

Abstract
Introduction
Based on its role as a mitotic regulatory kinase, overexpressed and associated with aneuploidy in cancer, small molecule inhibitors have been developed for Aurora-A (AURKA) kinase. In preclinical and clinical assessments, these agents have shown efficacy in inducing stable disease or therapeutic response. In optimizing the use of Aurora-A inhibitors, it is critical to have robust capacity to measure the kinase activity of Aurora-A in tumors.
Areas covered
we provide an overview of molecular mechanisms of mitotic and non-mitotic activation of Aurora-A kinase, and interaction of Aurora-A with its regulatory partners. Typically, Aurora-A activity is measured by use of phospho-antibodies targeting an auto-phosphorylated T288 epitope. However, recent studies have identified alternative means of Aurora-A activation control, including allosteric regulation by partners, phosphorylation on alternative activating residues (S51, S98), dephosphorylation on inhibitory sites (S342), and T288 phosphorylation by alternative kinases such as Pak enzymes. Additional work has shown that the relative abundance of Aurora-A partners can affect the activity of Aurora-A inhibitors, and that Aurora-A activation also occurs in interphase cells.
Expert opinion
Taken together, this work suggests the need for comprehensive analysis of Aurora-A activity and expression of Aurora-A partners in order to stratify patients for likely therapeutic response.
Keywords: Allosteric activation, AURKA, Aurora-A, cancer, NEDD9, PAK, phosphorylation, targeted therapy, TPX2
1. Introduction
As cancer treatment is increasingly informed by knowledge of the precise oncogenic lesions that drive tumor growth, the dependence of medical oncology on protein-targeted therapies continues to increase. Many tumors are “addicted” to the continuous activity of oncogenic proteins for their survival [1], and some of these proteins have emerged as valuable drug targets. At present, the majority of targeted therapies are designed to inhibit enzymes, and particularly kinases, so that loss of activity in an addicted cell results in tumor cell death. Effective application of targeted kinase inhibitors in the clinic requires the identification and validation of settings in which the therapeutic target is functionally important, typically in an active state, and also requires the ability to confirm that therapeutic agents are inhibiting the target within tumors. In pre-clinical and clinical settings, the assessment of the activation state of the target kinase and its inhibition by a targeted drug is often most conveniently performed using antibodies to an auto-phosphorylated epitope on the target kinase. In some cases, where antibodies to such epitopes are not available, the phosphorylation of a direct downstream substrate of the kinase of interest is measured. For instance, phosphorylation of MEK1/2 on S218/S222 is often used as a surrogate reporter of RAF activity [2]. Information regarding target activation helps inform the accurate assessment of clinical response to targeted drugs.
In this article, we focus on an under-appreciated issue that has the potential to confound the clinical application of valuable targeted drugs. This is the possibility that some kinases can be activated in multiple different ways, which can confound standard means of detecting and/or inhibiting kinase activity. Historically, many oncogenic enzymes, including kinases, were identified as auto-activating (typically due to activating mutations in the catalytic domain), or alternatively, as activated by protein-protein interaction with a single upstream partner that conferred allosteric changes supporting auto-phosphorylation. However, as the mapping of protein signaling networks becomes more complete, this relatively simple paradigm is often challenged. Increasingly, drug targets of considerable therapeutic interest are found to be activated in multiple ways, by diverse upstream factors. Hence, classic clinical indicators of target activation may not apply, depending on the upstream activation mechanism. As a specific example of this issue, we focus on one important therapeutic target: the oncogenic Aurora-A kinase, an evolutionarily conserved serine/threonine kinase essential for mitotic progression.
Aurora-A is overexpressed in many tumors arising from breast, colon, ovary, and other tissues, and functions as an oncogene when exogenously expressed in numerous cell line models [3–7]. Overexpression of Aurora-A causes supernumerary centrosomes and multipolar spindles arising as consequence of failed cytokinesis, which leads to aneuploidy [8]. High Aurora-A expression in cancer patients is an independent predictive and prognostic marker associated with resistance to taxanes [9] and decreased survival [10]. Aurora-A directly interacts with important oncogenes and tumor suppressor genes: it phosphorylates Src [11], stabilizes N-myc [12], and phosphorylates and down-regulates the major tumor suppressor p53 [13]. P53, in turn, negatively regulates Aurora-A via both transcriptional and posttranslational mechanisms [14]. Conversely, p53 inactivation, common in solid tumors, supports induction of Aurora-A, with concomitant aneuploidy and chromosomal instability [15]. Consequently, Aurora-A has been a popular target for development of anti-cancer agents, with the Aurora-A inhibitor alisertib now being tested in multiple late stage clinical trials [16, 17].
In the preclinical studies, because available biosamples are typically formalin-fixed, paraffin-embedded (FFPE) tissues, Aurora-A activity is commonly reported based on measurement of auto-phosphorylation on residue T288 in the activation or T-loop. Many assessments of alisertib and other Aurora-A inhibitors have been based on analysis of T288 phosphorylation. In the past few years, several confounding issues – activation uncoupled to T288 phosphorylation, non-mitotic activities of Aurora-A, and poor antibody quality – have emerged in studies of the signaling activity of Aurora-A. However, perhaps because of the silo effect that frequently separates work in biochemistry and structural biology from preclinical and clinical drug development, many of these issues are not typically considered in clinical efforts.
Appreciation of these issues, coupled with accurate understanding of how well Aurora-A-targeting drugs inhibit their target, is essential in designing effective clinical strategies. In this paper, we describe the mitotic and non-mitotic function of Aurora-A, discuss activation of Aurora-A dependent on or independent of T288 phosphorylation, and highlight the role of other kinases and phosphatases regulating activity of Aurora-A. We then place this work in the context of a discussion of alisertib, the most advanced and clinically effective of the Aurora-A targeting agents.
2. Mitotic activation of Aurora-A: the important role of T288 autophosphorylation
In its well-validated role as a mitotic regulator (reviewed in Nikonova et al. [8]), Aurora-A accumulates at the centrosome in G2, and becomes highly active at the G2/M transition. Aurora-A contributes to centrosome maturation by recruiting γ-tubulin, centrosomin, and other centrosomal proteins to the pericentriolar mass [18, 19]. Mitotic entry is catalyzed by the kinase activity of cyclin-dependent kinase 1 (CDK1) in complex with cyclin B1 [20]. Aurora-A phosphorylates the CDK-activating phosphatase CDC25B, and supports the activation of the CDK1/cyclin B1 complex to allow mitotic entry [21]. Reciprocally, active CDK1 in complex with another cyclin (B2) promotes further mitotic activation of Aurora-A [22]. In additional pro-mitotic activities, Aurora-A phosphorylation of the BRCA1 protein reduces G2/M checkpoint controls [23], and Aurora-A phosphorylation of the RAS family protein RALA regulates mitochondrial fusion, which is important for equal post-mitotic segregation of mitochondria between daughter cells [24].
Aurora-A remains active through the M phase, supports functioning of the centrosomes as bipolar microtubule organizing centers, and coordinates chromosome segregation. Beginning in prophase, Aurora-A propagates from the centrosome to the spindle, and at metaphase and later localizes to the midzone, regulating spindle dynamics [25]. In all metazoans assessed to date, mutation or depletion of Aurora-A causes formation of spindles with abnormally organized poles, including characteristic monopolar structures, and weak, sparse, or short astral microtubules [8]. Before cytokinesis, the APC/Cdh1 complex ubiquitinates Aurora-A and targets it for proteasomal degradation at the midbody; failure to degrade Aurora-A is associated with failed cytokinesis [25]. In addition to proteasomal degradation, Aurora-A is mitotically SUMOylated, which may contribute to its localization control [26].
Aurora-A activation is frequently reported based on measurement of phosphorylation on residue T288 in the activation loop (all numbering based on the human Aurora-A) [27]. The adjacent residue, T287, is sometimes phosphorylated [28], and partially redundant with T288, following a model established for other kinases [29]. However, one interesting recent study has shown that while phosphorylation of T288 or T287 alone activates Aurora-A, phosphorylation of both together can inhibit kinase activity by competition for a binding site, clearly indicating the need for more study [30]. Figure 1 shows an alignment of the entire activation loop of Aurora A and Aurora B sequences from several species. T288 is conserved in all forms of Aurora-A and Aurora-B; T287 is less conserved in Aurora-A, and absent in Aurora-B. The activation loop of almost all kinases begins with a conserved “DFG motif” (sequence Asp-Phe-Gly) and ends with a sequence similar to APE (Ala-Pro-Glu; Aurora A and B have Pro-Pro-Glu). The conformation of the loop is different in active and inactive kinases, and is loosely categorized by the position of the DFG sequence of the activation loop, sometimes referred to as DFG-in for active kinases and DFG-out for some inactive kinases. In most inactive kinases, the activation loop can be located in several different positions, depending on the inhibitor bound. For Aurora A, one of these is shown in Figure 2A. This structure has been referred to as DFG-up by Dodson et al. since the Phe of the DFG loop points upwards far into the N-terminal domain in an unusual way [31]. Many Aurora A structures exhibit this DFG-up conformation [8].
Figure 1.

A. Sequence alignment of the activation loop of Aurora-A and a related protein, Aurora-B from various species. Completely conserved residues are marked in blue; the T288 phosphorylation site is shown in red.
Figure 2.


A. Drug-inhibited, inactive form of Aurora-A catalytic domain; colored blue to red from amino- to carboxy-terminus [119]. The inhibitor, a 2–4 bisanilinopyrimidine (pink) is shown in sticks. The activation loop is shown in magenta, and is in a position to block the active site. The Phe275 of the DFG (Asp-Phe-Gly) loop is shown in stick figures and is in a “DFG-up” configuration [31]. B. Phosphorylated active Aurora-A catalytic domain bound to TPX2 (beige ribbon). The activation loop is in an active DFG-in position with phosphorylation on T287 and T288 (marked TPO287 and TPO288) shown in sticks. Phe275 of the activation loop DFG motif and Asp256 of the HRD motif in the active site, which binds substrate Ser and Thr OH groups, are also shown in sticks. The structure includes bound ADP and two magnesium atoms (yellow spheres). Ser342, an additional regulatory phosphorylation site of Aurora-A discussed in the text, is also marked.
While Aurora-A is capable of auto-phosphorylation on T288 [32], a number of proteins directly associate with Aurora-A and regulate this process. The structural basis for activation by one partner, TPX2, was the first to be identified and has been the most thoroughly characterized [33–38]. Activation of the GTPase Ran at the time of nuclear envelope breakdown releases TPX2 from an importin inhibitory complex, allowing its binding to Aurora-A. This promotes a conformational change in Aurora-A associated with T288 autophosphorylation, and moves the activation loop to an extended position away from the active site. The Phe of DFG now points downwards and underneath the C-helix of the N-terminal domain in an active conformation. This is shown in Figure 2B, which depicts the Aurora-A kinase domain with bound ATP, two magnesium ions in the active site, and phosphorylation of residues T287 and T288 of the activation loop. TPX2 is shown as a beige ribbon, bound to the N-terminal domain far from the active site. In this position the activating loop is protected from dephosphorylation by negative Aurora-A regulators, and is available for interaction with Aurora-A substrates [37–43]. The order of TPX2 binding and Aurora-A phosphorylation in vivo is not known, though it has been proposed that low levels of auto-phosphorylation of Aurora-A occur at centrosomes in the early stages of mitosis, followed by allosteric activation by TPX2 promoting high levels of actions as the centrosome assembles the spindle microtubules [32]. The TPX2 interaction also helps to target Aurora-A to mitotic spindles, proximal to substrates [41].
Assessment of Aurora-A kinase activity based on use of antibodies to the phosphorylated T288 epitope can provide useful information, but particularly in recent years has some associated issues. One issue is limitation in the quality of available commercial reagents for phospho-T288 Aurora-A. Commercially available T288 phospho-antibodies have been shown to cross-react with a family member, Aurora-B, under some conditions [44] (see Figure 1). While it is difficult to publish negative results, in inter-laboratory communications, it has been noted by numerous groups (including our group) that the quality of antibodies to phospho-T288 Aurora-A has become variable. In particular, reactivity of commercial antibodies against murine phospho-T288 Aurora-A has deteriorated since ~2010, for applications including Western blotting, immunohistochemistry, and immunofluorescence, as documented by the fact that very few publications have appeared in the last two years using antibody to murine phospho-T288 Aurora-A to study endogenous Aurora-A protein. This can limit preclinical studies of Aurora-A inhibitors, particularly when using mouse cancer models and cell lines. This may reflect the fact that the mouse activation site sequence uniquely contains RRTT288M, instead of the RRTT288L found in most vertebrates (Figure 1), with this change reducing the affinity of the antibody for the epitope. For human tissue, while much better performance is obtained, most phospho-T288 antibodies have multiple cross-reacting bands in Western blots, raising some cautions as to the interpretation of immunohistochemistry assessments. Aside from reagent quality, a more important issue in using antibody to phospho-T288 Aurora-A to gauge activity of this protein is the increasing abundance of publications indicating the activation of Aurora-A is not only based on auto-phosphorylation on T288. Rather, Aurora-A can be activated through other pathways and T288 can be phosphorylated by other kinases, as discussed below.
3. Activation of Aurora-A exclusive of T288 auto-phosphorylation
While most literature addressing Aurora-A activation focuses on the T-loop phosphorylation site T288, one of the earliest studies of mitotic activation of Aurora-A, using a Xenopus system, showed that active Aurora-A is also phosphorylated on residue S51 and lacks phosphorylation on S342 (all numbering based on the human Aurora-A) [27]. Subsequent studies confirmed these phosphorylation sites in mitosis and meiosis, and identified additional sites of mitotic phosphorylation on S53/S54, S66/S67, S89, and S98 [27, 45–49]. These phosphorylation sites are functionally important.
First, auto-phosphorylation of Aurora on residue S342 on the αG helix of the C-terminal domain (see Figure 2B) limits Aurora-A activity [27, 50, 51], and provides a mechanism for fine regulation of active Aurora-A in mitosis. As shown in Xenopus models, an S342D mutation (mimicking constitutive phosphorylation of serine) completely blocks Aurora-A activity [27, 51], while an S342A mutation renders Aurora-A resistant to inhibition following DNA damage [52]. Autophosphorylation of Aurora-A on residue S342, in its turn, is induced by glycogen synthase kinase 3 (GSK-3) placing a “priming” phosphorylation of Aurora-A on residues S283 and S284 located on the activation loop just prior to T288 [51]. Serine to alanine substitutions at the S283 and S284 priming sites prevent their phosphorylation, and results in a constitutively active form of Aurora-A dephosphorylated on S342 residue. Substitution of the serine with aspartic acid at the priming sites mimics their constitutive phosphorylation and results in a completely inactive Aurora-A autophosphorylated on S342 [51]. As evidence of the importance of this activation control mechanism, Sarkissian et al. showed the absence of phosphorylation at S342 predicted the activity of Aurora-A better than did phosphorylation at T288 [51].
Additional mitotic regulation of Aurora-A is provided by binding of Ca2+-liganded calmodulin (CaM) to the unstructured amino-terminal domain of Aurora-A [46]. Ca2+/CaM binding induced Aurora-A autophosphorylation on residue S51 and nearby residues S53/S54, S66/S67 and S98 within the disordered amino-terminal region of Aurora-A (residues 1–121), as detected by mass spectroscopy [46]. This analysis did not detect T288 phosphorylation, although it was not established whether this phosphorylation failed to occur, or whether the peptide containing T288 was poorly detectable by mass spectrometry. Mutation within or proximal to five of the Ca2+/CaM phosphorylation sites (S51, S53, S54, S66 and S61) disrupted Aurora-A binding to CaM and to at least one co-activator, NEDD9, and disrupted Aurora-A functions associated with mitotic progression, resulting in a high frequency of mitotic catastrophe at metaphase-anaphase transition; whereas mutation of an adjacent site S98A nonspecifically impaired Aurora-A function [45, 46]. Importantly, mitotic phosphorylation on S51 had been suggested to protect Aurora-A from degradation until the end of mitosis [27, 53], as discussed further below.
More recently, nucleophosmin (B23) was identified as an additional Aurora-A activating partner [47]. Nucleophosmin, colocalizing with Aurora-A at the centrosome, induces Aurora-A auto-phosphorylation on residue S89, but not T288. Phosphorylation of S89 was necessary for mitotic phosphorylation of the Aurora-A substrate CDC25B in vivo, suggesting it is required for at least some Aurora-A-dependent mitotic phosphorylation events [47].
Importantly, even the well-studied Aurora-A activator TPX2 was recently shown to exert some of its activity through mechanisms not involving the induction of T288 auto-phosphorylation. In elegant studies, Dodson and Bayliss dissected the kinase activity of T288-unphosphorylatable (T288A mutant) Aurora-A with and without TPX2, and T288-phosphorylated Aurora-A with and without TPX2 [42]. Unexpectedly, TPX2 binding increased the catalytic activity of T288A Aurora-A 15-fold, while T288 phosphorylation also increased the activity of Aurora-A: the effect TPX2 binding and Aurora-A autophosphorylation bound together was the exact sum of their individual contributions to catalysis. The authors proposed a revised model of Aurora-A activation, in which the first step was a reduction in the mobility of the activation loop, which could be induced by either TPX2 binding or T288 phosphorylation. These very important results suggests that T288-unphosphorylated Aurora-A bound to the mitotic spindle by TPX2 is catalytically active and that measurement of T288 phosphorylation is not invariably a surrogate for the Aurora-A activation state [42].
4. Activation control of Aurora-A at the centrosome by interaction with additional kinases, phosphatases, and scaffolding proteins
At the centrosome, Aurora-A associates with the scaffolding factors CEP192, NEDD9, Ajuba, PAK kinase family members, and nucleophosmin [35, 47, 54–56], as well as activating proteins Arpc1b, LIM1 kinase, phosphatase inhibitor I-2, BORA, TACC3, and TPX2 [33, 56–60]. For some of the Aurora-A interacting proteins, such as CEP192, the activating function is thought to involve recruitment to the centrosome, increasing local concentration of Aurora-A [35, 61, 62], while others, such as NEDD9, do not affect centrosomal recruitment [54]. It is important to note that in cancer amplification or overexpression of Aurora-A gene leads to abnormal localization of Aurora-A protein through the cytoplasm, or even in the nuclear compartment. It is reasonable to speculate that activation of non-centrosomal Aurora-A in cancer may be subject to different constraints than centrosomal Aurora-A in non-transformed cells, but to date, the topic has not been rigorously addressed. This is worthy of consideration, as a number of Aurora-A activating proteins discussed below have been shown to be overexpressed or activated in a subset of tumors, and to have pro-oncogenic function that may or may not be related to the control of Aurora-A activity.
4.1 Protein interactions that activate Aurora-A
In brief review of some of the known, cancer-relevant Aurora-A activators (Figure 3), binding of the scaffolding protein NEDD9 to multiple sites on Aurora-A likely protects Aurora-A from degradation and contributes to a conformational change that enhances activation [63]. Depletion of NEDD9 does not affect Aurora-A accumulation at the centrosome, but blocks the T288 phosphorylation and activation of Aurora-A at mitotic entry. Overexpression of NEDD9 induces Aurora-A hyperactivation and T288 phosphorylation, and produces cells with both multipolar spindles and supernumerary centrosomes and failure of cytokinesis [54, 64]. NEDD9 is commonly overexpressed in cancer, and promotes invasion and metastasis [63, 65].
Figure 3.
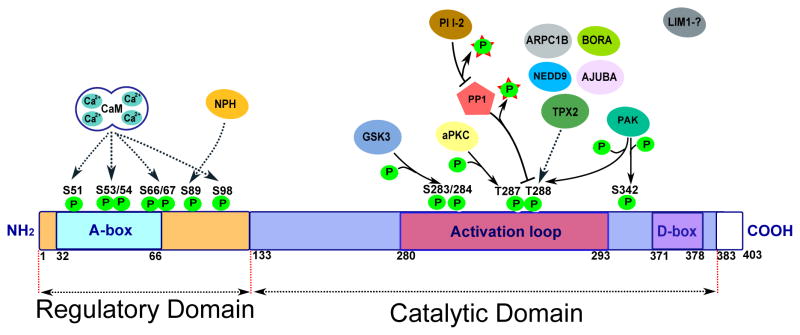
Activating and inactivating interactions and phosphorylation sites of Aurora-A. See text for details.
The LIM-domain centrosomal scaffolding protein Ajuba binds Aurora-A and has been reported to induce its autophosphorylation on T288 [56]. This interaction was shown to be important for activation of the cyclin-B/CDK1 complex and for commitment of cells to mitosis. Depletion of Ajuba prevents activation of Aurora-A at centrosomes in late G2 phase and inhibits mitotic entry [56]. However, several studies suggest a more complicated interpretation of Ajuba activity. In genetic studies in Drosophila, Sabino et al show that Ajuba does not affect Aurora-A activation per se, but retention of activated Aurora-A at the centrosome [66]. Another study has indicated that mutations in Aurora-A that disrupt N-terminal and C-terminal domain interactions (AurA-K250G and AurA-D294G/Y295G) disrupt Aurora-A activation [67], and also eliminate binding to Ajuba, although other work by the same group suggests Ajuba may influence intramolecular dynamics of Aurora-A, impacting auto-phosphorylation [68, 69].
The centrosomal protein Arpc1b interacts with the N-terminal domain of Aurora-A and likely changing the conformation of the Aurora-A active site, causing an increase in T288 phosphorylation. Overexpression of Arpc1b leads to abnormal centrosomal amplification, whereas depletion of Arpc1b drastically reduces the ability of cells to enter the cell cycle; this is accompanied by failure to accumulate active Aurora-A at the centrosome at the G2/M transition [58].
LIM kinase 1 (LIMK1) colocalizes with Aurora-A at the centrosome between early prophase through anaphase and phosphorylates Aurora-A. The target of LIMK1 is not T288, but has otherwise not been determined [70].
Phosphatase inhibitor I-2 is an activating partner of Aurora-A with two distinct domains, each of which separately influences Aurora-A activation: a C-terminal motif causes allosteric activation of Aurora-A (independent of T288 phosphorylation), while a N-terminal motif inhibits the action of protein phosphatase 1 in dephosphorylating T288 [57].
A recently described Aurora-A activator, BORA, does not localize to centrosomes. BORA is a nuclear protein, but is released from the nucleus to the cytoplasm upon entry into mitosis, where it can bind and activate Aurora-A, facilitating T288 autophosphorylation in vitro [59]. Most likely BORA acts downstream of other proteins that interact with Aurora-A before nuclear envelope breakdown occurs, and at least one study suggests the primary activity of BORA is more important for the activity of additional mitotic kinases such as PLK1, under physiological conditions within cultured cells [71].
PAK kinases are particularly confounding for the use of T288 phosphorylation as a gauge of Aurora-A activation. PAK kinases are a family of 6 paralogous serine-threonine kinases that are frequently activated in cancer and promote aggressive tumor growth [72]. PAK1, PAK2 and PAK3 each were shown to bind Aurora-A and induce phosphorylation of the T288 activation loop site[55]. Intriguingly, in the same study, they were shown to also phosphorylate the inhibitory S342 site [55], implying PAK kinases might similarly be imposing activating and inactivating signals on Aurora-A. Because of this duality of action, and because PAK kinases independently phosphorylate the T288 residue, gauge of Aurora-A activity based on measurement of T288 Aurora-A is likely to be inaccurate in tumors expressing high levels of PAKs.
4.2 Protein interactions that inhibit Aurora-A
The list of currently known Aurora-A inhibitors (Figure 3) is significantly shorter than activators; it includes protein phosphatases 1, 2A, and 6 (PP1, PP2A, PP6), p53 and Gadd45a. Dephosphorylation of T288 by PP1 limits Aurora-A activity [73]. As mentioned above, phosphatase inhibitor I-2 opposes PP1 in this function, thus enhancing Aurora-A activity [57]. Intriguingly, some evidence suggests that the binding site of PP1 is adjacent to the S342 negative regulatory site, corresponding to an RVEF motif at residues 343–354; and that mutation of these residues so as to eliminate PP1 binding resulted in a derivative of Aurora-A kinase that was hyper-phosphorylated, but also inactive [73]. These data suggest that PP1 may be influencing Aurora-A in a complex way, similar to PAK kinases, and suggesting caution in evaluating Aurora-A activity in tumors with anomalous function of PP1. More recently, the PP6 holoenzyme has been described as the major negative regulator of Aurora-A activation, inhibiting the stability of the Aurora-A/TPX2 complex [74].
Finally, the centrosomal population of the p53 tumor suppressor inhibits the kinase activity of Aurora-A and suppresses oncogenic transformation of cells induced by overexpression of Aurora-A [14, 75]. Gadd45a, a DNA damage-inducible protein that is regulated by tumor suppressors p53 and BRCA1, physically associates with Aurora-A, strongly inhibits Aurora-A kinase activity and antagonizes Aurora-A induced centrosome amplification [76].
It is likely that there are additional regulators of Aurora-A activity that have not yet been defined. This is a fertile area for further investigation.
5. Relation of Aurora-A phosphorylation to total protein expression and susceptibility to inhibition
Some of the auto-phosphorylation events and protein interactions discussed above do not only affect Aurora-A kinase activity: some are clearly documented as affecting the resistance of the protein to degradation. In brief background, while the majority of solid tumors have elevated Aurora-A protein levels, only a minority has Aurora-A gene amplification, implying that posttranscriptional mechanisms of Aurora-A stabilization are very important in cancer [8]. At the end of mitosis, Aurora-A is dephosphorylated, polyubiquitinated by the anaphase-promoting complex/cyclosome (APC/C) and targeted for degradation by the proteasome. APC/C-dependent degradation of Aurora-A requires substrate recognition subunits CDH20 and CDH1 [77]. Overexpression of CDH1 reduces Aurora-A levels, whereas CDH1 knockdown or mutation of the Aurora-A CDH1–binding site results in elevated Aurora-A expression [25, 78].
Two degradation-targeting sequences on Aurora-A mediate destruction by the APC/C complex: the carboxy-terminal D-box (destruction box) and an amino-terminal A-box [53, 65, 79]. Phosphorylation of Aurora-A on S51 in the A-box inhibits CDH1-APC/C–mediated ubiquitination and consequent degradation [78]. The Aurora-A inhibitor PP1, noted above, removes the S51 and T-loop T288 phosphorylations, enhancing the destruction process [8].
Several proteins are involved in the regulation of AURKA stability either by direct deubiquitination of Aurora-A (USP2a) [80] or through interference with Aurora-A ubiquitination by APC/C (NEDD9, PUM2, TPX2, LIMK2, PHDL1) [40, 65, 81–83]. As an example, NEDD9 associates with the Aurora-A through mitosis, supports its T288 phosphorylation, and hampers the ability of the APC/C complex to ubiquitinate Aurora-A by preventing CDH1 binding [65]. Deletion or mutation of NEDD9 dramatically decreases Aurora-A protein level and kinase activity. Phosphorylation of NEDD9 by Aurora-A serves as a negative feedback loop to regulate the levels of active Aurora-A. The abundance of NEDD9 in epithelial cancers, and dephosphorylation of NEDD9 by the PP2A phosphatase [84] creates a constant supply of unphosphorylated NEDD9 that can stabilize and activate Aurora-A [65].
The above examples imply that clinical measurements of Aurora-A activity or Aurora-A inhibitor activity based on T288 phosphorylation at minimum need to be normalized to levels of total Aurora-A, given phosphorylation is influencing Aurora-A processing by the cellular degradation machinery. As an additional complication, two recent studies have indicated that increased interaction of activating partners with Aurora-A (based on amplification or overexpression in cancer) can reduce the effectiveness of the small molecule Aurora-A inhibitor alisertib (discussed below) [65, 85]. No detailed biophysical analysis has as yet been performed to analyze this resistance mechanism. Potentially, resistance reflects allosteric changes in the configuration of Aurora-A that affect the T-loop, or disrupt the interaction of dephosphorylated Aurora-A with the CDH1/CDH20 destruction-targeting proteins. Investigation is clearly needed.
6. Activation of Aurora-A in non-mitotic contexts
Typically, detection of a positive signal for Aurora-A T288 phosphorylation is ascribed to cells in mitosis. However, multiple recent studies have shown more diverse, non-mitotic functions of Aurora-A regulating protrusion and resorption of cellular cilia [86], participating in cellular calcium signaling [46, 87], and orchestrating remodeling of the microtubular cytoskeleton during neurite extension [28]. Given the recent nature of these discoveries, the degree to which interphase Aurora-A activity contributes to measurements of total cellular Aurora-A activity is not clear. The kinetics of Aurora-A activation are clearly different in the interphase comparing to those in mitosis. Further, inactivation of Aurora-A in interphase does not appear to involve kinase degradation.
In a brief summary of findings, most mammalian cells have a single non-motile cilium that extends from a perimembrane basal body and acts as a receiver for extracellular mechanical and chemical cues. Loss of cilia from the cell surface has been linked to more aggressive phenotypes in transformed cells and many types of cancer [88]. However, for some tumor types, such as medulloblastoma, ciliary dynamics are more complicated, with cilia required for tumor induction by the Hedgehog-responsive protein Smoothened, which localizes to cilia, but prohibitive for induction by Gli2, a downstream transducer of Hedgehog signaling [89]. Beyond the Hedgehog pathway, PDGFR-α signals from cilia [90], while cilia-dependent signaling pathways include mTOR, VHL, TSC, and WNT [91], all highly relevant to cancer.
The ciliary basal body differentiates from the centrosome (an important site of action for Aurora-A) in G0/G1 phase, but redifferentiates to a centrosome later in the cell cycle. Protrusion and resorption of cilia is cell cycle regulated, with some cilia resorbed as quiescent cells move from G0 to G1 (or in early G1 in cycling cells) and all cilia resorbed prior to mitosis [92]. Ciliary resorption is controlled by Aurora-A activation at the basal body of the cilium [86]. Stimuli leading to ciliary disassembly activate Aurora-A at the basal body in G0/G1 cells – a time when many of the canonical Aurora-A-activating factors were not thought to be active. This activation lasts for approximately an hour, is reflected by T288 phosphorylation, and depends on at least one Aurora-A mitotic partner, NEDD9. After ciliary resorption, Aurora-A ceases to be active (as judged by T288 autophosphorylation and measurements of in vitro kinase activity), but is not targeted for degradation. These data raise the interesting possibility that inhibition of T288-phosphorylated Aurora-A may be differently regulated, and have different effects, in tumors that depend on cilia-localized oncogenic signaling.
Defects in cilia are strongly associated with clinically important “ciliopathies”, including polycystic kidney disease (PKD), nephronophthisis, Joubert Syndrome and others [93]. The mitotic activation of Aurora-A by Ca2+/CaM binding, noted above, was first detected in the context of interphase signaling in pre-clinical models of PKD. The ciliopathy PKD is associated with defects in intracellular calcium signaling by the cilia-localized PKD2 calcium channel. Plotnikova et al found that numerous stimuli that transiently increase cytoplasmic Ca2+ dramatically induce Aurora-A activation with extremely rapid kinetics. Ca2+-induced Aurora-A activity peaks within 1 minute of stimulation, returns to baseline within 5 minutes, occurs in interphase cells, and is not associated with Aurora-A degradation. Activation of Aurora-A depended on a direct interaction between the N-terminal domain of Aurora-A with Ca2+/CaM resulting in autophosphorylation of Aurora-A on S51, S66, and S98 [46] (Figure 4A). The S51/S53 phosphorylation, first reported in mitotic cells [48], suggested that CaM might also be relevant to the mitotic activation of the kinase [46]. Targeted mutations or drugs disrupting Aurora-A binding to Ca2+/CaM inhibited Aurora-A activation not only in ciliary resorption, but also in mitosis, CaM was shown to co-localize with Aurora-A throughout mitosis [45], and chelation of calcium was shown to reduce not only CaM-Aurora-A but also Aurora-A NEDD9 interactions. These data imply Aurora-A activity may be affected in complex ways by drug treatments that affect calcium signaling, and that these activity changes will not be reflected in measurement of T288 phosphorylation. Further, Aurora-A directly phosphorylates and negatively regulates the activity of the cilia-associated PKD2 calcium channel. This provides an interesting connection between Aurora-A function and the pathology of PKD that may be relevant to the clinical use of Aurora-A inhibitors [87].
Figure 4.

A. Interaction of Aurora-A with Ca2+/calmodulin (CaM) results in autophosphorylation of Aurora-A on S51, S66, and S98; additional interaction with NEDD9 results in autophosphorylation of Aurora-A on T288. Phosphorylated Aurora-A orchestrates resorption of cilia. B. Atypical protein kinase C (aPKC) phosphorylates Aurora-A at T287, inducing autophosphorylation of Aurora-A at T288, which facilitates binding between Aurora-A and TPX2. Aurora-A bound to TPX2 phosphorylates NDEL1 on S251 residue. Active NDEL1 regulates neurite extension.
Aurora-A, TPX2, and two additional binding partners - atypical protein kinase C (aPKC), and NDEL1 - are crucial for the regulation of neuronal microtubule organization and remodeling of the cytoskeleton during neurite extension [28, 94] (Figure 4B). aPKC phosphorylates Aurora-A at T287, inducing autophosphorylation of Aurora-A at T288, which facilitates binding between Aurora-A and TPX2. Aurora-A bound to TPX2 subsequently phosphorylates NDEL1. Active forms of Aurora-A, TPX2 and NDEL1 colocalize and co-immunoprecipitate in vitro. Importantly, phosphorylated Aurora-A and NDEL1 localizations overlap in an area surrounding the centrosome, which may later determine neuron polarity. Suppression of aPKC, Aurora-A or TPX2, or disruption of NDEL1 results in a significant decrease in the frequency of microtubule emanation from the microtubule organizing center of neurons and severe impairment of neurite extension [28, 94]. Further, the potential interaction between Aurora-A, aPKC and other key centrosomal proteins may mediate microtubule dynamics determining the cell polarity in the neuron [95]. Thus, Aurora-A activation determines normal post-mitotic neuron differentiation. The degree to which Aurora-A inhibitors act in brain tissue versus brain tumors is currently not known; nor is the potential role for phosphorylation control of Aurora-A activity through S342, S98, or other non-canonical sites in interphase contexts.
7. Expert Opinion
Aurora-A inhibitors were envisioned as a new class of anti-mitotic agents, potentially more active and less toxic than chemotherapeutic mitotic inhibitors. In tests to date, alisertib has had some efficacy, particularly in hematologic malignancies compared to solid tumors, and with some benefits in pre-treated patients. To improve the efficacy of alisertib, combination approaches with chemotherapy or other targeted agents are under study. Overall, Aurora-A inhibitors remain highly promising, but are not yet clinically optimized. The identification of multiple interphase settings in which Aurora-A is activated and has functional roles have provided one form of complication in assessment of Aurora-A molecular and clinical activity.
To understand the cell and tumor response to alisertib, it is important to be able to correlate growth inhibition with inhibition of the enzymatic target. There have been many preclinical and clinical studies of Aurora-A activation, involving many tissue types (e.g. [100–119]). Typically, in the pre-clinical studies, the activity of Aurora-A inhibitors was assessed using antibodies to the phospho-T288 epitope. Few studies have taken into account the level of total Aurora-A to prove that that the decreased T288 phosphorylation was due to inhibition of phosphorylation, and not to Aurora A degradation or down-regulation. Gold standard approaches, including the use of mass spectrometry with immunoprecipitated Aurora-A to fully evaluate phosphorylation profile, or the evaluation of the activity of immunoprecipitated Aurora-A against multiple substrates by in vitro kinase assay, were not utilized. Although a small number of studies looked at expression of NEDD9 or p53 or cancer-relevant proteins downstream of Aurora-A, the expression of Aurora-A partners known to regulate protein activity was typically not profiled. In clinical studies, the biological effect of alisertib was commonly evaluated using markers of cell proliferation, such as mitotic index, in the tumor samples or skin biopsies. While total levels of Aurora-A were measured by immunohistochemistry or assessment of gene amplification, direct assessment of how well alisertib is inhibiting kinase activity of Aurora-A by measuring Aurora-A phosphorylation or kinase activity was typically not performed. Similarly, expression of Aurora-A partners is almost never assessed in clinical specimens. Clearly, acquisition of such information has the potential to greatly illuminate the response profile of individuals treated with alisertib or other Aurora-A inhibitors.
As clinical development of Aurora-A inhibitors continues, the growing evidence, summarized above, that indicates multiple factors contributing to Aurora-A activation, should be taken into account in correlate studies for trials. Aurora-A inhibitors that are now in clinical trials all work by blocking T288 auto-phosphorylation in the activation loop. The facts that Aurora-A has multiple other phosphorylation sites modulating its mitotic and non-mitotic activity (S51, S53/S54, S66/S67, S89, S98, and S342 residues), and that alternative kinases (e.g. PAK) have been reported to phosphorylate Aurora-A on T288, together suggest that it is imperative to develop and apply antibody reagents to some of these additional phospho-residues. Aurora-A activation without phosphorylation is possible as well, when binding of Aurora-A activating partner TPX-2 changes the conformation of Aurora-A active center resulting in kinase activity [42]. Hence, some effort should be applied to developing surrogate kinase assays, measuring phosphorylation of other proteins dependent on active Aurora-A. Other biomarkers may reflect stability of downstream factors dependent on Aurora-A phosphorylation. For example, in mouse models of neuroblastoma driven by N-Myc amplification, alisertib disrupted the Aurora-A/N-Myc complex and promoted degradation of N-Myc; this, in turn, inhibited N-Myc-dependent transcription, correlating with tumor regression and prolonged survival. Amplification of the N-Myc oncogene commonly drives neuroendocrine tumors such as neuroblastoma, small cell lung carcinoma, and neuroendocrine prostate cancer. As no targeted inhibitors for N-Myc exist, destabilization of N-Myc by targeting Aurora-A could both be potentially valuable therapeutically, and serve as a proxy measurement for kinase inhibition [117].
The tight spatial and temporal control of Aurora-A activation in normal cells involves the multiple partner proteins discussed above [33, 35, 42, 47, 54–59, 73]. Expression of TPX2 and NEDD9 has already been shown to affect Aurora-A kinase stability, and biological activity of Aurora-A kinase inhibitors. Clearly, these and other proteins regulating Aurora-A activation are excellent candidates for biomarkers that may be able to stratify patients for likely response to Aurora-A targeting agents. However, because of the complexity of control of Aurora-A regulation, identification of biomarkers may be difficult. For example, in models of CML and Philadelphia chromosome positive ALL, alisertib showed cytotoxic effects irrespective of p53 status, contrary to expectation [118]. Nevertheless, trial outcomes emphasize the need for biomarker development. For example, alisertib has significant activity in a small sub-population (10%) of patients with platinum resistant ovarian cancer, resulting in durable partial responses lasting from half a year to almost a year. This is a clinically meaningful outcome in a very treatment refractory patient population; a biomarker is clearly needed to identify those patients who will respond. One possibility is the development of a quantitative RT-PCR or tumor tissue microarray-based immunohistochemistry assay to track the expression or activity of Aurora-A interacting partners such as NEDD9, PAK1, TPX2, and others. Some of Aurora-A partner proteins may be appropriate as targets for co-inhibition with Aurora-A. For example, PAK kinases are currently active targets of inhibitor development, with some promising results in preclinical testing [72]; combination of PAK inhibitors with alisertib would be of considerable interest. If there is one consistent lesson emerging from the field of systems biology, it is that inhibition of a single target, no matter how promising, is likely to be insufficient for cancer therapy except in the most unusual cases. Understanding the complexity of Aurora-A regulation and function is essential for designing new and effective targeted therapies and therapeutic combinations. Certainly, knowledge of structural and molecular biology can accelerate clinical development.
Article highlight box.
Many valuable clinical targets are kinases. A robust ability to select patients for use of kinase inhibiting drugs, and to assess activity of kinase inhibitors in patients is essential for optimal clinical performance of these drugs.
The Aurora-A kinase, essential for mitosis, is a valuable clinical target in cancer. Alisertib and other Aurora-A inhibitors targeting the catalytic site are now being tested in multiple clinical trials.
Typically, Aurora-A activity is assessed through use of antibodies targeting a T288 phospho-epitope in the activation loop that is auto-phosphorylated under many conditions of Aurora-A activation.
A growing number of biophysical and biochemical studies have identified non-canonical means of Aurora-A activation, including auto-phosphorylation on alternative sites, phosphorylation by other kinases, and allosteric changes imposed by protein-protein interactions. Many of the proteins affecting Aurora-A activation are themselves variably expressed and active in cancer.
We provide an overview of the implications of these recent findings for the utilization and assessment of Aurora-A inhibitors in further clinical development.
Acknowledgments
The authors were supported by R21CA181287 and R01CA63366 (to EAG), R01 GM084453 (to RLD), and P30 CA006927 (to Fox Chase Cancer Center). We thank Ilya Serebriiskii for valuable comments on the paper.
Abbreviations
- FFPE
formalin-fixed, paraffin-embedded
- CDK1
cyclin-dependent kinase 1
- CaM
calmodulin
- Ca2+/CaM
Ca2+-liganded calmodulin
- LIMK1
LIM domain kinase 1
- PP1
protein phosphatase 1
- PP6
protein phosphatase 6
- APC/C
anaphase-promoting complex/cyclosome
- D-box
destruction box
- PKD
polycystic kidney disease
- aPKC
atypical protein kinase C
- ALL
acute lymphoblastic leukemia
- CML
chronic myelogenous leukemia
- MPM-2
mitotic protein monoclonal #2
- PHH3
phospho-histone H3
References
- 1.Weinstein IB. Cancer. Addiction to oncogenes--the Achilles heal of cancer Science. 2002;297:63–4. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 2.Matallanas D, Birtwistle M, Romano D, et al. Raf family kinases: old dogs have learned new tricks. Genes Cancer. 2011;2:232–60. doi: 10.1177/1947601911407323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **3.Meraldi P, Honda R, Nigg EA. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53−/− cells. EMBO J. 2002;21:483–92. doi: 10.1093/emboj/21.4.483. Mechanistic explanation of Aurora-A activity in promoting genomic instability. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **4.Anand S, Penrhyn-Lowe S, Venkitaraman AR. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell. 2003;3:51–62. doi: 10.1016/s1535-6108(02)00235-0. Early demonstration of the relationship of Aurora-A expression and drug response. [DOI] [PubMed] [Google Scholar]
- 5.Vader G, Lens SM. The Aurora kinase family in cell division and cancer. Biochim Biophys Acta. 2008;1786:60–72. doi: 10.1016/j.bbcan.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 6.Zhang D, Hirota T, Marumoto T, et al. Cre-loxP-controlled periodic Aurora-A overexpression induces mitotic abnormalities and hyperplasia in mammary glands of mouse models. Oncogene. 2004;23:8720–30. doi: 10.1038/sj.onc.1208153. [DOI] [PubMed] [Google Scholar]
- 7.Tatsuka M, Katayama H, Ota T, et al. Multinuclearity and increased ploidy caused by overexpression of the aurora- and Ipl1-like midbody-associated protein mitotic kinase in human cancer cells. Cancer Res. 1998;58:4811–6. [PubMed] [Google Scholar]
- 8.Nikonova AS, Astsaturov I, Serebriiskii IG, et al. Aurora A kinase (AURKA) in normal and pathological cell division. Cell Mol Life Sci. 2013;70:661–87. doi: 10.1007/s00018-012-1073-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agnese V, Bazan V, Fiorentino FP, et al. The role of Aurora-A inhibitors in cancer therapy. Ann Oncol. 2007;18 (Suppl 6):47–52. doi: 10.1093/annonc/mdm224. [DOI] [PubMed] [Google Scholar]
- 10.Nadler Y, Camp RL, Schwartz C, et al. Expression of Aurora A (but not Aurora B) is predictive of survival in breast cancer. Clin Cancer Res. 2008;14:4455–62. doi: 10.1158/1078-0432.CCR-07-5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ratushny V, Pathak HB, Beeharry N, et al. Dual inhibition of SRC and Aurora kinases induces postmitotic attachment defects and cell death. Oncogene. 2012;31:1217–27. doi: 10.1038/onc.2011.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Otto T, Horn S, Brockmann M, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell. 2009;15:67–78. doi: 10.1016/j.ccr.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 13.Liu Q, Kaneko S, Yang L, et al. Aurora-A abrogation of p53 DNA binding and transactivation activity by phosphorylation of serine 215. J Biol Chem. 2004;279:52175–82. doi: 10.1074/jbc.M406802200. [DOI] [PubMed] [Google Scholar]
- 14.Wu CC, Yang TY, Yu CT, et al. p53 negatively regulates Aurora A via both transcriptional and posttranslational regulation. Cell Cycle. 2012;11:3433–42. doi: 10.4161/cc.21732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Carcer G, Malumbres M. A centrosomal route for cancer genome instability. Nat Cell Biol. 2014;16:504–6. doi: 10.1038/ncb2978. [DOI] [PubMed] [Google Scholar]
- 16.Friedberg JW, Mahadevan D, Cebula E, et al. Phase II study of alisertib, a selective Aurora A kinase inhibitor, in relapsed and refractory aggressive B- and T-cell non-Hodgkin lymphomas. J Clin Oncol. 2014;32:44–50. doi: 10.1200/JCO.2012.46.8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matulonis UA, Sharma S, Ghamande S, et al. Phase II study of MLN8237 (alisertib), an investigational Aurora A kinase inhibitor, in patients with platinum-resistant or -refractory epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. Gynecol Oncol. 2012;127:63–9. doi: 10.1016/j.ygyno.2012.06.040. [DOI] [PubMed] [Google Scholar]
- 18.Abe Y, Ohsugi M, Haraguchi K, et al. LATS2-Ajuba complex regulates gamma-tubulin recruitment to centrosomes and spindle organization during mitosis. FEBS Lett. 2006;580:782–8. doi: 10.1016/j.febslet.2005.12.096. [DOI] [PubMed] [Google Scholar]
- 19.Mori D, Yano Y, Toyo-oka K, et al. NDEL1 phosphorylation by Aurora-A kinase is essential for centrosomal maturation, separation, and TACC3 recruitment. Mol Cell Biol. 2007;27:352–67. doi: 10.1128/MCB.00878-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nurse P. Universal control mechanism regulating onset of M-phase. Nature. 1990;344:503–8. doi: 10.1038/344503a0. [DOI] [PubMed] [Google Scholar]
- 21.Dutertre S, Cazales M, Quaranta M, et al. Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to the G2-M transition. J Cell Sci. 2004;117:2523–31. doi: 10.1242/jcs.01108. [DOI] [PubMed] [Google Scholar]
- 22.Nam HJ, van Deursen JM. Cyclin B2 and p53 control proper timing of centrosome separation. Nat Cell Biol. 2014;16:538–49. doi: 10.1038/ncb2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ouchi M, Fujiuchi N, Sasai K, et al. BRCA1 phosphorylation by Aurora-A in the regulation of G2 to M transition. J Biol Chem. 2004;279:19643–8. doi: 10.1074/jbc.M311780200. [DOI] [PubMed] [Google Scholar]
- 24.Kashatus DF, Lim KH, Brady DC, et al. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat Cell Biol. 2011;13:1108–15. doi: 10.1038/ncb2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Floyd S, Pines J, Lindon C. APC/C Cdh1 targets aurora kinase to control reorganization of the mitotic spindle at anaphase. Curr Biol. 2008;18:1649–58. doi: 10.1016/j.cub.2008.09.058. [DOI] [PubMed] [Google Scholar]
- 26.Perez de Castro I, Aguirre-Portoles C, Martin B, et al. A SUMOylation Motif in Aurora-A: Implications for Spindle Dynamics and Oncogenesis. Front Oncol. 2011;1:50. doi: 10.3389/fonc.2011.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **27.Littlepage LE, Wu H, Andresson T, et al. Identification of phosphorylated residues that affect the activity of the mitotic kinase Aurora-A. Proc Natl Acad Sci U S A. 2002;99:15440–5. doi: 10.1073/pnas.202606599. Important study of regulatory phosphorylation sites on Aurora-A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *28.Mori D, Yamada M, Mimori-Kiyosue Y, et al. An essential role of the aPKC-Aurora A-NDEL1 pathway in neurite elongation by modulation of microtubule dynamics. Nat Cell Biol. 2009;11:1057–68. doi: 10.1038/ncb1919. Identification of Aurora-A activation in a post-mitotic setting. [DOI] [PubMed] [Google Scholar]
- 29.Diaz B, Barnard D, Filson A, et al. Phosphorylation of Raf-1 serine 338-serine 339 is an essential regulatory event for Ras-dependent activation and biological signaling. Mol Cell Biol. 1997;17:4509–16. doi: 10.1128/mcb.17.8.4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rowan FC, Richards M, Bibby RA, et al. Insights into Aurora-A kinase activation using unnatural amino acids incorporated by chemical modification. ACS Chem Biol. 2013;8:2184–91. doi: 10.1021/cb400425t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *31.Dodson CA, Kosmopoulou M, Richards MW, et al. Crystal structure of an Aurora-A mutant that mimics Aurora-B bound to MLN8054: insights into selectivity and drug design. Biochem J. 2010;427:19–28. doi: 10.1042/BJ20091530. Structural analysis of drug binding specificity for inhibitors of Aurora-A kinase. [DOI] [PubMed] [Google Scholar]
- 32.Zorba A, Buosi V, Kutter S, et al. Molecular mechanism of Aurora A kinase autophosphorylation and its allosteric activation by TPX2. Elife. 2014;3:e02667. doi: 10.7554/eLife.02667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **33.Bayliss R, Sardon T, Vernos I, Conti E. Structural basis of Aurora-A activation by TPX2 at the mitotic spindle. Mol Cell. 2003;12:851–62. doi: 10.1016/s1097-2765(03)00392-7. Structural insights into Aurora-A activation by one of its most important activators. [DOI] [PubMed] [Google Scholar]
- 34.Zhao B, Smallwood A, Yang J, et al. Modulation of kinase-inhibitor interactions by auxiliary protein binding: crystallography studies on Aurora A interactions with VX-680 and with TPX2. Protein Sci. 2008;17:1791–7. doi: 10.1110/ps.036590.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clark MA, Acharya RA, Arico-Muendel CC, et al. Design, synthesis and selection of DNA-encoded small-molecule libraries. Nat Chem Biol. 2009;5:647–54. doi: 10.1038/nchembio.211. [DOI] [PubMed] [Google Scholar]
- 36.Bibby RA, Tang C, Faisal A, et al. A cancer-associated aurora A mutant is mislocalized and misregulated due to loss of interaction with TPX2. J Biol Chem. 2009;284:33177–84. doi: 10.1074/jbc.M109.032722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *37.Eyers PA, Erikson E, Chen LG, Maller JL. A novel mechanism for activation of the protein kinase Aurora A. Curr Biol. 2003;13:691–7. doi: 10.1016/s0960-9822(03)00166-0. Study of allosteric activation of Aurora-A by TPX2. [DOI] [PubMed] [Google Scholar]
- *38.Tsai MY, Wiese C, Cao K, et al. A Ran signalling pathway mediated by the mitotic kinase Aurora A in spindle assembly. Nat Cell Biol. 2003;5:242–8. doi: 10.1038/ncb936. Connection of the Ran GTPase to control of Aurora-A activation. [DOI] [PubMed] [Google Scholar]
- 39.Eyers PA, Maller JL. Regulation of Xenopus Aurora A activation by TPX2. J Biol Chem. 2004;279:9008–15. doi: 10.1074/jbc.M312424200. [DOI] [PubMed] [Google Scholar]
- 40.Giubettini M, Asteriti IA, Scrofani J, et al. Control of Aurora-A stability through interaction with TPX2. J Cell Sci. 2011;124:113–22. doi: 10.1242/jcs.075457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kufer TA, Sillje HH, Korner R, et al. Human TPX2 is required for targeting Aurora-A kinase to the spindle. J Cell Biol. 2002;158:617–23. doi: 10.1083/jcb.200204155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *42.Dodson CA, Bayliss R. Activation of Aurora-A kinase by protein partner binding and phosphorylation are independent and synergistic. J Biol Chem. 2012;287:1150–7. doi: 10.1074/jbc.M111.312090. Mechanistic evaluation of the interaction of different mechanisms for Aurora-A activation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu X, Wang X, Xiao Z, et al. Two TPX2-dependent switches control the activity of Aurora A. PLoS One. 2011;6:e16757. doi: 10.1371/journal.pone.0016757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Product Data Sheet Purified anti-Aurora A (Aurora 2)-Phosphorylated (Thr288) Antibody. http://www.biolegend.com/purified-anti-aurora-a-aurora-2-phosphorylated-thr288-antibody-2176.html.
- 45.Plotnikova OV, Nikonova AS, Loskutov YV, et al. Calmodulin activation of Aurora-A kinase (AURKA) is required during ciliary disassembly and in mitosis. Mol Biol Cell. 2012;23:2658–70. doi: 10.1091/mbc.E11-12-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **46.Plotnikova OV, Pugacheva EN, Dunbrack RL, Golemis EA. Rapid calcium-dependent activation of Aurora-A kinase. Nat Commun. 2010;1:64. doi: 10.1038/ncomms1061. Identification of Aurora-A activation by calmodulin in interphase cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **47.Reboutier D, Troadec MB, Cremet JY, et al. Nucleophosmin/B23 activates Aurora A at the centrosome through phosphorylation of serine 89. J Cell Biol. 2012;197:19–26. doi: 10.1083/jcb.201107134. Identification of Aurora-A activation by nucleophosmin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haydon CE, Eyers PA, Aveline-Wolf LD, et al. Identification of novel phosphorylation sites on Xenopus laevis Aurora A and analysis of phosphopeptide enrichment by immobilized metal-affinity chromatography. Mol Cell Proteomics. 2003;2:1055–67. doi: 10.1074/mcp.M300054-MCP200. [DOI] [PubMed] [Google Scholar]
- 49.Pascreau G, Delcros JG, Morin N, et al. Aurora-A kinase Ser349 phosphorylation is required during Xenopus laevis oocyte maturation. Dev Biol. 2008;317:523–30. doi: 10.1016/j.ydbio.2008.02.053. [DOI] [PubMed] [Google Scholar]
- 50.Lawrence HR, Martin MP, Luo Y, et al. Development of o-chlorophenyl substituted pyrimidines as exceptionally potent aurora kinase inhibitors. J Med Chem. 2012;55:7392–416. doi: 10.1021/jm300334d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sarkissian M, Mendez R, Richter JD. Progesterone and insulin stimulation of CPEB-dependent polyadenylation is regulated by Aurora A and glycogen synthase kinase-3. Genes Dev. 2004;18:48–61. doi: 10.1101/gad.1136004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krystyniak A, Garcia-Echeverria C, Prigent C, Ferrari S. Inhibition of Aurora A in response to DNA damage. Oncogene. 2006;25:338–48. doi: 10.1038/sj.onc.1209056. [DOI] [PubMed] [Google Scholar]
- **53.Littlepage LE, Ruderman JV. Identification of a new APC/C recognition domain, the A box, which is required for the Cdh1-dependent destruction of the kinase Aurora-A during mitotic exit. Genes Dev. 2002;16:2274–85. doi: 10.1101/gad.1007302. Identification that N-terminal phosphorylation sites govern Aurora-A stability. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *54.Pugacheva EN, Golemis EA. The focal adhesion scaffolding protein HEF1 regulates activation of the Aurora-A and Nek2 kinases at the centrosome. Nat Cell Biol. 2005;7:937–46. doi: 10.1038/ncb1309. First identification of the pro-metastatic factor HEF1 (NEDD9) as an Aurora-A activator. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *55.Zhao ZS, Lim JP, Ng YW, et al. The GIT-associated kinase PAK targets to the centrosome and regulates Aurora-A. Mol Cell. 2005;20:237–49. doi: 10.1016/j.molcel.2005.08.035. Identification of Pak kinase as an Aurora-A activator. [DOI] [PubMed] [Google Scholar]
- *56.Hirota T, Kunitoku N, Sasayama T, et al. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell. 2003;114:585–98. doi: 10.1016/s0092-8674(03)00642-1. Identification of the Ajuba scaffolding protein as an Aurora-A activator. [DOI] [PubMed] [Google Scholar]
- *57.Satinover DL, Leach CA, Stukenberg PT, Brautigan DL. Activation of Aurora-A kinase by protein phosphatase inhibitor-2, a bifunctional signaling protein. Proc Natl Acad Sci U S A. 2004;101:8625–30. doi: 10.1073/pnas.0402966101. Identification of PPI-2 as an Aurora-A activator. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *58.Molli PR, Li DQ, Bagheri-Yarmand R, et al. Arpc1b, a centrosomal protein, is both an activator and substrate of Aurora A. J Cell Biol. 2010;190:101–14. doi: 10.1083/jcb.200908050. Identification of Arpc1b as an Aurora-A activator. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *59.Hutterer A, Berdnik D, Wirtz-Peitz F, et al. Mitotic activation of the kinase Aurora-A requires its binding partner Bora. Dev Cell. 2006;11:147–57. doi: 10.1016/j.devcel.2006.06.002. Identification of Bora as an Aurora-A activator. [DOI] [PubMed] [Google Scholar]
- *60.Pascreau G, Delcros JG, Cremet JY, et al. Phosphorylation of maskin by Aurora-A participates in the control of sequential protein synthesis during Xenopus laevis oocyte maturation. J Biol Chem. 2005;280:13415–23. doi: 10.1074/jbc.M410584200. Identification of TACC3/maskin as an Aurora-A activator. [DOI] [PubMed] [Google Scholar]
- 61.Sloane DA, Trikic MZ, Chu ML, et al. Drug-resistant aurora A mutants for cellular target validation of the small molecule kinase inhibitors MLN8054 and MLN8237. ACS Chem Biol. 2010;5:563–76. doi: 10.1021/cb100053q. [DOI] [PubMed] [Google Scholar]
- *62.Joukov V, De Nicolo A, Rodriguez A, et al. Centrosomal protein of 192 kDa (Cep192) promotes centrosome-driven spindle assembly by engaging in organelle-specific Aurora A activation. Proc Natl Acad Sci U S A. 2010;107:21022–7. doi: 10.1073/pnas.1014664107. Identification of Cep192 as an Aurora-A activator. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nikonova AS, Gaponova AV, Kudinov AE, Golemis EA. CAS proteins in health and disease: An update. IUBMB Life. 2014 doi: 10.1002/iub.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dadke D, Jarnik M, Pugacheva EN, et al. Deregulation of HEF1 impairs M-phase progression by disrupting the RhoA activation cycle. Mol Biol Cell. 2006;17:1204–17. doi: 10.1091/mbc.E05-03-0237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **65.Ice RJ, McLaughlin SL, Livengood RH, et al. NEDD9 depletion destabilizes Aurora A kinase and heightens the efficacy of Aurora A inhibitors: implications for treatment of metastatic solid tumors. Cancer Res. 2013;73:3168–80. doi: 10.1158/0008-5472.CAN-12-4008. Determination that expression of the Aurora-A activator Nedd9 regulates response to Aurora-A inhibitors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sabino D, Brown NH, Basto R. Drosophila Ajuba is not an Aurora-A activator but is required to maintain Aurora-A at the centrosome. J Cell Sci. 2011;124:1156–66. doi: 10.1242/jcs.076711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bai M, Ni J, Shen S, et al. Aurora-A kinase-inactive mutants disrupt the interaction with Ajuba and cause defects in mitotic spindle formation and G2/M phase arrest in HeLa cells. BMB Rep. 2014 doi: 10.5483/BMBRep.2014.47.11.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bai M, Ni J, Shen S, et al. Two newly identified sites in the N-terminal regulatory domain of Aurora-A are essential for auto-inhibition. Biotechnol Lett. 2014;36:1595–604. doi: 10.1007/s10529-014-1516-3. [DOI] [PubMed] [Google Scholar]
- 69.Bai M, Ni J, Wu J, et al. A novel mechanism for activation of Aurora-A kinase by Ajuba. Gene. 2014;543:133–9. doi: 10.1016/j.gene.2014.03.048. [DOI] [PubMed] [Google Scholar]
- 70.Ritchey L, Ottman R, Roumanos M, Chakrabarti R. A functional cooperativity between Aurora A kinase and LIM kinase1: implication in the mitotic process. Cell Cycle. 2012;11:296–309. doi: 10.4161/cc.11.2.18734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seki A, Coppinger JA, Jang CY, et al. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science. 2008;320:1655–8. doi: 10.1126/science.1157425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Radu M, Semenova G, Kosoff R, Chernoff J. PAK signalling during the development and progression of cancer. Nat Rev Cancer. 2014;14:13–25. doi: 10.1038/nrc3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *73.Katayama H, Zhou H, Li Q, et al. Interaction and feedback regulation between STK15/BTAK/Aurora-A kinase and protein phosphatase 1 through mitotic cell division cycle. J Biol Chem. 2001;276:46219–24. doi: 10.1074/jbc.M107540200. Identification of PP1 as an Aurora-A inhibitor. [DOI] [PubMed] [Google Scholar]
- 74.Zeng K, Bastos RN, Barr FA, Gruneberg U. Protein phosphatase 6 regulates mitotic spindle formation by controlling the T-loop phosphorylation state of Aurora A bound to its activator TPX2. J Cell Biol. 2010;191:1315–32. doi: 10.1083/jcb.201008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen SS, Chang PC, Cheng YW, et al. Suppression of the STK15 oncogenic activity requires a transactivation-independent p53 function. EMBO J. 2002;21:4491–9. doi: 10.1093/emboj/cdf409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shao S, Wang Y, Jin S, et al. Gadd45a interacts with aurora-A and inhibits its kinase activity. J Biol Chem. 2006;281:28943–50. doi: 10.1074/jbc.M600235200. [DOI] [PubMed] [Google Scholar]
- *77.Castro A, Arlot-Bonnemains Y, Vigneron S, et al. APC/Fizzy-Related targets Aurora-A kinase for proteolysis. EMBO Rep. 2002;3:457–62. doi: 10.1093/embo-reports/kvf095. Identification of APC as a primary agent targeting Aurora-A for destruction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Crane R, Kloepfer A, Ruderman JV. Requirements for the destruction of human Aurora-A. J Cell Sci. 2004;117:5975–83. doi: 10.1242/jcs.01418. [DOI] [PubMed] [Google Scholar]
- 79.Yu X, Minter-Dykhouse K, Malureanu L, et al. Chfr is required for tumor suppression and Aurora A regulation. Nat Genet. 2005;37:401–6. doi: 10.1038/ng1538. [DOI] [PubMed] [Google Scholar]
- 80.Shi Y, Solomon LR, Pereda-Lopez A, et al. Ubiquitin-specific cysteine protease 2a (USP2a) regulates the stability of Aurora-A. J Biol Chem. 2011;286:38960–8. doi: 10.1074/jbc.M111.231498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Huang YH, Wu CC, Chou CK, Huang CY. A translational regulator, PUM2, promotes both protein stability and kinase activity of Aurora-A. PLoS One. 2011;6:e19718. doi: 10.1371/journal.pone.0019718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Johnson EO, Chang KH, Ghosh S, et al. LIMK2 is a crucial regulator and effector of Aurora-A-kinase-mediated malignancy. J Cell Sci. 2012;125:1204–16. doi: 10.1242/jcs.092304. [DOI] [PubMed] [Google Scholar]
- 83.Johnson EO, Chang KH, de Pablo Y, et al. PHLDA1 is a crucial negative regulator and effector of Aurora A kinase in breast cancer. J Cell Sci. 2011;124:2711–22. doi: 10.1242/jcs.084970. [DOI] [PubMed] [Google Scholar]
- 84.Zheng M, McKeown-Longo PJ. Cell adhesion regulates Ser/Thr phosphorylation and proteasomal degradation of HEF1. J Cell Sci. 2006;119:96–103. doi: 10.1242/jcs.02712. [DOI] [PubMed] [Google Scholar]
- *85.Chowdhury A, Chowdhury S, Tsai MY. A novel Aurora kinase A inhibitor MK-8745 predicts TPX2 as a therapeutic biomarker in non-Hodgkin lymphoma cell lines. Leuk Lymphoma. 2012;53:462–71. doi: 10.3109/10428194.2011.619018. Identification of TPX2 expression as a regulator of clinical response to Aurora-A inhibitors. [DOI] [PubMed] [Google Scholar]
- 86.Pugacheva EN, Jablonski SA, Hartman TR, et al. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell. 2007;129:1351–63. doi: 10.1016/j.cell.2007.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Plotnikova OV, Pugacheva EN, Golemis EA. Aurora A kinase activity influences calcium signaling in kidney cells. J Cell Biol. 2011;193:1021–32. doi: 10.1083/jcb.201012061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yuan K, Frolova N, Xie Y, et al. Primary cilia are decreased in breast cancer: analysis of a collection of human breast cancer cell lines and tissues. J Histochem Cytochem. 2010;58:857–70. doi: 10.1369/jhc.2010.955856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Han YG, Kim HJ, Dlugosz AA, et al. Dual and opposing roles of primary cilia in medulloblastoma development. Nat Med. 2009;15:1062–5. doi: 10.1038/nm.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schneider L, Clement CA, Teilmann SC, et al. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr Biol. 2005;15:1861–6. doi: 10.1016/j.cub.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 91.Seeger-Nukpezah T, Little JL, Serzhanova V, Golemis EA. Cilia and cilia-associated proteins in cancer. Drug Discov Today Dis Mech. 2013;10:e135–e42. doi: 10.1016/j.ddmec.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Plotnikova OV, Golemis EA, Pugacheva EN. Cell cycle-dependent ciliogenesis and cancer. Cancer Res. 2008;68:2058–61. doi: 10.1158/0008-5472.CAN-07-5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jackman M, Lindon C, Nigg EA, Pines J. Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat Cell Biol. 2003;5:143–8. doi: 10.1038/ncb918. [DOI] [PubMed] [Google Scholar]
- 94.Yamada M, Hirotsune S, Wynshaw-Boris A. The essential role of LIS1, NDEL1 and Aurora-A in polarity formation and microtubule organization during neurogensis. Cell Adh Migr. 2010;4:180–4. doi: 10.4161/cam.4.2.10715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lefkowitz GK, Gleeson JG. Aurora A moonlights in neurite extension. Nat Cell Biol. 2009;11:1053–4. doi: 10.1038/ncb0909-1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **96.Manfredi MG, Ecsedy JA, Chakravarty A, et al. Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin Cancer Res. 2011;17:7614–24. doi: 10.1158/1078-0432.CCR-11-1536. Charactization of the biological activity of the lead compound for Aurora-A inhibition. [DOI] [PubMed] [Google Scholar]
- 97.Do TV, Xiao F, Bickel LE, et al. Aurora kinase A mediates epithelial ovarian cancer cell migration and adhesion. Oncogene. 2014;33:539–49. doi: 10.1038/onc.2012.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhou N, Singh K, Mir MC, et al. The investigational Aurora kinase A inhibitor MLN8237 induces defects in cell viability and cell-cycle progression in malignant bladder cancer cells in vitro and in vivo. Clin Cancer Res. 2013;19:1717–28. doi: 10.1158/1078-0432.CCR-12-2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sehdev V, Katsha A, Ecsedy J, et al. The combination of alisertib, an investigational Aurora kinase A inhibitor, and docetaxel promotes cell death and reduces tumor growth in preclinical cell models of upper gastrointestinal adenocarcinomas. Cancer. 2013;119:904–14. doi: 10.1002/cncr.27801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sehdev V, Peng D, Soutto M, et al. The aurora kinase A inhibitor MLN8237 enhances cisplatin-induced cell death in esophageal adenocarcinoma cells. Mol Cancer Ther. 2012;11:763–74. doi: 10.1158/1535-7163.MCT-11-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Brewer Savannah KJ, Demicco EG, Lusby K, et al. Dual targeting of mTOR and aurora-A kinase for the treatment of uterine Leiomyosarcoma. Clin Cancer Res. 2012;18:4633–45. doi: 10.1158/1078-0432.CCR-12-0436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Carol H, Boehm I, Reynolds CP, et al. Efficacy and pharmacokinetic/pharmacodynamic evaluation of the Aurora kinase A inhibitor MLN8237 against preclinical models of pediatric cancer. Cancer Chemother Pharmacol. 2011;68:1291–304. doi: 10.1007/s00280-011-1618-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Patel AV, Eaves D, Jessen WJ, et al. Ras-driven transcriptome analysis identifies aurora kinase A as a potential malignant peripheral nerve sheath tumor therapeutic target. Clin Cancer Res. 2012;18:5020–30. doi: 10.1158/1078-0432.CCR-12-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Muscal JA, Scorsone KA, Zhang L, et al. Additive effects of vorinostat and MLN8237 in pediatric leukemia, medulloblastoma, and neuroblastoma cell lines. Invest New Drugs. 2013;31:39–45. doi: 10.1007/s10637-012-9831-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gorgun G, Calabrese E, Hideshima T, et al. A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood. 2010;115:5202–13. doi: 10.1182/blood-2009-12-259523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mahadevan D, Stejskal A, Cooke LS, et al. Aurora A inhibitor (MLN8237) plus vincristine plus rituximab is synthetic lethal and a potential curative therapy in aggressive B-cell non-Hodgkin lymphoma. Clin Cancer Res. 2012;18:2210–9. doi: 10.1158/1078-0432.CCR-11-2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Qi W, Spier C, Liu X, et al. Alisertib (MLN8237) an investigational agent suppresses Aurora A and B activity, inhibits proliferation, promotes endo-reduplication and induces apoptosis in T-NHL cell lines supporting its importance in PTCL treatment. Leuk Res. 2013;37:434–9. doi: 10.1016/j.leukres.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Qi W, Cooke LS, Liu X, et al. Aurora inhibitor MLN8237 in combination with docetaxel enhances apoptosis and anti-tumor activity in mantle cell lymphoma. Biochem Pharmacol. 2011;81:881–90. doi: 10.1016/j.bcp.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tomita M, Mori N. Aurora A selective inhibitor MLN8237 suppresses the growth and survival of HTLV-1-infected T-cells in vitro. Cancer Sci. 2010;101:1204–11. doi: 10.1111/j.1349-7006.2010.01499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kelly KRSR, Mahalingam D, et al. The novel orally active aurora A kinase inhibitor MLN8237 is highly active in preclinical models of acute myeloid leukemia and significantly increases the efficacy of cytarabine. Blood (ASH Annual Meeting Abstracts) 2009;114:abstr 2087. [Google Scholar]
- 111.Kelly KR, Ecsedy J, Medina E, et al. The novel Aurora A kinase inhibitor MLN8237 is active in resistant chronic myeloid leukaemia and significantly increases the efficacy of nilotinib. J Cell Mol Med. 2011;15:2057–70. doi: 10.1111/j.1582-4934.2010.01218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Infante JDE, Cohen RB, et al. Phase I study of the safety, pharmacokinetics (PK), and pharmacodynamics (PD) of MLN8237, a selective aurora A kinase inhibitor, in the United States. Eur J Cancer Suppl. 2008;6:90. (abstr 280) [Google Scholar]
- 113.Dees EC, Cohen RB, von Mehren M, et al. Phase I study of aurora A kinase inhibitor MLN8237 in advanced solid tumors: safety, pharmacokinetics, pharmacodynamics, and bioavailability of two oral formulations. Clin Cancer Res. 2012;18:4775–84. doi: 10.1158/1078-0432.CCR-12-0589. [DOI] [PubMed] [Google Scholar]
- 114.Cervantes A, Elez E, Roda D, et al. Phase I pharmacokinetic/pharmacodynamic study of MLN8237, an investigational, oral, selective aurora a kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2012;18:4764–74. doi: 10.1158/1078-0432.CCR-12-0571. [DOI] [PubMed] [Google Scholar]
- 115.Cervantes-Ruiperez BH, Cohen RB, et al. Pharmacokinetic and pharmacodynamic results from two phase I studies of the investigational selective aurora A kinase (AAK) inhibitor MLN8237: Exposure-dependent AAK inhibition in human tumors. J Clin Oncol. 2010;28(suppl):abstr 3031. [Google Scholar]
- 116.Kelly KR, Shea TC, Goy A, et al. Phase I study of MLN8237-investigational Aurora A kinase inhibitor-in relapsed/refractory multiple myeloma, Non-Hodgkin lymphoma and chronic lymphocytic leukemia. Invest New Drugs. 2013 doi: 10.1007/s10637-013-0050-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Brockmann M, Poon E, Berry T, et al. Small molecule inhibitors of aurora-a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer Cell. 2013;24:75–89. doi: 10.1016/j.ccr.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang MHJ, Sells T, et al. In vivo characterization of the aurora A kinase inhibitor MLN8237 in subcutaneous and disseminated models of human cancer. Proc Am Assoc Cancer Res. 2009;49:abstr 5646. [Google Scholar]
- 119.Aliagas-Martin I, Burdick D, Corson L, et al. A class of 2,4-bisanilinopyrimidine Aurora A inhibitors with unusually high selectivity against Aurora B. J Med Chem. 2009;52:3300–7. doi: 10.1021/jm9000314. [DOI] [PubMed] [Google Scholar]