G). None of the genes showed much difference across the disease sites of cancerous and non-cancerous tissue though p16 (Fig. 4F), it showed the smallest p-value of 0.072, which is not considered as significant by our criterion.
Fig. 4.
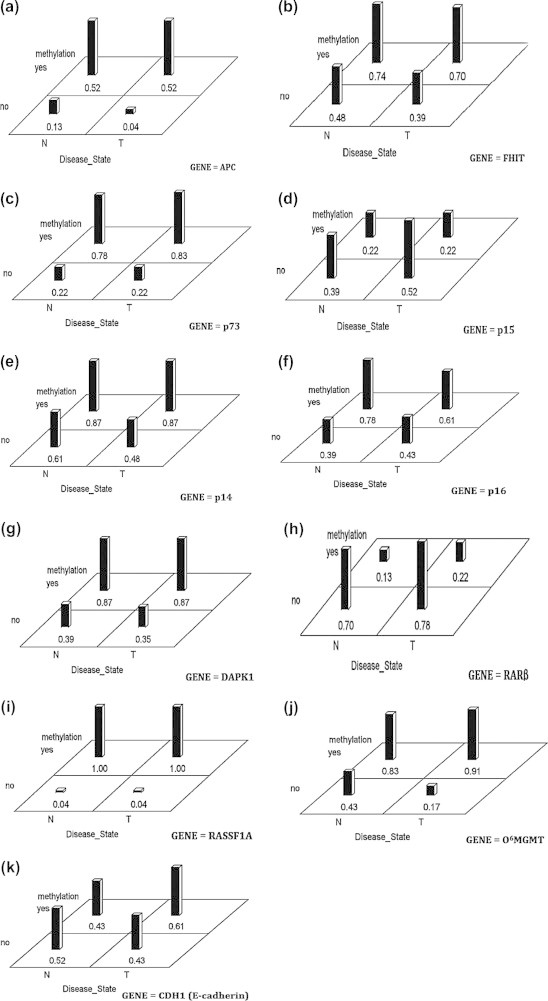
Differences across methylation profiles between HCC* cases and their ANT# samples with 0.0045 as a cut-off for significance. ∗ HCC = T. # ANT = N.
Analysis of DNA methylation status across the disease groups
Across group differences among the four groups enrolled (HCC, CAH, ASC, NHT) were analyzed using binary logistic regression in PROC LOGISTIC for each gene. Our results indicate that there is a significant interaction between disease state (groups) and DNA methylation of genes (Fig. 5A–K,). As shown in Fig. 4A, there is a significant group effect for APC (ASC group is different from HCC Group, p-value = 0.0006). As can be seen from the graph, the interaction is explained by the fact that there is a bigger difference between methylation and un-methylation for the CH group than any of the other groups especially the NHT. For DAPK1 (Fig. 5G), there is a marginal group effect, not significant by our corrected level of p value = 0.004 (NHT is different fromHCC p value = 0.007) and RARβ (Fig. 5H) (NHT is different fromHCC Group p-value = 0.007). In contrast, there are significant methylation effects for APC (p-value <0.0001), FHIT (p-value <0.0001), p15, (p-value = 0.003), p14 (p-value <0.0001), DAPK1 (p-value <0.0001), RARβ (p-value <0.0001) and E-cadherin (p-value <0.0001).
Fig. 5.
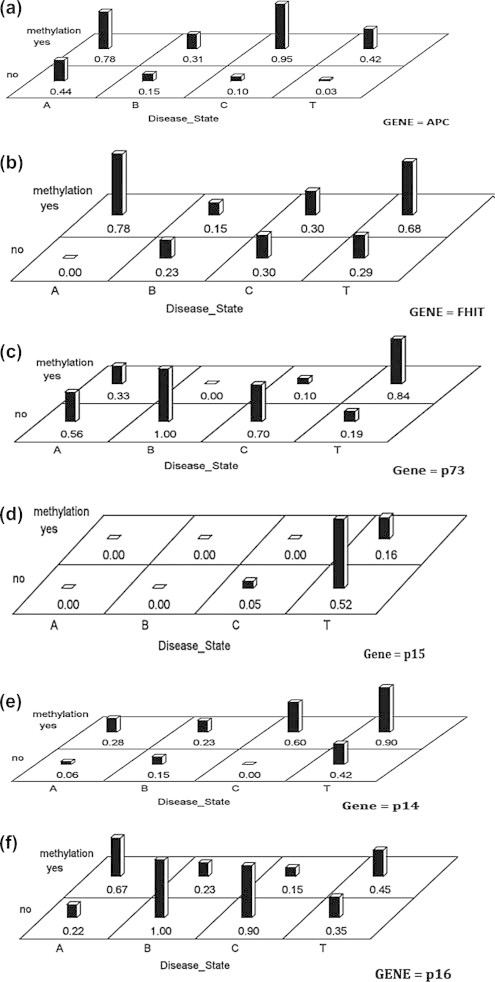
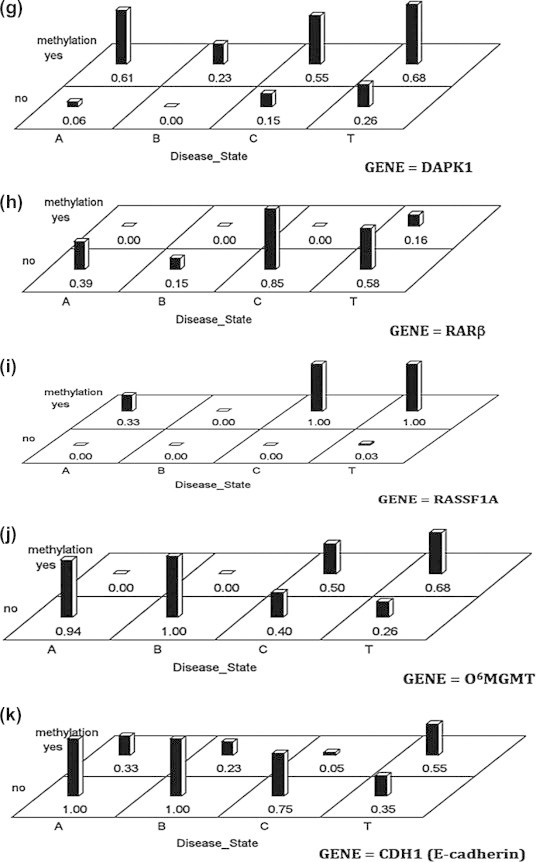
Differences in the methylation frequency among the four studied groups. (T = HCC, C = CAH with cirrhosis, A = asymptomatic carrier and B = normal hepatic tissue).
Analysis of methylation coordination
Coordination of methylation at the 11 tested genes was analyzed by the Mann–Whitney U test through comparing the status of each gene (M or U) with the MI calculated with the remaining genes. A summary of methylation results and concordance tests of each locus in HCC patients is shown in Table 4 and Fig. 2. The combined effect of the studied methylated genes as biomarkers for diagnosis of HCC and CAH has also been studied. When all significant variables were entered into the stepwise logistic regression, only APC, p73, p14, O6MGMT independently affected the classification of cases into HCC and CH as in Table 5. These four genes combined give an accuracy of 89.9%, sensitivity 83.9% and specificity 94.7%.
Table 4.
Summary of methylation specific PCR results and concordance tests of each locus in HCC samples.
| Factor | Concordance | Kappa# | p-Value⁎ |
|---|---|---|---|
| n = 31 | |||
| n (%) | |||
| APC | 28 (90.3) | 0.803 | <0.001 |
| FHIT | 24 (77.4) | 0.497 | 0.006 |
| P15 | 31 (100.0) | 1.000 | <0.001 |
| P73 | 18 (58.1) | −0.248 | 0.150 |
| P14 | 31 (100.0) | 1.000 | <0.001 |
| P16 | 24 (77.4) | 0.558 | 0.001 |
| DAPK | 22 (71.0) | 0.318 | 0.076 |
| RARβ | 27 (87.1) | 0.431 | 0.012 |
| RASSF | 31 (100.0) | – | – |
| O6O6-MGMT | 26 (83.9) | 0.640 | <0.001 |
| CDH1 | 22 (71.0) | 0.425 | 0.016 |
– Numbers are too small for a valid statistical analysis.
Kappa measure of agreement.
p-Values ⩽ 0.05 are considered significant.
Table 5.
Stepwise logistic regression for HCC.
| Parameter | Regression estimate | p-Value | Odds ratio | 95% CI for OR | |
|---|---|---|---|---|---|
| APC | −3.606 | 0.003 | 0.027 | 0.003 | 0.287 |
| p73 | 3.671 | 0.001 | 39.302 | 4.752 | 325.017 |
| P14 | 3.638 | 0.009 | 38.014 | 2.492 | 579.829 |
| O6-MGMT | 2.589 | 0.014 | 13.311 | 1.685 | 105.132 |
Immunohistochemistry
Protein expression of two of APC and O6MGMT was assessed in 20 NHT samples, 20 HCC and 20 CH tissues as well as in an additional set of samples including 40 NHT, 52 CH and 107 HCC tissue samples for confirmation of the methylation results. In the original set, cytoplasmic immunostaining for the APC protein was detected in 11 (55%) NHT, with loss of staining in 10 (50%) CH, and 15 (75%) HCC. As for the confirmatory set, we were able to detect cytoplasmic immunostaining for the APC protein in 20 (50%) NHT, with loss of staining in 30 (57.7%) CH, and 77 (72%) HCC tissues. On the other hand, nuclear immunostaining for O6MGMT protein was detected in 13 (65%) NHT with loss of expression in 11(55%) CH and 16 (80%) HCC of the original set. While in the confirmatory set O6MGMT protein were lost in 26 (50%) CH and 70 (65.4%) HCC cases (Fig. 6).
Fig. 6.
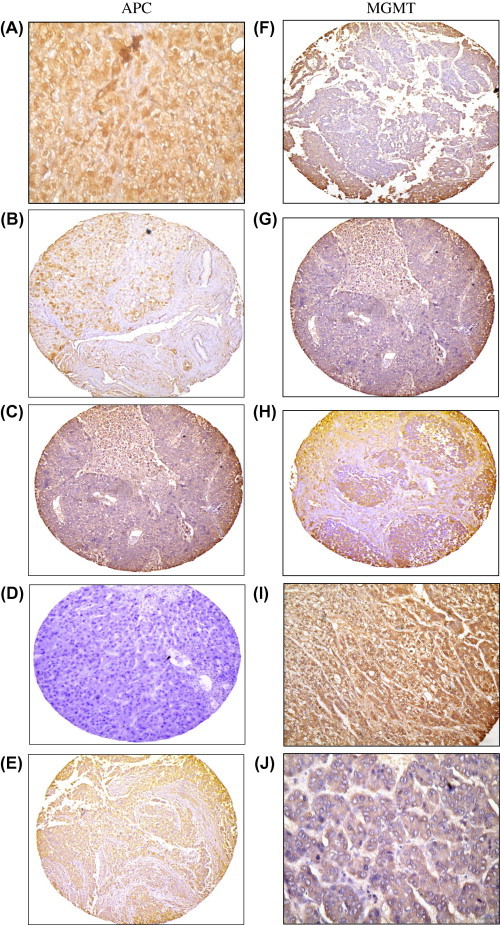
(A) Normal hepatic tissue sample showing positive cytoplasmic immunostaining for APC (X200) B: A case of HCV induced chronic hepatitis showing mild focal cytoplasmic immunostaining for APC (X100). C: A case of HCV induced chronic hepatitis with cirrhosis negative for APC (X100). D: A case of HCV-associated HCC negative for APC (X100). E: A case of HCV-associated HCC with positive cytoplasmic immunostaining for APC (X40). (F) Normal hepatic tissue negative for MGMT (X100). (G): A case of HCV-induced chronic hepatitis with cirrhosis negative for MGMT (X100). (H) A case of HCV-induced chronic hepatitis with cirrhosis positive for MGMT immunostaining (X100). (I) A case of HCV-induced HCC with marked cytoplasmic immunostaining for MGMT(X200). (J) A case of HCV-induced HCC showing faint cytoplasmic immunostaining for MGMT(X200).
Discussion
Changes in DNA methylation patterns of TSGs play a role in the development and progression of many tumor types. However, data regarding HCC show wide variability in the results that could be attributed to several factors including the underlying etiologic factor(s) [18,19]. Our study is the first to assess the role of DNA PM of a well selected panel of genes in clinical samples obtained from a cohort of patient population infected with HCV/genotype 4 in an attempt to understand their impact on disease progression.
We have previously reported a high methylation frequency of APC, FHIT, CDH1 and p16 in the plasma and tissues of 28 HBV and HCV-associated HCC patients from Egypt [15]. Therefore, we sought to confirm this data in a larger cohort of HCV- genotype 4 infected patients, including asymptomatic carriers, CH with cirrhosis and HCC using 11 genes that are commonly hypermethylated in several tumor types. We determined several differentially methylated genes both in liver tissues and PBL that represent the progression from NHT to CH and HCC in HCV genotype 4-infected persons. We also identified a panel of genes (APC, p73, p14, O6MGMT) that can independently affect the classification of cases into HCC and CH with 89.9% accuracy, 83.9% sensitivity and 94.7% specificity.
A high methylation frequency was reported for all studied genes, except for p15, in the PBL and tissues with increasing MI as the disease progresses. Our data regarding the p15 gene confirms our previous study where p15 methylation was reported in 14.2% only of HCC cases [15]. Within the studied groups, the methylation frequency of p14, p73, RASSF1A and O6MGMT was significantly higher in HCC and their ANT compared to CH and the NHT samples whereas PM of APC was significantly higher in CH patients. This applied to PBL and tissues except for RASSF1A and O6MGMT, where the difference in methylation frequency in PBL was statistically insignificant. The high methylation frequency reported here confirms the results of some previous studies including that of Archer [20] who found a high methylation frequency of their studied genes in HCV-associated HCC compared to HBV-associated cases or to NHT. They concluded that the virus may induce accelerated hypermethylation even in the early stages of infection, which can subsequently lead to tumor development.
RASSF1A, a candidate TSG, frequently shows hypermethylation and loss of heterozygosity with consequent gene silencing in several human cancers [21]. In HCC, PM of RASSF1A gene was reported in 78–95% of the studied cases [22–24]. Our results are comparable to these studies since RASSF1A methylation was detected in all HCC and in 68.4% CH cases (second only to APC). Our results are also comparable to Gioia et al. [25] who reported an increase in RASSF1A methylation with progression from regenerative conditions (cirrhosis) to hepatocellular nodules and HCC as well as with Chan et al. [26] who reported RASSF1A methylation in the blood and tissues of HCC patients.
Within the identified panel of genes that independently affected the classification of cases into HCC and CH in this study, p14 showed a high methylation frequency in HCC cases. Our data confirms those of Anzola et al. [27] and Yang et al. [28] who demonstrated that p14 PM is associated with the pathogenesis of HCC and suggested that inactivation of p14 through PM could be an important mechanism for HCV-induced HCC. The fact that we were able to detect PM of the p14 gene in NHT as well as in CH with almost the same frequency suggests that it might be an early event during the cascade of HCV-induced HCC. In contrast, p16PM did not show a similar profile suggesting that p14 and p16 are regulated by different promoters [29].
Our results show an increasing frequency of p16 PM from NHT to HCC which is in agreement with the some previous studies of Vivekanandan and Torbenson [30].
Similar to p14, O6MGMT plays an important role in cyto-protection by preventing DNA damage and triggering DNA repair mechanisms [31]. Because O6MGMT methylation is a hallmark of specific cancers, it is perhaps not surprising to find a consistent PM in O6MGMT in both CH and HCC tissues. Our results show a significant increase in the frequency of O6MGMTPM from CH (26%) to HCC (67.7%) providing an evidence that this gene could differentiate between CH and HCC. Literature reviews also revealed varied frequencies of O6MGMT PM in HCC ranging from 0% [32], to 22–39% [33]. The variability in the results could be attributed to several factors including the sensitivity of the PCR, the primer sequences and the differences in CpG sites, the etiological factors contributing to HCC and the geographical differences. The significant association reported here between O6MGMT hypermethylation and HCV infection is also comparable to previously published data [22,33].
Our results also show a significant difference in the methylation frequency of APC and CDH1 between CH and HCC cases where APC was more frequent in the first group and CDH1 in the second group. Methylation of APC and CDH1 genes has been previously reported by Yang et al. [28] who demonstrated that PM of APC and CDH1 are more frequent in HBV- and HCV-positive HCC than in HBV- and HCV-negative cases. PM of these genes was also reported by other investigators [34]. Nomoto et al. [32] reported APC PM in 88.2% of the NHT compared to 21.6% in chronic hepatitis with cirrhosis and 82.4% in HCC. They claimed that loss of APC in cirrhotic and inflammatory cases could possibly be attributed to the presence of inflammatory cells and fibroblasts. In contrast, we reported a high methylation frequency of the APCPM in CH patients, both in blood and tissues. The difference between our results and those of Nomoto et al. [32] could be attributed to (a) their smaller sample size (19 cases); (b) samples of CH and cirrhosis were obtained from the HCC cases and not from separate patients or (c) a possibly different etiology since viral infection was not mentioned in their study.
Finally, PM of thep73 was also reported in 83.9% of the HCC cases assessed in the current study compared to 21.1% of CH and none of the NHT. Thus p73 PM could be used to differentiate between CH and HCC cases even in patient’s blood. PM of the p73 was reported in some previous studies on HCC and CH [34].
Conclusion
We conclude that aberrant DNA PM of multiple cancer-related genes is associated with different stages of disease progression from hepatitis to HCC since PM of p73, p14, O6-MGMT was associated with HCC whereas aberrant PM of APC was more common in CH. APC PM could be used as a maker for early detection of HCV-induced chronic active hepatitis. In our study the (APC, p73, p14, O6-MGMT) panel independently affected the classification of cases into HCC and CH with high accuracy (89.9%), sensitivity (83.9%) and specificity (94.7%). Moreover, detection of PM of certain genes in PBL is a highly sensitive and specific, noninvasive indicates that blood could be used, as efficiently as tissue biopsies, to assess PM which could help in the follow-up of chronic hepatitis patients and possibly for early detection of HCC, especially when using well-selected panel.
To study the combined effect of the different markers on the diagnosis of HCC compared to CAH. All significant variables were entered into the stepwise logistic regression. The above variables are the ones which independently affects the classification of cases into HCC and CAH. These four variables combined will give an accuracy of 89.9%, sensitivity 83.9% and specificity 94.7%.
Conflict of interest
The author has declared no conflict of interest.
Footnotes
Peer review under responsibility of Cairo University.

References
- 1.El-Serag H.B. Epidemiology of hepatocellular carcinoma in USA. Hepatol Res. 2007;2:S88–S94. doi: 10.1111/j.1872-034X.2007.00168.x. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A., Bray F., Center M.M., Ferlay J., Ward E., Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 3.Anwar W.A., Khaled H.M., Amra H.A., El-Nezami H., Loffredo C.A. Changing pattern of hepatocellular carcinoma (HCC) and its risk factors in Egypt: possibilities for prevention. Mutat Res. 2008;659(1–2):176–184. doi: 10.1016/j.mrrev.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 4.Lau W., Lai E.C.H. Hepatocellular carcinoma: current management and recent advances. Hepatobiliary Pancreat Dis Int. 2008;7(3):237–257. [PubMed] [Google Scholar]
- 5.Lehman E.M., Wilson M.L. Epidemiology of hepatitis viruses among hepatocellular carcinoma cases and healthy people in Egypt: a systematic review and meta-analysis. Int J Cancer. 2009;124(3):690–697. doi: 10.1002/ijc.23937. [DOI] [PubMed] [Google Scholar]
- 6.Baylin S.B., Herman J.G. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet. 2000;16:168–174. doi: 10.1016/s0168-9525(99)01971-x. [DOI] [PubMed] [Google Scholar]
- 7.Feinberg A.P. Cancer epigenetics takes center stage. Proc Natl Acad Sci USA. 2001;98:392–394. doi: 10.1073/pnas.98.2.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang C., Li H., Zhou G., Zhang Q., Zhang T., Li J. Transcriptional silencing of the TMS1/ASC tumour suppressor gene by an epigenetic mechanism in hepatocellular carcinoma cells. J Pathol. 2007;212(2):134–361. doi: 10.1002/path.2173. [DOI] [PubMed] [Google Scholar]
- 9.Teodoridis J.M., Hardie C., Brown R. CpG island methylator phenotype (CIMP) in cancer: causes and implications. Cancer Lett. 2008;268(2):177–355. doi: 10.1016/j.canlet.2008.03.022. [DOI] [PubMed] [Google Scholar]
- 10.Tsou J.A., Galler J.S., Wali A., Ye W., Siegmund K.D., Groshen S. DNA methylation profile of 28 potential marker loci in malignant mesothelioma. Lung Cancer. 2007;58(2):220–230. doi: 10.1016/j.lungcan.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lehman E.M., Wilson M.L. Epidemiology of hepatitis viruses among hepatocellular cases and healthy people in Egypt: a systematic review and meta-analysis. Int J Cancer. 2009;24:690–697. doi: 10.1002/ijc.23937. [DOI] [PubMed] [Google Scholar]
- 12.Hamilton S.R., Aaltonen L.A. World health organization classification of tumors. IARC Press; Lyon: 2000. (Pathology and genetics of tumors of digestive system). p. 157–202. [Google Scholar]
- 13.Green FL, Page DL, Fleming ID, Fritz AG, Balch CM, Haller DG, et al., editors. AJCC Cancer Staging Manual, 6th ed. New York: Springer; 2002.
- 14.Ishak K., Baptista A., Bianchi L., Callea F., Degroote J., Denk F. Histopathological staging and grading of chronic hepatitis. J Hepatol. 1995;22:696–699. doi: 10.1016/0168-8278(95)80226-6. [DOI] [PubMed] [Google Scholar]
- 15.Iyer P., Zekri A.R., Hung C.W., Schiefelbein E., Ismail K., Hablas S. Concordance of DNA methylation pattern in plasma and tumor DNA of Egyptian hepatocellular carcinoma patients. Exp Mol Pathol. 2010;88:107–111. doi: 10.1016/j.yexmp.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gutiérrez M.I., Siraj A.K., Bhargava M., Ozbek U., Banavali S., Chaudhary M.A. Concurrent methylation of multiple genes in childhood ALL: correlation with phenotype and molecular subgroup. Leukemia. 2003;17:1845–1850. doi: 10.1038/sj.leu.2403060. [DOI] [PubMed] [Google Scholar]
- 17.Bahnassy A.A., Zekri A.R., Loutfy S.A., Mohamed W.S., Abdel Moneim A., Salem E.S. The role of cyclins and cyclin dependent kinases in development and progression of hepatitis C virus-genotype 4-associated hepatitis and hepatocellular carcinoma. Exp Mol Pathol. 2011;91:643–652. doi: 10.1016/j.yexmp.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 18.Berdasco M., Esteller M. Aberrant epigenetic landscape in cancer: how cellular identity goes away. Dev Cell. 2010;19:698–711. doi: 10.1016/j.devcel.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 19.Zaina S., Pérez-Luque E.L., Lund G. Genetics talks to epigenetics? The interplay between sequence variants and chromatin structure. Curr Genom. 2010;11:359–367. doi: 10.2174/138920210791616662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Archer K.J. High-throughput assessment of CpG site methylation for distinguishing between HCV-cirrhosis and HCV-associated hepatocellular carcinoma. Mol Genet Genom. 2010;283(4):341–349. doi: 10.1007/s00438-010-0522-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hesson L.B., Cooper W.N., Latif F. The role of RASSF1A methylation in cancer. Dis Markers. 2007;23:73–87. doi: 10.1155/2007/291538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lia Z., Zhangb H., Yangb J., Haoc T., Li S. Promoter hypermethylation of DNA damage response genes in hepatocellular carcinoma. Cell Biol Int. 2012;36:427–432. doi: 10.1042/CBI20100851. [DOI] [PubMed] [Google Scholar]
- 23.Zhong S., Yeo W., Tang M.W., Wong N., Lai P.B., Johnson P.J. Intensive hypermethylation of the CpG island of Ras association domain family 1A in hepatitis B virus-associated hepatocellular carcinomas. Clin Cancer Res. 2003;9:3376–3382. [PubMed] [Google Scholar]
- 24.Tischoff I., Markwarth A., Witzigmann H., Uhlmann D., Hauss J., Mirmohammadsadegh A. Allele loss and epigenetic inactivation of 3p21.3 in malignant liver tumors. Int J Cancer. 2005;115:684–689. doi: 10.1002/ijc.20944. [DOI] [PubMed] [Google Scholar]
- 25.Gioia S., Bianchi P., Destro A., Grizzi F., Malesci A., Laghi L. Quantitative evaluation of RASSF1A methylation in the non-lesional, regenerative and neoplastic liver. BMC Cancer. 2006;89(6):1471–2407. doi: 10.1186/1471-2407-6-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan K.C., Lai P.B., Mok T.S., Chan H.L., Ding C., Yeung S.W. Quantitative analysis of circulating methylated DNA as a biomarker for hepatocellular carcinoma. Clin Chem. 2008;54:1528–1536. doi: 10.1373/clinchem.2008.104653. [DOI] [PubMed] [Google Scholar]
- 27.Anzola M., Cuevas N., López-Martinez M., Saiz A., Burgos J.J. Martinez de Pancorbo M. p14ARF gene alterations in human hepatocellular carcinoma. Eur J Gastroenterol Hepatol. 2004;16:19–26. doi: 10.1097/00042737-200401000-00004. [DOI] [PubMed] [Google Scholar]
- 28.Yang B., Guo M., Herman J.G., Clark D.P. Aberrant promoter methylation profiles of tumor suppressor genes in hepatocellular carcinoma. Am J Pathol. 2003;163(3):1101–1107. doi: 10.1016/S0002-9440(10)63469-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Esteller M., Tortola S., Toyota M., Capella G., Peinado M.A., Baylin S.B. Hypermethylation-associated inactivation of p14 (ARF) is independent of p16 (INK4a) methylation and p53 mutational status. Cancer Res. 2000;60:129–133. [PubMed] [Google Scholar]
- 30.Vivekanandan P., Torbenson M. Epigenetic instability is rare in fibrolamellar carcinomas but common in viral-associated hepatocellular carcinomas. Modern Pathol. 2008;21:670–675. doi: 10.1038/modpathol.2008.32. [DOI] [PubMed] [Google Scholar]
- 31.Lenz G., Hutter G., Hiddemann W., Dreyling M. Promoter methylation and expression of DNA repair genes hMLH1 and O6-MGMT in acute myeloid leukemia. Ann Hematol. 2004;83:628–633. doi: 10.1007/s00277-004-0925-0. [DOI] [PubMed] [Google Scholar]
- 32.Nomoto S., Kinoshita T., Kato K., Otani S., Kasuya H., Takeda S. Hypermethylation of multiple genes as clonal markers in multicentric hepatocellular carcinoma. Br J Cancer. 2007;97:1260–1265. doi: 10.1038/sj.bjc.6604016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsukura S., Soejima H., Nakagawachi T., Yakushiji H., Ogawa A., Fukuhara M. CpG methylation of O6-MGMT and hMLH1 promoter in hepatocellular carcinoma associated with hepatitis viral infection. Br J Cancer. 2003;88:521–529. doi: 10.1038/sj.bjc.6600743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsumura T., Makino R., Mitamura K. Frequent downregulation of E-cadherin by genetic and epigenetic changes in the malignant progression of hepatocellular carcinomas. Clin Cancer Res. 2001;20017:594–599. [PubMed] [Google Scholar]