

ABSTRACT
Mutations in inositol polyphosphate 5-phosphatase E (INPP5E) cause the ciliopathies known as Joubert and MORM syndromes; however, the role of INPP5E in ciliary biology is not well understood. Here, we describe an interaction between INPP5E and AURKA, a centrosomal kinase that regulates mitosis and ciliary disassembly, and we show that this interaction is important for the stability of primary cilia. Furthermore, AURKA phosphorylates INPP5E and thereby increases its 5-phosphatase activity, which in turn promotes transcriptional downregulation of AURKA, partly through an AKT-dependent mechanism. These findings establish the first direct link between AURKA and phosphoinositide signaling and suggest that the function of INPP5E in cilia is at least partly mediated by its interactions with AURKA.
KEY WORDS: AURKA, INPP5E, Primary cilium
INTRODUCTION
Primary cilia are dynamic structures, which grow and resorb in response to cues arising from the cell division cycle and the environment (Seeley and Nachury, 2010). Ciliary dysfunction underlies the pathogenesis of human ciliopathies (Hildebrandt et al., 2011), a group of inherited developmental disorders that include Joubert Syndrome (JBTS), polycystic kidney disease (PKD), Bardet-Biedel Syndrome and others. Recent studies have shown that mutations in inositol polyphosphate-5-phosphatase E (INPP5E) cause Joubert Syndrome (Bielas et al., 2009) and MORM (mental retardation, truncal obesity, retinal dystrophy and micropenis) (Jacoby et al., 2009), thereby linking INPP5E function to primary cilia.
INPP5E is best known for its role in hydrolyzing phosphatidylinositol (3,4,5)-trisphosphate [PtdIns(3,4,5)P3] and phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2] to form PtdIns(3,4)P2 and phosphatidylinositol 4-phosphate [PtdIns(4)P], respectively, creating second messengers that mediate cell responses to various stimuli (Conduit et al., 2012). INPP5E localizes to the primary cilium, and Inpp5e−/− mice develop ciliopathic phenotypes including polycystic kidneys, polydactyly and neural tube closure defects (Jacoby et al., 2009). They also have abnormally shortened cilia and exhibit decreased stability of pre-established cilia after induction of ciliary disassembly (Jacoby et al., 2009). These observations suggest that INPP5E also stabilizes cilia structure and length in growth-factor-stimulated quiescent cells. INPP5E interacts with several ciliary and centrosomal proteins (ARL13B, PDE6D and CEP164) that mediate INPP5E targeting to the cilium (Humbert et al., 2012). However, none of these proteins have been linked to regulation of the stability of primary cilia. Indeed the mechanism by which loss of INPP5E leads to decreased cilia stability is very poorly understood.
Aurora kinase A is an important regulator of mitosis, where it is associated with centrosome maturation and assembly and stability of the spindle body. However, a distantly related Chlamydomonas ortholog, CALK, regulates the resorption of the flagella (a structure that is in some respects analogous to the mammalian cilium) in response to altered ionic conditions or cues for mating (Pan et al., 2004). Chlamydomonas has proven invaluable in predicting the function of cilia-associated genes, and these observations suggest an important role for AURKA in the regulation of cilia disassembly. Indeed, in mammalian cells, AURKA localizes to the basal body of the cilium and induces ciliary resorption in response to growth factor stimulation (Pugacheva et al., 2007). In this way, it plays a central role as a regulator of cilia stability. Furthermore, AURKA-mediated phosphorylation of the polycystic kidney disease protein PKD2 also influences intracellular Ca2+ levels through a cilia-based mechanism (Plotnikova et al., 2011). Aside from AURKA, there are a number of other molecules that are thought to regulate ciliary resorption, including the NIMA-related kinase (NRK) family of proteins found in the highly ciliated organism Tetrahymena (Wloga et al., 2008). NRKs induce the resorption of specific sets of cilia on the surface of the organism. Cilia resorption and accelerated G1-S progression has also been shown to be regulated in mammalian cells as a result of insulin-like growth factor 1 (IGF-1)-mediated activation of its receptor, by way of non-canonical Gβγ signaling and T94-phosphorylated Tctex-1 present at the base of the cilium (Yeh et al., 2013). Despite these findings, the mechanistic basis for cilium disassembly and, importantly, the inter-relationships between the cell signaling pathways involved, remain to be characterised.
In this paper, we report a direct functional interaction between INPP5E and AURKA, and we demonstrate that this interaction is important for regulating ciliary stability under normal and pathological conditions. These studies establish the first direct link between AURKA and phosphoinositide signaling; observations that will be crucial to better understanding the diseases associated with dysregulation of both proteins and the cell signaling pathways they control.
RESULTS
AURKA directly interacts with INPP5E
Given their apparent functional overlap in regulating cilia stability, we first asked whether INPP5E and AURKA themselves interact. Co-immunoprecipitation experiments in transfected kidney cells showed that overexpressed HA-tagged INPP5E bound to both full-length RFP-tagged AURKA and its non-catalytic N-terminal regulatory domain, but not to the AURKA catalytic domain alone (Fig. 1A). Reciprocal interactions were also confirmed between co-overexpressed AURKA and HA-tagged INPP5E (supplementary material Fig. S1A). We also established that endogenous AURKA and overexpressed INPP5E co-immunoprecipitated from murine inner medullary collecting duct (IMCD3) cells (Fig. 1B). We then demonstrated that the interaction between these proteins was direct, as purified recombinant AURKA robustly co-immunoprecipitated with purified recombinant GST–INPP5E (Fig. 1C). These preliminary studies established that INPP5E interacts with AURKA, most likely through direct binding with the AURKA regulatory domain.
Fig. 1.

AURKA directly interacts with INPP5E. (A) RFP-fused full-length (FL) AURKA and catalytic (cat; amino acids 132–403) and non-catalytic (non cat; amino acids 1–131) domains were coexpressed with HA–INPP5E in HEK293 cells and immunoprecipitated (IP) with anti-RFP antibody. Immunoprecipitates were western blotted with the antibodies indicated. (B) Endogenous AURKA was immunoprecipitated from IMCD3 cells expressing HA–INPP5E and blotted as indicated. Immunoglobulin G (IgG) was used as negative control for immunoprecipitation. (C) AURKA was immunoprecipitated from the in vitro mixture containing GST–INPP5E or GST only and recombinant His–AURKA, and immunoprecipitates were probed by western blotting (anti-GST, left panel). Starting material is shown on the right (Input). The asterisk indicates the heavy chain of IgG. (D) Anti-AURKA antibody was used to immunoprecipitate endogenous AURKA from whole IMCD3 cell lysates expressing vector alone, FLAG-tagged full-length INPP5E and INPP5E deletion mutants, followed by western blotting as indicated. Molecular masses are indicated in kDa. The graph indicates the ratio of precipitated to total INPP5E, quantified from analysis of blots from three independent experiments. Data show the mean±s.e.m.; *P<0.001 compared to full-length INPP5E (Student's t-test). (E) Schematic representation of INPP5E variants used for AURKA binding studies. The proline-rich domain (PRD) is indicated in blue, inositol polyphosphate phosphatase catalytic (IPPc) domain in red, CaaX motif is in light blue and the SH3 domain is in yellow. The extent to which each construct localizes to the cilium and binds to AURKA is indicated on the right. +, weak binding; ++, medium binding; +++, strong binding; −, absence of binding. (F) Immunofluorescent detection of AURKA (green) and INPP5E (red) in IMCD3 cells demonstrating basal body localization of AURKA and ciliary axoneme localization of INPP5E, overlapping with AURKA signal at basal end of cilium (arrowheads). 4′,6-diamidino-2-phenylindole (DAPI) staining indicates the nucleus (blue). Scale bar: 10 µm. Insets show a magnification of the basal body and cilium.
INPP5E consists of a proline-rich domain that encompasses two SH3 binding sites, the inositol polyphosphate phosphatase catalytic domain (IPPc) and a CaaX motif with an adjacent ciliary-targeting sequence (Humbert et al., 2012). We mapped the minimal domain required for interaction with AURKA to the IPPc region, although the presence of the SH3 binding sites and CaaX domain enhanced binding (Fig. 1D,E). This is consistent with previous studies showing that the presence of the SH3 domain in NEDD9 is able to mediate its binding to AURKA (Pugacheva and Golemis, 2005). Whether the SH3 and CaaX sites enhance binding by creating additional contact points between the proteins, by enhancing colocalization or by contributing to proper protein conformation remains to be determined.
INPP5E localized principally to the ciliary axoneme and, to a lesser extent, to the basal end of cilia, consistently showing spatial overlap with AURKA (Fig. 1F; supplementary material Fig. S1B). Importantly, we observed negligible colocalization of the proteins in any other subcellular compartment. To examine whether the ciliary localization of INPP5E was dependent on AURKA activity, we employed a specific AURKA inhibitor, C1368 (Zhang et al., 2006; Plotnikova et al., 2011), but this did not alter INPP5E localization (supplementary material Fig. S1C,D). Basal body localization of AURKA also was unchanged in the absence of INPP5E (Fig. 3A).
Fig. 3.

AURKA levels are regulated by INPP5E. (A) Immunofluorescence detection of AURKA (green) or T288-phosphorylated AURKA (phAURKA, green), acetylated α-tubulin (cilia, red) and DAPI (DNA, blue) in primary fibroblasts from Inpp5e+/+ (WT) and Inpp5e−/− (−/−) embryos with or without serum as indicated. Arrowheads indicate primary cilia. Scale bar: 10 µm. Insets show magnifications of the centrosome and cilium. The graphs show the relative intensity of phosphorylated (ph)AURKA or AURKA in the basal body from three triplicate assays measuring 50 cells per assay. Data show the mean±s.e.m.; *P = 0.003, **P = 0.013 (for phosphorylated AURKA) and *P = 0.0012, **P = 0.014 (for AURKA; Student's t-test). (B) Western blot analysis of Ser473-phosphorylated AKT and AURKA in Inpp5e+/+ and Inpp5e−/− embryonic fibroblasts. Graphs indicate the ratio of either AURKA, phosphorylated AKT or AKT to actin loading control, from analysis of three blots. Data show the mean±s.e.m.; *P<0.05. (C) IMCD3 cells were transfected with INPP5E-specific siRNA (siINPP5E) or scrambled control siRNA (Scr). Graphs indicate the ratio of AURKA to tubulin loading control, quantified from three blots. Data show the mean±s.e.m.; *P = 0.0045. (D) IMCD3 cells were transfected and western blotted as indicated. Graphs indicate the ratio of AURKA to tubulin loading control, quantified from three blots; Data show the mean±s.e.m.; *P≤0.0032 (compared with pcDNA). (E) mRNA expression levels of Aurka were determined from Inpp5e+/+ and Inpp5e−/− embryonic fibroblast cultures by real-time RT-PCR, and data are presented as the fold change relative to the wild-type control. Data show the mean±s.e.m.; *P = 0.017. (F) Plasmids expressing HA–INPP5E were transfected into HEK293 cells, and lysates were collected at the indicated time-points. Western blotting was performed with the indicated antibodies. Molecular masses are indicated in kDa. (G) AURKA levels and the relative phosphorylation of AKT and INPP5E were quantified from the results of the experiment presented in F. Data are expressed as the mean±s.e.m. (three experiments). (H) IMCD3 cells were siRNA transfected then treated for 12 h with 10 µM AKT inhibitor MK2206 or DMSO. Lysates were collected at the indicated time-points and western blotted as indicated. (I) The ratio of AURKA or phosphorylated AKT to loading control actin under different conditions following AUKRA inhibition, as quantified from analysis of three blots. Data show the mean±s.e.m.; *P<0.01.
Interactions between AURKA and INPP5E affect the activity of both proteins
AURKA phosphorylates a wide range of protein substrates relevant to the regulation of ciliary disassembly and to other canonical AURKA functions, such as mitotic regulation (Nikonova et al., 2013). To examine whether INPP5E is a substrate for AURKA, we performed an in vitro kinase assay using recombinant proteins. Recombinant activated AURKA was able to phosphorylate INPP5E (Fig. 2A), and the presence of INPP5E in the reaction did not affect AURKA activity on a control substrate, recombinant histone H3 (HH3) (Fig. 2A). However, the reverse was not the case, as incubation with recombinant AURKA enhanced INPP5E phosphatase activity towards PtdIns(3,4,5)P3 in vitro (Fig. 2B; supplementary material Fig. S2A). This enhancement required AURKA catalytic activity, as inhibition of AURKA with its inhibitor C1368 in INPP5E-transfected HEK293 cells caused a dose-dependent decrease in 5-phosphatase activity (Fig. 2C; supplementary material Fig. S2B–D).
Fig. 2.

Interactions between AURKA and INPP5E affect the activity of both proteins. (A) In vitro kinase assay. The upper panel indicates γ-[32P]ATP signal corresponding to phosphorylated GST–INPP5E (∼110 kDa), auto-phosphorylated (ph)AURKA (∼50 kDa) and phosphorylated histone H3 (phHH3, ∼16 kDa). The lower panel shows input proteins western blotted as indicated. (B) Recombinant active AURKA activates the hydrolysis of PtdIns(3,4,5)P3 by INPP5E (n = 3 for each sample). Data show the mean±s.e.m.; *P = 0.014 (Student's t-test). (C) Quantification of INPP5E 5-phosphatase activity against PtdIns(3,4,5)P3 upon AURKA inhibition with the indicated concentrations of C1368 in HEK293 cells overexpressing HA–INPP5E or empty vector. Data show the mean±s.e.m.; *P<0.01 (Student's t-test). Results were normalized to those of the vector-expressing cells to reduce background caused by the presence of other phosphatases in the cells. Non-normalized data are shown in supplementary material Fig. S2D. (D) HEK293 cells were co-transfected with plasmids expressing the indicated proteins. Cell lysates were separated and probed with the antibodies indicated. Blots from three independent experiments were quantified and the relative phosphorylation of AURKA was calculated (lower panel). Data show the mean±s.e.m.; *P = 0.002 (Student's t-test). (E) HEK293 cells were co-transfected with the indicated plasmids. AURKA was immunoprecipitated (IP) and a fraction of the sample was used in an in vitro kinase assay to detect AURKA auto-phosphorylation and AURKA activity towards HH3. A second fraction was used to assess AURKA immunoprecipitation with HA–INPP5E. The kinase dead AURKA mutant K162D was used as a negative control for specificity of AURKA phosphorylation. (F) Graphs indicate the ratio of phosphorylated HH3 to total HH3 (upper panel) and of immunoprecipitated INPP5E to input in cell lysates. Data were quantified from three blots and show the mean±s.e.m.; *P<0.05 (Student's t-test). (G,H) Western blot analysis of lysates (G) and immunoprecipitates (H) from HEK293 cells co-transfected with RFP–AURKA and either wild-type (WT) INPP5E or the indicated INPP5E JBTS mutants. Plasmid V5-LacZ was used as a negative control for immunoprecipitation. (I) Western blot analysis of precipitates and lysates from HEK293 cells co-transfected with RFP–AURKA and the indicated phosphatases. Molecular masses are indicated in kDa.
These observations indicate that, in isolation, direct interaction with INPP5E does not affect the activation of AURKA. We next wished to determine whether this was the case in cells. A common mechanism for AURKA activation is through auto-phosphorylation of T288 (Walter et al., 2000). Although T288 AURKA phosphorylation was not demonstrably altered by interactions between recombinant proteins in vitro (Fig. 2A), we did observe marked increases in AURKA auto-phosphorylation upon coexpression of INPP5E and AURKA in cells (Fig. 2D), as well as concomitant increases in phosphorylation of the AURKA substrate HH3 (Fig. 2E,F). These results indicate that activation of AURKA is not induced directly by INPP5E, but instead results from downstream INPP5E-mediated signaling events. Interestingly, the capacity for INPP5E to activate AURKA in this context appeared to be dependent on its 5-phosphatase activity, as a catalytically inactive variant of INPP5E, D480N (Horan et al., 2007), did not increase AURKA T288 phosphorylation (Fig. 2D–F). For both AURKA and INPP5E, protein–protein interaction appeared to be dependent on catalytic function, as the capacity of D480N-INPP5E to bind to AURKA was also abrogated, and a kinase-dead version of AURKA (K162D) (Cheetham et al., 2002) was less able to bind INPP5E (Fig. 2E,F). To confirm that this change in binding was derived from altered activity and not from a specific conformational change associated with the D480N mutation, we tested whether a range of Joubert Syndrome (JBTS) missense mutations in INPP5E could influence AURKA binding and activity. Importantly, these changes are in different parts of the 5-phosphatase domain of the protein but all are thought to disrupt the phosphatase activity of INPP5E towards PtdIns(3,4,5)P3 (Bielas et al., 2009). Overexpression of V5-tagged wild-type and mutant versions of INPP5E demonstrated that JBTS missense mutants were unable to induce AURKA phosphorylation (Fig. 2G) or to interact with AURKA (Fig. 2H). Given the disparate nature of these changes, our findings suggested that both the activation of AURKA by INPP5E and the physical interaction of the proteins relied upon the capacity of INPP5E to dephosphorylate or bind to PtdIns(3,4,5)P3.
Given these observations, we hypothesized that the changes in activity and interaction might be mediated in some way by the relative levels of INPP5E metabolites. To investigate this possibility, we coexpressed AURKA with the 4-phosphatase INPP4B, which reduces levels of PtdIns(3,4)P2, the product of PtdIns(3,4,5)P3 hydrolysis mediated by INPP5E (Kisseleva et al., 2002). In doing so, we blocked the activation of AURKA but did not change the interaction between AURKA and INPP5E (Fig. 2I), indicating that only activation required the presence of PtdIns(3,4)P2.
AURKA levels are regulated by INPP5E
To establish the relevance of INPP5E–AURKA interactions in a physiological setting, we obtained murine embryonic fibroblasts (MEFs) from Inpp5e−/− embryos, which phenocopy those mouse models described previously (Jacoby et al., 2009). AURKA localises to the ciliary basal body in mammalian cells, where it regulates ciliary disassembly through interactions with NEDD9 and other proteins (Pugacheva et al., 2007). Increases in phosphorylation of AURKA are linked to increased cilia disassembly (Pugacheva et al., 2007; Plotnikova et al., 2012), whereas Inpp5e−/− MEFs have ciliary disassembly defects (Jacoby et al., 2009). In serum-starved primary fibroblasts from Inpp5e−/− mice, we observed an increase in the level of phosphorylated AURKA compared with that of wild-type cells. This differential was exacerbated upon treatment with serum to induce ciliary disassembly (Fig. 3A). Unexpectedly, analysis of total AURKA levels in Inpp5e−/− MEFs (Fig. 3B) indicated that the increase in the absolute levels of AURKA phosphorylation was not a consequence of specific activation of the protein. Instead, it was apparent that this was actually a reflection of increases in the total levels of AURKA protein within the cell. As further evidence that INPP5E regulates AURKA protein levels, depletion of INPP5E in IMCD3 cells by small interfering (si)RNA increased the amount of AURKA protein (Fig. 3C; supplementary material Fig. S3A). Conversely, overexpression of INPP5E (but not a catalytically inactive mutant) decreased AURKA protein levels (Fig. 3D).
One mechanism by which AURKA protein levels are regulated is through proteasome-mediated degradation of AURKA (Walter et al., 2000), but cycloheximide block-chase experiments indicated that this was not affected by INPP5E (supplementary material Fig. S3B). Instead, we found that Aurka mRNA expression in Inpp5e−/− fibroblasts was significantly increased compared with that of wild-type cells (Fig. 3E). Previous studies have shown that Aurka expression is regulated by AKT, a downstream mediator of phosphoinositide signaling (Liu et al., 2008). Decreased AKT phosphorylation has been reported in cell lines overexpressing Inpp5e (Kisseleva et al., 2002), and depletion of INPP5E or expression of JBTS mutants lacking 5-phosphatase activity in HEK293 cells results in elevated levels of phosphorylated (phospho)-AKT (Bielas et al., 2009). INPP5E primarily regulates AKT activity through hydrolysis of PtdIns(3,4,5)P3, which is essential for the effective activation of AKT (Kisseleva et al., 2002). Consistent with these previous findings, we found that Inpp5e−/− MEFs had increased levels of steady-state AKT and phospho-AKT (Fig. 3B), suggesting that increases in AURKA were mediated by AKT signaling. To test this hypothesis, we examined the dynamic changes in AURKA levels at different time-points following INPP5E overexpression. Overexpression of INPP5E decreased the amount of AURKA in a time-dependent manner and was accompanied by decreased AKT phosphorylation (Fig. 3F,G). To further evaluate how INPP5E governs Aurka expression through a phospho-AKT-dependent mechanism, we examined AURKA levels in IMCD3 cells treated with siINPP5E or scrambled control siRNA with or without the well-validated AKT inhibitor MK2206 (Pal et al., 2010). Inhibition of AKT phosphorylation with MK2206 abolished the increase in AURKA levels upon siINPP5E transfection, in a time-dependent manner (Fig. 3H,I; supplementary material Fig. S3C,D). We obtained similar results using another AKT inhibitor, AKTiX (Bertotti et al., 2009) (supplementary material Fig. S3E–I). These results confirmed that INPP5E regulates AURKA protein levels by transcriptional mechanisms that are mediated, at least in part, by AKT activity.
Functional dissection of the role of the AURKA–INPP5E interaction in primary cilia
Increases in cellular levels of phospho-AURKA are associated with an increase in cilia disassembly (Pugacheva et al., 2007; Plotnikova et al., 2012) and similar defects are observed in Inpp5e−/− cells (Bielas et al., 2009). To determine whether there was a shared functional basis to these phenotypes, we serum starved fibroblasts from wild-type and Inpp5e−/− mice and profiled cilia disassembly upon the addition of serum in the presence or absence of an AURKA inhibitor (C1368) or vehicle control (DMSO). This experiment confirmed the reduced number of ciliated Inpp5e−/− MEFs at 2–4 h after serum application compared with that of wild-type controls (Bielas et al., 2009) and showed that AURKA inhibition rescued the ciliary disassembly defects associated with loss of INPP5E (Fig. 4A). We obtained similar results using another AURKA inhibitor, MLN8237 (supplementary material Fig. S4A), and also in experiments depleting INPP5E in the IMCD3 cell line (supplementary material Fig. S4B). Thus, inhibition of AURKA is able to restore normal cilia stability in cells lacking INPP5E, demonstrating an opposing role for the two proteins in controlling cilia stability.
Fig. 4.

Functional role of the interaction between AURKA and INPP5E. (A) Cilia in Inpp5e+/+ (WT) and Inpp5e−/− (−/−) primary mouse embryonic fibroblasts (72 h under low-serum conditions) were induced to disassemble in the presence of 2 µM AURKA inhibitor (C1368) or DMSO control. All images show merged panels of the ciliary marker acetylated α-tubulin (green), the centrosome and basal body marker γ-tubulin (red) and DAPI (blue). Arrowheads indicate primary cilia. Scale bars: 10 µm. The percentage of ciliated cells at times before and after the indicated treatments are indicated in the graph on the right. Three triplicate assays were undertaken, counting 100 cells per assay. Data show the mean±s.e.m.; *P<0.01; ns, not significant (Student's t-test). (B) Immunofluorescence images of siRNA-treated (siINPP5E) IMCD3 cells with 2 µM C1368 AURKA inhibitor or DMSO control, grown in 3D Matrigel culture. Scr, scrambled control RNA; AcTub, acetylated tubulin (green, cilia); E-cadh, E-cadherin (red, cell membrane); DAPI, DNA staining (blue). (C) Quantitative analysis of spheroid numbers per well. Data show the mean±s.e.m.; *P<0.01 (Student's t-test).
Primary cilia are also known to play a central role in mediating cell polarity and lumen formation in cultured kidney cells (and in renal cysts). In addition to other ciliopathy features, Inpp5e−/− mice also develop renal cysts, a phenotype that is frequently associated with primary cilium defects (Jacoby et al., 2009). Taking into account the previously described role for AURKA in PKD (Plotnikova et al., 2011) and in the regulation of primary cilia stability (Pugacheva et al., 2007; Plotnikova et al., 2012), we evaluated the impact of altered AURKA function in a cellular model of INPP5E-dependent cyst formation. To examine whether the AURKA–INPP5E complex impacted upon these features, we depleted INPP5E from IMCD3 cells and grew them in a three-dimensional (3D) Matrigel matrix. The resulting cell colonies failed to form spheroids and instead developed as clusters of cells with irregular or non-existent lumens, randomly oriented shortened cilia and a lack of polarized expression of E-cadherin (Fig. 4B). These features are a common characteristic of cyst-lining cells (Wilson, 2004). To examine whether these phenotypes were a consequence of INPP5E-mediated dysregulation of AURKA, we treated the cultures with the AURKA inhibitor C1368. Doing so resulted in a striking rescue of spheroid formation and an increase in the length of luminal cilia (Fig. 4B,C). Taken together, these findings indicate that AURKA plays a crucial part in mediating the ciliary defects observed in Inpp5e−/− mice and suggest that manipulation of AURKA activity might influence cyst formation.
DISCUSSION
We have described a complex inter-relationship between the 5-phosphatase INPP5E and the AURKA kinase – an interaction that regulates the activity of both proteins at the primary cilium. We propose that hydrolysis of PtdIns(3,4,5)P3 by INPP5E alters the levels of different phosphoinositide species and, in so doing, mediates the functional interaction between INPP5E and AURKA, resulting in the activation of the latter protein through auto-phosphorylation (Fig. 5A). The mechanism by which this occurs remains to be determined. Activated AURKA binds to and phosphorylates INPP5E, thereby increasing its phosphatase activity and the hydrolysis of PtdIns(3,4,5)P3, establishing a feed-forward loop that downregulates AKT activity and a feed-back loop that reduces AURKA transcription (Fig. 5B). Both AURKA (Fig. 1F; Pugacheva et al., 2007) and AKT (Schneider et al., 2010) are detected at the base of primary cilia, and INPP5E is detected along the ciliary axoneme, extending to the base (Fig. 1F; Bielas et al., 2009), suggesting that the crosstalk between these proteins might occur within primary cilia. Importantly, the only significant colocalization between the two proteins was detected at the ciliary basal body. However, further studies are required to precisely define the location and dynamics of this interaction.
Fig. 5.
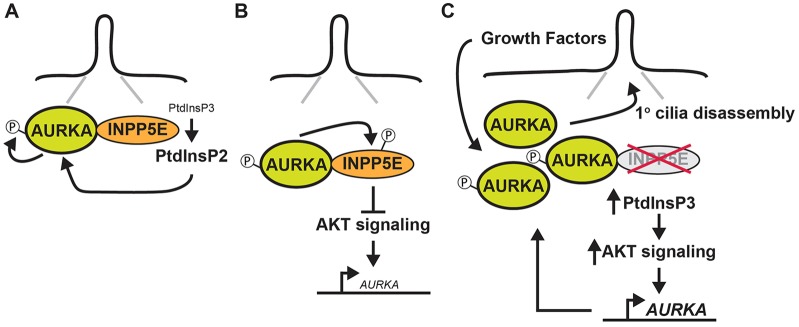
A model for reciprocal interactions between INPP5E and AURKA. (A) At the primary cilium we propose that hydrolysis of PtdIns(3,4,5)P3 by INPP5E alters the levels of different phosphoinositide species and, in so doing, mediates the functional interaction between INPP5E and AURKA, resulting in the activation of the latter protein through auto-phosphorylation. (B) Activated AURKA binds to and phosphorylates INPP5E, increasing its phosphatase activity and reducing AURKA transcription (small font) downstream of AKT. (C) When INPP5E is deleted or mutated, increased AKT signaling elevates AURKA transcription (large font), resulting in a generalized accumulation of AURKA protein at the cilia. In response to physiological stimuli, we propose that the increased pool of AURKA at this site drives the abnormal cilia disassembly phenotype observed in cells lacking INPP5E.
We show that when INPP5E is deleted or mutated (as in Joubert Syndrome), changes in the total level of AKT increase AURKA gene transcription, resulting in a generalized accumulation of AURKA protein at cilia. In response to physiological stimuli (e.g. growth factors), we propose that the increased pool of AURKA at this site drives the abnormal cilia disassembly phenotype observed in Inpp5e−/− MEFs (Fig. 5C). One prediction of this model is that inhibiting AURKA should be able to rescue defects caused by loss of INPP5E. We have observed exactly this effect in the context of cilia stability defects in response to growth factor signaling and in rescuing the defective cystogenesis apparent upon INPP5E deletion or knockdown. Given the complex nature of this interaction, understanding the consequences of perturbation in vivo will require further investigation.
The work presented here reveals a novel function of AURKA, suggesting that it acts as both a target and a regulator of phosphoinositide signaling. These findings have considerable implications for our understanding of the mechanisms that drive the development of disease phenotypes in the ciliopathies and, in particular, it provides a link between phosphoinositide signaling and the development of renal cysts. Mutations in INPP5E give rise to two clinically distinct diseases – Joubert Syndrome and MORM. The former condition is caused principally by point mutations in the catalytic domain of the protein that alter the phosphatase activity of the enzyme (Bielas et al., 2009). The latter disease is instead caused by C-terminal truncating mutations that affect ciliary localization, but do not appear to alter the phosphatase activity of the enzyme (Jacoby et al., 2009). Given that the interactions between AURKA and INPP5E are significantly regulated by catalytic activity, it is possible that the clinical differential between these diseases might in some manner be shaped by the interactions of INPP5E with AURKA. More broadly, a number of cancers are characterized by hyper-activation of phosphoinositide signaling (Jaiswal et al., 2009) and increases in AURKA protein levels and activity (Gautschi et al., 2008). The complex functional interaction between INPP5E and AURKA described in this report will therefore be important to investigate in this context.
MATERIALS AND METHODS
Plasmids, cell culture and inhibitors
AURKA and catalytically inactive AURKA (K162R) were expressed from pcDNA3.1-mRFP vector (Plotnikova et al., 2012), murine wild-type INPP5E, INPP5E D480N and INPP4B were expressed from pcDNA3.1-HA, and FLAG–INPP5E was expressed from pcDNA3.1-Flag. V5-tagged human wild-type INPP5E and INPP5E containing Joubert syndrome (JBTS) missense mutations were cloned into pLenti6.2/V5-DEST (Life Technologies). pLenti6.2/V5-DEST-LacZ (V5-LacZ) was used as negative control for immunoprecipitation. FLAG–INPP5E and truncated variants were as described previously (Humbert et al., 2012). Murine embryonic fibroblasts (MEFs) were harvested from embryonic day (E)12.5 embryos. The embryos were harvested, the heart and digestive system were discarded and the rest was minced, washed and placed in a 10-ml conical tube to incubate in TrypLE trypsin (Gibco Life Technologies) for 15 min at 37°C with constant shaking at 150 rpm, then centrifuged and washed in PBS buffer (Gibco Life Technologies) (Xu, 2005). MEFs and HEK293 cells were maintained in DMEM with 10% fetal bovine serum (FBS). IMCD3 cells were grown in DMEM/F-12 supplemented with 10% FBS (Gibco Life Technologies). All media were supplemented with 100 U/ml penicillin, and 100 µg/ml streptomycin (Life Technologies).
For spheroid formation assays, IMCD-3 cells were trypsinized, washed with PBS and resuspended in DMEM/F12 supplemented with 2% FBS and 2% growth-factor-deleted Matrigel (BD Bioscience). Lab-Tek chamber slides (8-well) were treated with 40 µl of 100% Matrigel and incubated at 37°C for 15 min to allow the Matrigel to solidify. Approximately 5000 cells/well were layered over the bed of Matrigel and grown at 37°C for 4 days to form spheroids. We transiently transfected HEK293 and IMCD3 cells with constructs or siRNAs using Lipofectamine 2000 reagent (Life Technologies) according to the manufacturer's instructions (2 µg of DNA was transfected using 8 µl of Lipofectamine 2000). RNA oligonucleotide duplexes targeted to INPP5E (siRNA SMARTpool M-041109-00-0005) were purchased from Thermo Scientific, along with scrambled negative controls. After transfection of siRNAs, the degree of depletion of INPP5E was determined by real time PCR. In this study, we used 10 µM MK2206 (Life Research Pty Ltd) and 3 µM AKTiX (Merck Millipore Pty Ltd) to inhibit AKT for 12 h and the indicated concentration of MLN8237 (Selleck Chemicals) or C1368 (Sigma-Aldrich) to inhibit AURKA.
Immunofluorescence
Cells growing on coverslips or in chamber slides were fixed with 4% paraformaldehyde (10 min), permeabilized with 1% Triton X-100 in PBS, blocked in 1×PBS with 3–5% bovine serum albumin (BSA), and incubated with antibodies using standard protocols. Alternatively, to maximize clear signals of phospho-AURKA at centrosomes, cells were fixed in cold methanol (−20°C) for 2 min, blocked and incubated with antibody. Primary antibodies included mouse anti-AURKA (1∶200, BD Biosciences), rabbit polyclonal anti-phospho-AURKA-T288 (1∶100, Cell Signaling Technology), anti-acetylated-α-tubulin monoclonal antibody (1∶300, clone 6-11B-1, Sigma-Aldrich; and 1∶200, clone K(Ac)40, Biomol International, Enzo Life Sciences), rabbit anti-INPP5E (1∶150, Protein Tech), mouse anti-γ-tubulin mAb (1∶200, Sigma-Aldrich) and rabbit anti-E-cadherin (1∶400, Life Technologies). Secondary antibodies labeled with Alexa Fluor 488 and Alexa Fluor 568, and 4′,6-diamidino-2-phenylindole (DAPI, to stain DNA) were from Life Technologies. Confocal microscopy was performed using a Nikon C1 Spectral confocal microscope (Nikon) equipped with a numerical aperture (NA) 1.40, oil immersion, 60× Plan Apo objective (Nikon). Images were acquired at room temperature using EZ-C1 3.8 software (Nikon) and analyzed using MetaMorph (Molecular Devices) and Photoshop, version CS2 (Adobe) software. Adjustments to brightness and contrast were minimal and were applied to the whole image.
Fluorescence intensity measurements
Relative intensity was measured as the fluorescence intensity normalized to that of the wild type under the ‘no serum’ condition. Basal body or centriole fluorescence intensity was calculated by subtracting background fluorescence from total integrated intensity. Measurement of integrated intensity was carried out in the basal body region using summed projections of acquired z-stacks. We measured 50–100 cells for each experimental condition. Quantification of fluorescence intensity was performed using MetaMorph software (Molecular Devices). Exposure times were constant for each individual experiment. To measure all statistical differences, a Student's t-test was performed using Excel (Microsoft, Redmond, WA).
Western blotting and immunoprecipitation
For western blotting and immunoprecipitation, mammalian cells were disrupted in RIPA lysis buffer supplemented with a protease and phosphate inhibitor cocktail (Roche). Whole-cell lysates were used either directly for SDS-PAGE or for immunoprecipitation. Immunoprecipitation samples were incubated overnight with antibody at 4°C, and subsequently incubated for 2 h with protein A/G–Sepharose (Pierce), washed and resolved by SDS-PAGE. Western blotting was performed using standard procedures, and blots were visualized by chemiluminescence using the Immobilon Western Chemiluminescent HRP Substrate (Millipore). Primary antibodies included mouse anti-AURKA (1∶2000, BD Biosciences), anti-phospho-AURKA-Th288 (1∶1000), anti-HA mAb (1∶1000), anti-Histone H3 (1∶2000), anti-phospho-Histone-H3-Ser10 (1∶4000), anti-AKT (1∶4000), anti-phospho-AKT-Ser473 (1∶3000), anti-β-tubulin (1∶5000), anti-GST mAb (1∶10,000, Cell Signaling Technology), anti-RFP (1∶5000, Santa Cruz Biotechnology), mAB anti-V5 (1∶10,000, Life Technologies) and anti-β-actin mAb (1∶1000, AC15, Sigma). Polyclonal anti-AURKA agarose-immobilized conjugate (Bethyl) was used for immunoprecipitations (1∶500). Secondary anti-mouse and anti-rabbit horseradish-peroxidase-conjugated antibodies (GE Healthcare) were used for visualization of western blots. Image analysis was performed using NIH ImageJ image processing and analysis software (National Institutes of Health), with signal intensity normalized to β-actin, tubulin or total protein level.
Biochemical assays
To assess phosphorylation of INPP5E by AURKA, an in vitro kinase assay was performed using GST-fused INPP5E purified from COS-7 cells and recombinant active AURKA (Millipore) or overexpressed RFP–AURKA immunoprecipitated from HEK293 cells in kinase buffer (40 mM MOPS, 1 mM EDTA pH 7.2) with the addition of 1× Mg/ATP cocktail (Millipore) and γ-[32P]ATP (Perkin Elmer). Histone H3 (Millipore) and GST were used as positive and negative controls for AURKA phosphorylation, respectively, using standard methods. Parallel aliquots without γ-[32P]ATP were processed for SDS-PAGE and western blotting. To analyze 5-phosphatase activity of INPP5E, an in vitro kinase reaction was performed as described previously and then PtdIns(3,4,5)P3 hydrolysis was measured by adding purified substrate [0.98 µg of PtdIns(3,4,5)P3 in the presence of 8 µg phosphatidylserine (Echelon Inc.)] and phosphatase buffer (50 mM MOPS pH 6.5, 200 mM NaCl, 10 mM EDTA) to the kinase reaction mix at 37°C for 10 min, followed by incubation with Malachite Green (Echelon Inc.) for an additional 10–20 min (Vandeput et al., 2006). Released inorganic phosphate was measured at 650 nm, compared against a standard curve. Assays were performed in triplicate. For measurement of 5-phosphatase activity of INPP5E in cell lysates, HEK293 cells were transiently transfected with constructs encoding HA–INPP5E or HA. At 24 h after transfection, cells were treated with 4 µM or 8 µM C1368 or DMSO for 3 h and then cells were harvested and lysed in RIPA buffer, and PtdIns(3,4,5)P3 5-phosphatase assays were performed.
Quantitative RT-PCR
Quantitative RT-PCR was performed in an ABI 7500 Real-Time PCR Cycler and analyzed using Applied Biosystems SDS software.
Protein stability studies
Approximately 2×107 HEK293 cells were plated onto 10-cm dishes. After 12 h, fresh medium containing cycloheximide (50 µg/ml) was added for 12 h. At the indicated time intervals, cells were lysed in RIPA buffer with protease inhibitors (Roche).
Statistical analysis
Statistical comparisons were made using a two-tailed Student's t-test. Experimental values are reported as the mean±s.e.m. Differences in mean values were considered significant at P<0.05. All calculations of statistical significance were made using the InStat (GraphPad Software) or Excel (Microsoft) software.
Supplementary Material
Acknowledgments
We are grateful to Erica Golemis and Carol Wicking for helpful discussions about the manuscript. We thank the Monash Micro Imaging Platform for their assistance, Lisa Ooms for help with phosphatase assays and David James (University of Sydney, Australia) and Dominic Ng (Bio21 Institute, Melbourne, Australia) for providing AKT and AURKA inhibitors, respectively.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
O.V.P. contributed to research design, conducted experiments, performed data analysis, prepared figures and wrote the paper; S.S. commented on the manuscript and contributed to research design; D.L.C. contributed to the paper writing; S.C., S.H. and J.M.D. provided reagents; C.A.M. commented on manuscript, research design and provided reagents; I.M.S. contributed to research design, manuscript writing and data analysis.
Funding
This work was supported by a National Health and Medical Research Council project grant [grant number APP1046174]. I.M.S. acknowledges support from an Australian Research Council Future Fellowship; and a Monash University Fellowship.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.161323/-/DC1
References
- Bertotti A., Burbridge M. F., Gastaldi S., Galimi F., Torti D., Medico E., Giordano S., Corso S., Rolland-Valognes G., Lockhart B. P. <et al. (2009). Only a subset of Met-activated pathways are required to sustain oncogene addiction. Sci. Signal. 2, ra80 10.1126/scisignal.2000643 [DOI] [PubMed] [Google Scholar]
- Bielas S. L., Silhavy J. L., Brancati F., Kisseleva M. V., Al-Gazali L., Sztriha L., Bayoumi R. A., Zaki M. S., Abdel-Aleem A., Rosti R. O. <et al. (2009). Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat. Genet. 41, 1032–1036 10.1038/ng.423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheetham G. M., Knegtel R. M., Coll J. T., Renwick S. B., Swenson L., Weber P., Lippke J. A., Austen D. A. (2002). Crystal structure of aurora-2, an oncogenic serine/threonine kinase. J. Biol. Chem. 277, 42419–42422 10.1074/jbc.C200426200 [DOI] [PubMed] [Google Scholar]
- Conduit S. E., Dyson J. M., Mitchell C. A. (2012). Inositol polyphosphate 5-phosphatases; new players in the regulation of cilia and ciliopathies. FEBS Lett. 586, 2846–2857 10.1016/j.febslet.2012.07.037 [DOI] [PubMed] [Google Scholar]
- Gautschi O., Heighway J., Mack P. C., Purnell P. R., Lara P. N., Jr and Gandara D. R. (2008). Aurora kinases as anticancer drug targets. Clin. Cancer Res. 14, 1639–1648 10.1158/1078-0432.CCR-07-2179 [DOI] [PubMed] [Google Scholar]
- Hildebrandt F., Benzing T., Katsanis N. (2011). Ciliopathies. N. Engl. J. Med. 364, 1533–1543 10.1056/NEJMra1010172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horan K. A., Watanabe K., Kong A. M., Bailey C. G., Rasko J. E., Sasaki T., Mitchell C. A. (2007). Regulation of FcgammaR-stimulated phagocytosis by the 72-kDa inositol polyphosphate 5-phosphatase: SHIP1, but not the 72-kDa 5-phosphatase, regulates complement receptor 3 mediated phagocytosis by differential recruitment of these 5-phosphatases to the phagocytic cup. Blood 110, 4480–4491 10.1182/blood-2007-02-073874 [DOI] [PubMed] [Google Scholar]
- Humbert M. C., Weihbrecht K., Searby C. C., Li Y., Pope R. M., Sheffield V. C., Seo S. (2012). ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. Proc. Natl. Acad. Sci. USA 109, 19691–19696 10.1073/pnas.1210916109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby M., Cox J. J., Gayral S., Hampshire D. J., Ayub M., Blockmans M., Pernot E., Kisseleva M. V., Compère P., Schiffmann S. N. <et al. (2009). INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat. Genet. 41, 1027–1031 10.1038/ng.427 [DOI] [PubMed] [Google Scholar]
- Jaiswal B. S., Janakiraman V., Kljavin N. M., Chaudhuri S., Stern H. M., Wang W., Kan Z., Dbouk H. A., Peters B. A., Waring P. <et al. (2009). Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell 16, 463–474 10.1016/j.ccr.2009.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisseleva M. V., Cao L., Majerus P. W. (2002). Phosphoinositide-specific inositol polyphosphate 5-phosphatase IV inhibits Akt/protein kinase B phosphorylation and leads to apoptotic cell death. J. Biol. Chem. 277, 6266–6272 10.1074/jbc.M105969200 [DOI] [PubMed] [Google Scholar]
- Liu X., Shi Y., Woods K. W., Hessler P., Kroeger P., Wilsbacher J., Wang J., Wang J. Y., Li C., Li Q. <et al. (2008). Akt inhibitor a-443654 interferes with mitotic progression by regulating aurora a kinase expression. Neoplasia 10, 828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikonova A. S., Astsaturov I., Serebriiskii I. G., Dunbrack R. L., Jr and Golemis E. A. (2013). Aurora A kinase (AURKA) in normal and pathological cell division. Cell. Mol. Life Sci. 70, 661–687 10.1007/s00018-012-1073-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S. K., Reckamp K., Yu H., Figlin R. A. (2010). Akt inhibitors in clinical development for the treatment of cancer. Expert Opin. Investig. Drugs 19, 1355–1366 10.1517/13543784.2010.520701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J., Wang Q., Snell W. J. (2004). An aurora kinase is essential for flagellar disassembly in Chlamydomonas. Dev. Cell 6, 445–451 10.1016/S1534-5807(04)00064-4 [DOI] [PubMed] [Google Scholar]
- Plotnikova O. V., Pugacheva E. N., Golemis E. A. (2011). Aurora A kinase activity influences calcium signaling in kidney cells. J. Cell Biol. 193, 1021–1032 10.1083/jcb.201012061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikova O. V., Nikonova A. S., Loskutov Y. V., Kozyulina P. Y., Pugacheva E. N., Golemis E. A. (2012). Calmodulin activation of Aurora-A kinase (AURKA) is required during ciliary disassembly and in mitosis. Mol. Biol. Cell 23, 2658–2670 10.1091/mbc.E11-12-1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugacheva E. N., Golemis E. A. (2005). The focal adhesion scaffolding protein HEF1 regulates activation of the Aurora-A and Nek2 kinases at the centrosome. Nat Cell Biol. 7, 937–946 10.1038/ncb1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugacheva E. N., Jablonski S. A., Hartman T. R., Henske E. P., Golemis E. A. (2007). HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 129, 1351–1363 10.1016/j.cell.2007.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider L., Cammer M., Lehman J., Nielsen S. K., Guerra C. F., Veland I. R., Stock C., Hoffmann E. K., Yoder B. K., Schwab A. <et al. (2010). Directional cell migration and chemotaxis in wound healing response to PDGF-AA are coordinated by the primary cilium in fibroblasts. Cell. Physiol. Biochem. 25, 279–292 10.1159/000276562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley E. S., Nachury M. V. (2010). The perennial organelle: assembly and disassembly of the primary cilium. J. Cell Sci. 123, 511–518 10.1242/jcs.061093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandeput F., Backers K., Villeret V., Pesesse X., Erneux C. (2006). The influence of anionic lipids on SHIP2 phosphatidylinositol 3,4,5-trisphosphate 5-phosphatase activity. Cell. Signal. 18, 2193–2199 10.1016/j.cellsig.2006.05.010 [DOI] [PubMed] [Google Scholar]
- Walter A. O., Seghezzi W., Korver W., Sheung J., Lees E. (2000). The mitotic serine/threonine kinase Aurora2/AIK is regulated by phosphorylation and degradation. Oncogene 19, 4906–4916 10.1038/sj.onc.1203847 [DOI] [PubMed] [Google Scholar]
- Wilson P. D. (2004). Polycystic kidney disease. N. Engl. J. Med. 350, 151–164 10.1056/NEJMra022161 [DOI] [PubMed] [Google Scholar]
- Wloga D., Rogowski K., Sharma N., Van Dijk J., Janke C., Eddé B., Bré M. H., Levilliers N., Redeker V., Duan J. <et al. (2008). Glutamylation on alpha-tubulin is not essential but affects the assembly and functions of a subset of microtubules in Tetrahymena thermophila. Eukaryot. Cell 7, 1362–1372 10.1128/EC.00084-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J. (2005). Preparation, culture, and immortalization of mouse embryonic fibroblasts. Curr. Protoc. Mol. Biol. 70, 28.1.1–28.1.8 10.1002/0471142727.mb2801s70 [DOI] [PubMed] [Google Scholar]
- Yeh C., Li A., Chuang J. Z., Saito M., Cáceres A., Sung C. H. (2013). IGF-1 activates a cilium-localized noncanonical Gβγ signaling pathway that regulates cell-cycle progression. Dev. Cell 26, 358–368 10.1016/j.devcel.2013.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Liu Y., Gao F., Ding Q., Cho C., Hur W., Jin Y., Uno T., Joazeiro C. A., Gray N. (2006). Discovery of EGFR selective 4,6-disubstituted pyrimidines from a combinatorial kinase-directed heterocycle library. J. Am. Chem. Soc. 128, 2182–2183 10.1021/ja0567485 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.