

Abstract
Chromatin conformation plays a major role in all cellular decisions. We showed previously that P53 pro-apoptotic target promoters are enriched with H3K9me3 mark and induction of P53 abrogates this repressive chromatin conformation by down-regulating SUV39H1, the writer of this mark present on these promoters. In the present study, we demonstrate that in response to P53 stabilization, its pro-apoptotic target promoters become enriched with the H3K4me3 epigenetic mark as well as its readers, Wdr5, RbBP5 and Ash2L, which were not observed in response to SUV39H1 down-regulation alone. Overexpression of Ash2L enhanced P53–dependent apoptosis in response to chemotherapy, associated with increased P53 pro–apoptotic gene promoter occupancy and target gene expression. In contrast, pre–silencing of Ash2L abrogated P53's ability to induce the expression of these transcriptional targets, without affecting P53 or RNAP II recruitment. However, Ash2L pre–silencing, under the same conditions, resulted in reduced RNAP II ser5–CTD phosphorylation on these same pro-apoptotic target promoters, which correlated with reduced promoter occupancy of TFIIB as well as TFIIF (RAP74). Based on these findings, we propose that Ash2L acts in concert with P53 promoter occupancy to activate RNAP II by aiding formation of a stable transcription pre–initiation complex required for its activation.
Keywords: P53, Ash2L, RNAP II, Ser5–CTD, H3K4me3, Transcription, Apoptosis
Introduction
Chromatin conformation plays a fundamental role in regulating gene transcription and silencing, as well as DNA repair 1. Genome wide studies have shown that the H3K9me3 (histone H3 lys9 trimentylation) enrichment represents a transcriptionally repressive chromatin conformation, whereas H3K4me3 (histone H3 lys4 trimethylation) is localized mainly at the TSS (transcription start site) of the promoter regions 2-4. As a consequence of epigenetic modifications and chromatin remodeling, components of the Pre-Initiation Complex (PIC) can be either recruited or altered allowing transcription initiation 5.
P53 is stabilized in response to a variety of cellular stresses and binds to its responsive DNA elements and drives the transcription of its target genes to initiate P53–dependent cellular responses ranging from reversible cell cycle arrest to senescence or apoptosis 6, 7. We have shown previously that in the absence of P53 induction, H3K9me3 is enriched at P53 target promoters, and P53 overcomes this barrier by reducing the level of the writer of this repressive mark, SUV39H1, by both transcriptional and post–translational mechanisms 8. In relation to H3K4 methylation mechanisms, previous studies have demonstrated DNA damage induced accumulation of H3K4me3 on the P21 promoter 9, which is a well-known P53 target gene 10. In human cells, H3K4 methylation is catalyzed by the SET1/MLL family of histone methyl transferases (HMTs). Biochemical studies have shown that similar to yeast prototype Set1 11, human SET1/MLL complexes possess both unique as well as common subunits (WDR5, RbBP5, DPY30 as well as ASH2L) 12. However, the enrichment of H3K4me3 on P53 pro-apoptotic promoters, as well as the roles played by any of the common or unique MLL subunits on the P53-mediated pro-apoptotic gene expression and apoptosis is not well established.
Previous studies have shown that a variety of general transcriptional cofactors including TAF6, TAF9, TRAP80 and TFIIH 13 physically interact with P53. In addition, P53 itself is able to assist in the recruitment of various PIC components, including TFIIA, TFIIH as well as TBP and its associated factors to its target promoters 5. How the histone modifications affect the recruitment of these general transcription factors on P53 pro-apoptotic target promoters is also not well understood.
In this present study, we show that in response to P53 induction, P53–dependent pro–apoptotic target promoters become enriched with the H3K4me3 histone mark as well as by the common MLL subunit proteins, Wdr5, RbBP5 and Ash2L. More importantly, we establish that Ash2L (absent, small, or homeotic – like Drosophila), which is a common component of H4K4 methylation complexes regulates P53–dependent pro–apoptotic response by favoring the formation of a stable PIC on P53 pro–apoptotic target promoters, which leads to RNAP II activation.
Results
P53 induction enriches for the H3K4me3 mark on its pro–apoptotic target promoters
High resolution profiling of histone modifications in the human genome has indicated H3K4me3 enrichment in promoter regions of active genes 14-18 and active enhancers 19. To investigate whether P53 activation influences the enrichment of this histone mark on P53–target promoters, we performed ChIP (Chromatin Immunoprecipitation) analysis, both in the absence and presence of induced P53. Up regulation of P53, by treating B5/589 cells with MI–219 (Fig. 1a), by removing tetracycline from EJ–P53 (tet off) culture medium (Supplementary Fig. 1a) or by treating HCT116 cells with Nutlin3a (Supplementary Fig. 1b) or with etoposide (Supplementary Fig. 1c) led to a marked increase in P53 occupancy on the promoters of several of its known target genes. Previous studies have shown that P53 promoter occupancy results in a concomitant increase in H3K4me3 enrichment on the P21 promoter 9, 20. In agreement with these studies, we observed H3K4me3 enrichment at the P21 TSS (transcription start site) with P53 induction (Fig. 1b, Supplementary Fig. 1d, e, f). We also observed that up regulation of P53 by the same approaches enriched for H3K4me3 at the TSS of P53 pro-apoptotic as well as MDM2 promoters, as shown for B5/589 cells (Fig. 1b), EJ–P53 cells (Supplementary Fig. 1d) and HCT116 cells treated either with nutlin3a (Supplementary Fig. 1e) or with etoposide (Supplementary Fig. 1f).
Figure 1. P53 induction establishes the H3K4me3 histone mark on its target promoters.
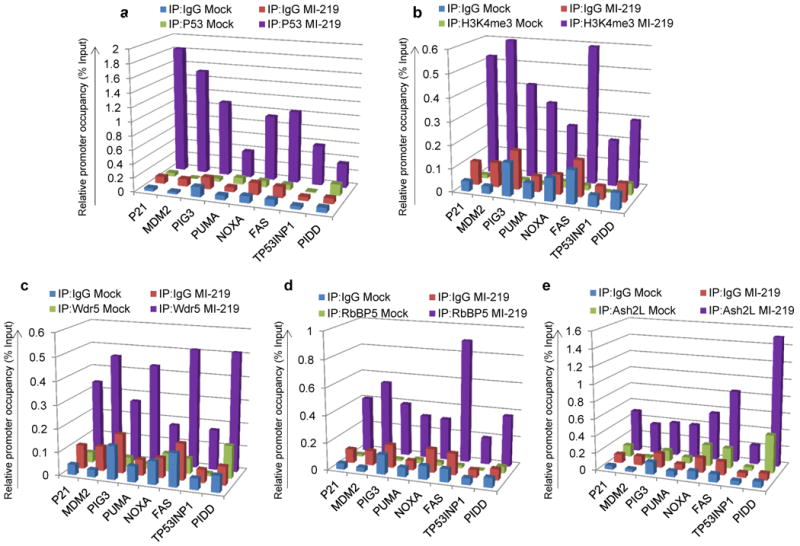
a, b, c, d, e, ChIP analysis showing P53 (a), H3K4me3 (b), Wdr5 (c), RbBP5 (d) and Ash2L (e) occupancy on P53 target promoters in B5/589 cells treated with MI–219 for 16 h. The target sequences were detected by quantitative real-time PCR analysis of eluted DNA. Relative promoter occupancy over the % input is shown in the form of a bar diagram. Each ChIP experiment was repeated at least 3 times and a representative experimental result is shown.
We previously demonstrated that P53–target promoters exist in a transcriptionally repressive chromatin conformation as measured by H3K9me3 enrichment on these promoters and that H3K9me3 loss is essential for P53–promoter occupancy 8. Since SUV39H1 knockdown in the absence of induced P53 was sufficient to down regulate H3K9me3 mark enrichment on P53 pro–apoptotic target promoters 8, we tested whether reduction in SUV39H1 level alone was sufficient to result in H3K4me3 mark enrichment on these same promoters. In HCT116 cells stably expressing doxycycline–inducible sh–SUV39H1 8, doxycycline addition to the culture medium reduced SUV39H1 expression with a concomitant increase in P21 expression as previously reported 8 (data not shown). Similarly, SUV39H1 silencing induced H3K4me3 enrichment on the P21 TSS, but failed to induce H3K4me3 mark enrichment on any of the P53 pro–apoptotic target promoters analyzed (Supplementary Fig. 2a). These results argued that loss of SUV39H1–mediated H3K9me3 mark reduction on P53 pro–apoptotic target promoters by itself was not sufficient for H3K4me3 mark establishment, which required functional P53 up regulation and/ or P53 promoter occupancy.
A functional H3K4 methyltransferase complex contains at least four common structural components, Wdr5, RbBP5, DPY30 and Ash2L, in addition to a catalytic subunit 21-24. There were no detectable changes in expression of Wdr5, RbBP5, DPY30 or Ash2L in response to P53 induction in EJ-P53 or B5/589 cells as tested at both mRNA (Supplementary Fig. 3a,b) and protein (Supplementary Fig. 3c,d) levels. However, we observed enhanced occupancy of Wdr5 (Fig. 1c; Supplementary Fig. 4a, 5a), RbBP5 (Fig. 1d; Supplementary Fig. 4b, 5b) as well as Ash2L (Fig. 1e; Supplementary Fig. 4c, 5c) at the TSS of all the P53–target promoters analyzed in response to P53 induction either in B5/589 cells treated with MI–219 or in EJ–P53 cells following tetracycline regulatable p53 induction or in HCT116 cells treated with etoposide. Of note, SUV39H1 knockdown by itself, in the absence of induced P53, was not sufficient to recruit Wdr5, RbBP5 or Ash2L on to P53 pro-apoptotic target promoters analyzed (Supplementary Fig. 2b; data not shown).
Figure 5. Ash2L is required for efficient Serine5-CTD phosphorylation on P53 target promoters, in response to P53 induction.
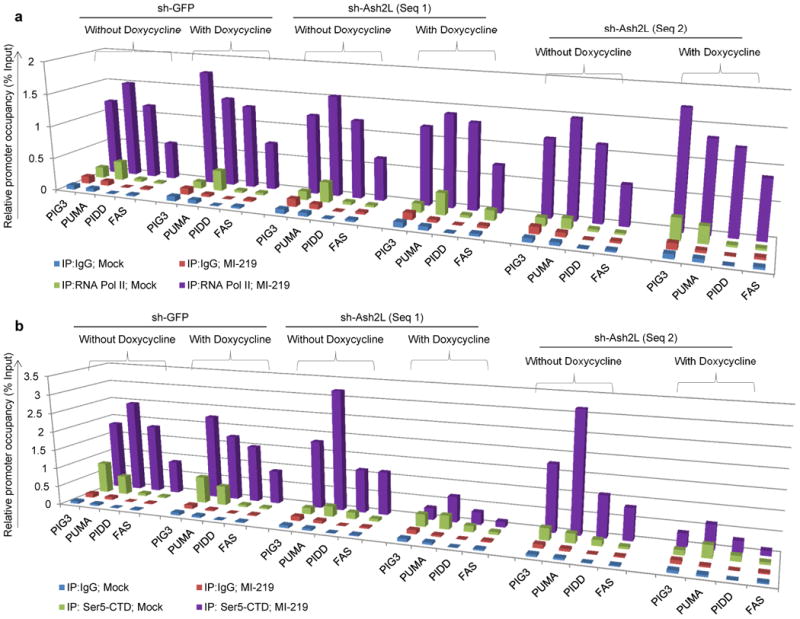
a, b, ChIP analysis showing occupancy of RNAP II (a) and Serine5–CTD (b) on P53 pro–apoptotic target promoters in B5/589 cells pre–silenced for either GFP (non-specific control) or Ash2L (2 independent sequences) expression. The cells were cultured in the presence of doxycycline for 48 h followed by MI–219 treatment for another 16 h. The target sequences were detected by quantitative real-time PCR analysis of eluted DNA. Relative promoter occupancy over the % input is shown in the form of a bar diagram. All the experiments were repeated at least 3 times a representative result is shown.
Overexpression of Ash2L enhances the P53 pro–apoptotic response
Ash2L functions along with Wdr5 and RbBP5 to form a sub-module of the Setd1A, Setd1B, and MLL methyltransferase complexes 23. The Ash2L, RbBP5, Wdr5, DPY30 sub-module has been shown to possess an intrinsic methyltransferase activity toward histone H3K4 in vitro independent of an MLL, although none of these proteins contain a catalytic SET domain 25. Furthermore, depletion of Ash2L in cell lines leads to globally decreased levels of H3K4me3, whereas levels of H3K4 methylation remain unchanged in the absence of MLL 21, 23, 26. Furthermore, MLL complexes reconstituted in vitro, but lacking Ash2L exhibit reduced di– and trimethyl transferase activity towards recombinant histone H3 21, 25, 27, suggesting that Ash2L is an important contributor in establishing as well as maintaining the H3K4me3 histone mark on the promoters.
Since we observed enrichment of H3K4me3 as well as increased occupancy of Ash2L on the P53 pro–apoptotic target promoters in response to P53 induction, and since Ash2L is essential in maintaining H3K4me3, we next investigated the role of Ash2L in P53–dependent transactivation of its pro-apoptotic target genes and induction of apoptosis. Both HCT116 and B5/589 cells overexpressing Ash2L showed an increased pro–apoptotic response in comparison to their vector controls as analyzed by PI (propidium iodide) staining (Fig. 2a; Supplementary Fig. 6a) following treatment with etoposide, a topoisomerase II inhibitor 28, or paclitaxel, which interferes with the normal breakdown of microtubules during cell division 29. The increased apoptotic response observed in Ash2L overexpressing cells was associated with increased P53 pro–apoptotic target gene expression, as measured at mRNA levels in B5/589 and HCT116 cells treated with increasing doses of etoposide and Nutlin3a respectively (Fig. 2b; Supplementary Fig. 6b) and as measured at protein levels in B5/589 cells treated with increasing doses of etoposide (Fig. 2c). These effects correlated well with enhanced P53 pro–apoptotic promoter occupancy, as demonstrated in B5/589 cells treated with MI–219 (Fig. 2d) and HCT116 cells treated with Nutlin3a (Supplementary Fig. 7a), associated in each case with further enrichment of the H3K4me3 mark on the TSS of these same P53 pro-apoptotic target promoters (Fig. 2e; Supplementary Fig. 7b).
Figure 2. Ash2L overexpression enhances P53–dependent apoptosis by enhancing P53–promoter occupancy.
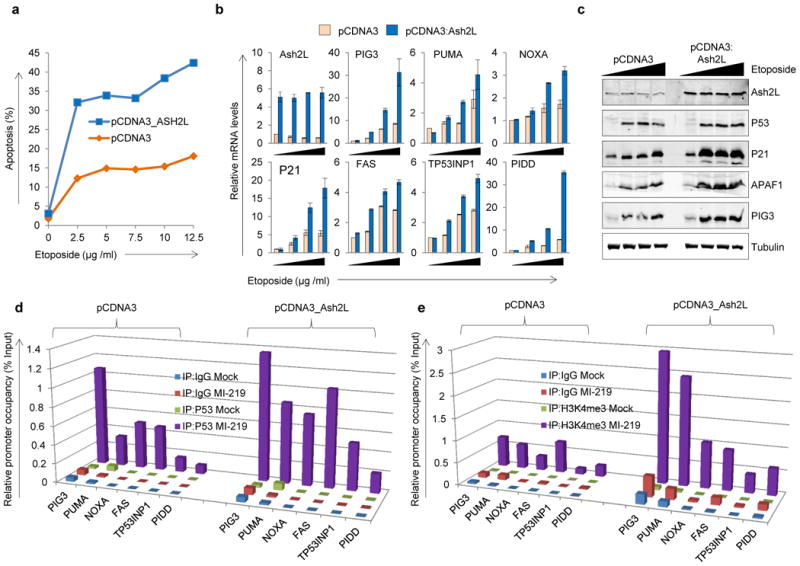
a, Propidium iodide (PI) staining of HCT116 cells overexpressing Ash2L and treated for 48 h with indicated doses of etoposide. The % of cells undergoing apoptosis (less than 2N content of DNA) is shown in the form of a line diagram. b, Real-time quantitative PCR of B5/589 cells overexpressing Ash2L and treated with 0, 2.5, 5 or 7.5 μg / ml of etoposide for 48 h. mRNA expression levels are normalized to relative levels of 18S RNA. c, Western blot analysis of B5/589 cells overexpressing Ash2L and treated with 0, 2.5, 5 or 7.5 μg / ml of etoposide for 48 h. Tubulin blot was used as a lysate control. d, e, ChIP analysis showing P53 occupancy (d) and H3K4me3 enrichment (e) on P53 target promoters in B5/589 control or Ash2L overexpressing cells and treated with MI–219 for 16 h. The target sequences were detected by quantitative real-time PCR analysis of eluted DNA. Relative promoter occupancy over the % input is shown in the form of bar diagram. All the experiments were repeated at least 3 times and a representative experimental result is shown.
Ash2L silencing decreases P53–dependent transcription without altering P53 promoter occupancy
To assess the impact of Ash2L silencing on P53–dependent pro–apoptotic target gene expression, we generated B5/589 and HCT116 cells stably expressing doxycycline–inducible Ash2L shRNA utilizing 2 independent shRNA's. shRNA targeting GFP was used as a negative control in all the experiments to rule out non-specific shRNA effects. Doxycycline addition to the culture medium led to a decrease in Ash2L expression in sh-Ash2L but not in sh-GFP cells, as measured in both the cell lines at mRNA (Fig. 3a, Supplementary Fig. 8a) and protein levels (Fig. 3b, Supplementary Fig. 8b). Ash2L silencing by itself did not affect the basal expression of P53 or its pro–apoptotic target genes as measured at both mRNA and protein levels (Fig. 3a, b, Supplementary Fig. 8a, b). Moreover, addition of MI–219 to B5/589, or etoposide treatment of HCT116 cells stabilized similar amounts of P53, irrespective of whether Ash2L was pre–silenced or not (Fig. 3b; Supplementary Fig. 8b). But to our surprise, stabilized P53 protein was less efficient in transactivating its target genes in Ash2L pre–silenced cells, as measured at both mRNA (Fig. 3a; Supplementary Fig. 8a) and protein levels (Fig. 3b; Supplementary Fig. 8b). Further, this reduced P53–transactivation ability correlated with diminished P53–dependent apoptosis, as measured in HCT116 cells treated with increasing doses of etoposide (Fig. 3c) as well as in B5/589 cells treated with increasing paclitaxol concentration (Supplementary Fig. 8c). Of note, we did not detect a significant decrease in P53 pro–apoptotic promoter occupancy in Ash2L pre–silenced cells, as measured in B5/589 cells treated with MI–219 (Fig. 4a) or HCT116 cells treated with Nutlin3a (data not shown). Confirming previous studies indicating that Ash2L is important for H3K4 methylation 21, 25, 27, we observed reduced H3K4me3 enrichment at the TSS of these promoters in response to P53 induction with Ash2L pre–silencing, in MI–219 or Nutlin3a induced B5/589 and HCT116 cells, respectively (Fig. 4b; data not shown). Thus, reduced P53 pro–apoptotic target gene expression correlated with the reduced levels of H3K4me3 enrichment at the TSS of P53 pro–apoptotic target promoters rather than with the level of P53 recruitment to these same promoters. Under these same conditions, we did not observe any significant reduction in Wdr5 promoter occupancy on any of the P53 target promoters analyzed (Supplementary Fig. 9). We observed reduced P21 transcript levels under these conditions (Fig. 3a,b; Supplementary Fig. 8a), but failed to detect a reduced cell cycle arrest, consistent with our previous findings that a low level of P21 is sufficient to induce efficient cell cycle arrest 8.
Figure 3. Ash2L silencing decreases P53–dependent target gene expression and apoptosis.
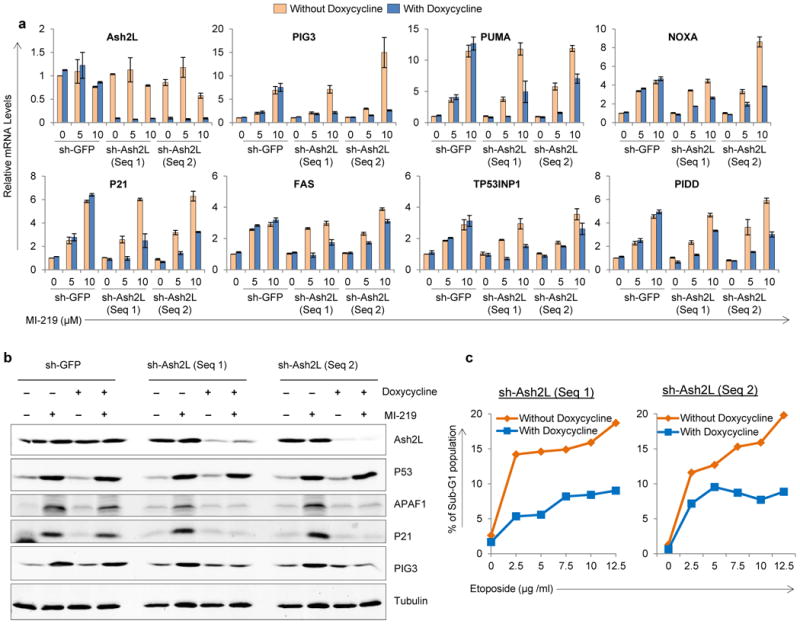
a, Real-time quantitative PCR analysis of B5/589 cells stably transduced with inducible sh-GFP or sh–Ash2L (2 sequences) and cultured in the presence of doxycycline for 48 h followed by either treating cells with 0, 5 and 10 μM of MI–219 for another 16 h. mRNA expression levels are normalized to relative levels of 18S RNA. The expression of sh-GFP untreated sample was normalized to one and all the other values are shown relative to this. b, Western blot analysis of B5/589 cells stably transduced with inducible sh-GFP or sh–Ash2L (2 sequences) and cultured in the presence of doxycycline for 48 h followed by treating cells with 10 μM of MI–219 for another 16 h. Tubulin blot was used as a lysate control. c, Propidium iodide (PI) staining in HCT116 cells stably transduced with inducible sh–Ash2L (2 sequences). The cells were cultured in the presence of doxycycline for 48 h followed by treatment with increasing doses of etoposide for another 48 h.
Figure 4. Ash2L silencing does not alter P53 promoter occupancy.
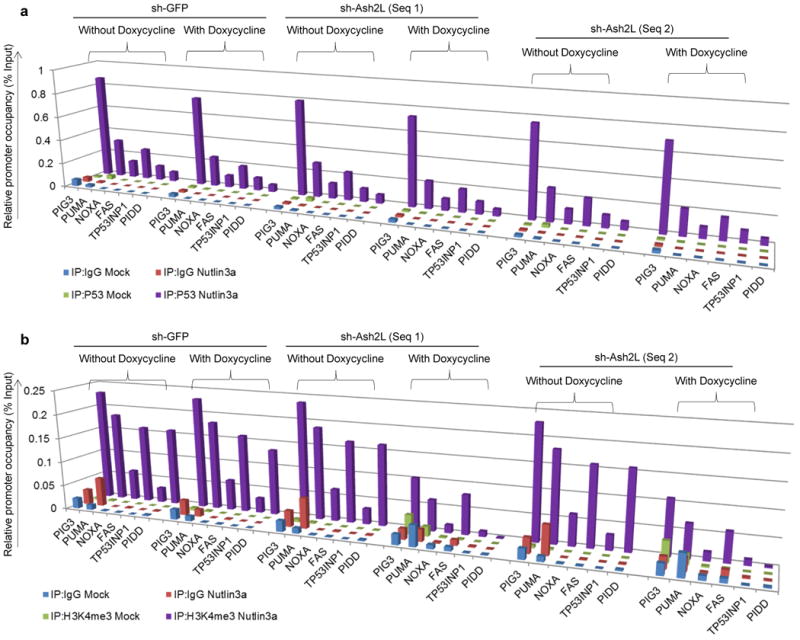
a, b, ChIP analysis showing P53 occupancy (a) and H3K4me3 enrichment (b) on P53 pro–apoptotic target promoters in B5/589 pre–silenced for either GFP (non-specific control) or Ash2L (2 independent sequences) expression. The cells were cultured in the presence of doxycycline for 48 h followed by MI–219 treatment for another 16 h. The target sequences were detected by quantitative real-time PCR analysis of eluted DNA. Relative promoter occupancy over the % input is shown in the form of bar diagram. All the experiments were repeated at least 3 times and a representative experimental result is shown.
Ash2L pre–silencing inhibits RNAP II initiation of P53–dependent pro–apoptotic gene transcription
In eukaryotes, all mRNA (messenger RNA) as well as some snRNA (small nuclear RNA) and snoRNA (small nucleolar RNA) are transcribed by RNAP II 30. Since we observed reduced P53–dependent expression of its target genes in Ash2L pre-silenced cells, we investigated RNAP II occupancy on the TSS of the P53 pro–apoptotic target promoters in response to P53 induction. As shown previously 31, we observed RNAP II occupancy on P21 and MDM2 TSS both in the absence and presence of P53 induction (data not shown). However, P53 pro–apoptotic target promoters, PIG3, PUMA, PIDD and FAS (CD95/APO–1), showed RNAP II occupancy only in response to P53 induction as observed in B5/589 cells treated with MI–219 (Fig. 5a) or HCT116 cells treated with Nutlin3a (Supplementary Fig. 10a). Of note, RNAP II occupancy was comparable in each case in control or Ash2L pre–silenced cells (Fig. 5a; Supplementary Fig. 10a), suggesting that Ash2L silencing did not act by altering RNAP II recruitment on to the TSS of these P53 pro–apoptotic target promoters.
Rpb1 is the largest subunit of the RNAP II and the CTD (c–terminal domain) of mammalian Rpb1 contains 52 multiple tandemly repeated heptapeptides with a consensus sequence T1S2P3T4S5P6S7 30. Post Translational Modifications (PTMs) that occur on the CTD, generate distinct information that is crucial at different steps of the transcription cycle 30. These include phosphorylation of serine, tyrosine and threonine residues, cis–trans isomerization of the two proline residues, as well as more recently identified arginine methylation 32 and serine / threonine glycosylation 33. CTD serine–5 phosphorylation occurs at the beginning of the transcription cycle and decreases towards the end, whereas serine–2 and serine–7 phosphorylation's are high towards the end of the transcription unit 34. Further, serine–5 CTD phosphorylation has been shown to be a mark for transcription initiation and promoter clearance 35. Since we observed reduced P53–dependent expression of its pro–apoptotic targets without any detectable changes in either P53 or RNAP II occupancy in response P53 induction in Ash2L pre–silenced cells, we next measured levels of ser5–CTD at each of these promoters. Whereas we observed ser5–CTD enrichment at the TSS of P53 pro–apoptotic target genes in response to P53 induction in control cells, Ash2L pre–silencing completely abrogated ser5–CTD occupancy under the same conditions in B5/589 cells treated with MI–219 (Fig. 5b) and in HCT116 cells treated with Nutlin3a (Supplementary Fig. 10b). There were no detectable changes in total cellular protein expression levels of either total RNAP II or ser5–CTD under any of these conditions (Supplementary Fig. 11). All of these results indicated that Ash2L is not essential for either P53 or RNAP II recruitment on to P53 pro–apoptotic target promoters but is absolutely required for RNAP II ser5–CTD phosphorylation on these promoters.
Ash2L is essential for stable pre–initiation complex formation on P53 pro–apoptotic target promoters in response to P53 induction
Present understanding of transcriptional initiation suggests that RNAP II does not directly recognize its target promoters and employs a host of GTFs in order to perform this function 36. We did not detect any changes in cellular TFIIF, TFIIB or TAF1 protein expression with or without P53 induction, both in the presence or absence of Ash2L pre–silencing (Supplementary Fig. 11). Recent studies have also suggested that P53–dependent transcription initiation is distinct from transcription of housekeeping genes 37. Thus, we compared GTFs occupancy on P53 pro–apoptotic target promoters in Ash2L pre-silenced cells with or without P53 induction. In the absence of P53 induction, we failed to detect TFIIB and TFIIF occupancy on P53 pro–apoptotic promoters in both B5/589 and HCT116 cells. However, P53 induction in response to MI–219 or Nutlin3a treatment in B5/589 and HCT116 cells respectively, led to P53 pro–apoptotic promoter occupancy by both TFIIB and TFIIF (Fig. 6a,b; Supplementary Fig. 12a,b). Of note, these same GTFs failed to be recruited on these same promoters in response to P53 induction in these cells pre–silenced for Ash2L under the same conditions (Fig. 6a,b; Supplementary Fig. 12a,b). All of these results strongly suggest that Ash2L pre–silencing inhibits P53–dependent expression of its pro–apoptotic target genes by blocking a stable PIC formation, which is required for RNAP II ser5–CTD phosphorylation and activation.
Figure 6. Ash2L is required for stable PIC formation on P53 pro–apoptotic target promoters, in response to P53 induction.
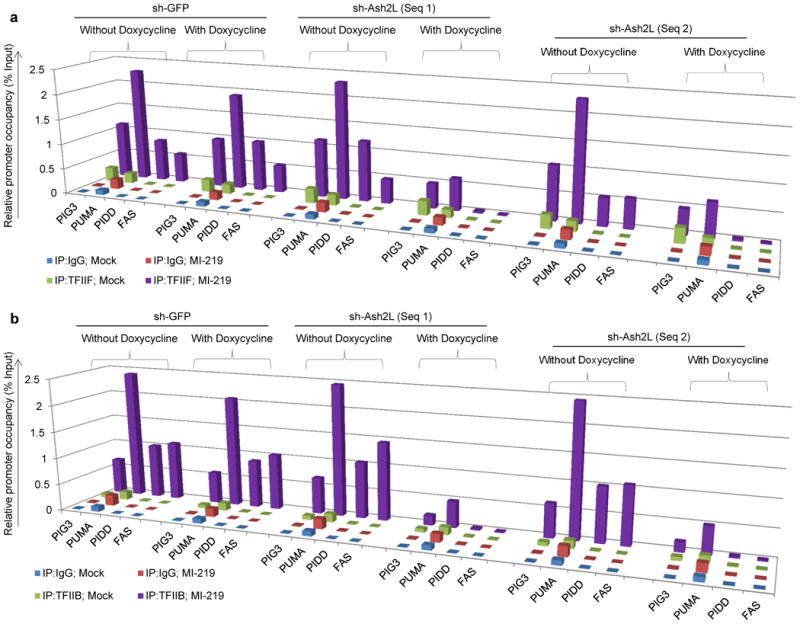
a, b, ChIP analysis showing occupancy of TFIIB (a) and TFIIF (b) on P53 pro–apoptotic target promoters in B5/589 cells pre–silenced for either GFP (non-specific control) or Ash2L (2 independent sequences) expression. The cells were cultured in the presence of doxycycline for 48 h followed by MI–219 treatment for another 16 h. The target sequences were detected by quantitative real-time PCR analysis of eluted DNA. Relative promoter occupancy over the % input is shown in the form of a bar diagram. All the experiments were repeated at least 3 times a representative result is shown.
Discussion
H3K9me3 and H3K4me3 histone marks signify inactive and active transcription, respectively 2-4. Previous studies have shown that both P53 pro-arrest and pro-apoptotic target promoters are enriched with H3K9me3 repressive histone conformation in unstressed cells 8, 38, 39, and that activated P53 overcomes this repressive chromatin conformation barrier by down-regulating SUV39H1 8, the writer of this mark 4. While SUV39H1 down regulation was alone sufficient to reduce the H3K9me3 mark on both P53 pro–arrest and pro–apoptotic target promoters 8, this resulted in increased expression of P21, but not of pro–apoptotic target genes.
In the present study, we observed that in response to SUV39H1 silencing alone, only p21, but none of the P53 pro-apoptotic promoters exhibited the H3K4me3 active histone mark enrichment, even though H3K9me3 was lost on all of these promoters 8. In case of the p21 promoter, SUV39H1 silencing alone led to H3K4me3 enrichment even in HCT116 P53 -/- cells (data not shown), indicating that P53 presence and /or occupancy was not required for enrichment of this mark. These results indicate that loss of the repressive chromatin conformation (H3K9me3) by itself is sufficient for H3K4me3 enrichment and P21 induction, while the activation of P53 pro-apoptotic targets is more complex and requires additional steps. In the present study, we focused our attention on how P53 promotes H3K4me3 enrichment on its pro-apoptotic promoters and the role of H3K4me3 reader, Ash2L, in modulating the P53 pro-apoptotic response.
All known H3K4 methyl transferase complexes contain four common structural components Wdr5, RbBP5, DPY30 and Ash2L, in addition to a catalytic subunit 21-24. In agreement with our H3K4me3 ChIP data, we demonstrated that Ash2L, as well as Wdr5 and RbBP5 were recruited to P53 pro-apoptotic promoters specifically in response to P53 induction. We observed further that cells overexpressing Ash2L exhibited increased P53 pro-apoptotic promoter occupancy and target gene expression associated with an enhanced P53 apoptotic response. In addition, such cells showed enhanced H3K4me3 enrichment on these same P53 pro–apoptotic target promoters in response to P53 induction. In contrast, upon Ash2L pre-silencing, P53 induction resulted in complete abrogation of H3K4me3 mark and reduced P53 pro–apoptotic gene expression without affecting P53 occupancy on these same promoters. Under the same conditions, we also did not observe any reduction in Wdr5 promoter occupancy, which is another critical component of the H3K4me3 methylation complex. Thus Ash2L is not essential for Wdr5 promoter recruitment, but is required for efficient H3K4 trimethylation of P53 target pro-apoptotic promoters. These findings also argue that Ash2L-dependent H3K4me3 enrichment is not required for P53 to occupy its pro-apoptotic promoters, but its enrichment favors P53 promoter occupancy.
The process of transcription involves initiation, elongation and termination. In Drosophila, it has been shown that H3K4me3 is responsible for the generation of a chromatin structure at active promoters that ensures optimal RNAP II release into productive elongation 40. The Rpb1 CTD undergoes extensive PTMs and has important functions in each of these processes 30, 34, 41. In mammals, this CTD consists of 52 T1S2P3T4S5P6S7 consensus repeats. RNAP II is unable to recognize its target promoters directly and employs a host of GTFs in order to perform this function 36. Initiation of transcription starts with hypo–phosphorylated CTD that assembles with GTFs including TFIIA, TFIIB, TFIID, TFIIE, TFIIF and TFIIH at the promoter 36. Lauberth et al reported that H3K4me3 interacts with TAF3 and directs global TFIID recruitment to active genes, some of which are p53 targets 42. We observed that recruitment of the total RNAP II in response to P53 induction, like P53 itself, was not affected by Ash2L pre-silencing. However, under the same conditions, P53 pro–apoptotic target promoter occupancy by TFIIB and TFIIF was abrogated in response to Ash2L pre–silencing. These results indicate that RNAP II does not require these GTFs (TFIIB and TFIIF) for its recruitment on P53 pro–apoptotic target promoters in response to P53 induction and is independent of H3K4me3 mark enrichment as well.
Many studies have suggested that TFIIB and TFIIF complexes need to bind to the core promoter prior to RNAP II recruitment 36. In fact, TFIIB plays a central role in transcription complex assembly at gene promoters as well as in the events that lead to transcription initiation 43. While, serine–65 phosphorylation of TFIIB is essential for productive transcription of several housekeeping genes 44, it has been recently shown that TFIIB becomes dephosphorylated in response to genotoxic stress leading to general transcription attenuation, but favoring P53 target gene transcription 37. These findings suggest that P53's requirement for general transcription factors differs from that of other housekeeping genes. Further, there are reports suggesting that TFIIF is not required for transcription initiation, but is essential to stabilize TFIIB required in early elongation 45. Our findings that RNAP II recruitment on P53–target promoters occurs without TFIIB and TFIIF occupancy are consistent with the concept that the mechanism of PIC formation is different on P53 pro–apoptotic target promoters analyzed from that of typical housekeeping genes. P53 and Ash2L have been reported to form a protein complex 9. Several studies have implicated P300 as an essential coactivator of P53 46-49, and P300 has been reported to act synergistically with the H3K4me3 methyl transferase, Set1, to transcriptionally activate P53 target genes 20. In contrast, Kasper et al. reported that CBP and p300 are dispensable for p53 to transactivate p21 and mdm2 promoters 50. Of note, Ash2L has been shown to physically interact with P300 20, and P300 has been established to link P53 to the RNAPII holo-enzyme 51. Thus, our results support the concept that Ash2L bridges P53 promoter occupancy with PIC complex assembly in concert with P300.
P53 is a key controller of cell death by regulating the transcription of a number of pro–apoptotic genes 5, 52, 53. Many cancer therapies exploit the P53 pathway to suppress cancer cell growth by stimulating P53–mediated apoptosis 54, and chromatin conformation plays a major role in transcription 55, 56. We utilized a tet-inducible shRNA-mediated approach to knock-down Ash2L expression to evaluate its role in P53 pro-apoptotic gene induction. The tet system provides very low basal expression and high maximal expression after induction, making it suitable for efficient knock-down of gene expression. However, this system can exhibit leaky expression and/or toxicity if high tetracycline levels are used. We utilized rtTA based induction, which is known to overcome such problems 57. Finally, it is conceivable that rtTA might lead to non-desired gene expression. We utilized tet-inducible sh-GFP as a control to rule out such possibilities. Different cell lines and methods of p53 induction were used by us to propose a model (Fig. 7) in which P53 activation leads to occupancy of components of the H3K4me3 complex and enrichment of the H3K4me3 mark on its pro–apoptotic target promoters. Further, Ash2L facilitates the recruitment of basal transcriptional machinery including TFIIB and TFIIF on P53 pro–apoptotic target promoters in response to P53 induction. While RNAP II occupies these same pro–apoptotic promoters in response to P53 induction irrespective of the Ash2L occupancy, Ash2L is absolutely required for serine5–CTD phosphorylation and the initiation of transcription, perhaps through modulation of recruitment of other transcription cofactors. Taken together, our findings indicate that Ash2L functions to bridge P53 pro–apoptotic promoter occupancy with RNAP II activation by favoring stable PIC formation. While we analyzed H3K4me3 enrichment as well as its reader's occupancy on P53 target pro-apoptotic promoters in response to P53 induction, P53 has other classes of transcriptional targets, including down regulated (e.g., Arf), metabolic (e.g., TIGAR, GLS2), DNA-damage repair (e.g., TRIM22, Gadd45) as well as microRNA genes (e.g., miR125b, miR-34) 52, 53. Whether similar chromatin modulators regulate P53's transactivation of these sets of gene targets is not yet known and will be of interest for future studies.
Figure 7. Role of Ash2L in P53–dependent pro–apoptotic target gene expression.
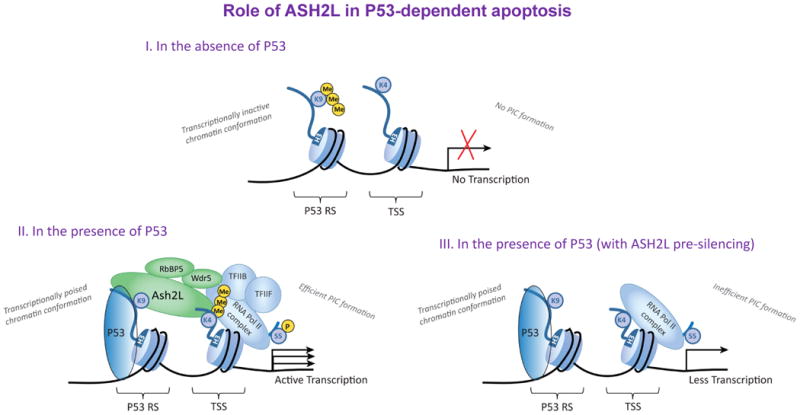
Schematic diagram illustrating the role of Ash2L in P53–dependent pro–apoptotic target gene expression. I) In the absence of P53 induction, P53 RS (response sequence) is enriched with the H3K9me3 repressive mark, and the TSS (transcription start site) lacks detectable H3K4me3, indicating non–permissive chromatin conformation for active transcription. II) In response to P53 induction, H3K9me3 is lost on the P53 RS in a SUV39H1–dependent manner8, and the TSS acquires the H3K4me3 mark and its readers, Wdr5, RbBP5 and Ash2L. RNAP II is also recruited on to the promoter and becomes ser5–CTD phosphorylated in response to P53 induction, leading to efficient PIC (pre–initiation complex) formation. III) In Ash2L pre–silenced cells, P53 induction results P53 and RNAP II recruitment on to the promoter similar to that of non–silenced cells, but there is no detectable H3K4me3 enrichment or occupancy by its readers at the TSS. Further, RNAP II ser5–CTD phosphorylation is compromised in Ash2L pre–silenced cells, leading to inefficient PIC formation. Ash2L thereby acts in concert with P53 promoter occupancy to activate RNAP II by aiding formation of a stable transcription pre–initiation complex required for its activation.
Materials and Methods
Plasmids, Cell Lines and Treatments
The Tet–pLKO puro vector expressing Ash2L shRNA was generated by cloning the shRNA sequences into the Tet–inducible pLKO.1 vector was obtained from Addgene (#21915). The sequences of shRNA's used will be provided upon request. HCT116 cells stably expressing doxycycline–inducible shSUV39H1 was described previously 8. A full length cDNA construct expressing Ash2L were procured from Open Biosystems (#MHS4768–99609425) and cloned into pCDNA3 vector using EcoRI and XhoI enzymes. B5/589 immortalized breast epithelial cells and HCT116 colon carcinoma cells were cultured in RPMI–1640 containing 10 % FBS and 5 ng ml–1 of human EGF and DMEM containing 10 % FBS respectively, at 37 °C. EJ–P53 bladder carcinoma cells that express tetracycline–regulatable P53 was cultured in DMEM containing 10 % FBS, 750 μg ml–1 G418, 100 μg ml–1 hygromycin and 1 μg ml–1 tetracycline 58. Nutlin3a (Cayman chemicals #10004372; Ann Arbor, MI, USA) and MI–219 59 were used for 16 h at a concentration of 20 μM and 10 μM, respectively 59, 60. Etoposide was purchased from Sigma, St. Louis, MO, USA; (#E1383), and used at the indicated concentrations for 48 h.
RNA Extraction and Real–time PCR analysis
Total RNA was extracted from cultured cells by Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. First –strand cDNA synthesis was performed using Superscript II RT (Invitrogen), and SYBR Green (Roche; #04887352001)–based quantitative PCR was carried out using gene–specific primer sets. 18S gene specific primers were used as an internal control and all the mRNA expression levels were normalized to 18S expression levels. Sequences of the primers used for real–time PCR are shown in supplementary table.
Western blotting and Flow Cytometry
Western blotting was performed as described previously 8, 61, with the exception that Alexa Fluor secondary antibodies were used. All blots were developed using the Odyssey fluorescence image scanner and the band intensities were quantified using LI–COR software. Cell cycle analysis was performed using PI (propidium iodide) staining as described previously 8, using the CycleTEST PLUS DNA reagent kit (BD Biosciences, San Jose, CA, USA; # 340242). The following antibodies were used in the study: anti–P53 (SCBT; sc–6243), anti–P21 (BD; #556431), anti–APAF1 (R & D; #MAB828), anti–PIG3 (abcam; #ab64798), anti–PUMA (CST; #4976); anti–Ash2L (Bethyl Labs; #A300-108A), anti–H3K4me3 (abcam; #ab1012), anti–Wdr5 (abcam; #ab56919); anti–RbBP5 (Bethyl Labs; #A300-109A), anti–RNAP II (SCBT; sc–899); anti–Serine5–CTD (abcam; #ab5131–50); anti–TAFII (SCBT; sc–735); anti–TFIIB (SCBT; sc–225); anti–TFIIF (SCBT; sc–235); anti–Tubulin (Sigma; #T5168), Alexa Fluor 680 goat anti–mouse IgG (Molecular Probes; #A21057) and Alexa Fluor goat anti–rabbit IgG (Molecular Probes; #A21076).
Quantitative Chromatin Immunoprecipitation (ChIP) Assay
ChIP assay was performed as described previously 8. 2.5 X 106 cells were taken for ChIP assay. The whole cell extract was pre–cleared using 60 μl of Protein A/G PLUS – agarose beads (SCBT; sc–2003) and incubated overnight with anti–P53, anti–H3K4me3, anti–Wdr5, anti–RbBP5, anti–Ash2L or control IgG antibodies. 1 μg of antibody was used for 100 μg of total lysate. Protein G agarose beads pre–blocked with salmon sperm DNA (to reduce non–specific DNA binding) was purchased from Millipore, Saint Charles, MO, USA (#16–2001) and 60 μl of this solution was used for each immunoprecipitation. 10 % of chromatin from each sample was removed before immunoprecipitation and used for PCR amplification (input). Both Immunoprecipitated and whole cell extract (input) were treated with RNaseA, proteinase K and DNA were purified using DNeasy Blood and Tissue kit (Qiagen, Valencia, CA, USA; #69504). Quantitative real–time PCR was performed on this DNA to identify the amount of target sequence. The list of primers used is given in supplementary table.
Generation of stable cell lines
B5/589, HCT116 cells were infected with Tet–pLKO–shAsh2L lentivirus (both sequence 1 and sequence 2) and selected for puromycin (2 μg ml–1) resistance. Stable puromycin resistant clones were pooled to generate doxycycline–inducible shRNA cell lines. Ash2L overexpressing clones were generated in HCT116 and B5/589 cells by transfecting cells with pCDNA3–Ash2L (under the control of the CMV promoter) or by infecting with VIRSP–Ash2L (under the control of the mPGK promoter) and selecting with G418 (800 μg ml–1) or puromycin (2 μg ml–1) respectively. Stable resistant cells were pooled and used for the experiments.
Supplementary Material
Acknowledgments
The authors would like to thank S. W. Lee (MGH, Harvard) for helpful discussions. This research was supported by grants from the National Cancer Institute (P01CA080058) and Breast Cancer Research Foundation (SAA). The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the US National Institutes of Health.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 2.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 3.Schmid CD, Bucher P. ChIP-Seq data reveal nucleosome architecture of human promoters. Cell. 2007;131:831–832. doi: 10.1016/j.cell.2007.11.017. author reply 832-833. [DOI] [PubMed] [Google Scholar]
- 4.Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 5.Beckerman R, Prives C. Transcriptional regulation by p53. Cold Spring Harb Perspect Biol. 2010;2:a000935. doi: 10.1101/cshperspect.a000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laptenko O, Prives C. Transcriptional regulation by p53: one protein, many possibilities. Cell Death Differ. 2006;13:951–961. doi: 10.1038/sj.cdd.4401916. [DOI] [PubMed] [Google Scholar]
- 7.Mandinova A, Lee SW. The p53 pathway as a target in cancer therapeutics: obstacles and promise. Sci Transl Med. 2011;3:64–61. doi: 10.1126/scitranslmed.3001366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mungamuri SK, Benson EK, Wang S, Gu W, Lee SW, Aaronson SA. p53-mediated heterochromatin reorganization regulates its cell fate decisions. Nat Struct Mol Biol. 2012;19:478–484. S471. doi: 10.1038/nsmb.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee J, Kim DH, Lee S, Yang QH, Lee DK, Lee SK, et al. A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4. PNAS. 2009;106:8513–8518. doi: 10.1073/pnas.0902873106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 11.Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. 2012;81:65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vermeulen M, Timmers HT. Grasping trimethylation of histone H3 at lysine 4. Epigenomics. 2010;2:395–406. doi: 10.2217/epi.10.11. [DOI] [PubMed] [Google Scholar]
- 13.Bieging KT, Attardi LD. Deconstructing p53 transcriptional networks in tumor suppression. Trends Cell Biol. 2011 doi: 10.1016/j.tcb.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 15.Bernstein BE, Humphrey EL, Erlich RL, Schneider R, Bouman P, Liu JS, et al. Methylation of histone H3 Lys 4 in coding regions of active genes. PNAS. 2002;99:8695–8700. doi: 10.1073/pnas.082249499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim TH, Barrera LO, Zheng M, Qu C, Singer MA, Richmond TA, et al. A high-resolution map of active promoters in the human genome. Nature. 2005;436:876–880. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roh TY, Cuddapah S, Zhao K. Active chromatin domains are defined by acetylation islands revealed by genome-wide mapping. Genes Dev. 2005;19:542–552. doi: 10.1101/gad.1272505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roh TY, Cuddapah S, Cui K, Zhao K. The genomic landscape of histone modifications in human T cells. PNAS. 2006;103:15782–15787. doi: 10.1073/pnas.0607617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pekowska A, Benoukraf T, Zacarias-Cabeza J, Belhocine M, Koch F, Holota H, et al. H3K4 tri-methylation provides an epigenetic signature of active enhancers. EMBO J. 2011 doi: 10.1038/emboj.2011.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang Z, Chen WY, Shimada M, Nguyen UT, Kim J, Sun XJ, et al. SET1 and p300 act synergistically, through coupled histone modifications, in transcriptional activation by p53. Cell. 2013;154:297–310. doi: 10.1016/j.cell.2013.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, et al. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13:713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- 22.Han Z, Guo L, Wang H, Shen Y, Deng XW, Chai J. Structural basis for the specific recognition of methylated histone H3 lysine 4 by the WD-40 protein WDR5. Molecular cell. 2006;22:137–144. doi: 10.1016/j.molcel.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 23.Steward MM, Lee JS, O'Donovan A, Wyatt M, Bernstein BE, Shilatifard A. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nature structural & molecular biology. 2006;13:852–854. doi: 10.1038/nsmb1131. [DOI] [PubMed] [Google Scholar]
- 24.Trievel RC, Shilatifard A. WDR5, a complexed protein. Nature structural & molecular biology. 2009;16:678–680. doi: 10.1038/nsmb0709-678. [DOI] [PubMed] [Google Scholar]
- 25.Patel A, Dharmarajan V, Vought VE, Cosgrove MS. On the mechanism of multiple lysine methylation by the human mixed lineage leukemia protein-1 (MLL1) core complex. J Biol Chem. 2009;284:24242–24256. doi: 10.1074/jbc.M109.014498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ng HH, Robert F, Young RA, Struhl K. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell. 2003;11:709–719. doi: 10.1016/s1097-2765(03)00092-3. [DOI] [PubMed] [Google Scholar]
- 27.Southall SM, Wong PS, Odho Z, Roe SM, Wilson JR. Structural basis for the requirement of additional factors for MLL1 SET domain activity and recognition of epigenetic marks. Mol Cell. 2009;33:181–191. doi: 10.1016/j.molcel.2008.12.029. [DOI] [PubMed] [Google Scholar]
- 28.Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol. 2010;17:421–433. doi: 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fu Y, Li S, Zu Y, Yang G, Yang Z, Luo M, et al. Medicinal chemistry of paclitaxel and its analogues. Curr Med Chem. 2009;16:3966–3985. doi: 10.2174/092986709789352277. [DOI] [PubMed] [Google Scholar]
- 30.Egloff S, Dienstbier M, Murphy S. Updating the RNA polymerase CTD code: adding gene-specific layers. Trends Genet. 2012;28:333–341. doi: 10.1016/j.tig.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 31.Gomes NP, Espinosa JM. Differential regulation of p53 target genes: it's (core promoter) elementary. Genes Dev. 2010;24:111–114. doi: 10.1101/gad.1893610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sims RJ, 3rd, Rojas LA, Beck D, Bonasio R, Schuller R, Drury WJ, 3rd, et al. The C-terminal domain of RNA polymerase II is modified by site-specific methylation. Science. 2011;332:99–103. doi: 10.1126/science.1202663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang DW, Rodriguez-Molina JB, Tietjen JR, Nemec CM, Ansari AZ. Emerging Views on the CTD Code. Genet Res Int. 2012;2012:347214. doi: 10.1155/2012/347214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Egloff S, Murphy S. Cracking the RNA polymerase II CTD code. Trends Genet. 2008;24:280–288. doi: 10.1016/j.tig.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 35.Bartkowiak B, Greenleaf AL. Phosphorylation of RNAPII: To P-TEFb or not to P-TEFb? Transcription. 2011;2:115–119. doi: 10.4161/trns.2.3.15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas MC, Chiang CM. The general transcription machinery and general cofactors. Crit Rev Biochem Mol Biol. 2006;41:105–178. doi: 10.1080/10409230600648736. [DOI] [PubMed] [Google Scholar]
- 37.Shandilya J, Wang Y, Roberts SG. TFIIB dephosphorylation links transcription inhibition with the p53-dependent DNA damage response. PNAS. 2012;109:18797–18802. doi: 10.1073/pnas.1207483109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng H, Chen L, Pledger WJ, Fang J, Chen J. p53 promotes repair of heterochromatin DNA by regulating JMJD2b and SUV39H1 expression. Oncogene. 2013 doi: 10.1038/onc.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bosch-Presegue L, Raurell-Vila H, Marazuela-Duque A, Kane-Goldsmith N, Valle A, Oliver J, et al. Stabilization of Suv39H1 by SirT1 Is Part of Oxidative Stress Response and Ensures Genome Protection. Molecular cell. 2011;42:210–223. doi: 10.1016/j.molcel.2011.02.034. [DOI] [PubMed] [Google Scholar]
- 40.Ardehali MB, Mei A, Zobeck KL, Caron M, Lis JT, Kusch T. Drosophila Set1 is the major histone H3 lysine 4 trimethyltransferase with role in transcription. EMBO J. 2011;30:2817–2828. doi: 10.1038/emboj.2011.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buratowski S. The CTD code. Nat Struct Biol. 2003;10:679–680. doi: 10.1038/nsb0903-679. [DOI] [PubMed] [Google Scholar]
- 42.Lauberth SM, Nakayama T, Wu X, Ferris AL, Tang Z, Hughes SH, et al. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell. 2013;152:1021–1036. doi: 10.1016/j.cell.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deng W, Roberts SG. TFIIB and the regulation of transcription by RNA polymerase II. Chromosoma. 2007;116:417–429. doi: 10.1007/s00412-007-0113-9. [DOI] [PubMed] [Google Scholar]
- 44.Wang Y, Fairley JA, Roberts SG. Phosphorylation of TFIIB links transcription initiation and termination. Curr Biol. 2010;20:548–553. doi: 10.1016/j.cub.2010.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cabart P, Ujvari A, Pal M, Luse DS. Transcription factor TFIIF is not required for initiation by RNA polymerase II, but it is essential to stabilize transcription factor TFIIB in early elongation complexes. PNAS. 2011;108:15786–15791. doi: 10.1073/pnas.1104591108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Avantaggiati ML, Ogryzko V, Gardner K, Giordano A, Levine AS, Kelly K. Recruitment of p300/CBP in p53-dependent signal pathways. Cell. 1997;89:1175–1184. doi: 10.1016/s0092-8674(00)80304-9. [DOI] [PubMed] [Google Scholar]
- 47.Lill NL, Grossman SR, Ginsberg D, DeCaprio J, Livingston DM. Binding and modulation of p53 by p300/CBP coactivators. Nature. 1997;387:823–827. doi: 10.1038/42981. [DOI] [PubMed] [Google Scholar]
- 48.Thomas A, White E. Suppression of the p300-dependent mdm2 negative-feedback loop induces the p53 apoptotic function. Genes Dev. 1998;12:1975–1985. doi: 10.1101/gad.12.13.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grossman SR, Perez M, Kung AL, Joseph M, Mansur C, Xiao ZX, et al. p300/MDM2 complexes participate in MDM2-mediated p53 degradation. Mol Cell. 1998;2:405–415. doi: 10.1016/s1097-2765(00)80140-9. [DOI] [PubMed] [Google Scholar]
- 50.Kasper LH, Lerach S, Wang J, Wu S, Jeevan T, Brindle PK. CBP/p300 double null cells reveal effect of coactivator level and diversity on CREB transactivation. EMBO J. 2010;29:3660–3672. doi: 10.1038/emboj.2010.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- 52.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 54.Murray-Zmijewski F, Slee EA, Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev Mol Cell Biol. 2008;9:702–712. doi: 10.1038/nrm2451. [DOI] [PubMed] [Google Scholar]
- 55.Jaskelioff M, Peterson CL. Chromatin and transcription: histones continue to make their marks. Nat Cell Biol. 2003;5:395–399. doi: 10.1038/ncb0503-395. [DOI] [PubMed] [Google Scholar]
- 56.Grewal SI, Elgin SC. Transcription and RNA interference in the formation of heterochromatin. Nature. 2007;447:399–406. doi: 10.1038/nature05914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hosoda H, Miyao T, Uchida S, Sakai S, Kida S. Development of a tightly-regulated tetracycline-dependent transcriptional activator and repressor co-expression system for the strong induction of transgene expression. Cytotechnology. 2011;63:211–216. doi: 10.1007/s10616-011-9335-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sugrue MM, Shin DY, Lee SW, Aaronson SA. Wild-type p53 triggers a rapid senescence program in human tumor cells lacking functional p53. PNAS. 1997;94:9648–9653. doi: 10.1073/pnas.94.18.9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. PNAS. 2008;105:3933–3938. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 61.Mungamuri SK, Yang X, Thor AD, Somasundaram K. Survival signaling by Notch1: mammalian target of rapamycin (mTOR)-dependent inhibition of p53. Cancer Res. 2006;66:4715–4724. doi: 10.1158/0008-5472.CAN-05-3830. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.