

SUMMARY
Competition between synapses contributes to activity-dependent refinement of the nervous system during development. Does local competition between neighboring synapses drive circuit remodeling during experience-dependent plasticity in the cerebral cortex? Here, we examined the role of activity-mediated competitive interactions in regulating dendritic spine structure and function on hippocampal CA1 neurons. We found that high-frequency glutamatergic stimulation at individual spines, which leads to input-specific synaptic potentiation, induces shrinkage and weakening of nearby unstimulated synapses. This heterosynaptic plasticity requires potentiation of multiple neighboring spines, suggesting that a local threshold of neural activity exists beyond which inactive synapses are punished. Notably, inhibition of calcineurin, IP3Rs, or group I mGluRs blocked heterosynaptic shrinkage without blocking structural potentiation, and inhibition of CaMKII blocked structural potentiation without blocking heterosynaptic shrinkage. Our results support a model in which activity-induced shrinkage signal, and not competition for limited structural resources, drives heterosynaptic structural and functional depression during neural circuit refinement.
INTRODUCTION
Plasticity of neuronal structure, such as the growth and retraction of individual dendritic spines, is thought to support experience-dependent neural circuit remodeling (Bosch and Hayashi, 2012; Holtmaat and Svoboda, 2009). Indeed, as neural circuits are modified during learning, their optimization and fine-tuning involves the weakening and loss of superfluous synaptic connections. Manipulations leading to experience-dependent plasticity of neuronal circuits also increase the rate of spine shrinkage and elimination (Holtmaat et al., 2006; Tschida and Mooney, 2012; Xu et al., 2009; Yang et al., 2009). Yet it remains unclear how neural activity drives the selective shrinkage and loss of individual dendritic spines in response to sensory experience.
Several studies have established that activity-dependent spine shrinkage and elimination are associated with long-term depression (LTD) of synaptic transmission (Nagerl et al., 2004; Zhou et al., 2004), which can occur at individual dendritic spines via an input- and synapse-specific mechanism (Oh et al., 2013) or via a spreading depression (Hayama et al., 2013; Wiegert and Oertner, 2013). In this study, we hypothesized that competitive interactions with neighboring synapses may also play a major role in the structural plasticity associated with synaptic weakening, as it is well-established that stimuli that induce long-term potentiation (LTP) of synaptic strength in one population of synapses can induce heterosynaptic LTD at inactive synapses on the same cell (Abraham and Goddard, 1983; Coussens and Teyler, 1996; Lo and Poo, 1991; Lynch et al., 1977; Scanziani et al., 1996). Intriguingly, ultrastructural studies have shown that LTP-inducing theta-burst stimuli lead to increased spine sizes and decreased spine densities in the hippocampus (Bourne and Harris, 2011) and motor skill training leads to increased numbers of multiple-synapse boutons and decreased size of neighboring spines in the cerebellum (Lee et al., 2013), suggesting that heterosynaptic plasticity may also operate at the level of synaptic structure.
Here, we used two-photon glutamate uncaging and time-lapse imaging of dendritic spines and fluorescently labeled surface AMPA receptors to investigate the role of competitive interactions between synapses in driving structural and functional synaptic plasticity. We show that high-frequency stimulation of individual dendritic spines, which leads to input-specific synaptic potentiation, induces shrinkage and synaptic weakening of nearby unstimulated spines. Heterosynaptic structural plasticity was restricted to local dendritic segments and only came into play following strengthening of multiple neighboring synapses, indicating a local activity threshold that when exceeded leads to shrinkage of nearby inactive spines. Furthermore, heterosynaptic shrinkage requires calcineurin, IP3R, and group I mGluR activation, but not CaMKII-dependent structural potentiation of stimulated spines. Our data support a model in which activation of a cluster of synapses leads to the generation of an activity-induced signal that acts through calcineurin and IP3Rs to drive the shrinkage and depression of nearby inactive synapses.
RESULTS
Structural potentiation of multiple spines on a single dendritic segment induces structural depression of nearby unstimulated spines
To directly test whether competition between neighboring spines could contribute to the spine shrinkage and loss observed during experience-dependent plasticity, we examined whether activity-dependent structural potentiation of dendritic spines leads to shrinkage of nearby inactive spines. We used two-photon glutamate uncaging to stimulate multiple individual dendritic spines with a high-frequency uncaging protocol (HFU; 30 pulses of 1 ms duration at 2 Hz) that induces long-term spine enlargement (Hill and Zito, 2013) and monitored the consequences on the size of nearby unstimulated spines. Remarkably, we found that long-term structural potentiation of a cluster of spines (on average 6) induced long-lasting heterosynaptic shrinkage (21 ± 4% decrease) of a nearby unstimulated spine on the same dendritic segment (Figure 1A–D). Unstimulated spines shrank to a similar extent without regard to their initial size (Figure S1A). Shrinkage of unstimulated spines was not due to glutamate spillover from the clustered glutamate uncaging because unstimulated spines on dendrites exposed to a shifted HFU stimulus, which released the same amount of glutamate at a similar distance away without causing structural potentiation of neighboring spines, did not shrink (Figure 1A, B; Figure S1B). Together, these results demonstrate that potentiation of multiple spines on a single dendritic segment can lead to shrinkage of inactive spines via heterosynaptic interactions.
Figure 1. Structural potentiation of multiple spines on a single dendritic segment induces structural depression of nearby unstimulated spines.
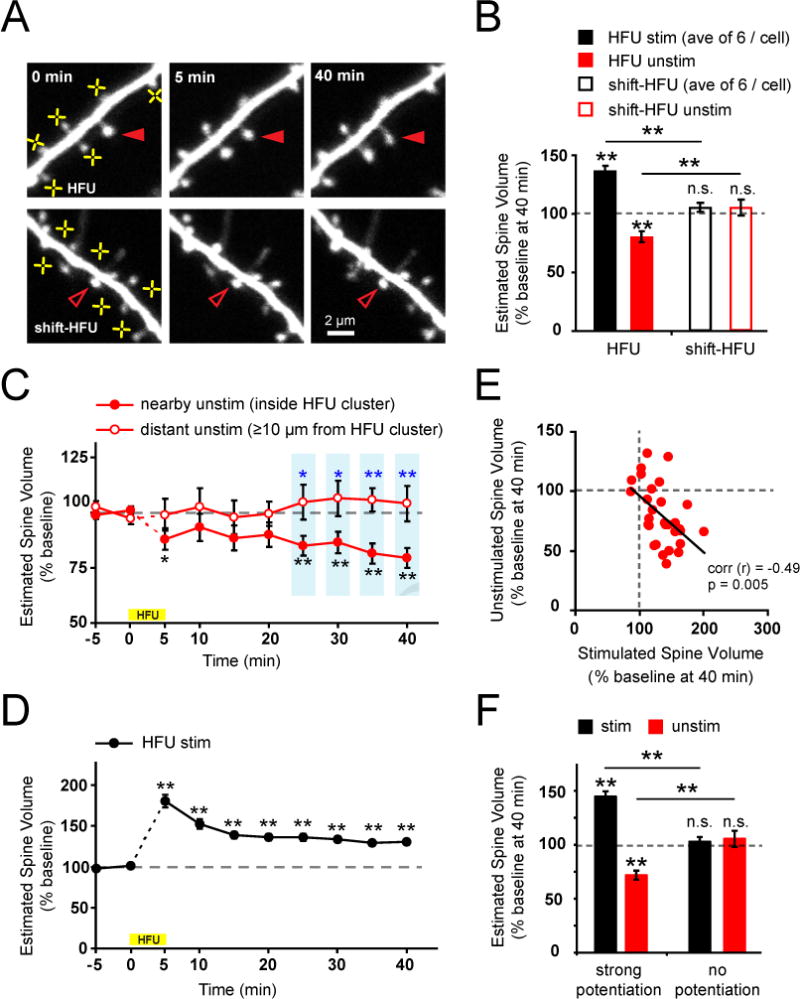
(A) Images of dendrites from EGFP-transfected CA1 neurons (13–18 DIV) exposed to high frequency uncaging (HFU; yellow crosses). A nearby unstimulated spine (filled red arrowheads) shrank following HFU-stimulation of multiple neighboring spines. In contrast, heterosynaptic shrinkage was not observed at an inactive spine (open red arrowheads) following shifted HFU stimulation.
(B) Structural potentiation of multiple neighboring spines (black bar; 31 cells, average 6 spines per cell; p < 0.01) decreased the size of nearby unstimulated spines compared with baseline (red bar; 31 spines; p < 0.01); in contrast, neither neighboring (open black bar; 15 cells, average 8 spines per cell; p = 0.1) nor unstimulated spines (open red bar, 15 spines; p = 0.3) showed changes in size following shifted HFU stimulation.
(C) Time-course of heterosynaptic spine shrinkage. Unstimulated spines inside the cluster (3.2 ± 0.6 μm from HFU stimulated spines; filled red circles; 31 spines) of stimulated neighbors shrank as compared to baseline (p < 0.01) or to distant unstimulated spines (≥ 10 μm from HFU stimulated spines; p < 0.05; blue asterisks) which did not shrink (open red circles; 12 spines; p > 0.3 at all post-HFU time points).
(D) Time-course of homosynaptic spine enlargement. Stimulated spines (black circles; 43 cells, average 6.3 ± 0.1 stimulated spines per cell) increased in size in response to HFU stimulation (p < 0.01 at all post-HFU time points).
(E) An inverse correlation was found between the magnitude of structural potentiation of stimulated spines (average of all stimulated spines) and the magnitude of shrinkage of inside cluster unstimulated spines on the same dendrites (31 cells; r = −0.49, p < 0.01).
(F) When HFU-induced structural potentiation of neighboring spines was successful (left black bar; 24 cells, p < 0.01), inside cluster unstimulated spines shrank (left red bar; p < 0.01); however, shrinkage of inside cluster unstimulated spines was not observed (right red bar; 7 cells, p = 0.49) when HFU did not lead to potentiation of neighboring spines (HFU failure, right black bar, p = 0.53).
If competitive interactions drive spine shrinkage, we would expect an inverse correlation between the degree of structural enhancement and the extent of heterosynaptic shrinkage. Such an inverse correlation has been shown between homosynaptic LTP and heterosynaptic LTD (Royer and Pare, 2003). As expected, we observed a significant inverse correlation between homosynaptic spine enlargement and heterosynaptic spine shrinkage (Figure 1E; Figure S1C, D). Indeed, a decrease in size (28 ± 4% decrease) of unstimulated spines was observed only when the neighboring cluster of stimulated spines successfully (>115% average increase) underwent structural potentiation (Figure 1F). These data strongly support that competitive interactions between neighboring spines lead to heterosynaptic spine shrinkage.
Heterosynaptic spine shrinkage is tightly coupled to synaptic weakening
To address whether heterosynaptic spine shrinkage is accompanied by synaptic weakening, we combined glutamate uncaging with two-photon imaging of surface AMPARs fused with superecliptic pHluorin (SEP; Miesenbock et al., 1998). Previous studies have reported that SEP-GluA2 fluorescence is a reliable marker of activity-dependent AMPAR endocytosis (Ashby et al., 2004; Lin and Huganir, 2007) and that LTP-inducing stimuli increase SEP-GluA2 levels in dendritic spines (Kopec et al., 2006). Therefore, we cotransfected CA1 neurons with tDimer-dsRed and SEP-GluA2 and examined both structural and functional heterosynaptic plasticity.
Following induction of heterosynaptic structural plasticity, we monitored the consequences on synaptic strength using SEP-GluA2 fluorescence (Figure 2). As in Figure 1, we observed that unstimulated spines decreased in size in response to long-term structural potentiation of a cluster of neighboring spines. Associated with spine structural plasticity, we found that SEP-GluA2 fluorescence decreased at unstimulated spines and increased at HFU-stimulated spines (Figure 2A, B). Notably, SEP-GluA2 expression levels were positively correlated with spine size (Figure S2A), and increases and decreases in SEP-GluA2 and tDimer-dsRed fluorescence were highly correlated in both stimulated and unstimulated spines (Figure 2C, D). Strong inverse correlations were observed in both structural and functional heterosynaptic plasticity (Figure S2B, C). Thus, potentiation of multiple neighboring spines leads to heterosynaptic spine shrinkage and functional depression at nearby unstimulated spines.
Figure 2. Heterosynaptic spine shrinkage is tightly coupled to synaptic weakening.
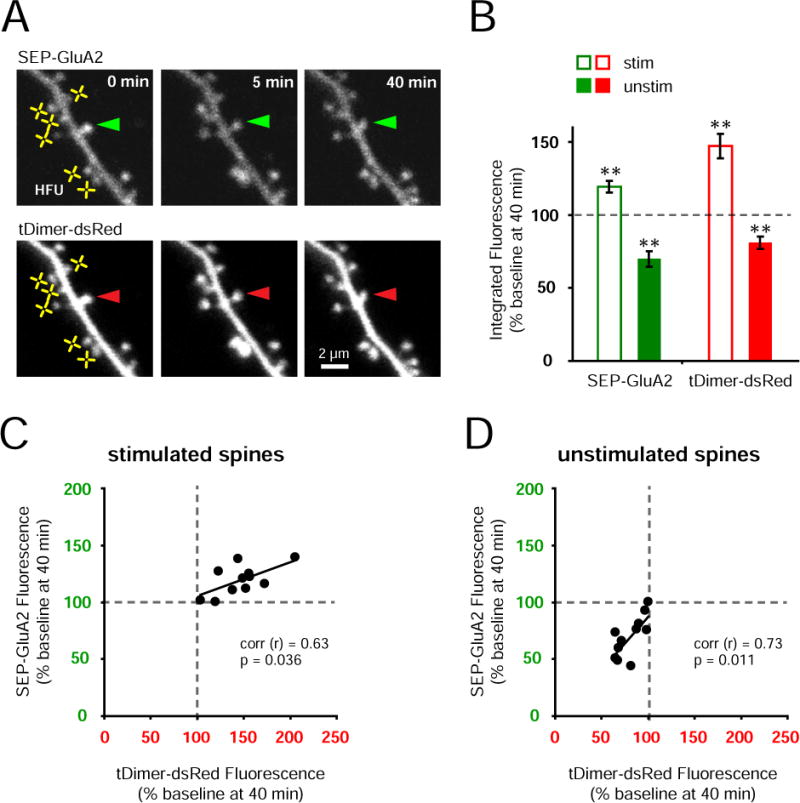
(A) Images of a dendrite from a CA1 neuron cotransfected with SEP-GluA2 and tDimer-dsRed. Fluorescence of SEP-GluA2 (top row) and tDimer-dsRed (bottom row) decreased in an unstimulated spine (arrowheads) following HFU-stimulation (yellow crosses) of multiple neighboring spines.
(B) SEP-GluA2 fluorescence increased (open green bar; 11 cells, average 6 spines per cell; p < 0.01) along with spine size (open red bar; p < 0.01) in HFU-stimulated spines; SEP-GluA2 fluorescence decreased (solid green bar; 11 spines; p < 0.01) along with spine size (solid red bar; p < 0.01) in nearby unstimulated spines.
(C) Increases in tDimer-dsRed and SEP-GluA2 fluorescence intensity were tightly correlated in stimulated spines (r = 0.63; p < 0.05) in response to HFU.
(D) Decreases in tDimer-dsRed and SEP-GluA2 fluorescence intensity were tightly correlated in unstimulated spines (r = 0.73; p < 0.05) following potentiation of multiple neighboring spines.
Heterosynaptic spine shrinkage requires close physical proximity to multiple potentiated spines
What are the constraints on the stimulation paradigms that induce heterosynaptic spine shrinkage? We examined how many dendritic spines needed to potentiate in order to trigger heterosynaptic shrinkage of nearby unstimulated spines. Further analysis of the data from HFU at multiple spines (average 6) revealed that heterosynaptic spine shrinkage occurred only after the structural potentiation of more than three spines (Figure 3A). Indeed, following structural potentiation of a single spine or three spines, unstimulated spines did not shrink (Figure 3B, C). These results suggest that a minimum of four structurally potentiated spines is required for heterosynaptic shrinkage.
Figure 3. Heterosynaptic spine shrinkage requires close physical proximity to multiple potentiated spines.
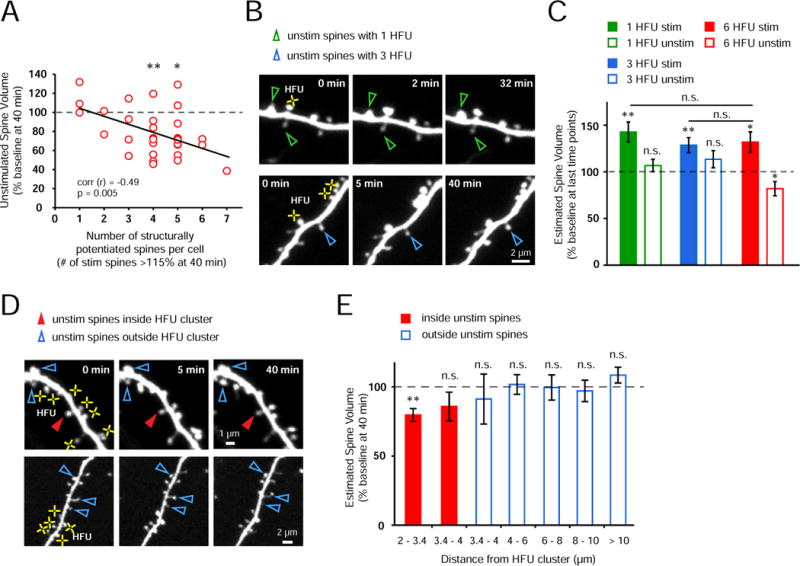
(A) Shrinkage of unstimulated spines was not observed on those cells for which HFU led to structural potentiation (> 115% of baseline at 40 min) of less than four spines; in contrast, when four or more spines potentiated, unstimulated spines shrank (4 spines, p < 0.01; 5 spines, p < 0.05). Notably, an inverse correlation was found between the number of potentiated spines and the magnitude of shrinkage of unstimulated spines (31 cells, r = −0.49, p = 0.005).
(B) Images of a dendrite from an EGFP-transfected neuron exposed to one (top row) and three (bottom row) HFU (yellow crosses). Neither single nor triple HFU induced shrinkage of nearby unstimulated spines.
(C) Single (green bar; 25 cells; 1 spine per cell; p < 0.01) or triple (blue bar; 10 cells, 3 spines per cell; p < 0.01) HFU increased the size of stimulated spines; however, nearby unstimulated spines did not shrink (open green bar, 50 spines, p = 0.32; open blue bar, 10 spines, p = 0.17). Importantly, the magnitude of spine enlargement by single and triple HFU was indistinguishable (single, p = 0.56; triple, p = 0.81) from that observed to induce shrinkage of unstimulated spines (red bar; 11 cells, 6 spines per cell; p < 0.05).
(D) Images of dendrites from EGFP-transfected CA1 neurons exposed to multiple HFU stimuli (yellow crosses). An unstimulated spine located within the cluster of HFU-stimulated spines (filled red arrowheads) decreased in size; in contrast, neither unstimulated spines located outside, but directly adjacent to the HFU-stimulated cluster (top row), nor distant unstimulated spines (bottom row) shrank.
(E) Unstimulated spines located closest (2–3.4 μm) to and inside the HFU cluster decreased in size (red bar; 21 spines; p < 0.01); whereas those located inside the cluster but 3.4–4 μm from stimulated spines did not shrink (red bar; 9 spines; p = 0.19). Unstimulated spines located outside of the HFU-stimulated cluster did not shrink (3.4–4 μm, 6 spines, p = 0.62; 4–6 μm, 30 spines, p = 0.9; 6–8 μm, 23 spines, p = 0.89; 8–10 μm, 22 spines, p = 0.63; > 10 μm, 25 spines, p = 0.2). “inside unstim” data from Fig. 1B.
To address the spatial constraints on heterosynaptic structural plasticity, we examined the relationship between heterosynaptic shrinkage and average distance to the stimulated neighboring spines. We found that unstimulated spines within the cluster that underwent heterosynaptic shrinkage were on average closer (< 3.4 μm) to the stimulated spines than those that did not shrink inside the cluster (3.4–4 μm) and outside the cluster (> 3.4 μm; Figure 3D, E). Furthermore, we found that heterosynaptic shrinkage was not related to the magnitude of potentiation of the nearest stimulated spines (Figure S3). Together, our data suggest that heterosynaptic regulation is mediated locally on individual dendritic segments and strongly support a local activity threshold that when exceeded leads to punishment of nearby inactive synapses.
Heterosynaptic spine shrinkage requires calcineurin, IP3Rs, and group I mGluRs, but not CaMKII
What might constitute a local heterosynaptic shrinkage mechanism? To address whether the shrinkage of inactive spines depends on competition with neighboring stimulated spines for limited structural resources, or whether it is caused by spread of an activity-induced shrinkage signal, we first examined Ca2+/calmodulin-dependent protein kinase II (CaMKII). Inhibition of CaMKII blocks LTP and long-lasting spine enlargement (Lee et al., 2009; Matsuzaki et al., 2004). If an activity-dependent shrinkage-inducing signal, and not competition for limited resources, drives heterosynaptic spine shrinkage, then blocking structural potentiation per se would not be expected to block heterosynaptic spine shrinkage. Indeed, we found that bath-application of KN62 blocked HFU-induced spine enlargement without preventing heterosynaptic shrinkage of inactive spines (Figure 4A, B). Thus, CaMKII-mediated spine enlargement is not necessary for heterosynaptic spine shrinkage, suggesting that an activity-mediated shrinkage signal, rather than competition for limited structural resources, drives spine shrinkage during heterosynaptic structural plasticity.
Figure 4. Heterosynaptic spine shrinkage requires signaling through calcineurin, IP3Rs, and group I mGluRs, but not CaMKII.
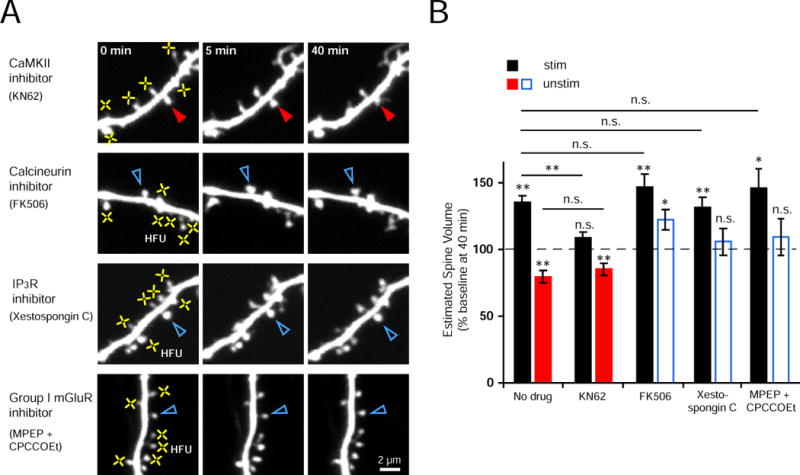
(A) Images of dendrites from EGFP-transfected neurons exposed to multiple HFU (yellow crosses) in the presence of inhibitors of CaMKII (KN62, 10 μM), calcineurin (FK506, 2 μM), IP3Rs (Xesto C, 1 μM), or group I mGluRs (MPEP, 15 μM and CPCCOEt, 45 μM).
(B) Inhibition of CaMKII with KN62 blocked structural potentiation of stimulated spines (black bar; 13 cells; p = 0.067) but did not block heterosynaptic shrinkage (red bar; 13 spines; p < 0.01). In contrast, inhibition of calcineurin with FK506 (blue bar; 13 spines, p < 0.05), IP3Rs with Xesto C (blue bar; 11 spines, p = 0.58), or group I mGluRs with MPEP and CPCCOEt (blue bar; 10 spines, p = 0.51) blocked heterosynaptic shrinkage without affecting HFU-induced spine enlargement (black bars; FK506, 13 cells, p < 0.01; Xesto C, 11 cells, p < 0.01; MPEP and CPCCOEt, 10 cells, p < 0.05), which was not different from that observed without drug (far left black bar; vs. FK506, p = 0.24; vs. Xesto C, p = 0.66; vs. MPEP and CPCCOEt, p = 0.49). “No drug” data from Fig. 1B.
If competition for limited structural resources does not drive heterosynaptic spine shrinkage, then it should be possible to observe structural potentiation of multiple stimulated spines in the absence of heterosynaptic shrinkage of unstimulated spines. To test this hypothesis, we examined the role of calcineurin. Calcineurin is a Ca2+/calmodulin-dependent protein phosphatase that is required for LTD and spine shrinkage, but not for LTP (Mulkey et al., 1994; Pontrello et al., 2012; Zhou et al., 2004), and therefore could inhibit an activity-induced shrinkage signal without blocking structural potentiation of HFU-stimulated spines. Notably, we found that heterosynaptic spine shrinkage was abolished in the presence of a calcineurin inhibitor, FK506, despite that HFU-stimulated spines (on average six) underwent normal structural potentiation (Figure 4A, B). Thus, calcineurin signaling is necessary for heterosynaptic spine shrinkage, most likely through a mechanism that involves an activity-dependent shrinkage-inducing signal generated from stimulated spines.
How might calcineurin in the unstimulated spine be activated to promote spine shrinkage? One possibility is that HFU-stimulation could elevate calcium levels on a local dendritic segment, leading to activation of calcineurin localized in the unstimulated spine. In fact, inositol 1,4,5-trisphosphate receptor (IP3R)-dependent propagation of calcium waves in the dendrite is required for heterosynaptic LTD (Nishiyama et al., 2000). We therefore examined the role of IP3R and the upstream group I metabotropic glutamate receptors (mGluRs) in heterosynaptic spine shrinkage. We found that bath-application of Xestospongin C, a selective IP3R inhibitor, or MPEP and CPCCOEt, group I mGluR-specific antagonists, blocked heterosynaptic spine shrinkage without affecting structural potentiation of HFU-stimulated spines (Figure 4A, B). Importantly, the size of distant (4 – 10 μm from HFU) unstimulated spines was not altered by KN62, FK506, Xestospongin C, or MPEP and CPCCOEt (Figure S4). Together, our data strongly support an activity-induced shrinkage signal that is mediated by calcineurin, IP3Rs, and group I mGluRs to drive heterosynaptic spine shrinkage.
DISCUSSION
Here, we show that competition between neighboring synapses drives spine shrinkage and synaptic weakening on dendrites of hippocampal pyramidal neurons. Our finding that activity-dependent potentiation of a cluster of synapses reliably leads to the shrinkage and weakening of nearby inactive synapses provides a mechanism by which synapses that are not used in a regular manner would remain immature or become eliminated while robustly active neighboring synapses would strengthen and grow (Bourne and Harris, 2011; Buffelli et al., 2003; Lee et al., 2013).
How does synaptic competition lead to spine shrinkage? Several sources have argued that competition for limited structural resources could drive heterosynaptic adjustments in synaptic weights (Fonseca et al., 2004; Miller, 1996) and parallel changes in synaptic morphology (Ramiro-Cortes et al., 2014). For example, key structural components of the synapse such as PSD-95, which has been associated with spine stability, could be redistributed by diffusion to growing synapses at the expense of their neighbors (Gray et al., 2006; Tsuriel et al., 2006). However, we found that heterosynaptic shrinkage persisted when activity-dependent spine growth was blocked by inhibiting CaMKII, demonstrating that growth of neighboring spines is not necessary to drive heterosynaptic spine shrinkage. In addition, unstimulated spines did not shrink in the presence of inhibitors of calcineurin, IP3Rs, or group I mGluRs, despite normal growth at HFU-stimulated spines, demonstrating that structural potentiation of neighboring spines does not by itself induce shrinkage of inactive spines. Together, our results support a model in which a shrinkage signal generated in response to vigorous activity at neighboring synapses, rather than competition for limited resources, leads to heterosynaptic spine shrinkage and depression.
Activity-induced growth of at minimum four spines was necessary to drive heterosynaptic spine shrinkage. Why might heterosynaptic shrinkage require activation of multiple spines? An attractive hypothesis is that widespread and strong activation of multiple glutamatergic inputs is required to generate a sustained calcium elevation that spreads on local dendritic segments (Zhai et al., 2013) by calcium propagation involving IP3Rs, leading to activation of calcineurin at nearby inactive spines. Alternatively, calcineurin activated in the HFU-stimulated spines (Fujii et al., 2013) could diffuse into adjacent regions of the dendrite, only reaching levels sufficiently high to induce spine shrinkage following activation of several neighboring spines.
What determines the spatial constraints on heterosynaptic plasticity? The limited range for heterosynaptic spine shrinkage in our studies may be determined by the extent of spread of calcium released from internal stores, which should be 3 – 10 μm (Malinow et al., 1994; Zhai et al., 2013), or, alternatively, by the range of diffusion of activated calcineurin. Our observation that heterosynaptic shrinkage was limited to nearby inactive spines is consistent with several electrophysiological studies on heterosynaptic depression (Lo and Poo, 1991; Royer and Pare, 2003). In contrast, some examples of heterosynaptic depression of synaptic currents can occur over relatively long distances (several-hundred microns) possibly via intercellular diffusible signals (Abraham and Goddard, 1983; Chen et al., 2013; Coussens and Teyler, 1996; Huang et al., 2008; Lynch et al., 1977; Scanziani et al., 1996). Because our experiments utilized glutamate uncaging on a single dendritic segment, thus bypassing the presynaptic terminals, we conclude that the heterosynaptic shrinkage and depression observed in our studies occurs locally via a postsynaptic mechanism involving calcium wave propagation and calcineurin activation.
How might heterosynaptic plasticity contribute to experience-dependent circuit remodeling? Several studies demonstrate that synaptic potentiation occurs in a spatially clustered manner both in vitro (De Roo et al., 2008; Losonczy et al., 2008) and in vivo (Fu et al., 2012; Makino and Malinow, 2011). Heterosynaptic shrinkage and depression could drive compensatory, local homeostatic plasticity on individual dendritic segments in response to local strengthening of neighboring synapses on dendrites, thus acting to constrain total synaptic weights within stable physiological ranges (Turrigiano, 2008; Vitureira and Goda, 2013). Alternatively, heterosynaptic competition could drive the selective weakening of inactive synapses during experience-dependent neural circuit refinement. Thus, heterosynaptic shrinkage and depression could play a fundamental role in modifying synaptic structure and function in vivo via both Hebbian and homeostatic mechanisms (Goldberg et al., 2002).
EXPERIMENTAL PROCEDURES
Preparation and transfection of organotypic slice cultures
Organotypic hippocampal slice cultures were prepared from P6–P7 Sprague-Dawley rats, as described (Stoppini et al., 1991), and transfected 2–3 days (EGFP; Clontech) or 3–4 days (tDimer-dsRed and SEP-GluA2; Kopec et al., 2006) prior to imaging using biolistic gene transfer (180 psi). 20 μg of EGFP or 10 μg of tDimer-dsRed and 16 μg of SEP-GluA2 were coated onto 6–7 mg of gold particles.
Time-lapse two-photon imaging
CA1 pyramidal neurons (13–18 DIV) at depths of 20–50 μm were imaged using a custom two-photon microscope with a pulsed Ti::sapphire laser (Mai Tai, Spectra Physics) tuned to 930 nm (EGFP: 0.5–1.5 mW, tDimer-dsRed and SEP-GluA2: 2–2.5 mW at the sample). The microscope and data acquisition were controlled with ScanImage (Pologruto et al., 2003). For each neuron, image stacks (512 × 512 pixels; 0.02 μm / pixel) with 1 μm z-steps were collected from one segment of secondary or tertiary basal dendrites 30–80 μm from the soma. Dendrites were imaged at 5–6 min intervals at 30°C in recirculating artificial cerebrospinal fluid (ACSF; in mM: 127 NaCl, 25 NaHCO3, 1.2 NaH2PO4, 2.5 KCl, 25 D-glucose, aerated with 95%O2/5%CO2, ~310 mOsm, pH 7.2) with 2 mM CaCl2, 0 mM MgCl2, 2.5 mM MNI-glutamate, and 0.001 mM TTX.
High-Frequency Uncaging (HFU) stimulus
Uncaging of MNI-glutamate was achieved as described (Zito et al., 2009). In brief, laser pulses were delivered by parking the beam at a point ~0.5 μm from the center of the spine head. For multiple-HFU experiments, HFU consisted of 30 pulses (720 nm; 10–12 mW at the sample) of 1 ms duration delivered at 2 Hz. For single-HFU experiments, HFU consisted of 60 pulses (720 nm; 8–9 mW at the sample) of 2 ms duration delivered at 2 Hz. To avoid confounds due to glutamate spillover, we chose unstimulated spines that were located at least 1.5 μm away from nearest stimulated spines. One dendritic region of interest (ROI) was stimulated per cell.
Image analysis
Estimated spine volume and SEP-GluA2 expression level were measured from background-subtracted and bleed-through-corrected green (EGFP or SEP-GluA2) and red (tDimer-dsRed) fluorescence images using the integrated pixel intensity of a boxed region surrounding the spine head, as described (Woods et al., 2011). For multiple-HFU experiments, all spines stimulated with HFU, one unstimulated spine inside the HFU cluster, and one to three unstimulated spines outside the HFU cluster were analyzed per cell; for single-HFU experiments, one stimulated spine and two unstimulated neighboring spines were analyzed per cell. Less than 15% average growth of stimulated spines was considered as HFU failure (12 / 61 cases). All images shown are maximum projections of 3D stacks after applying a median filter (3 × 3) to the raw image data.
Pharmacology
Stocks were prepared at 1000X (or greater) by dissolving TTX (Calbiochem) and MPEP in water; FK506, KN62, Xestospongin C, and CPCCOEt (Tocris) in DMSO. All drugs were applied at least 20 min prior to HFU stimulation.
Statistics
All statistics were calculated across cells. Error bars represent standard error of the mean and significance was set at p = 0.05 (student’s two-tailed t-test). Correlation was examined by Pearson’s correlation. Single and double asterisks indicate p < 0.05 and p < 0.01, respectively.
Supplementary Material
Highlights.
Local competition between hippocampal synapses leads to changes in synaptic structure
Structural potentiation of multiple spines drives shrinkage of nearby inactive spines
Heterosynaptic spine shrinkage is tightly coupled to synaptic weakening
Heterosynaptic spine shrinkage requires activation of calcineurin, IP3Rs, and mGluRs
Acknowledgments
We thank J. Culp for help with experiments; T. Hill for expert programming; H.J. Cheng, L. Borodinsky, J. Hell, and members of the Zito lab for valuable discussion; and H.B. Kwon and I. Stein for critical reading of the manuscript. This work was supported by the NIH (NS062736), the Whitehall Foundation (2014-05-99), and a Burroughs Wellcome Career Award in the Biomedical Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
W.C.O. and K.Z. conceived and designed the study. W.C.O. performed and analyzed all experiments except 3 HFU and mGluR inhibition experiments, which were performed and analyzed by L.K.P. W.C.O. prepared the figures and wrote the first draft of the paper. All authors revised the manuscript.
References
- Abraham WC, Goddard GV. Asymmetric relationships between homosynaptic long-term potentiation and heterosynaptic long-term depression. Nature. 1983;305:717–719. doi: 10.1038/305717a0. [DOI] [PubMed] [Google Scholar]
- Ashby MC, De La Rue SA, Ralph GS, Uney J, Collingridge GL, Henley JM. Removal of AMPA receptors (AMPARs) from synapses is preceded by transient endocytosis of extrasynaptic AMPARs. J Neurosci. 2004;24:5172–5176. doi: 10.1523/JNEUROSCI.1042-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch M, Hayashi Y. Structural plasticity of dendritic spines. Curr Opin Neurobiol. 2012;22:383–388. doi: 10.1016/j.conb.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne JN, Harris KM. Coordination of size and number of excitatory and inhibitory synapses results in a balanced structural plasticity along mature hippocampal CA1 dendrites during LTP. Hippocampus. 2011;21:354–373. doi: 10.1002/hipo.20768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffelli M, Burgess RW, Feng G, Lobe CG, Lichtman JW, Sanes JR. Genetic evidence that relative synaptic efficacy biases the outcome of synaptic competition. Nature. 2003;424:430–434. doi: 10.1038/nature01844. [DOI] [PubMed] [Google Scholar]
- Chen J, Tan Z, Zeng L, Zhang X, He Y, Gao W, Wu X, Li Y, Bu B, Wang W, et al. Heterosynaptic long-term depression mediated by ATP released from astrocytes. Glia. 2013;61:178–191. doi: 10.1002/glia.22425. [DOI] [PubMed] [Google Scholar]
- Coussens CM, Teyler TJ. Long-term potentiation induces synaptic plasticity at nontetanized adjacent synapses. Learn Mem. 1996;3:106–114. doi: 10.1101/lm.3.2-3.106. [DOI] [PubMed] [Google Scholar]
- De Roo M, Klauser P, Muller D. LTP promotes a selective long-term stabilization and clustering of dendritic spines. PLoS Biol. 2008;6:e219. doi: 10.1371/journal.pbio.0060219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca R, Nagerl UV, Morris RG, Bonhoeffer T. Competing for memory: hippocampal LTP under regimes of reduced protein synthesis. Neuron. 2004;44:1011–1020. doi: 10.1016/j.neuron.2004.10.033. [DOI] [PubMed] [Google Scholar]
- Fu M, Yu X, Lu J, Zuo Y. Repetitive motor learning induces coordinated formation of clustered dendritic spines in vivo. Nature. 2012;483:92–95. doi: 10.1038/nature10844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii H, Inoue M, Okuno H, Sano Y, Takemoto-Kimura S, Kitamura K, Kano M, Bito H. Nonlinear decoding and asymmetric representation of neuronal input information by CaMKIIalpha and calcineurin. Cell Rep. 2013;3:978–987. doi: 10.1016/j.celrep.2013.03.033. [DOI] [PubMed] [Google Scholar]
- Goldberg J, Holthoff K, Yuste R. A problem with Hebb and local spikes. Trends Neurosci. 2002;25:433–435. doi: 10.1016/s0166-2236(02)02200-2. [DOI] [PubMed] [Google Scholar]
- Gray NW, Weimer RM, Bureau I, Svoboda K. Rapid redistribution of synaptic PSD-95 in the neocortex in vivo. PLoS Biol. 2006;4:e370. doi: 10.1371/journal.pbio.0040370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayama T, Noguchi J, Watanabe S, Takahashi N, Hayashi-Takagi A, Ellis-Davies GC, Matsuzaki M, Kasai H. GABA promotes the competitive selection of dendritic spines by controlling local Ca2+ signaling. Nat Neurosci. 2013;16:1409–1416. doi: 10.1038/nn.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill TC, Zito K. LTP-induced long-term stabilization of individual nascent dendritic spines. J Neurosci. 2013;33:678–686. doi: 10.1523/JNEUROSCI.1404-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- Holtmaat A, Wilbrecht L, Knott GW, Welker E, Svoboda K. Experience-dependent and cell-type-specific spine growth in the neocortex. Nature. 2006;441:979–983. doi: 10.1038/nature04783. [DOI] [PubMed] [Google Scholar]
- Huang Y, Yasuda H, Sarihi A, Tsumoto T. Roles of endocannabinoids in heterosynaptic long-term depression of excitatory synaptic transmission in visual cortex of young mice. J Neurosci. 2008;28:7074–7083. doi: 10.1523/JNEUROSCI.0899-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopec CD, Li B, Wei W, Boehm J, Malinow R. Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci. 2006;26:2000–2009. doi: 10.1523/JNEUROSCI.3918-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KJ, Park IS, Kim H, Greenough WT, Pak DT, Rhyu IJ. Motor skill training induces coordinated strengthening and weakening between neighboring synapses. J Neurosci. 2013;33:9794–9799. doi: 10.1523/JNEUROSCI.0848-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Escobedo-Lozoya Y, Szatmari EM, Yasuda R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature. 2009;458:299–304. doi: 10.1038/nature07842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DT, Huganir RL. PICK1 and phosphorylation of the glutamate receptor 2 (GluR2) AMPA receptor subunit regulates GluR2 recycling after NMDA receptor-induced internalization. J Neurosci. 2007;27:13903–13908. doi: 10.1523/JNEUROSCI.1750-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo YJ, Poo MM. Activity-dependent synaptic competition in vitro: heterosynaptic suppression of developing synapses. Science. 1991;254:1019–1022. doi: 10.1126/science.1658939. [DOI] [PubMed] [Google Scholar]
- Losonczy A, Makara JK, Magee JC. Compartmentalized dendritic plasticity and input feature storage in neurons. Nature. 2008;452:436–441. doi: 10.1038/nature06725. [DOI] [PubMed] [Google Scholar]
- Lynch GS, Dunwiddie T, Gribkoff V. Heterosynaptic depression: a postsynaptic correlate of long-term potentiation. Nature. 1977;266:737–739. doi: 10.1038/266737a0. [DOI] [PubMed] [Google Scholar]
- Makino H, Malinow R. Compartmentalized versus global synaptic plasticity on dendrites controlled by experience. Neuron. 2011;72:1001–1011. doi: 10.1016/j.neuron.2011.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Otmakhov N, Blum KI, Lisman J. Visualizing hippocampal synaptic function by optical detection of Ca2+ entry through the N-methyl-D-aspartate channel. Proc Natl Acad Sci U S A. 1994;91:8170–8174. doi: 10.1073/pnas.91.17.8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Miller KD. Synaptic economics: competition and cooperation in synaptic plasticity. Neuron. 1996;17:371–374. doi: 10.1016/s0896-6273(00)80169-5. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Endo S, Shenolikar S, Malenka RC. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- Nagerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004;44:759–767. doi: 10.1016/j.neuron.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Nishiyama M, Hong K, Mikoshiba K, Poo MM, Kato K. Calcium stores regulate the polarity and input specificity of synaptic modification. Nature. 2000;408:584–588. doi: 10.1038/35046067. [DOI] [PubMed] [Google Scholar]
- Oh WC, Hill TC, Zito K. Synapse-specific and size-dependent mechanisms of spine structural plasticity accompanying synaptic weakening. Proc Natl Acad Sci U S A. 2013;110:E305–312. doi: 10.1073/pnas.1214705110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, Svoboda K. ScanImage: flexible software for operating laser scanning microscopes. Biomed Eng Online. 2003;2:13. doi: 10.1186/1475-925X-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontrello CG, Sun MY, Lin A, Fiacco TA, DeFea KA, Ethell IM. Cofilin under control of beta-arrestin-2 in NMDA-dependent dendritic spine plasticity, long-term depression (LTD), and learning. Proc Natl Acad Sci U S A. 2012;109:E442–451. doi: 10.1073/pnas.1118803109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramiro-Cortes Y, Hobbiss AF, Israely I. Synaptic competition in structural plasticity and cognitive function. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130157. doi: 10.1098/rstb.2013.0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer S, Pare D. Conservation of total synaptic weight through balanced synaptic depression and potentiation. Nature. 2003;422:518–522. doi: 10.1038/nature01530. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Malenka RC, Nicoll RA. Role of intercellular interactions in heterosynaptic long-term depression. Nature. 1996;380:446–450. doi: 10.1038/380446a0. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Tschida KA, Mooney R. Deafening drives cell-type-specific changes to dendritic spines in a sensorimotor nucleus important to learned vocalizations. Neuron. 2012;73:1028–1039. doi: 10.1016/j.neuron.2011.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuriel S, Geva R, Zamorano P, Dresbach T, Boeckers T, Gundelfinger ED, Garner CC, Ziv NE. Local sharing as a predominant determinant of synaptic matrix molecular dynamics. PLoS Biol. 2006;4:e271. doi: 10.1371/journal.pbio.0040271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitureira N, Goda Y. Cell biology in neuroscience: the interplay between Hebbian and homeostatic synaptic plasticity. J Cell Biol. 2013;203:175–186. doi: 10.1083/jcb.201306030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegert JS, Oertner TG. Long-term depression triggers the selective elimination of weakly integrated synapses. Proc Natl Acad Sci U S A. 2013;110:E4510–4519. doi: 10.1073/pnas.1315926110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods GF, Oh WC, Boudewyn LC, Mikula SK, Zito K. Loss of PSD-95 enrichment is not a prerequisite for spine retraction. J Neurosci. 2011;31:12129–12138. doi: 10.1523/JNEUROSCI.6662-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Yu X, Perlik AJ, Tobin WF, Zweig JA, Tennant K, Jones T, Zuo Y. Rapid formation and selective stabilization of synapses for enduring motor memories. Nature. 2009;462:915–919. doi: 10.1038/nature08389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Pan F, Gan WB. Stably maintained dendritic spines are associated with lifelong memories. Nature. 2009;462:920–924. doi: 10.1038/nature08577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai S, Ark ED, Parra-Bueno P, Yasuda R. Long-distance integration of nuclear ERK signaling triggered by activation of a few dendritic spines. Science. 2013;342:1107–1111. doi: 10.1126/science.1245622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Zito K, Scheuss V, Knott G, Hill T, Svoboda K. Rapid functional maturation of nascent dendritic spines. Neuron. 2009;61:247–258. doi: 10.1016/j.neuron.2008.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.