

Abstract
Pemphigus vulgaris (PV) is a prototypic tissue-specific autoantibody-mediated disease in which anti-desmoglein 3 (Dsg3) immunoglobulin G (IgG) autoantibodies cause life-threatening blistering. We characterized the autoimmune B-cell response over 14 patient-years in two patients with active and relapsing disease, then in one of these patients after long-term remission induced by multiple courses of rituximab (anti-CD20 antibody). Characterization of the anti-Dsg3 IgG+ repertoire by antibody phage display (APD) and PCR indicated that 6 clonal lines persisted in patient 1 (PV3) over 5.5 years, with only one new clone detected. Six clonal lines persisted in patient 2 (PV1) for 4 years, of which 5 persisted for another 4.5 years without any new clones detected. However, after long-term clinical and serologic remission, ~11 years after initial characterization, we could no longer detect any anti-Dsg3 clones in PV1 by APD. Similarly, in another PV patient, ~4.5 years after a course of rituximab that induced long-term remission, anti-Dsg3 B-cell clones were undetectable. These data suggest that in PV a given set of non-tolerant B-cell lineages causes autoimmune disease and that new sets do not frequently or continually escape tolerance. Therapy such as rituximab, aimed at eliminating these aberrant sets of lineages, may be effective for disease because new ones are unlikely to develop.
Introduction
In PV anti-Dsg3 IgG autoantibodies cause loss of keratinoctye adhesion resulting in severe blistering (Amagai et al., 1994; Mahoney et al., 1999; Mascaro et al., 1997; Payne et al., 2004; Payne et al., 2005). Conventional therapy consists of corticosteroids and other immunosuppressives (e.g., mycophenolate mofetil) (Hammers et al., 2013; Kasperkiewicz et al., 2012). Recently, the use of rituximab to deplete CD20+ B cells has been shown to be effective in achieving clinical and serological remission (Ahmed et al., 2006; Joly et al., 2007; Leshem et al., 2013; Lunardon et al., 2012). However, with both therapies, many patients who achieve clinical remission subsequently relapse (Almugairen et al., 2013). In this study we asked why patients relapse. Specifically, is there an ongoing escape of new IgG+ anti-Dsg3 B-cell clones from tolerance or do the same B-cell clones persist over time? The first possibility suggests a basic defect in tolerance to Dsg3 that allows new lineages of IgG+ anti-Dsg3 B-cell clones to continually develop over time in these patients; while the second suggests that a time-limited event caused a set of IgG+ anti-Dsg3 B-cell clones to escape tolerance and if these could be eliminated disease might be cured.
We used antibody phage display to characterize IgG+ B-cell clones to address this question. Antibody phage display (APD) is a very sensitive screening method for finding antigen-specific immunoglobulins because it has the capacity for screening up to 108 independent monoclonal antibodies in large phage libraries that are selected by multiple rounds of binding to an antigen followed by amplification (Barbas III, 2001; Hammers and Stanley, 2014). Previous studies have shown that APD can be used to detect anti-Dsg antibodies in both PV and pemphigus foliaceus (PF), and that the specificity of these autoantibodies is mostly carried by the IgG heavy chain (Ishii et al., 2008; Payne et al., 2005; Yamagami et al., 2009; Yamagami et al., 2010). By sequencing the variable heavy chain repertoire the clonal relatedness of heavy chains (and the B cells from which they are derived) can be determined by shared complementarity determining region 3 (CDR3) nucleotide (or their deduced amino acid) sequences (Yamagami et al., 2010).
Results
Patient characteristics
Details of the patients, given in Table S1, are summarized below.
Patient PV3 was first seen in 2006 and treated over 5.5 years with prednisone and mycophenolate mofetil. He went into complete clinical remission off therapy twice (i.e., absence of new or established lesions with the patient off any systemic therapy for at least 2 months; see Murrell et al. (2008)) but disease recurred each time. His B-cell response (some sequences reported previously by Yamagami et al. (2010)) was analyzed in 2006 (initial analysis, designated PV3) and ~5.5 years later (analysis designated PV3a; Fig. 1a). The second patient’s B-cell response was characterized at initial presentation in 2002 (designated PV1; sequences previously reported by Payne et al. (2005)), then again 4 years later after routine therapy (PV1a). Additional studies were performed after three courses of rituximab (each 2 g over 2 weeks), at which time his anti-Dsg3 IgG serum titer was indeterminate and shortly after which disease recurred (PV1b); then after a 22 month clinical and serologic remission following a fourth course of rituximab (PV1c; ~11 years after first studied) (Fig. 1b). Both these patients had mucocutaneous PV with all relapses involving cutaneous lesions. Such patients usually have anti-Dsg1 IgG in addition to anti-Dsg3 (Ishii et al., 1997; Mahoney et al., 1999; Shirakata et al., 1998). Although we did not check anti-Dsg1 at all time points, patient PV3 had a Dsg1 ELISA index value of 147 on final relapse (black box Fig. 1), and PV1 had Dsg1 ELISA index values of 96 when first seen (PV1 red box Fig 1) and of 0.85 at the last time point studied (PV1c red box Fig. 1).
Figure 1. APD libraries: timeline of construction and panning against Dsg3.

(a, b) Serum anti-Dsg3 ELISA index values are indicated by boxes. Above horizontal dotted line are positive values. Red boxes indicate blood used for APD library construction. Periods of complete clinical remission off therapy (CROT) are shaded in turquoise. (c) Enrichment for Dsg3-binding was measured by phage ELISA of the libraries amplified after sequential pannings (further explained in methods). Shown is the ratio of the optical density (O.D.) value from rounds P2–P4 relative to the O.D. of the unpanned P0 library (with a value of 1), expressed as arbitrary units (A.U.).
A third PV patient, designated COL-JA, was in clinical and serological remission off therapy for 56 months (anti-Dsg3/1 serum IgG titers negative) after a single course of rituximab (4x 375 mg/m2, over 1 month) when we analyzed her anti-Dsg3 B-cell response.
Analysis of IgG+ anti-Dsg3 B-cell clones over time in patients PV3 and PV1 with active and remitting disease
We constructed APD-libraries from mRNA from blood mononuclear cells by cloning a PCR-amplified repertoire of IgG-variable heavy (VH) and light chains (VL) into the M13-based phagemid vector pComb3X that allows expression of a single chain variable fragment (scFv) antibody on the surface of individual phage (which contain the corresponding cDNA inside) (Barbas III, 2001). Multiple rounds of panning on immobilized Dsg3 allow for increasing enrichment of phage that express anti-Dsg3 antibodies from the library, if they are present, with each round. We monitored enrichment of the APD-libraries by using a Dsg3-ELISA developed with anti-M13 antibodies (Fig. 1c). Patients with active, recurrent or impending disease (PV3/3a; PV1/1a/1b) showed enrichment with multiple pannings. To further characterize the autoimmune B-cell response, we cloned and genetically analyzed monoclonal anti-Dsg3 antibodies from phage that enriched on panning (Fig. 2, Table 1). Individual anti-Dsg3 clonal lineages were identified by their shared VH-CDR3 signatures formed during somatic VDJ-recombination in pre-B cells. By APD in patient PV3, we detected three anti-Dsg3 clonal lines (I–III; Fig. 2a). In the same patient, we detected two of these original lines (I, II) and four new lines (IV–VII) 5.5 years later. Because we routinely sample limited number of clones from each round of panning, it is conceivable that clones are present but not detected by APD. Therefore we used PCR with VH-CDR1- or 2- and 3-specific primers to determine if clones not detected by APD at one time point (but that were detected at another time by APD) in both our PV3 and PV3a libraries were actually present (Fig. 3). We verified the PCR amplification by nucleotide sequencing of all amplicons. By combining APD and PCR we could detect all anti-Dsg3 clonal lines at both time points except for clone IV, which we found only in relapse (Fig. 2a). We conclude that non-tolerant clonal anti-Dsg3 B-cell lineages persisted in this PV patient over years with only one possibly new clone forming. A similar study in patient PV1 had analogous findings (Fig. 2b). At the times when this patient had active, remitting and relapsing disease, six anti-Dsg3 B-cell clonal lines were detected by APD and PCR; all these clones were present in PV1a 4 years later, and five were detected another 4.5 years later (PV1b). No new clonal lines were detectable over these 8.5 years.
Figure 2. Dsg3-specific IgG+ B-cell clonal lineages in patients (a) PV3 and (b) PV1.

Each clonal lineage is indicated by circle and Roman numeral, grouped in gray-shaded vertical rectangles by time. Clones identified by PCR are outlined in black, others were determined by APD. VH/VL genes used for the VH/VL-chains are shown to the left of each clone. For each clone isolated by APD, VH-chains varying by somatic mutations (SM) are indicated to the right of the circle (with the Arabic number corresponding to the Roman numeral of each clone and small letters indicating distinct SM-patterns; e.g., 2a/2b indicate VH-chains with the same VH-CDR3, differing by SM throughout the rest of the VH-chain). Number of actual mutations (excluding VH-CDR3- and IgG heavy chain framework 4-regions) compared to germline are indicated in parenthesis. ND: not detected by APD or PCR.
Table 1.
Characterization of IgG anti-Dsg3 B-cell lineages in pemphigus patients PV3 and PV1.
| Pat. | B-cell clone (specificity)1 | Translation of the VH-CDR3 (#aa)2 | IMGT VH gene3 | IMGT VL genes4 | |
|---|---|---|---|---|---|
| PV3 | PV3a | ||||
| PV3 | I (D3) | ARDLGGFDFDY (11) | 1-46*03/01 | λ2-8*01 |
λ2-8*01 λ2-14*02 |
| II (D3) |
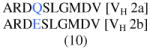
|
1-46*03/01 | λ2-14*01 | λ2-8*01 | |
| III (D3) | ARINYYDGSGHHSDADYM (18) | 5-a*03 | λ1-51*01 | (n/a) | |
| IV (D3) | AKGMHDIVVMIGSTYYYYYGLDV (23) | 3-23*01 | (n/a) | λ3-10*01 | |
| V (D3) | TREGPGYSSGSFHV (14) | 4-59*03 | (n/a) | κ3-11*01 | |
| VI (D3) | AKGYGDDHDASDI (13) | 3-30*18/03 | (n/a) | κ1-39*01/κ1D-39*01 | |
| VII (D3) | ARDRGVLFRNFFDL (14) | 1-2*02/04/05 | (n/a) | κ3-15*01 | |
| Pat. | B-cell clone (specificity)1 | Translation of the VH-CDR3 (#aa)2 | IMGT VH gene3 | IMGT VL genes4 | ||
|---|---|---|---|---|---|---|
| PV1 | PV1a | PV1b | ||||
| PV15 | I (D3) |
|
1-69*06 |
λ1-44*01 λ1-47*01/02 |
λ1-44*01 λ1-47*02 |
λ1-47*02 |
| II (D3) | ASGGVVDFDH (10) | 3-7*01 | λ2-23*01/03 | (n/a) | (n/a) | |
| III (D3) |
|
1-18*04 | (n/a) | κ4-1*01 | κ4-1*01 | |
| IV (D3) | AREALVYYDSSGYYKGGFDY (20) | 3-30*04 | κ3-11*01/02 | (n/a) | (n/a) | |
| V (D31) | ARGWHRTGFRGYPSHWYFDL (20) | 4-4*02 | λ1-47*02 | λ1-47*02 | λ1-47*02 | |
| VI (D31) | ARGGDYSGWYNFDY (14) | 1-69*09 | λ3-9*01 λ3-1*01 |
λ3-9*01 | (n/a) | |
We obtained clones specific for Dsg3 (D3) and for both Dsg3 and Dsg1 (D31) by panning APD-libraries on Dsg3.
B-cell lineages were defined by shared VH-CDR3 deduced amino acid (aa) sequences (allowing one aa change, presumably due to somatic mutation; blue).
Gene usage was determined by matching VH/VL-nucleotide sequences to the IMGT database. Note that VH/VL-gene usage, as detected by APD, in anti-Dsg3 antibodies is not completely random but in many cases, for the same clonal VH chain the VL gene for the paired VL-chain is the same over time (see PV3-PV3a clone I, PV1-PV1a-PV1b clone I, PV1a-PV1b clone III, PV1-PV1a-PV1b clone V, PV1-PV1a clone VI). This shows that although VH/VL-pairing in cloning the APD-library is theoretically random, only very specific light chains can pair with the VH-chains in making anti-Dsg3 antibodies. Bold typeface of VH/VL-genes indicates that these genes were found in pemphigus anti-Dsg3 antibodies by EBV-transformed memory B cell or human-mouse heterohybridoma in other studies (Di Zenzo et al., 2012; Qian et al., 2007). In addition, the VH/VL-gene pairing for PV3a clone VI (underlined) has previously been found by heterohybridoma cloning of pemphigus antibodies (Qian et al., 2007).
n/a indicates VL gene not determined because PCR, not APD, was used to detect the VH.
Library PV1c is not shown as no clones were detected by neither APD nor PCR.
Figure 3. PCR-strategy to identify patient-specific VH-CDR3s found by APD at one time point but not at another.
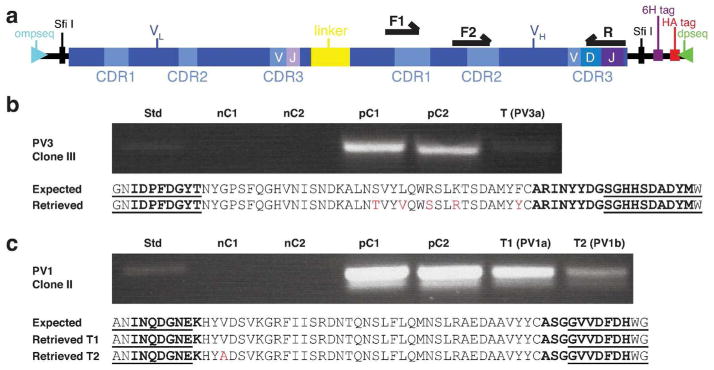
(a) PCR-template with forward primer from either CDR1 or CDR2 (F1, F2) and reverse primer from CDR3 (R). (b) PV3 clone III, isolated by APD in 2006, was not found by APD in PV3a (2012), but was identified by PCR (lane T(PV3a)). (c) PV1 clone II was found by APD in 2002, but only by PCR at later times (lanes T1(PV1a) and T2(PV1b)). Deduced amino acid (aa)-sequences from sequencing of PCR products shown below the gels (retrieved); compared to that of the corresponding APD-isolated clone (expected). Presumed somatic mutations (or PCR errors) are in red, VH-CDR2/3 regions in bolded font, primer-covered regions are underlined. Std, 200 bp standard; nC1, negative control (water); nC2, negative control (VH-cDNA from an unrelated PV patient); pC1, positive control (monoclonal phagemid DNA of clone); pC2, positive control (polyclonal plasmid DNA from APD-library from which clone of interest was identified by panning).
IgG+ anti-Dsg3 B-cell clones were not detectable after long-term clinical and serological remission induced by rituximab
No clones could be detected by either APD or PCR ~11 years after initial analysis, after patient PV1, treated with 4 courses of rituximab, maintained a clinical and serological remission off therapy for ~22 months (PV1c). To confirm this finding, we tested another patient treated with rituximab in complete clinical and serologic remission off therapy for ~56 months (COL-JA). In this patient no anti-Dsg3 clones could be detected by APD as well. These findings are in marked contrast to all active pemphigus patients we have tested by APD so far (3 PV, 2 PF), in whom we always found multiple anti-Dsg clonal lineages (Ishii et al. (2008); Payne et al. (2005); Yamagami et al. (2009); and unpublished). These findings indicate that even in some patients who have the potential to actually develop PV, if rituximab effectively eliminates the pathogenic clones, they no longer have detectable IgG+ anti-Dsg3 B cells that are escaping tolerance. Taken together with the persistence of the same autoimmune B-cell clones persisting for years in active and remitting disease, these data suggest that rituximab works, at least in some patients, by eliminating sets of established pathogenic clones that are not, or rarely, replaced by new sets of autoimmune B-cell clones.
Analysis of somatic hypermutation and variable light chain usage over time
Analyzing the nucleotide sequences encoding the anti-Dsg3 VH-chains over time allowed us to determine that affinity maturation was generally not an ongoing process in the autoimmune response of PV, because in most clones, the number of somatic mutations was stable over time (Fig. 2). Occasionally we found the exact VH-nucleotide sequence at different time points (VH 1c, 3a, 5a, 6a in patient PV1; 1a in PV3; Fig. 2). This was not from cross-contamination between libraries, because we used barcoded PCR primers to distinguish libraries (see Methods). These data also show that B cells producing identical VH-chains can persist for up to 8.5 years, and are not necessarily replaced by more somatically-mutated clones.
Furthermore, we analyzed the light chain usage of the anti-DSG3 clones found by APD (Table 1). Although when constructing libraries by APD, heavy and light chain pairing is theoretically random, these data show that with libraries made at different time points, for the same preserved heavy chain clones, certain light chain families are definitely favored for pairing.
Discussion
The basic findings of this study are that clonal lineages of IgG+ anti-Dsg3 B cells can persist up to 8.5 years even after rituximab therapy; that patients with recurrent disease maintain the same set of persistent B-cell clonal lineages over many years, and even maintain the same exact B-cell clone (i.e., with the same somatic mutations throughout the entire VH, e.g. PV3 I-1a, PV1 I-1c, II-3a, V-5a, VI-6a in Fig. 2); and that in PV patients new lines of IgG+ anti-Dsg3 B-cell clones do not continuously escape from tolerance, giving rise to new sets forming over time. There may have been one exception (clone IV in PV3a), however, we cannot rule out that this was a minor clone in PV3 that we could not detect or whether the cells that produced this antibody were not circulating at the time blood mononuclear cells were obtained for APD-library cloning. The data for all the other clones and time points suggest that there is not a basic defect in maintaining IgG+ B-cell tolerance to Dsg3 in PV patients that would allow new sets of anti-Dsg3 B-cell clonal lines to escape over time. In contrast, in SLE and MS, there is ongoing escape from peripheral tolerance at the mature naïve B-cell level (Kinnunen et al., 2013; Yurasov et al., 2006), which eventually may mature to IgG-mediated autoimmunity. Further data to support that there is not an ongoing defect permitting escape from tolerance of new IgG+ B-cell clones in PV is that elimination of all persistent clones detected prior to ablation by rituximab is associated with long-term serologic and clinical remission off therapy, which was also previously demonstrated by flow cytometry of IgG+ anti-Dsg3 B cells (Colliou et al., 2013). Our study confirms that finding by looking at the loss of the specific persistent anti-Dsg3 B-cell lines (identified by their VH-CDR3 sequences) with APD, a very sensitive technique for cloning rare antibody subsets (Barbas III, 2001; Hammers and Stanley, 2014). Although some of the original APD-cloned sequences of PV3 and PV1 at the first time point (PV3 and PV1 red boxes in Fig. 1) were reported previously (Payne et al., 2005; Yamagami et al., 2010), what is unique about this study is the characterization of the APD-derived clones over time.
Of course with any technique analyzing autoreactive B cells, some may be missed, but we have no reason to believe that the clones we found here are in any way not representative of the autoantibody B-cell clone repertoire as a whole. It is clear that the antigen-specific clonal lines we find are preserved over years. There is no reason to think we would find only clones that are preserved but not those that are not by these methods. In other words, we think it very unlikely that somehow APD would only miss finding newly emergent clones not present at previous time points. Finally, to study clones over time, APD is probably the most sensitive technique for picking up minor or rare clones because of the large number of antibody-bearing phage clones that can be screened.
The basic idea, that elimination of autoreactive B cells and regeneration of the B-cell repertoire from pro-B-cell precursors can reestablish tolerance has been suggested by previous studies. This conclusion has mostly been through studies of clonal expansions of B cells in disease with elimination of those expansions in remission. For example, in anti-myelin-associated glycoprotein neuropathy, in which IgM-antibodies mediate disease activity, response to CD20-ablative therapy, compared to non-responders, is associated with better elimination of clonally expanded IgM+ B cells (Maurer et al., 2012). Similarly, in three PV patients, assessment of VH-CDR3 length distributions (i.e., immunoscope analyses) indicated clonal expansions, and only the patient in complete remission showed normalization, suggesting elimination of aberrant, presumably pathologic, B-cell clones (Colliou et al., 2013; Mouquet et al., 2008). These studies suggested that elimination of B-cell clones in Dsg-autoantibody-mediated diseases is associated with remission but did not specifically trace ablation over time to study whether the same clones recur or, in fact, whether these or new clonal expansions arise in recurrence.
The finding that we cannot identify APD clones that bind Dsg3 in the two PV patients in long-term remission (PV1c and COL-JA) (Fig. 1) and other individuals without pemphigus (Yamagami et al. (2009), and unpublished) indicates that random VH/VL-pairing does not result in artifactual Dsg3-binding autoantibodies and may suggest that light chain receptor editing (Luning Prak et al., 2011) is not the basic mechanism by which humans maintain tolerance to the peripheral B-cell antigen Dsg3. Receptor editing would mean that during B-cell ontogeny, if a heavy-light chain pairing causes anti-Dsg reactivity, the light chain is ‘edited out’ in that autoimmune B cell, and another light chain is used in its place that does not result in an autoantibody. This tolerance mechanism would result in normals (with B-cell tolerance to Dsg) having potentially pathogenic heavy chains in their repertoire that just need the correct light chain pairing to form a pathogenic anti-Dsg antibody. However, in APD we allow any light chain to recombine with any heavy chain (Marzari et al., 2001; Qian et al., 2007) and we do not find anti-Dsg autoantibody formation. Although it is possible that there is no possible light chain in the entire B-cell repertoire to pair with a potentially pathogenic heavy chain to reform an anti-Dsg antibody, it is unlikely since we also show here, and have shown previously (Ishii et al., 2008; Payne et al., 2005; Yamagami et al., 2009), that several different light chains in pemphigus patients with clinically overt disease are capable of pairing to form such a Dsg3 antibody. However, further study would be necessary to rigorously validate this suggestion.
Although we have only been able to study three PV patients we feel the findings are relevant to understanding PV and its therapy. First of all, these are human immunology studies. The importance of studying human immunology has recently been highlighted (2013; Davis, 2008; Mestas and Hughes, 2004). Second, much of our insight into spontaneous autoimmune diseases is from inbred so-called autoimmune mouse strains (e.g., NOD mice for diabetes, NZB mice for systemic lupus erythematosus). In the same way, individual humans, even though of one genotype (like inbred mice with one genotype), allow insights into the immunology of spontaneously occurring human autoimmunity. Third, the major finding that in patients with PV new B-cell clonal lines rarely newly escape from tolerance was validated in two patients over several time points representing 14 patient years, and in a third patient in long-term remission. However, with the small number of patients studied, we cannot conclude that our findings would hold for all pemphigus patients.
Although rituximab clearly eliminates IgG+ anti-Dsg3 B-cell clones, it may have additional effects on maintaining tolerance through expansion of regulatory CD38high B cells (Bregs) that may down regulate Dsg3-specific CD4+ T helper (Th) cells which are also decreased after rituximab (Colliou et al., 2013; Eming et al., 2008). However, CD4+ Th cells may also be decreased because of a lack of Dsg3-specific B cells to present the autoantigen and activate them (Eming et al., 2008).
In summary, our findings show that the same clonal IgG+ anti-Dsg3 B-cell lines persist over 14 patient years in patients with active disease, even after complete clinical remission off therapy, but in two patients in long-term clinical and serological remission no IgG+ B-cell clones are detectable in the blood (11 and 15 years after active disease). It is possible that some clones are hidden (e.g., in the bone marrow; Hamza et al. (2012)) and that these may ultimately re-appear to cause relapse. If, indeed, these patients ultimately recur then we expect that the same clonal lines would be detected, however, so far, no recurrence has been seen. The implication of our findings is that for effective therapy, in at least some patients, and potential cure of PV, we should seek therapies adept at eliminating all existing anti-Dsg3 B-cell clones, e.g. higher doses or varying dose schedules of rituximab, and/or treating early disease (Colliou et al., 2013; Lunardon et al., 2012) at which time it may be easier to fully eliminate such clones, and new ones are unlikely to escape tolerance.
Materials & Methods
Further information is available in Supplemental Data.
Patients and clinical definitions
The study was conducted according to Declaration of Helsinki Principles. Written informed consent, approved by the Institutional Review Board of the University of Pennsylvania, was obtained from patients PV3 and PV1. From those two patients with a confirmed diagnosis of PV (see Supplemental Methods for details) ~50 ml blood was obtained to isolate peripheral blood mononuclear cells (PBMC). Clinical data, PBMC and serum from patient COL-JA were obtained from Pascal Joly, Rouen, France.
Anti-Dsg3/1 Serum ELISA
Titers of IgG antibodies against Dsg3/1 were determined with Mesacup Dsg3/1 ELISA kits (MBL), following the manufacturer’s instructions.
APD-library construction and selection of anti-Dsg3 expressing phage clones
Detailed methods for the APD-technique have been previously published (Barbas III, 2001; Payne et al., 2005). PBMC-RNA was extracted with RNeasy Midi Kits (Qiagen). Complementary DNA (cDNA) was generated from RNA with a SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen). We separately constructed IgGκ/IgGλ APD libraries using the phagemid vector pComb3X (Scripps Research Institute). During PCR amplification of IgG VH-transcripts from cDNA, the standard HSCG1234-B reverse primer was barcoded at the SpeI-site to distinguish phage clones derived from each APD-library in one patient (PV3: HSCG1234-B; PV3a: HSCG1234-E; PV1: HSCG1234-B; PV1a: HSCG1234-C; PV1b: HSCG1234-D; PV1c: HSCG1234-G; COL-JA: HSCG1234-H; see Table S2). Care was taken to prevent cross-contamination between libraries (see Supplemental Methods). Phagemid libraries of at least 108 independent transformants were electroporated into XL-1 Blue E.coli (Agilent), and recombinant phage were purified, after addition of VCSM13 helper phage (Stratagene), from culture supernatants using PEG-precipitation. The diversity of each unpanned library was determined by random sampling of ~20 phage clones, phagemid extraction with a plasmid preparation system (QIAGEN), and Sanger sequencing for VH/VL-gene usage and VH/L-CDR3 sequences (obtained sequences were subjected to an online database search at IMGT/V-QUEST and vbase2, accessible at http://www.imgt.org and http://www.vbase2.org). Equal numbers of unpanned P0-IgGκ and P0-IgGλ transformants were combined to select for Dsg3-binders during four rounds of ag-guided selection (panning) on immobilized Dsg3 substrate (MBL). Resulting polyclonal phage pools were diluted to 1:1,000 and 1:5,000 and tested for specificity towards Dsg3 by ELISA using HRP-conjugated anti-M13 antibody (1:5,000; GE Healthcare) for developing on Dsg3 substrate. If these pools of phage bind Dsg3, the peroxidase-conjugated anti-phage antibody renders a change in color, indicating enrichment of antigen-specific phage; for comparison, we calculated the ratio of the optical density (O.D.) value from panning rounds P2–P4 relative to the O.D. of the unpanned (P0) phage library (which was set=1; ratio expressed as arbitrary units, A.U.). In case of Dsg3-specific enrichment of phage pools, individual phage monoclonals were isolated from those panning rounds with positive readings (usually rounds 2–4; ~40 for each panned library). Obtained phagemids were analyzed for shared VH-CDR3 signatures, VH/VL-gene usage, and somatic mutations (IMGT, vbase2). Additionally, all individual monoclonals were re-tested on Dsg3 substrate for binding, using the anti-M13 ELISA detailed above.
Clone-specific PCR
To determine if B-cell clones identified at time point 1 by APD were present at time point 2 in the same patient (if they could not be found at that latter time point by APD), we obtained polyclonal pComb3X-scFv phagemid DNA as PCR template from the unpanned (P0) or once panned (P1) library made at time point 2. The VH-coding segments in the inserts were further PCR-amplified using ompseq and dpseq sequencing primers annealing up- and downstream to the insert (Fig. 3; primers at 20 μM; 5′ 94°C, then 30 cycles of 15″ 94°C, 15″ 66°C, 90″ 72°C, then 10′ 72°C). In some cases, gel-purified VH-DNA (used for construction of the APD library at time point 2) was used instead of the latter PCR product. On these templates, a second PCR with primers specific for the VH-CDR2/3 regions (designated F-CDR2 and R-CDR3) was performed (alternatively, primers were designed to anneal to the VH-CDR1 and VH-CDR3 regions). The sequences of these primers were determined from the clones isolated by APD from time point 1 and are given in Table S2. All nested PCR products were gel-purified (Qiagen), TOPO-TA cloned (Invitrogen), and Sanger sequenced.
Supplementary Material
Acknowledgments
This research was supported by a research fellowship from the DFG (HA6736/1-1) to C.M.H. and by grants R01-AR052672 and P30-AR057217 from the NIAMS, NIH to J.R.S. We thank Pascal Joly, Department of Dermatology, Inserm U905, Institute for Research and Innovation in Biomedicine, Rouen University Hospital, University of Normandy, F-76031 Rouen, France, for providing us clinical data, PMBC and serum from patient COL-JA; and Eline T. Luning Prak for reviewing the manuscript.
Footnotes
Conflict of Interest
The authors state no conflict of interest.
References
- Of men, not mice. Nat Med. 2013;19:379. doi: 10.1038/nm.3163. [DOI] [PubMed] [Google Scholar]
- Ahmed AR, Spigelman Z, Cavacini LA, et al. Treatment of pemphigus vulgaris with rituximab and intravenous immune globulin. N Engl J Med. 2006;355:1772–9. doi: 10.1056/NEJMoa062930. [DOI] [PubMed] [Google Scholar]
- Almugairen N, Hospital V, Bedane C, et al. Assessment of the rate of long-term complete remission off therapy in patients with pemphigus treated with different regimens including medium- and high-dose corticosteroids. J Am Acad Dermatol. 2013;69:583–8. doi: 10.1016/j.jaad.2013.05.016. [DOI] [PubMed] [Google Scholar]
- Amagai M, Hashimoto T, Shimizu N, et al. Absorption of pathogenic autoantibodies by the extracellular domain of pemphigus vulgaris antigen (Dsg3) produced by baculovirus. J Clin Invest. 1994;94:59–67. doi: 10.1172/JCI117349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbas CF., III . Phage Display: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. [Google Scholar]
- Colliou N, Picard D, Caillot F, et al. Long-term remissions of severe pemphigus after rituximab therapy are associated with prolonged failure of desmoglein B cell response. Sci Transl Med. 2013;5:175ra30. doi: 10.1126/scitranslmed.3005166. [DOI] [PubMed] [Google Scholar]
- Davis MM. A prescription for human immunology. Immunity. 2008;29:835–8. doi: 10.1016/j.immuni.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Zenzo G, Di Lullo G, Corti D, et al. Pemphigus autoantibodies generated through somatic mutations target the desmoglein-3 cis-interface. J Clin Invest. 2012;122:3781–90. doi: 10.1172/JCI64413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eming R, Nagel A, Wolff-Franke S, et al. Rituximab exerts a dual effect in pemphigus vulgaris. J Invest Dermatol. 2008;128:2850–8. doi: 10.1038/jid.2008.172. [DOI] [PubMed] [Google Scholar]
- Hammers CM, Lunardon L, Schmidt E, et al. Contemporary management of pemphigus. Expert Opinion on Orphan Drugs. 2013;1:295–314. [Google Scholar]
- Hammers CM, Stanley JR. Antibody phage display: technique and applications. J Invest Dermatol. 2014;134:e17. doi: 10.1038/jid.2013.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza N, Bootsma H, Yuvaraj S, et al. Persistence of immunoglobulin-producing cells in parotid salivary glands of patients with primary Sjogren’s syndrome after B cell depletion therapy. Ann Rheum Dis. 2012;71:1881–7. doi: 10.1136/annrheumdis-2011-201189. [DOI] [PubMed] [Google Scholar]
- Ishii K, Amagai M, Hall RP, et al. Characterization of autoantibodies in pemphigus using antigen-specific enzyme-linked immunosorbent assays with baculovirus-expressed recombinant desmogleins. J Immunol. 1997;159:2010–7. [PubMed] [Google Scholar]
- Ishii K, Lin C, Siegel DL, et al. Isolation of pathogenic monoclonal anti-desmoglein 1 human antibodies by phage display of pemphigus foliaceus autoantibodies. J Invest Dermatol. 2008;128:939–48. doi: 10.1038/sj.jid.5701132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joly P, Mouquet H, Roujeau JC, et al. A single cycle of rituximab for the treatment of severe pemphigus. N Engl J Med. 2007;357:545–52. doi: 10.1056/NEJMoa067752. [DOI] [PubMed] [Google Scholar]
- Kasperkiewicz M, Schmidt E, Zillikens D. Current therapy of the pemphigus group. Clin Dermatol. 2012;30:84–94. doi: 10.1016/j.clindermatol.2011.03.014. [DOI] [PubMed] [Google Scholar]
- Kinnunen T, Chamberlain N, Morbach H, et al. Specific peripheral B cell tolerance defects in patients with multiple sclerosis. J Clin Invest. 2013;123:2737–41. doi: 10.1172/JCI68775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leshem YA, Hodak E, David M, et al. Successful treatment of pemphigus with biweekly 1-g infusions of rituximab: a retrospective study of 47 patients. J Am Acad Dermatol. 2013;68:404–11. doi: 10.1016/j.jaad.2012.08.010. [DOI] [PubMed] [Google Scholar]
- Lunardon L, Tsai KJ, Propert KJ, et al. Adjuvant rituximab therapy of pemphigus: a single-center experience with 31 patients. Arch Dermatol. 2012;148:1031–6. doi: 10.1001/archdermatol.2012.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luning Prak ET, Monestier M, Eisenberg RA. B cell receptor editing in tolerance and autoimmunity. Ann N Y Acad Sci. 2011;1217:96–121. doi: 10.1111/j.1749-6632.2010.05877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney MG, Wang Z, Rothenberger K, et al. Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Invest. 1999;103:461–8. doi: 10.1172/JCI5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzari R, Sblattero D, Florian F, et al. Molecular dissection of the tissue transglutaminase autoantibody response in celiac disease. J Immunol. 2001;166:4170–6. doi: 10.4049/jimmunol.166.6.4170. [DOI] [PubMed] [Google Scholar]
- Mascaro JM, Jr, Espana A, Liu Z, et al. Mechanisms of acantholysis in pemphigus vulgaris: role of IgG valence. Clin Immunol Immunopathol. 1997;85:90–6. doi: 10.1006/clin.1997.4408. [DOI] [PubMed] [Google Scholar]
- Maurer MA, Rakocevic G, Leung CS, et al. Rituximab induces sustained reduction of pathogenic B cells in patients with peripheral nervous system autoimmunity. J Clin Invest. 2012;122:1393–402. doi: 10.1172/JCI58743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172:2731–8. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- Mouquet H, Musette P, Gougeon ML, et al. B-cell depletion immunotherapy in pemphigus: effects on cellular and humoral immune responses. J Invest Dermatol. 2008;128:2859–69. doi: 10.1038/jid.2008.178. [DOI] [PubMed] [Google Scholar]
- Murrell DF, Dick S, Ahmed AR, et al. Consensus statement on definitions of disease, end points, and therapeutic response for pemphigus. J Am Acad Dermatol. 2008;58:1043–6. doi: 10.1016/j.jaad.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne AS, Hanakawa Y, Amagai M, et al. Desmosomes and disease: pemphigus and bullous impetigo. Curr Opin Cell Biol. 2004;16:536–43. doi: 10.1016/j.ceb.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Payne AS, Ishii K, Kacir S, et al. Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest. 2005;115:888–99. doi: 10.1172/JCI24185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, Diaz LA, Ye J, et al. Dissecting the anti-desmoglein autoreactive B cell repertoire in pemphigus vulgaris patients. J Immunol. 2007;178:5982–90. doi: 10.4049/jimmunol.178.9.5982. [DOI] [PubMed] [Google Scholar]
- Shirakata Y, Amagai M, Hanakawa Y, et al. Lack of mucosal involvement in pemphigus foliaceus may be due to low expression of desmoglein 1. J Invest Dermatol. 1998;110:76–8. doi: 10.1046/j.1523-1747.1998.00085.x. [DOI] [PubMed] [Google Scholar]
- Yamagami J, Kacir S, Ishii K, et al. Antibodies to the desmoglein 1 precursor proprotein but not to the mature cell surface protein cloned from individuals without pemphigus. J Immunol. 2009;183:5615–21. doi: 10.4049/jimmunol.0901691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagami J, Payne AS, Kacir S, et al. Homologous regions of autoantibody heavy chain complementarity-determining region 3 (H-CDR3) in patients with pemphigus cause pathogenicity. J Clin Invest. 2010;120:4111–7. doi: 10.1172/JCI44425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurasov S, Tiller T, Tsuiji M, et al. Persistent expression of autoantibodies in SLE patients in remission. J Exp Med. 2006;203:2255–61. doi: 10.1084/jem.20061446. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.