

Abstract
Oligodendrocyte progenitor cells (OPCs) have the ability to divide or to arrest growth and differentiate into myelinating oligodendrocytes in the developing brain. Due to their high number and the persistence of their proliferative capacity in the adult brain, OPCs are being studied as potential targets for myelin repair and also as potential source of brain tumors. This study addresses the molecular mechanisms regulating the transcriptional changes occurring at the critical transition between proliferation and cell cycle exit in cultured OPCs. Using bioinformatic analysis of existing datasets, we identified c-Myc as a key transcriptional regulator of this transition and confirmed direct binding of this transcription factor to identified target genes using chromatin immunoprecipitation. The expression of c-Myc was elevated in proliferating OPCs, where it also bound to the promoter of genes involved in cell cycle regulation (i.e. Cdc2) or chromosome organization (i.e. H2afz). Silencing of c-Myc was associated with decreased histone acetylation at target gene promoters and consequent decrease of gene transcripts. c-Myc silencing induced also a global increase of repressive histone methylation and premature nuclear peripheral chromatin compaction and promoted the progression of OPCs towards differentiation. We conclude that c-Myc is an important modulator of the transition between proliferation and differentiation of OPCs, although its decrease is not sufficient to induce progression into a myelinating phenotype.
Keywords: development, myelin, epigenetic, histone variant, CNS, glioma
INTRODUCTION
Oligodendrocyte progenitor cells (OPCs) have the ability to proliferate and generate alternative lineage cells. Because of their abundance in the adult brain, they are the subject of an intense debate due to their potential for repair but also due to the potential risk for transformation into cell-of-origin for glioblastoma, one of the most aggressive adult brain tumors (Lei et al., 2011). It is well accepted that transformation of progenitors requires: a proliferative advantage (typically by escaping the cell cycle regulatory circuitry, due to mutations, deletions or amplifications), changes in chromatin structure and aberrant transcriptional regulation of differentiation. Recent studies conducted in human GBM have identified striking similarities between the human “transcriptional signature” and that of animal models (Verhaak et al., 2010). Intriguingly, this “proneural signature” shared a substantial overlap with the OPC transcriptome. Thus, studies on the transcriptional regulation of the transition between proliferation and cell cycle exit and differentiation in OPCs are of high relevance for both repair and for a better understanding of human brain tumors. The decision of OPC to divide or exit the cell cycle and start differentiating relies on important key transcriptional regulators, including E2F1 and c-Myc. The ability of E2F1 to regulate gene expression is dependent on post-translational modifications of the molecule and its binding to the unphosphorylated retinoblastoma protein Rb (Dyson, 1998) (Nevins, 1998). In proliferating cells, Rb is highly phosphorylated due to the activity of cyclin dependent kinases (i.e. CDK2 and CDK4), bound to their positive regulators (i.e cyclins) (Sherr, 1995). Mitogens increase the bioavailability of cyclins, resulting in the formation of activated cyclinD-CDK4/6 and cyclinE/CDK2 complexes which sequentially phosphorylate Rb (Sherr, 1994) (Weinberg, 1995), and in turn free E2F1 from sequestration and allow expression of S-phase genes (Ruas and Peters, 1998) (Sherr and Roberts, 1999).
c-Myc is a very powerful proto-oncogene which has also been shown to play a critical role in cell reprogramming (Thier et al., 2012) (Song et al., 2012) and has been proposed to serve the function of “sensor” of mitogen and nutrient availability (Dang, 1999). It has been shown to bind DNA to specific sites and activate transcription (Amati et al., 2001) of enzymes regulating energy metabolism and synthesis of nucleic acids (Dang et al., 2009) (Sloan and Ayer, 2010). The Myc family of transcription factors includes proteins characterized by a basic DNA binding region and a leucine-zipper domain, responsible for dimerization with other components. The DNA-binding region of Myc recognizes the E box consensus sequences (i.e. CANNTG) on DNA and modulates activation or repression, depending on its binding partners (Blackwell et al., 1990) (Pelengaris et al., 2002). The leucine zipper recruits other proteins to form functional complexes. In the absence of the leucine zipper domain, for instance, c-Myc retains the ability to transform cells, even though it is unable to affect myoblast differentiation (La Rocca et al., 1994). c-Myc was also reported to modulate acetylation of large chromatin domains (Guccione et al., 2006) and to bind the histone acetyl-transferase GCN5 in cancer cells (McMahon et al., 2000), in a cell-specific and context-dependent fashion. Histone acetylation, which neutralizes the positive charge of the lysine side-chain, is thought to weaken histone-DNA interactions, thus destabilizing the chromatin structure and allowing activation of transcription. By recruiting histone acetyl-transferases (HATs) to chromatin, c-Myc is thought to modulate histone acetylation at multiple genomic locations (Frank et al., 2001) (Martinato et al., 2008). We and others have previously reported the importance of histone acetylation in proliferating OPCs and identified histone de-acetylation as required for the transition between proliferation and differentiation (Lyssiotis et al., 2007) (Marin-Husstege et al., 2002) (Shen et al., 2005) (He et al., 2007) (Shen et al., 2008). Given the proposed role of c-Myc in modulating cell cycle and histone acetylation in other cell types, we reasoned it may play an important role in the transcriptional regulation of the transition from proliferation to differentiation in OPCs, possibly by modulating gene expression, histone acetylation and chromatin reorganization and this is the focus of the current study.
EXPERIMENTAL PROCEDURES
Animals
Cnp-EGFP mice were provided by Dr. Gallo (Children's Hospital Washington, DC). Use of animals in this research was strictly compliant with the guidelines set forth by the US Public Health Service in their policy on Humane Care and Use of Laboratory Animals, and in the Guide for the Care and Use of Laboratory Animals. Mice were maintained under pathogen-free environment at Mount Sinai School of Medicine animal facility. All procedures received prior approval from the Institutional Animal Care and Use Committee. Timed pregnancy Sprague-Dawley rats and mice were purchased from Charles River Laboratory (Wilmington, MA). Animal handlings and experiments were performed according to the German animal protection laws (LANUV Nordrhein-Westfalen (AZ 8.87-51.05.20.10.262).
Cell culture and treatment
Mouse oligodendrocyte progenitors were isolated from P6-P8 C57Bl6 mice and cultured as previously described (Cahoy et al., 2008). Briefly, dissociated mouse forebrains were resuspended in panning buffer. To deplete microglia, the single-cell suspension was sequentially panned on BSL1 panning plates and then incubated on a PDGFRα plates. The adherent cells were trypsinized and plated onto poly-D-lysine coated plates. The cultures were maintained under proliferating conditions by addition of PDGFA (10ng/ml) and bFGF (20ng/ml) and then differentiated by adding L-3,3’,5-triiodothyronine sodium salt (T3 hormone, 45nM). The mouse oligodendrocyte precursor cell line Olineu (Jung et al., 1995) were grown on poly-ornithine-coated culture dishes. The immature Olineu cells were maintained in growth medium consisting of DMEM supplemented with 2 mM L-glutamine, 1 mM sodium pyruvate, 10 ng/ml biotin, 100 μg/ml apotransferrin, 100 μM putrescine, 20 nM progesterone, 30 nM sodium selenite, 5 μg/ml insulin, 1% horse serum, 100 U/ml penicillin and 100 μg/ml streptomycin. Differentiation was induced by switching the cells to a serum-free medium containing 45nM T3.
Tissue Collection and Sectioning
Cnp-EGFP mice were perfused intracardially with 4% paraformaldehyde in 0.1 M phosphate buffer. Brains were removed from the skulls, postfixed overnight, and cryopreserved by sequential immersion of 10%, 20% and 30% sucrose solution in 0.1M phosphate buffer pH7.4. Brains were then embedded in OCT (Fisher Scientific) and sectioned (1μm).
Immunohistochemistry
Cryostat brain sections from Cnp-EGFP mice at P2 and P21 were immunostained with antibody against c-Myc (Sc-764, Santa Cruz Biotechnology). Sections were incubated overnight at 4°C with antibody diluted in 0.1 M phosphate-buffered saline (pH 7.4) containing 0.5% Triton X-100 (vol/vol) and 10% normal goat serum (vol/vol). For secondary we used Alexa-fluor 546 goat antibody to rabbit IgG. Sections were incubated with secondary antibodies for 1h at 22-25°C, than washed and mounted on the slides.
Immunocytochemistry
Cells were grown on CC2-coated 8 well chambers (Lab-Tek) for all immunocytochemistry. For staining oligodendrocyte lineage markers, cells were rinsed gently with PBS and incubated live with O4 hybridoma supernatant (1:10) for 30 min at 37°C. Cells were then fixed with 1% paraformaldehyde for 20 min at room temperature and first incubated with pageing solution (PGBA plus 10% normal goat serum) for 60 min followed by incubation with secondary antibodies for 1 h at room temperature. For staining with rabbit polyclonal antibodies against the histone marks H3K9Ac (Abcam, ab4441) and H3K9me3 (Abcam, ab8898) cells were equilibrated with Triton X-100 containing pageing solution (PGBA plus 10% normal goat serum and 0.5% TX-100) for 30 min, then processed for the primary antibody staining followed by the appropriate secondary antibody. For staining with surface marker NG2 (Chemicon, Ab5320) cells were equilibrated in pageing solution (PGBA plus 10% normal goat serum) for 60 min, then processed for the primary antibody staining followed by the appropriate secondary antibody. For staining with chicken polyclonal antibody against GFP (Abcam, ab13970) cells were equilibrated with Triton X-100 containing pageing solution (PGBA plus 10% normal goat serum and 0.5% TX-100) for 30 min, then processed for the primary antibody staining followed by the appropriate secondary antibody. Cells were then counterstained with 4_, 6_-diamidino-2-phenylindole (DAPI; 1:20000; Invitrogen) to visualize cell nuclei.
Analysis of immunocytochemistry
Analysis of the immunocytochemistry on mOPCs (oligodendrocyte progenitors) and OLs (oligodendrocytes) in vitro was performed using the ImageJ software (NIH, Bethesda, MD, USA). The intensity value for histone marks staining was determined as pixel intensity/area. Only GFP labeled nuclei were analyzed. Both the confocal and the ImageJ settings were consistent throughout all the images analyzed. The pixel value intensity was obtained by analyzing integrated density function in ImageJ in arbitrary unit and then it was normalized to the scramble value. Both the length of the longest process by NG2 and the membrane extension by O4 was measured with ImageJ in artitrary unit and then normalized to the scramble.
RNA isolation and qRT-PCR analysis
Mouse primary oligodendrocyte progenitors and oligodendrocytes, or mouse tissue derived from corpus callosum at different time points were homogenized in Trizol Reagent and RNA was isolated following manufacturer's instruction and cleaned using RNeasy Mini kit (Qiagen, CA). 500 nanograms of total RNA was used in 20μl of reverse transcription reaction, using qScript cDNA SuperMix (Quanta Biosciences). qRT-PCR was performed using PerfeCTa SYBR Green FastMix (Quanta Biosciences) in Applied Biosystem 7900HT Sequence Detection PCR System. The melting curve of each sample was measured to ensure the specificity of the products. Data were normalized to the internal control 18S and analyzed using Pfaffl ΔΔCtmethod. Primers used in quantitative PCR are given in Table 1.
Table 1.
| Mouse RT Primers | For | Rev |
|---|---|---|
| c-Myc | TGAGGAGACACCGCCCAC | CAACATCGATTTCTTCCTCATCTTC |
| Mbp | ACACACGACAACTACCCATTATGG | AGAAATGGACTACTGGGTTTTCATCT |
| H2Az | TGTGGCTTCAAAGAAGCTATTG | TATCCACCAGAGTGGAAACAA |
| Cdc2 | GCCAGAGCGTTTGGAATACC | CAGATGTCAACCGGAGTGGAGTA |
| Pdgfrα | GGGGACAGACTGTGAGGTGT | ACTGCACTTGAGGCCCTTTA |
Primers for murine transcripts used in qPCR.
Western Blot
Nuclear extract proteins were derived from corpus callosum tissues and from Olineu cells using Nuclear Extract kit (Active Motif). 30 μg of nuclear extract were separated by SDS-PAGE and transferred onto a PVDF (Millipore) membrane using a buffer containing 25 mM Tris base, pH 8.3, 192 mM glycine, 20% (vol/vol) methanol for 1 h at 100 V at 4 °C. Western blot analysis was performed using anti-c-Myc (Sc-764, N-262, 1:500). Immunoreactive bands were visualized using horseradish peroxidase–conjugated secondary antibodies (Amersham), followed by chemiluminescence with ECL-Plus Western Blotting Detection System (Amersham). Equal protein loading was guaranteed by probing the blots with antibody against total H3.
Clustering and Ontology Analysis
The expression data for the time course of oligodendrocyte differentiation used for the clustering analysis was previously published (Swiss et al., 2011). Ontology analysis of each co-expressed group of genes was performed using the DAVID ontology software as previously described (Swiss et al., 2011) (Dennis et al., 2003) (Huang da et al., 2009) using the whole genome as the background dataset and an EASE score < 10−3 considered highly enriched ontology groups. ChEA analysis was performed by interrogating ChEA software (http://amp.pharm.mssm.edu/lib/chea.jsp) as described (Lachmann et al., 2010).
Transcription factor binding sites
The software MatInspector from Genomatix (Genomatix Software) was used to search the binding sites for c-Myc on the promoter regions of selected genes.
Chromatin immunoprecipitation
Chromatin was isolated from mouse OPC, either proliferating or differentiating (>10×106 cells), and from Olineu Cells, either proliferating or differentiating (>10×106 cells) as previously described (Liu et al., 2010). Cells were crosslinked in 1% formaldehyde for 10 min, lysed in nuclear lysis buffer (50mM TrisHCl pH8, 10mM EDTA, 1%SDS, protease inhibitor cocktail (Roche), phosphatase inhibitor cocktail (Roche) and sonicated using a Bioruptor (Diagenode) sonicator to produce an average size of 250–500 base pairs. Chromatin concentration was measured by its absorbance at 260 nm in the Nanodrop (1 U/ml of chromatin corresponds to 1=OD260) and diluted 1:10 with dilution buffer (NaCl 165 mM, SDS 0.01%, Triton X-100 1.1%, EDTA 1.2 mM, Tris-HCl 16.7 mM, pH 8.0). Chromatin was immunoprecipitated using protein A magnetic beads (Dynabeads-Invitrogen 100.01D) coated with the primary antibody. For c-Myc immunoprecipitation 4 μg of anti-c-Myc (Sc-764, Santa Cruz Biotechnology) for chromatin Unit was used. 2.5 Units of chromatin were used per condition. For histone marks immunoprecipitation 2 μg of anti-H3K9Ac (Abcam, ab4441) and 10 μl of anti-H3K14Ac (39599, Active Motif) were used for chromatin unit. 1 unit of chromatin was used per condition. The DNA recovered from chromatin, which was not immunoprecipitated, was used as input. A mock immunoprecipitation was set up as control (rabbit IgG, Santa Cruz, Sc2027). Following overnight immunoprecipitation, beads were washed twice consecutively with the following buffers: Lio-B (HEPES 50 mM, pH 8.0, NaCl 140 mM, Triton X-100 1%, Sodium deoxycholate 0.1%, EDTA 1 mM), Hio-B (HEPES 50 mM, pH 8.0, NaCl 500 mM, Triton X-100 1%, Sodium deoxycholate 0.1%, EDTA 1mM), LiCl (Tris-HCl 10mM, LiCl 250 mM, NP-40 0.5%, Sodium deoxycholate 0.5%, EDTA 1 mM) and TE (Tris-HCl 10 mM, pH 8.0, EDTA 1 mM). Immunoprecipitated chromatin and input DNA were reverse crosslinked in elution buffer (Tris-HCl 50mM, EDTA 10 mM, SDS 1%) in the presence of proteinase K (50 μg/ml) by shaking (1300 rpm) at 68°C using a thermomixer (Eppendorf) for 2h. DNA was purified using phenol-chloroform and precipitated overnight in ethanol at -20°C. DNA pellets were dissolved in 200 μl of ddH2O and amplified by quantitative PCR, using primers specific for selected genomic regions, as indicated in Table 2 for murine genes. Data are expressed as percentage over input.
Table 2.
| Mouse ChIP Primers | For | Rev |
|---|---|---|
| H2Az (−450) | GAGATCTGCGGACACGAAAG | GAGGGAGGAGCTGGAAGG |
| H2Az (−50) | GTTCTCCCATTGGCTGGAG | GGACCGCGATTCAAACTG |
| Cdc2 | ACAGAGCTCAAGAGTCAGTTGGC | CGCCAATCCGATTGCACGTAGA |
| Pdgfrα | GGGGACTTCATTTCCTGACA | AAAGTAAGCCCCAAGCTTCC |
| Mbp (promoter) | TTCAAGACCCCAGGAAGAAA | TTCTTTGGGTCTGCTGTGTG |
Primers for murine genes used in qPCR after ChIP.
Lentiviral shRNA infection
c-Myc transduction silencing particles were purchased from Sigma-Aldrich (TRCN0000042515). A bicistronic lentivirus encoding for GFP and for the shRNA construct was used. GFP expression marked the cells expressing the shRNA. ShRNA infection in primary OPCs was performed in cell suspension with a M.O.I of 5. In each infection, a lentivirus encoding a scrambled sequence (SHC002V) against non-mammalian gene target was used as a control. Polybrene was added at a final concentration of 2μg/ml to increase the infection efficiency. The virus/cell mixture was incubated in 37°C for 2 h, and plated at desired density in complete growth medium (PDGF, FGF2). After 48 hours infected cells were selected with puromycin (0.1μg/ml) and cells were finally harvested for analysis according to the experimental design.
Correlative light and electron microscopy
After shRNA infection, OPCs were plated on coated gridded glass bottom dishes (P35G-2-14-C-GRID or P35G-2-14-CGRD, MatTek Corporation). On this coverslip is etched an alphanumeric grid that enables cell relocation by light and electron microscopy. Correlative light and electron microscopy was then performed as described in (Hanson et al., 2010). Briefly, cells fixed with 4% glutaraldehyde (in 0.1M sodium cacodylate, pH 7.4) including 1 mMCaCl2 at 4°C for 20 min. Cells were then washed with 0.1 M sodium cacodylate, pH 7.4 at 4°C and maintained up to 2 weeks at 4°C in this buffer until laser scanning confocal microscopy (LSCM) imaging. An inverted Zeiss 780 laser scanning confocal microscope was used. Cells expressing GFP were identified at low magnification using the fluorescent light source. To orient the cells of interest with the gridded coverslip, bright-field and GFP LSCM single-plane images were taken using a 10x such that the grid was clearly visible. From these images, the location of the cells relative to the grid coordinates on the glass surface could be ascertained. After LSCM, the material was treated with 1% osmium tetroxide, 1.5 % potassium ferracyanide in 0.1M cacodylate buffer for 1 h at 4°C. The cells were then dehydrated in solutions of ethanol at increasing concentrations. After dehydration, cells were infiltrated with a 1:1 solution of resin (Embed 812 kit, Electron Microscopy Sciences) and 100% ethanol for 24 h at 4°C. Prior to adding the resin/ethanol, the approximate location of the cell of interest relative to the grid was noted on the coverslip. After infiltration overnight, the resin/ethanol was replaced with 1 mL layer of pure resin, which when solidified stabilizes the glass grid. Prior to solidification, an open ended embedding capsule with a 1× cm face (Electron Microscopy Sciences) was placed on the dish surrounding the cell of interest. The resin was then polymerized in a vacuum oven at of 65°C for 8-12 h. After the first layer was solidified, the capsule was topped off with more resin and put back in the oven for another 8–12 h. To separate the block from the dish, a hot plate was heated to 60°C and the dish was placed on a preheated hot plate for exactly 3 min and 30 s. The dish was removed from the hot plate and the capsule carefully dislodged free from the dish. Once separated, the block face retains all the cells and an imprint of the etched grid. The block was coarsely trimmed with a double-edged razorblade and a Diatome cryotrim 45° mesa trimming knife (Electron Microscopy Sciences) was used to finely trim the block around the cells of interests. Next, 70 nm serial ultrathin sections were cut from the block surface using a Reichardt Ultracut E ultramicrotome. The sample was dried on the grid and transferred to Hiraoka Staining Mats (Electron Microscopy Sciences) for heavy metal staining. Grids were stained with 3% uranyl acetate and Reynold's lead citrate, washed, and allowed to dry. For electron microscopy, cells were then viewed at low magnification and low beam to identify the cells of interest based on comparison of the cell morphology under the EM with the confocal GFP image. Serial sections of cells of interest were then imaged at 10-30.000× magnification.
Statistics
One-way ANOVAs were performed to determine significance for ChIP at different time points. Assuming that significant main effects were observed (p < 0.05), Bonferroni post hoc tests were used to compare selected groups. Student t tests were used for all other comparisons, including qPCR analysis. All values included in the figure legends represent mean ± S.E.M. The statistical significance is either described in figure legends, or indicated as asterisks (*). A p value of <0.05 was considered to be statistically significant (*p<0.05, **p < 0.01, ***p < 0.001).
RESULTS
Identification of c-Myc as a relevant transcription factor during oligodendrocyte progenitor differentiation
To define the relationship between cell cycle exit and transcriptional regulation of oligodendrocyte progenitor differentiation, we referred to the analysis of a previously reported comprehensive microarray dataset (Dugas et al., 2006), using a consensus clustering algorithm based on the temporal profile of gene expression (Swiss et al., 2011). This unbiased analysis of the oligodendrocyte transcriptome, revealed 12 distinct patterns of gene expression that were defined as clusters. Genes downregulated during the transition from proliferating to differentiating OPC differentiation were included in the first three clusters, which were enriched for gene categories related to: cell cycle (Ccnd1, Ccnd2, Cdc20, Pola1), RNA processing (E1f1A, EIf4EBP1, Rpl14, Rpl28, Rpl34, Rpl36, Hnrpa1, Hnrpa3), chromosomal reorganization (H2Afz, Dnmt1, Hmgn1, Hdac2, Uhrf1, Suv39H1) and transcription (Myc, Pdgfrα, Nfkb1) (Fig.1a). Due to the kinetics of expression of these transcripts, we reasoned that transcriptional regulators of each clusters could be identified by using a ChIP Enrichment Analysis (ChEA) (Lachmann et al., 2010), (Muller and Helin 2000), which is based on the statistical enrichment of ChIP-validated transcription factor binding sites within the gene list in each cluster. Interestingly, the transcription factor c-Myc was identified as the top ranking transcription factor for the first two clusters (Fig.1b). These data suggested that genes regulating cell cycle and chromatin organization are cMyc transcriptional targets.
Figure 1. Identification of c-Myc as a relevant transcription factor during oligodendrocyte differentiation.

(a) Heat Map showing the microarray signals of selected transcripts during a defined time course (Dugas et al., 2006) of primary oligodendrocyte progenitor cells (OPCs) differentiation. Only gene ontology categories that were enriched in the first three clusters of co-regulated genes are reported (cell cycle, RNA processing, chromosomal reorganization, transcription). These correspond to early downregulated genes, as previously described (Swiss et al., 2011). (b) ChIP Enrichment Analysis (ChEA) of the co-regulated gene list in clusters 1 and 2. Bar graphs identify the most significant transcription factors with ChIP-validated binding sites. C-Myc is the top ranking transcription factor regulating genes in cluster 1 and the second ranking transcription factor in cluster 2.
c-Myc is downregulated during oligodendrocyte differentiation
Low levels of c-Myc transcripts were detected in the developing corpus callosum, at the earliest points of detection of myelin gene transcripts, such as Myelin Basic Protein (Mbp) (Fig. 2a). c-Myc protein levels were similarly decreased in the developing white matter, as shown by western blot analysis (Fig. 2b). To define whether reduced c-Myc levels could be detected in oligodendrocyte lineage cells, we performed immunohistochemistry on brain sections obtained from CNPase-EGFP mice, which are characterized by EGFP expression restricted to oligodendrocyte-lineage cells (Yuan et al., 2002). Whereas c-Myc immunoreactivity was clearly detected in most GFP+ OPCs in the developing corpus callosum at P2, reduced co-labeling of c-Myc and GFP was detected at P21, consistent with the interpretation that c-Myc expression is elevated in OPCs and declines as they differentiate into myelin-forming oligodendrocytes (Fig. 2c).
Figure 2. c-Myc levels are downregulated during oligodendrocyte differentiation.

(a) Scatter-plot histograms depicting transcript levels of c-Myc and Myelin Basic Protein (Mbp) analyzed by quantitative RT-PCR of RNA samples isolated from the developing corpus callosum at the indicated time points. Each scatter point represents the expression level in an individual mouse. Values were normalized by the levels of 18S and shown as relative to the levels detected at P2. (b) Representative western blot showing c-Myc levels in nuclear extracts obtained from the developing white matter at the indicated time points. (c) c-Myc expression in the developing corpus callosum of CNPase-GFP mice stained for c-Myc (red) and EGFP (green) at P2 and P21. (d) Scatter-plot histograms depicting transcript levels of c-Myc and Mbp analyzed by quantitative RT-PCR of RNA samples isolated from mouse OPC cultured in mitogens (D0) or in differentiation conditions for 3 days (D3). Each scatter point represents expression from a distinct culture. Values were normalized by the levels of 18S and shown as relative to the levels detected at D0. (e) Representative western blot showing c-Myc levels in nuclear extracts obtained from Olineu cells cultured in serum (D0) or in differentiating conditions for 3 days (D3). P=postnatal day. Scale bar in (C) = 50 μm
c-Myc downregulation could also be detected in vitro, in cultured OPCs differentiated by withdrawal of mitogens and inclusion of thyroid hormone (T3) to the culture medium (Fig. 2d). Also in this case, the decreased levels of c-Myc were inversely correlated to the transcript levels of Mbp, which increased over time (Fig. 2d). We also validated the reduction of c-Myc at the protein level in Olineu cells differentiated in the presence of T3 (Fig. 2e). Taken together these results suggest that c-Myc levels are elevated in proliferating OPCs and progressively decline as the cells differentiate.
c-Myc binds the promoter of nucleosomal histones and cell cycle genes
To further validate the direct role of c-Myc as transcriptional regulator of co-expressed genes involved in cell cycle regulation and chromosomal reorganization, we used chromatin immunoprecipitation. The putative location of c-Myc binding sites on gene targets was determined using the MatInspector software from Genomatix. These regions were selected for the amplification of the DNA recovered after c-Myc immunoprecipitation of chromatin isolated from Olineu cells. We selected a representative gene from each of the functional categories identified by the bioinformatic approach and interpreted the presence of c-Myc binding on a putative target gene as further evidence of transcriptional regulation. Among the putative targets in the “chromosome organization” category, we selected the histone variant H2afz. Within the “cell cycle” category, we selected Cdc2 and within the “transcription” category, we selected the gene Pdgfrα. Chromatin immunoprecipitation detected reduced c-Myc binding in differentiating cells compared to proliferating cells, which nicely correlated with decreased H2afz transcripts (Fig. 3a-c).
Figure 3. c-Myc direct binding to the promoter of gene targets.

(a) MatInspector Analysis (Genomatix) showing c-Myc binding sites at the promoter of mouse H2afz gene. (b) Chromatin was isolated from murine Olineu cells either kept in proliferating conditions (D0) or cultured in differentiation conditions for 1 day (D1) and immunoprecipitated with antibodies specific for c-Myc. After reverse cross-linking, the bound DNA was amplified using the specific primer sets for the mouse promoter of H2Afz indicated by lines on top of the gene. A mock immunoprecipitation was used as negative control. The experiment was performed in three biological replicates. (c) The transcript value of H2afz was measured by qRT-PCR on RNA samples obtained from mouse OPCs in proliferating (D0) and differentiating (D3) conditions. The transcript levels were normalized by the levels of 18S and shown as relative to the levels detected at D0. (d) MatInspector Analysis (Genomatix) showing c-Myc binding sites at the promoter of mouse Cdc2 gene. (e) Chromatin was isolated from murine Olineu cells either kept in proliferating conditions (D0) or cultured in differentiation conditions for 1 day (D1) and immunoprecipitated with antibodies specific for c-Myc. After reverse cross-linking, the bound DNA was amplified using primer sets specific for the mouse promoter of Cdc2 and schematically shown as lines drawn on top of the gene. A mock immunoprecipitation was used as negative control. The experiment was performed in triplicate. (f) The transcript value of Cdc2 was measured by qRT-PCR on RNA samples obtained from mouse proliferating (D0) and differentiating (D3) OPC. The transcript levels were normalized by the levels of 18S and shown as relative to the levels detected at D0. (g) MatInspector Analysis (Genomatix) showing c-Myc binding sites at the promoter of mouse Pdgfrα gene. (h) Chromatin was isolated from murine Olineu cells either kept in proliferating conditions (D0) or cultured in differentiation conditions for 1 day (D1) and immunoprecipitated with antibodies specific for c-Myc. After reverse cross-linking, the bound DNA was amplified using primer sets specific for the mouse promoter of Pdgfrα and schematically shown as lines drawn on top of the gene. A mock immunoprecipitation was used as negative control. (i) The transcript value of Pdgfrα was measured by qRT-PCR on RNA samples obtained from mouse OPCs in proliferating (D0) and differentiating (D3) conditions. The transcript levels were normalized by the levels of 18S and shown as relative to the levels detected at D0. (mean ± s.e.m. n=3,*p<0.05 ** p<0.01, *** p<0.005).
Similarly, c-Myc binding to the promoter of Cdc2 gene decreased in differentiating cells and this correlated with diminished Cdc2 transcripts (Fig. 3d-f).
A different scenario was observed for Pdgfrα, whose gene product is characteristically used as marker of the progenitor state. MatInspector revealed the presence of a single c-Myc binding site within the Pdgfrα promoter (Figure 3g). Interestingly, the ChIP analysis did not detect differences in c-Myc binding to the Pdgfrα promoter between proliferating and differentiating cells (Fig. 3h), despite the fact that its transcripts were downregulated during OPC differentiation (Fig. 3i). These data suggested that transcriptional regulation of Pdgfrα is affected by components other than c-Myc.
To better validate the role of c-Myc in regulating the expression of its targets, we adopted a silencing approach. Primary mOPCs were transduced with a bicistronic lentivirus encoding either a sequence specific for c-Myc or a scrambled sequence (defined hereafter as “scrambled control”) and GFP, to allow visualization of the silenced cells (Fig. 4a). Silencing efficiency was assessed by measuring c-Myc transcripts (Fig. 4b), and resulted in decreased cell density (n=3) (Fig. 4c) and reduction of Pcna, a gene involved in cell cycle control (Fig. 4d). The levels of c-Myc target genes Cdc2 and H2afz were also reduced (Fig. 4d). At a molecular level, the decreased levels of Cdc2 and H2afz in silenced cells were associated with reduced acetylation of lysine residues K9 and K14 histone H3 at their promoters (Fig. 5a). Conversely, a not significant decrease of histone acetylation was detected at genomic locations corresponding to late differentiation genes as Mbp (Fig. 5b). Together, these data define c-Myc as an important regulator of histone acetylation at the promoter of target genes, encoding for regulators of the transition between proliferation and differentiation of OPCs.
Figure 4. Characterization of c-Myc silenced OPCs.

(a) Phase-contrast micrographs and GFP+ epi-fluorescence of mouse OPCs infected with a double-cistronic lentivirus expressing GFP and either a scrambled shRNA or shRNA specific for c-Myc. (b) Bar graphs indicate the transcript level of c-Myc measured by quantitative RT-PCR of RNA samples isolated from proliferating mOPCs 72h post-infection. Transcriptional data were normalized to the transcript levels of c-Myc measured in control cultures infected with the scrambled control. Note the c-Myc reduction in c-Myc silenced cells. (c) Bar graphs indicate cell density measured 72h post infection with either shRNA c-Myc or scrambled control. Cell density was measured as the total number of cells per area. The analysis was performed on three different c-Myc silenced cultures and values were expressed relative to scrambled control. (d) Bar graphs indicate the transcript levels of c-Myc target genes - Pcna, Cdc2 and H2Afz - measured by quantitative RT-PCR of RNA samples isolated from proliferating or early differentiating mOPCs, 72 hours post infection. Transcriptional data were normalized relative to scrambled control. Note the reduction of proliferation markers, cell cycle and c-Myc regulated genes upon shRNA c-Myc mediated silencing (mean ± s.e.m. n=3, ** p<0.01, *** p<0.005; scale bar in (A) = 20 μm).
Figure 5. Loss of c-Myc leads to decreased histone acetylation at its target gene promoters.
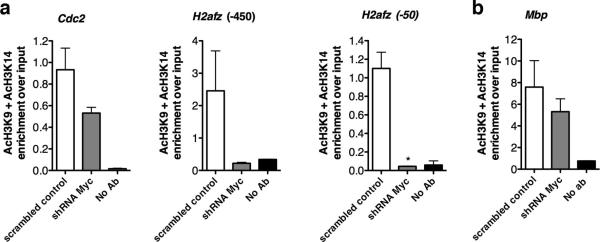
Chromatin was isolated from mouse OPCs 72h post-infection with either scramble shRNA c-Myc or scrambled control, and immunoprecipitated with antibodies specific for acetylated lysine 9 and lysine 14 on histone 3. After reverse cross-linking, the bound DNA was amplified using primer sets specific for the promoters of the c-Myc target genes (a) or for an independent gene, such as Mbp (b). A mock immunoprecipitation was used as negative control. Experiment was performed in duplicate.
To further define the effect of c-Myc on the progression of OPC towards a mature phenotype, we immunophenotypically characterized the c-Myc silenced and scrambled control cells, using antibodies specific for the progenitor marker NG2 and for specific lipid sulfatides recognized by O4 (Fig. 6a). Because lineage progression is characterized by the extension of cytoplasmic processes followed by elaboration of membrane sheets, we quantified these morphological changes in immunoreactive cells (fig. 6b). The analysis of 60 NG2+ cells per group revealed increased branching in proliferating OPCs (Fig. 6b) and a corresponding increase of O4+ membrane extension in differentiating cells (Fig. 6b). These morphological changes were associated with a tendency towards increased levels of late differentiation markers (i.e. Mbp, Sirt2), albeit not statistically significant (Fig. 6c). Together these data suggest that c-Myc silencing promotes OPC lineage progression, but it is not sufficient to drive the entire process of oligodendrocyte differentiation.
Figure 6. c-Myc silencing favors early oligodendrocyte lineage progression.

(a) c-Myc silenced cells were maintained either in proliferating condition (D0) or differentiated for 1 in the presence of T3 (D1). Proliferating mOPCs were stained for the progenitor marker NG2 (red) and for GFP (green) to visualize infected cells and counterstained with DAPI (blue). Differentiating mOPCs were stained for the progenitor marker NG2 (red) and for the lipid sulfatide recognized by the antibody O4 (red). (b) Bar graphs represent the average area occupied by NG2+ cytoplasmic processes at D0 and D1 and the O4+ membrane extension at D1, as measured by ImageJ in three independent experiments (n=3, mean ± s.e.m. *** p<0.005). (c) Effect of c-Myc silencing on the transcript levels of markers of oligodendrocyte differentiation (Mbp and Sirt2) as assessed by quantitative RT-PCR on RNA samples isolated from proliferating mOPCs after 72h post infection. Data were normalized to 18S and expressed as relative to scrambled controls. Scale bar in (a) = 20 μm.
c-Myc levels modulate chromatin changes in differentiating OPC
We previously discussed the role of c-Myc as an important modulator of histone acetylation at target genes with more modest contribution to overall histone acetylation.
Since histone deacetylation is one of the first modifications occurring during OPC differentiation and preceding the formation of compact chromatin (Liu and Casaccia, 2010), we asked whether c-Myc silencing increased a marker of compact chromatin (Fig. 7a). Immunocytochemical analysis of c-Myc silenced cells and scrambled controls revealed a statistically significant increase of heterochromatic foci, detected by the immunoreactivity with antibodies specific for H3K9me3 (Fig. 7b).
Figure 7. Increased heterochromatin marks upon c-Myc silencing.

(a) Confocal images of c-Myc silenced and scrambled infected mOPCs either kept proliferating (D0) or differentiated 24 hours in the presence of T3 (D1) and then stained for the heterochromatin histone mark H3K9me3 (red). Cells were also counterstained with GFP (green) to visualize infected cells and DAPI (blue) as nuclear counterstain. Inserts show a representative magnification of stained nuclei. (b) Bar graphs represent the average pixel intensity/area of H3K9me3 immunofluorescence as measured at D0 and D1. Values are expressed as relative to those measured in scrambled control in three independent experiments (n=3, mean ± s.e.m , *p<0.05, ***p<0.005; scale bar in (a) = 20 μm.
Finally, to further ascertain the changes in heterochromatin we performed electron microscopy on silenced cells using a method allowing for the visualization of fluorescent cells at the confocal microscopy followed by ultrastructural analysis (Fig. 8a) and quantification of the heterochromatin. A greater proportion of heterochromatin foci at the nuclear periphery was detected in c-Myc silenced cells as compared to scrambled shRNA infected cells (Fig. 8b).
Figure 8. Peripheral chromatin compaction upon c-Myc silencing.
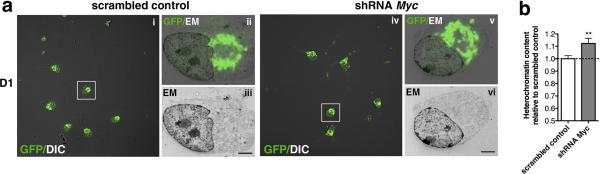
(a) Primary mouse OPCs were transduced with a double cistronic lentivirus encoding for GFP and for the shRNA (scrambled or against c-Myc sequence) and then differentiated for 1 day in the presence of T3. The same cell was imaged using a confocal microscope equipped with differential interference contrast (DIC) to detect silencing and a Hitachi 7800 electron microscope (EM) to visualize ultrastructure. All images were scaled to size and orientation to allow the overlay. Panels (i) and (iv) show low magnification DIC/GFP images. Higher magnification of the selected cells in (i) and (iv) are shown in panels (ii,iii) and (v,vi), respectively. Panels (ii) and (v) show the GFP/EM overlay whereas panels (iii) and (vi) show just the ultrastructure of the selected cells. (b) Bar graphs represent the average quantification of heterochromatin regions at the nuclear periphery at D1. Values were expressed as relative to those measured in scrambled control. Error bars represent s.e.m. (n=5-8 cells per group,**p<0.01). Scale bar 2 μm.
Together, these results suggest an important role for the transcription factor c-Myc in the maintenance of the progenitor state in multiple ways. C-Myc acts not only as transcription factor, responsible for direct binding to consensus sequences close to the TSS of genes, but also as recruiter of histone acetyltransferases to genes encoding for cell cycle and nucleosomal histone variants, which are characteristically associated with proliferating OPCs.
DISCUSSION
This study identifies c-Myc as an important transcriptional regulator of the transition from proliferating to differentiating OPCs. The importance of c-Myc in maintaining a proliferative and undifferentiated state is shown by a large number of evidence in several cell types. Another member of the Myc family, N-Myc has been described in neural stem cells and in neuroblastomas, where it similarly modulates the ability of neural precursors to proliferate and differentiate (Knoepfler et al., 2002) (Knoepfler et al., 2006) (Cetinkaya et al., 2007). C-Myc expression is high in proliferative cells and its deregulation and amplification has been associated with cancer transformation (Vita and Henriksson, 2006). Indeed, aberrant expression and activity of c-Myc has been described in gliomas (Hirvonen et al., 1994) where it has also been proposed to play a role in the maintenance of cancer stem cells (Wang et al., 2008). Previous studies in cultured OPC overexpressing c-Myc supported that c-Myc expression alone favored the establishment of immortalized cells (Barnett and Crouch, 1995), while the co-expression of c-Myc with the H-Ras oncogene was sufficient to induce transformation, as OPC expressing these molecules were capable of inducing gliomas when injected in the rat brain (Barnett et al., 1998).
Together these data were interpreted as resulting from the transcriptional activity of c-Myc in regulating proliferation as consequence to direct binding to consensus sequences. However more recent studies on the requirement of c-Myc for nuclear reprogramming of somatic cells (Takahashi and Yamanaka, 2006) (Thier et al., 2012) (Song et al., 2012) underscored the relevance of its chromatin-modifying properties and suggested that this molecule may have functions that go beyond transcriptional regulation of target genes. This idea was supported by genome-wide studies indicating that c-Myc is able to bind not only to its own recognition sequence, but also to genomic sites characterized by specific combinatorial histone codes, defined by activating trimethylation of lysine K4 and K27 together with hyperacetylation of nucleosomal histone H3 (Guccione et al., 2006). These findings implied that the transcriptional consequences of c-Myc dys-regulation might be far greater than just on genes bearing its consensus sequence. Our data in OPCs are consistent with a transcriptional role of c-Myc in modulating histone acetylation at target genes and affecting their expression. However we did not detect the prominent changes in global histone acetylation as suggested in other experimental systems.
Our study suggests that c-Myc directly binds to the promoter of genes encoding for cell cycle regulators (i.e. Cdc2) and histone variants (i.e. H2afz), and possibly favors the recruitment of histone acetyltransferases (Frank et al., 2003) (Vervoorts et al., 2003) (McMahon et al., 2000) (Liu et al., 2003), as indicated by the hyperacetylation detected at these loci in the presence of Myc and by the dramatic decrease of acetylated histones at the same loci in c-Myc silenced cells.
It is also conceivable, although not directly investigated in this study, that c-Myc might have additional transcriptional consequences due to the regulation of the histone variant H2Az, and possibly its incorporation into target promoters, as shown in human B-cell line (Martinato et al., 2008). The gene H2afz encodes for the nucleosomal H2Az histone variant, which has been shown to be characteristically positioned in genomic regions flanking transcription start sites (TSS) (Draker and Cheung, 2009) (Albert et al., 2007). H2Az is considered a component of euchromatic promoters, and proposed to play a role in regulating neural lineage specification (Creyghton et al., 2008).
Overall our data define c-Myc as important regulator of the maintenance of the proliferative and undifferentiated state in OPCs. Its silencing modulated the very early stages of oligodendrocyte lineage progression by reducing histone acetylation at target genes and inducing transcriptional changes consistent with decreased proliferation and inducing the formation of heterochromatin foci at the nuclear periphery. However decreasing its levels was not sufficient to induce differentiation of oligodendrocyte progenitors, even though it favored a morphologically more advanced phenotype characterized by extensive branching in proliferating conditions and membrane extensions in differentiating conditions. Its role in mature cells and the potential consequences on myelination remain to be thoroughly understood. This question has been approached several years ago, with the generation of transgenic mice overexpressing exons 2 and 3 of the human c-Myc from a fragment of the Mbp promoter (Orian et al., 1994). The phenotype of these mice was characterized by hypomyelination and aberrant postnatal development (Orian et al., 1997), which was eventually compensated by a population of cells that did not contain the transgene and were able to slowly proliferate and differentiate (Orian et al., 2001). A similar strategy used by a different group (Jensen and Celis, 1998), in contrast, showed a much more severe phenotype characterized by complete lack of myelin and severe cytotoxicity. The suggestion was that apoptosis would occur at a time coincident with the beginning of differentiation of OPC and that c-Myc overexpression is incompatible with terminal differentiation and maturation. Our study provides a solid support to the importance of c-Myc as an important transcriptional regulator in the oligodendrocyte lineage.
Figure 9. Proposed model for the role of c-Myc in oligodendrocyte progenitor cells.

(a) In proliferating OPCs, c-Myc activates transcription of target genes by favoring histone acetylation (AcH3K9 and AcH3K14) possibly by recruiting histone acetyl transferases (HAT) to the promoters of cell cycle genes (Cdc2) and chromatin components (H2Afz). (b) In differentiating OPCs, reduced levels of c-Myc result in decreased acetylation (H3K9Ac and H3K14Ac) at the promoters of the gene targets, thus leading to decreased expression. The effect of c-Myc decrease on peripheral nuclear chromatin compaction (black regions at the nuclear periphery) is also shown.
Acknowledgments
This work was supported by grants R01-NS52738 and R37-NS42925 to PC.
ABBREVIATIONS
- OPCs
oligodendrocyte progenitor cells
- PDGF-A
platelet derived growth factor
- T3
thyroid hormone
- MBP
myelin basic protein
- TSS
transcription start sites
- DAVID
database for annotation, visualization and integrated discovery
- HATs
histone acetyl-transferases
- HDAC
histone deacetylases
- ChIP
chromatin immunoprecipitation
- EM
electron microcopy
- GBM
glioblastoma multiforme
Footnotes
The authors declare no competing financial interests
REFERENCES
- Albert I, Mavrich TN, Tomsho LP, Qi J, Zanton SJ, Schuster SC, Pugh BF. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature. 2007;446:572–576. doi: 10.1038/nature05632. [DOI] [PubMed] [Google Scholar]
- Amati B, Frank SR, Donjerkovic D, Taubert S. Function of the c-Myc oncoprotein in chromatin remodeling and transcription. Biochimica et biophysica acta. 2001;1471:M135–145. doi: 10.1016/s0304-419x(01)00020-8. [DOI] [PubMed] [Google Scholar]
- Barnett SC, Crouch DH. The effect of oncogenes on the growth and differentiation of oligodendrocyte type 2 astrocyte progenitor cells. Cell growth & differentiation : the molecular biology journal of the American Association for Cancer Research. 1995;6:69–80. [PubMed] [Google Scholar]
- Barnett SC, Robertson L, Graham D, Allan D, Rampling R. Oligodendrocytetype-2 astrocyte (O-2A) progenitor cells transformed with c-myc and H-ras form high-grade glioma after stereotactic injection into the rat brain. Carcinogenesis. 1998;19:1529–1537. doi: 10.1093/carcin/19.9.1529. [DOI] [PubMed] [Google Scholar]
- Blackwell TK, Kretzner L, Blackwood EM, Eisenman RN, Weintraub H. Sequence-specific DNA binding by the c-Myc protein. Science. 1990;250:1149–1151. doi: 10.1126/science.2251503. [DOI] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cetinkaya C, Hultquist A, Su Y, Wu S, Bahram F, Pahlman S, Guzhova I, Larsson LG. Combined IFN-gamma and retinoic acid treatment targets the N-Myc/Max/Mad1 network resulting in repression of N-Myc target genes in MYCN-amplified neuroblastoma cells. Molecular cancer therapeutics. 2007;6:2634–2641. doi: 10.1158/1535-7163.MCT-06-0492. [DOI] [PubMed] [Google Scholar]
- Creyghton MP, Markoulaki S, Levine SS, Hanna J, Lodato MA, Sha K, Young RA, Jaenisch R, Boyer LA. H2AZ is enriched at polycomb complex target genes in ES cells and is necessary for lineage commitment. Cell. 2008;135:649–661. doi: 10.1016/j.cell.2008.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Molecular and cellular biology. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:6479–6483. doi: 10.1158/1078-0432.CCR-09-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G, Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome biology. 2003;4:P3. [PubMed] [Google Scholar]
- Draker R, Cheung P. Transcriptional and epigenetic functions of histone variant H2A.Z. Biochemistry and cell biology = Biochimie et biologie cellulaire. 2009;87:19–25. doi: 10.1139/O08-117. [DOI] [PubMed] [Google Scholar]
- Dugas JC, Tai YC, Speed TP, Ngai J, Barres BA. Functional genomic analysis of oligodendrocyte differentiation. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:10967–10983. doi: 10.1523/JNEUROSCI.2572-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson N. The regulation of E2F by pRB-family proteins. Genes & development. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- Frank SR, Parisi T, Taubert S, Fernandez P, Fuchs M, Chan HM, Livingston DM, Amati B. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO reports. 2003;4:575–580. doi: 10.1038/sj.embor.embor861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank SR, Schroeder M, Fernandez P, Taubert S, Amati B. Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes & development. 2001;15:2069–2082. doi: 10.1101/gad.906601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guccione E, Martinato F, Finocchiaro G, Luzi L, Tizzoni L, Dall' Olio V, Zardo G, Nervi C, Bernard L, Amati B. Myc-binding-site recognition in the human genome is determined by chromatin context. Nature cell biology. 2006;8:764–770. doi: 10.1038/ncb1434. [DOI] [PubMed] [Google Scholar]
- Hanson HH, Reilly JE, Lee R, Janssen WG, Phillips GR. Streamlined embedding of cell monolayers on gridded glass-bottom imaging dishes for correlative light and electron microscopy. Microscopy and microanalysis : the official journal of Microscopy Society of America, Microbeam Analysis Society, Microscopical Society of Canada. 2010;16:747–754. doi: 10.1017/S1431927610094092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Dupree J, Wang J, Sandoval J, Li J, Liu H, Shi Y, Nave KA, Casaccia-Bonnefil P. The transcription factor Yin Yang 1 is essential for oligodendrocyte progenitor differentiation. Neuron. 2007;55:217–230. doi: 10.1016/j.neuron.2007.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirvonen HE, Salonen R, Sandberg MM, Vuorio E, Vastrik I, Kotilainen E, Kalimo H. Differential expression of myc, max and RB1 genes in human gliomas and glioma cell lines. British journal of cancer. 1994;69:16–25. doi: 10.1038/bjc.1994.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Jensen NA, Celis JE. Proteomic changes associated with degeneration of myelin-forming cells in the central nervous system of c-myc transgenic mice. Electrophoresis. 1998;19:2014–2020. doi: 10.1002/elps.1150191121. [DOI] [PubMed] [Google Scholar]
- Jung M, Kramer E, Grzenkowski M, Tang K, Blakemore W, Aguzzi A, Khazaie K, Chlichlia K, von Blankenfeld G, Kettenmann H, et al. Lines of murine oligodendroglial precursor cells immortalized by an activated neu tyrosine kinase show distinct degrees of interaction with axons in vitro and in vivo. The European journal of neuroscience. 1995;7:1245–1265. doi: 10.1111/j.1460-9568.1995.tb01115.x. [DOI] [PubMed] [Google Scholar]
- Knoepfler PS, Cheng PF, Eisenman RN. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes & development. 2002;16:2699–2712. doi: 10.1101/gad.1021202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoepfler PS, Zhang XY, Cheng PF, Gafken PR, McMahon SB, Eisenman RN. Myc influences global chromatin structure. The EMBO journal. 2006;25:2723–2734. doi: 10.1038/sj.emboj.7601152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rocca SA, Crouch DH, Gillespie DA. c-Myc inhibits myogenic differentiation and myoD expression by a mechanism which can be dissociated from cell transformation. Oncogene. 1994;9:3499–3508. [PubMed] [Google Scholar]
- Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, Ma'ayan A. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics. 2010;26:2438–2444. doi: 10.1093/bioinformatics/btq466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei L, Sonabend AM, Guarnieri P, Soderquist C, Ludwig T, Rosenfeld S, Bruce JN, Canoll P. Glioblastoma models reveal the connection between adult glial progenitors and the proneural phenotype. PloS one. 2011;6:e20041. doi: 10.1371/journal.pone.0020041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Casaccia P. Epigenetic regulation of oligodendrocyte identity. Trends in neurosciences. 2010;33:193–201. doi: 10.1016/j.tins.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Sandoval J, Doh ST, Cai L, Lopez-Rodas G, Casaccia P. Epigenetic modifiers are necessary but not sufficient for reprogramming non-myelinating cells into myelin gene-expressing cells. PloS one. 2010;5:e13023. doi: 10.1371/journal.pone.0013023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Tesfai J, Evrard YA, Dent SY, Martinez E. c-Myc transformation domain recruits the human STAGA complex and requires TRRAP and GCN5 acetylase activity for transcription activation. The Journal of biological chemistry. 2003;278:20405–20412. doi: 10.1074/jbc.M211795200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyssiotis CA, Walker J, Wu C, Kondo T, Schultz PG, Wu X. Inhibition of histone deacetylase activity induces developmental plasticity in oligodendrocyte precursor cells. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:14982–14987. doi: 10.1073/pnas.0707044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Husstege M, Muggironi M, Liu A, Casaccia-Bonnefil P. Histone deacetylase activity is necessary for oligodendrocyte lineage progression. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2002;22:10333–10345. doi: 10.1523/JNEUROSCI.22-23-10333.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinato F, Cesaroni M, Amati B, Guccione E. Analysis of Myc-induced histone modifications on target chromatin. PloS one. 2008;3:e3650. doi: 10.1371/journal.pone.0003650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon SB, Wood MA, Cole MD. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Molecular and cellular biology. 2000;20:556–562. doi: 10.1128/mcb.20.2.556-562.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevins JR. Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell growth & differentiation : the molecular biology journal of the American Association for Cancer Research. 1998;9:585–593. [PubMed] [Google Scholar]
- Orian JM, Ahern AJ, Ayers MM, Levine JM, Tapp LD, Reynolds R. Disturbed oligodendrocyte development and recovery from hypomyelination in a c-myc transgenic mouse mutant. Journal of neuroscience research. 2001;66:46–58. doi: 10.1002/jnr.1196. [DOI] [PubMed] [Google Scholar]
- Orian JM, Mitchell AW, Marshman WE, Webb GC, Ayers MM, Grail D, Ford JH, Kaye AH, Gonzales MF. Insertional mutagenesis inducing hypomyelination in transgenic mice. Journal of neuroscience research. 1994;39:604–612. doi: 10.1002/jnr.490390512. [DOI] [PubMed] [Google Scholar]
- Orian JM, Slavin A, Ayers MM, Bernard CC. Delayed and incomplete myelination in a transgenic mouse mutant with abnormal oligodendrocytes. Journal of neuroscience research. 1997;50:809–820. doi: 10.1002/(SICI)1097-4547(19971201)50:5<809::AID-JNR17>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death. Nature reviews Cancer. 2002;2:764–776. doi: 10.1038/nrc904. [DOI] [PubMed] [Google Scholar]
- Ruas M, Peters G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochimica et biophysica acta. 1998;1378:F115–177. doi: 10.1016/s0304-419x(98)00017-1. [DOI] [PubMed] [Google Scholar]
- Shen S, Li J, Casaccia-Bonnefil P. Histone modifications affect timing of oligodendrocyte progenitor differentiation in the developing rat brain. The Journal of cell biology. 2005;169:577–589. doi: 10.1083/jcb.200412101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Sandoval J, Swiss VA, Li J, Dupree J, Franklin RJ, Casaccia-Bonnefil P. Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nature neuroscience. 2008;11:1024–1034. doi: 10.1038/nn.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ. The ins and outs of RB: coupling gene expression to the cell cycle clock. Trends in cell biology. 1994;4:15–18. doi: 10.1016/0962-8924(94)90033-7. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Mammalian G1 cyclins and cell cycle progression. Proceedings of the Association of American Physicians. 1995;107:181–186. [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes & development. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Sloan EJ, Ayer DE. Myc, mondo, and metabolism. Genes & cancer. 2010;1:587–596. doi: 10.1177/1947601910377489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B, Sun G, Herszfeld D, Sylvain A, Campanale NV, Hirst CE, Caine S, Parkington HC, Tonta MA, Coleman HA, et al. Neural differentiation of patient specific iPS cells as a novel approach to study the pathophysiology of multiple sclerosis. Stem cell research. 2012;8:259–273. doi: 10.1016/j.scr.2011.12.001. [DOI] [PubMed] [Google Scholar]
- Swiss VA, Nguyen T, Dugas J, Ibrahim A, Barres B, Androulakis IP, Casaccia P. Identification of a gene regulatory network necessary for the initiation of oligodendrocyte differentiation. PloS one. 2011;6:e18088. doi: 10.1371/journal.pone.0018088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Thier M, Worsdorfer P, Lakes YB, Gorris R, Herms S, Opitz T, Seiferling D, Quandel T, Hoffmann P, Nothen MM, et al. Direct conversion of fibroblasts into stably expandable neural stem cells. Cell stem cell. 2012;10:473–479. doi: 10.1016/j.stem.2012.03.003. [DOI] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervoorts J, Luscher-Firzlaff JM, Rottmann S, Lilischkis R, Walsemann G, Dohmann K, Austen M, Luscher B. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO reports. 2003;4:484–490. doi: 10.1038/sj.embor.embor821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vita M, Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer. Seminars in cancer biology. 2006;16:318–330. doi: 10.1016/j.semcancer.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Wang J, Wang H, Li Z, Wu Q, Lathia JD, McLendon RE, Hjelmeland AB, Rich JN. c-Myc is required for maintenance of glioma cancer stem cells. PloS one. 2008;3:e3769. doi: 10.1371/journal.pone.0003769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Yuan X, Chittajallu R, Belachew S, Anderson S, McBain CJ, Gallo V. Expression of the green fluorescent protein in the oligodendrocyte lineage: a transgenic mouse for developmental and physiological studies. Journal of neuroscience research. 2002;70:529–545. doi: 10.1002/jnr.10368. [DOI] [PubMed] [Google Scholar]