

Abstract
The core promoter and proximal precore regions are the most complex portions of the hepatitis B virus (HBV) genome. These regions cooperatively regulate viral replication and differentially regulate the synthesis of the viral proteins E, core, and X. Multiple mutations in these regions are associated with the persistency of viral infection and the development of cirrhosis and hepatocellular carcinoma (HCC). In South Korea, nearly all HBVs are classified as HBV genotype C2; the majority of these viruses have the basal core promoter double mutation, a precore stop mutation, or both. These mutations may play a role in the alteration of viral and clinical features, and abundant and complex mutations are particularly prevalent in the core promoter and proximal precore regions. We previously demonstrated that the accumulation of ≥ 6 mutations at eight key nucleotides located in these regions (G1613A, C1653T, T1753V, A1762T, G1764A, A1846T, G1896A, and G1899A) is a useful marker to predict the development of HCC regardless of advanced liver disease. In addition, certain mutation combinations were predominant in cases with ≥ 4 mutations. In cases with ≤ 5 mutations, a low Hepatitis B e antigen titer (< 35 signal to noise ratio) was indicative of HCC risk. Viral mutation data of the single HBV genotype C2 suggest that the combined effect of the number and pattern of mutations in the core promoter and proximal precore regions is helpful in predicting HCC risk.
Keywords: Hepatitis B virus, Point mutation, Hepatitis B virus X protein, Hepatocellular carcinoma, Cancer screening
Core tip: Multiple mutations in the core promoter and proximal precore regions of the hepatitis B virus (HBV) genome are associated with hepatocellular carcinoma (HCC), but mutations predictive of outcome in chronic HBV carriers have not been distinguished. In the Korean HBV genotype C2, the number of mutations at eight key nucleotides located in these regions (G1613A, C1653T, T1753V, A1762T, G1764A, A1846T, G1896A, and G1899A) is positively correlated with HCC. In addition, some selected mutation combinations among individuals with ≥ 4 mutations are predominant in the HCC group.
NATURAL HISTORY OF CHRONIC HEPATITIS B VIRUS INFECTION
Chronic hepatitis B virus (HBV) infection increases the risk of developing liver cirrhosis and hepatocellular carcinoma (HCC)[1-3]. The natural course of HBV infection involves three clinical phases: immune tolerance, immune eradication, and recovery. The phase of HBV infection is classified based on serum aminotransferase levels and HBV DNA titer, which represent hepatitis and viral replication, respectively[1-4]. When the HBV DNA titer is greater than 2000 IU/mL, host immune mechanisms trigger the flare-up of hepatitis and regulate hepatitis activities that bridge the gap between the virus replication phase and the development of liver cirrhosis or HCC. The majority of hepatitis patients are anicteric, and the process tends to repeat until HBV loses the capability to replicate in hepatocytes[5,6]. When serum viral loads are persistently less than 350 IU/mL, the progressive loss of HBV genomic activity and the inability to stimulate a host immune response are observed, indicating that the carrier is in the recovery phase[3]. Throughout chronic HBV infection, sex and age are important host factors for predicting HCC risk[2,7].
HISTOLOGICAL VIEWPOINT OF THE OUTCOME OF CHRONIC HBV INFECTION
Hepatitis is graded based on histological analysis of necro-inflammation and fibrosis, and severe hepatitis simultaneously promotes hepatic fibrogenesis and carcinogenesis[8]. For example, serial liver biopsy data indicate that chronic hepatitis B patients with severe necro-inflammation exhibited significantly poorer morbidity and mortality compared with patients with mild necro-inflammation[6]. The histological findings are occasionally paradoxical and indicate healing or aggravation of fibrosis. In cases of chronic hepatitis B with bridging hepatic necrosis, a feature of aggressive hepatitis, patients frequently recover after the flare-up, but fibrosis or cirrhosis remains[9]. These findings suggest that the degree of fibrosis, also referred to as the fibrosis stage, potentially reflects the sequential changes associated with progressive chronic liver disease and is a more efficient indicator of prognosis than the ongoing inflammation[3,7,10].
VIROLOGICAL VIEWPOINT OF THE OUTCOME OF CHRONIC HBV INFECTION
During the active hepatitis phase, the immune response significantly inhibits viral replication while simultaneously inducing mutation of the HBV genome, including so-called “escape mutants”. HBV DNA titers in serum and hepatocytes have been associated with a less favorable course due to either poor clearance of the virus or increased virus production, whereas the long-term prognosis of patients with a low viral load is generally good[2,11-14]. However, HBV DNA titers are dynamic and depend on the type of mutation and anti-viral immunity, and are intricately connected to changes in hepatitis B surface antigen and hepatitis B e antigen (HBeAg) levels[15-18]. In particular, serum HBeAg and HBV DNA levels are closely associated with A1762T and G1764A, which are known as the basal core promoter (BCP) double mutation (A1762T/G1764A), and G1896A, the precore stop mutation; both A1762T/G1764A and G1896A are associated with e-suppressive phenotypes as well as decreased HBV genome replication[13,15,16,19-21]. The precore stop mutation synergistically modulates the influence of the BCP double mutation on HBV replication[22]. These mutations tend to increase HBV persistence[22,23]. The relationship between viral loads and hepatitis flare-ups in the immune eradication phase is not clear, and persistent infection by mutant HBV may influence the progression of chronic hepatitis[2,12-15,20,24], prompting interest in the identification of viral mutations that affect the outcome of chronic HBV infection.
HBV GENOTYPES, THE BCP DOUBLE MUTATION, AND THE PRECORE STOP MUTATION
Eight distinct genotypes of HBV have been reported (denoted A-H); each genotype includes variants with less than 8% divergence among their DNA sequences[25,26]. HBV genotypes B and C are more closely associated with the development of HCC than other genotypes[15,20,23,27] and are characterized by a higher prevalence of the BCP double mutation and the precore stop mutation[20,25,27]. Thus, genotypes B and C are apparently aggressive with respect to the development of HCC. However, in South Korea, nearly all HBV cases are genotype C2 (Ce)[28-33]. Although highly prevalent in HCC (86%), the prevalence of the precore stop mutation does not differ significantly among chronic HBV carriers with or without HCC[32,34,35]. Most isolates of HBV genotype C2 in South Korea carry the T1858 mutation[32,33], which attenuates the stability of the secondary structure of the pregenome encapsidation signal (epsilon signal). In contrast, C1858 prevents the formation of G1896A[36]. The BCP double mutation is also not a significant factor because it is present in the majority of HBV genotype C2 strains in South Korea. For instance, the BCP double mutation is identified in 93.5% of HBeAg-negative bDNA-positive patients, 94.9% of HBeAg-negative bDNA-negative patients, and 74% of HBeAg-positive patients[32]. Despite the high prevalence of G1896A and BCP double mutations, the single C2 genotype of South Korea represents an intriguing model system in which to identify viral mutations with prognostic utility. Complex mutations in the core promoter and precore regions of the HBV genome are of particular interest.
CLINICAL FEATURES OF WILD TYPE HBV GENOTYPE C2
Because the literature regarding the clinical features of wild-type HBV genotype C2 is lacking, we analyzed this genotype in comparison with three mutation types using our published raw data (n = 442)[33,37]. The selected 109 patients consisted of four groups: wild-type (I, n = 29), precore stop mutation alone (II, n = 14), BCP double mutation only (III, n = 44), and the A1762T, G1764A and G1896A triple mutation (IV, n = 22). The proportion of patients classified as group I decreased dramatically among those over 40 years of age, whereas the other groups experienced a relative increase in the proportion of individuals over 40 years of age. The proportions of HBeAg-negative patients and patients with serum HBV DNA levels < 15000 IU/mL (or 6 log copies/mL) were reduced in groups I and III compared with groups II and IV (HBeAg negative, 3.4% and 15.9% vs 57.1% and 50%, respectively; HBV DNA < 15000 IU/mL, 7.1% and 16.2% vs 33.3% and 36.4%, respectively). These results suggest that the precore stop mutation is more closely associated with the attenuation of self-replication and HBeAg production than the BCP double mutation. In group I, active hepatitis, advanced liver disease, and HCC were uncommon regardless of age compared with groups II-IV. In addition, half of the cases remained inactive for a long period (i.e., greater than 5 years). In groups I-IV, active hepatitis was noted in 44.8%, 57.1%, 72.7%, and 54.5% of patients, respectively. Advanced liver disease was noted in 6.9%, 28.6%, 22.7%, and 18.2% of patients, respectively. HCC was reported in 3.4%, 14.3%, 13.6%, and 13.6% of patients, respectively. In groups II-IV, most of the patients with advanced liver disease and/or HCC were over the age of 40. Thus, the clinical features of wild-type HBV genotype C2 conversely reflect the aggressiveness and persistency of the mutant type, and the BCP double mutation is associated with the initiation of HCC regardless of age. Nevertheless, these three types of mutations are insufficient as viral markers for outcome prediction because their capacity to discriminate between high and low risk of HCC is minimal. However, other mutations are likely important, particularly among the BCP mutant type HBVs, and the potential combinations of mutations are abundant and complex.
KEY MUTATIONS IN THE CORE PROMOTER AND PROXIMAL PRECORE REGIONS OF THE HBV GENOME
The core promoter overlaps the distal part of the X gene, and the proximal precore includes the epsilon signal[38-40]. These two genetically distinct regions are the most complex portion of the HBV genome, which includes various functional gene clusters, such as enhancer II, the basal core promoter, the X-termination signal, two pregenomic RNA start points, the poly A signal, epsilon, and other important sequences[38-40]. These regions differentially regulate the synthesis of pregenomic and pre-C mRNAs of HBV and the production of HBeAg and hepatitis B core antigen (HBcAg), and co-operatively regulate viral replication[39-42]. Any single mutation can induce some form of inherent change that affects viral loads and the levels of HBeAg in serum and HBcAg and X protein in hepatocytes. These effects can subsequently modulate the immune response to viral antigens and enhance the carcinogenic effects of altered X proteins[40]. In Far East Asia, HBV genotypes B and C are predominant, and five mutations are prominent in the core promoter and proximal precore regions: G1613A, C1653T, T1753V, A1846T, and G1899A[43-47]. These select mutations are associated with the development of HCC when combined with the BCP double mutation. Many additional mutants have been reported in the literature, but most of these mutations are sporadic. Our data indicate that these mutations, together with A1762T, G1764A, and G1896A, are the most important frequent mutations in HBV genotype C2[33]. Considering the accumulation of mutations with time and the age of HCC patients, analyses must focus on the complexity of mutations associated with HCC risk, particularly in chronic HBV carriers greater than 40 years of age.
TRIPLE OR QUADRUPLE MUTATIONS INCLUDING THE BCP DOUBLE MUTATION
Although single G1613A or G1896A mutations are commonly noted in HBV genotype C2 in South Korea, single C1653T, T1753V, A1846T or G1899A mutations are rarely identified. Most of these mutations occur in combination with the BCP double mutation[37]. These results suggest that the BCP double mutation (A1762T/G1764A) may function as a starting point for the generation of viral variants harboring C1653T, T1753V, A1846T, and G1899A mutations. Therefore, it is not surprising that T1753V is more frequently linked to HBV genotype C than genotype B (19.2% vs 1.9%; P = 0.013)[15]. G1899A combined with the BCP double mutation is the single risk factor indicating HCC risk in Thailand and Tunisia, but the linkages between mutations are less clear in South Korea[33,37,45,48]. Our data indicate that while G1896A increases steadily with time, the accumulation of A1846T begins to increase during the quadruple phase of mutations. In contrast, the other mutations begin to accumulate at the triple phase[33,37]. Various specific quadruple mutations are superior to the BCP double mutation for determining HCC risk, whereas any individual triple mutation is not superior[33,37,43,49]. The combination of G1613A and C1653T is associated with HCC in HBV genotype C patients[49], whereas the combination of C1653T and T1753V is associated with HCC in HBV genotype B patients[43]. Among 15 different quadruple mutations containing the BCP double mutation, our analyses indicate that only five types are predominant [74.2% (72/97)], including the combinations (G1613A + C1653T), (C1653T + T1753V), (C1653T + G1896A), (T1753V + G1896A), and (A1846T + G1896A). These mutations account for 94.4% (34/36) of HCC cases that develop in the context of quadruple mutations. When exclusively compared with the BCP double mutation [HCC, 13.6% (6/44)], the prevalence of HCC among these five quadruple mutations was 46.2% (6/13, P = 0.02), 40% (2/5, P = 0.1821), 27.3% (6/20, P = 0.168), 66.7% (14/21, P = 0.00003) and 46.2% (6/13, P = 0.02), respectively. The respective odds ratios for HCC were 5.4286 (95%CI: 1.353-21.7821), 4.2222 (95%CI: 0.5797-30.7518), 2.7143 (95%CI: 0.7495-9.8293), 12.6667 (95%CI: 3.6262-44.2465) and 5.4286 (95%CI: 1.353-21.7821). The combination of C1766T and T1768A appears to enhance the carcinogenic effects of the X protein, but these mutations are rarely identified in South Korea[33,50]. Multivariate analyses of variables in relation to HCC indicate that mutation number is the only significantly independent viral factor[33]. These data indicate that complex mutations should be systematically evaluated as a function of the number of mutations.
THE UTILITY OF THE NUMBER OF MUTATIONS OF EIGHT KEY NUCLEOTIDES IN THE PREDICTION OF HCC
Although the development of HCC correlates with the accumulation of mutations, most studies have examined combinations of four mutations or less[43,51]. We analyzed the cumulative effects of complex mutations through a stratified analysis based on mutation number. The HCC rate in chronic HBV carriers increased linearly from wild-type to eight mutations as follows: 3.4% (1/29), 8.7% (2/23), 14.5% (8/55), 21.2% (21/99), 35.6% (36/101), 31.8% (21/66), 52.4% (22/42), 78.9% (15/19), and 75% (6/8), respectively (Y = 0.0917*X, r2 = 0.9199). Quadruple mutations were the most prevalent among the study subjects; double and triple mutations were most common in the non-HCC group, whereas multiple mutations, including more than four mutations, were predominant in the HCC group[33]. Compared with the BCP double mutation, the odds ratios (95%CI, P-value) of three to eight mutations were 1.7561 (0.6131-5.0301, 0.3250), 3.4286 (1.2623-9.3127, 0.0513), 2.7143 (0.9333-7.8939, 0.0820), 4.8571 (1.5527-15.1942, 0.0079), 30.0000 (5.3429-168.4491, 0.0000), and 24.0000 (2.4104-238.9646, 0.0023), respectively[33]. Based on these findings, we hypothesize that the number of mutations is a more sensitive predictor of HCC risk than any specific mutation[33]. In particular, cases with ≥ 6 mutations were associated with HCC with the greatest accuracy; the sensitivity, specificity, positive predictive value and negative predictive value were 44.0%, 97.3%, 94.3%, and 63.5%, respectively. The diagnostic efficiency of ≥ 6 mutations was comparable to that of alpha-fetoprotein (AFP), a specific biomarker for HCC diagnosis. The AUROC was 0.824 (95%CI: 0.759-0.890) for ≥ 6 mutations and 0.869 (95%CI: 0.812-0.925) for AFP[33].
SPECIFIC MUTATION COMBINATIONS ASSOCIATED WITH HCC
In a longitudinal cohort of 25 patients with serial serum samples spanning the years before and after HCC diagnosis, most of the patients with HCC (24/25, 96.0%) exhibited ≥ 4 mutations including the BCP double mutation years prior to HCC development; these patients also exhibited an equal or increasing number of mutations until HCC development[33]. In particular, some mutation combinations were specifically associated with HCC, and the core mutations differed little among combinations of four, six, and seven mutations. Although the (G1613A + C1653T), (C1653T + T1753V), (C1653T + G1896A), (T1753V + G1896A), and (A1846T + G1896A) double mutations were prominent in HCC patients with quadruple mutations, the addition of any single mutation did not improve the combined effect of an existing quadruple mutation[37]. With regard to six mutations, the (G1613A + C1653T + A1846T + G1896A) and (G1613A + C1653T + A1846T + G1899A) combinations were observed in half of the HCC patients, whereas additional mutations were sporadic[37]. Compared with the BCP double mutation alone, the prevalence and odds ratios were 71.4% (5/7, P = 0.0032) and 15.8333 (95%CI: 2.4843-100.9110) for the (G1613A + C1653T + A1846T + G1896A), respectively, and 83.3% (5/6, P = 0.0012) and 31.6667 (95%CI: 3.1331-320.0591) for the (G1613A + C1653T + A1846T + G1899A), respectively[37]. With regard to seven mutations, the combinations (G1613A + C1653T + T1753V + A1846T + G1896A) and (G1613A + C1653T + A1846T + G1896A + G1899A) were observed in 86.7% of the HCC group; the rate of HCC was 100% (6/6) for the former combination and 85.7% (6/7) for the latter[37]. These data suggest that the acquisition of a new mutation is not incidental; however, the new mutation potentially follows the rules of association and linkage between a mutation and an existing mutation combination.
ASSOCIATION BETWEEN LOW-TITER HBeAg AND A NUMBER OF KEY MUTATIONS
In the data analyses, we arbitrarily defined low-titer HBeAg as a signal-to-noise ratio of less than 35 as measured by ELISA (Abbott Laboratories, Diagnostic Division, Abbott Park, IL 60064, United States)[52]. Of 442 cases, 57.2% (n = 253) were HBeAg-positive. Of 132 HCC cases, 47% (n = 62) were classified as HBeAg-positive HCC[37]. The HBeAg-positive rate inversely correlated with the number of key mutations (96.6%, 65.2%, 76.4%, 64.6%, 46.5%, 40.9%, 47.6%, 47.4%, and 12.5% for 0 to 8 mutations, respectively). However, the proportion of low-titer HBeAg in the 253 HBeAg-positive cases positively correlated with mutation numbers (7.1%, 13.3%, 21.4%, 25%, 42.6%, 44.4%, 65%, 100%, and 100% of the HBeAg-positive cases for 0 to 8 mutations, respectively). More than half of the 62 HBeAg-positive HCC cases were classified as low-titer HBeAg (HCC cases with low-titer HBeAg/total HBeAg-positive HCC cases for 0 to 8 mutations were 1/1, 0/0, 3/5, 5/10, 9/15, 5/10, 7/11, 9/9, and 1/1, respectively). Notably, HCC patients infected by wild-type or BCP double mutant HBV were exclusively low-titer HBeAg-positive. These data suggest that the quantity of HBeAg is associated with HBV-related hepatocarcinogenesis.
CONCLUSION
Although extracting useful data regarding HBV mutations in South Korea has been difficult, the present analyses demonstrate that HBV genotype C2 is a good model to investigate the significance of viral mutations. Based on our previous two studies, we proposed the following hypothesis: the presence of ≥ 6 mutations is the most important viral factor in predictions of HCC risk in chronic HBV carriers infected by the BCP mutant virus (Figure 1). The number of mutations is positively correlated not only with advanced liver disease, but also with HCC independent of advanced liver disease (Figure 2). Although the eight key mutations can occur in various combinations, specific mutation combinations are predominant in the HCC group (Table 1). However, a low titer HBeAg (< 35 signal-to-noise ratio) is indicative of HCC risk for viruses containing ≤ 5 mutations or the BCP double mutation only (Table 2). Therefore, viral mutations and clinical features are complementary in the prediction of HCC risk.
Figure 1.
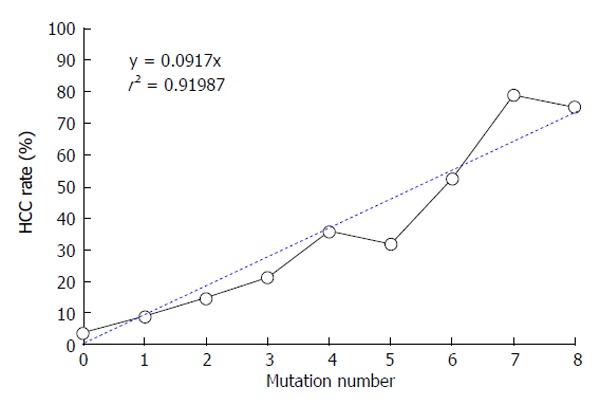
The number of mutations positively correlates with the rate of hepatocellular carcinoma: Pearson’s correlation = 0.9614 (95%CI: 0.8225-0.9921; P = 0.0000). HCC: Hepatocellular carcinoma.
Figure 2.
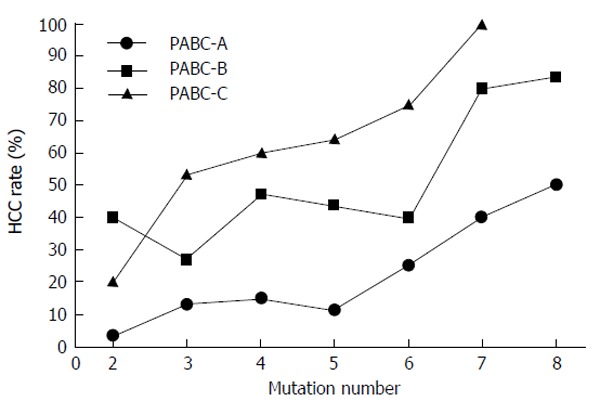
The number of mutations positively correlates with advanced liver disease, and advanced liver disease correlates with hepatocellular carcinoma. However, the number of mutations is correlated with hepatocellular carcinoma independent of advanced liver disease. We arbitrarily divided the clinical stages based on a combination of four laboratory parameters, including platelet counts, albumin levels, total bilirubin, and prothrombin time. The categories were defined according to PABC clinical staging: PABC-A exhibits normal values for the four parameters; PABC-B exhibits abnormal values for one or two biochemical parameter(s) in addition to abnormal platelet counts; and PABC-C exhibits abnormal values for all four laboratory parameters. Pearson’s correlation coefficient was 0.933 for PABC-A (95%CI: 0.6061-0.9903; P = 0.0021), 0.822 for PABC-B (95%CI: 0.1822-0.9729; P = 0.0231), and 0.938 for PABC-C (95%CI: 0.5285-0.9933; P = 0.0057). PABC: Platelet-albumin-bilirubin-coagulation ability (prothrombin time).
Table 1.
Hepatocellular carcinoma rate of specific mutation combinations and odds ratio in comparison with basal core promoter double mutation only
| Specific mutation combinations | HCC1 rate | OR | 95%CI | P-value |
| in combination with BCP2 double mutations | ||||
| Wild-type | 3.4% (1/29) | 0.23 | 0.0048-2.0622 | 0.2391 |
| BCP double mutations only [(A1762T + G1764A)]3 | 13.6% (6/44) | 13 | ||
| Dominant quadruple mutations | ||||
| (G1613A + C1653T) | 46.2% (6/13) | 5.4286 | 5.4286-1.3530 | 0.0200 |
| (C1653T + T1753V) | 40.0% (2/5) | 4.2222 | 4.2222-0.5797 | 0.1821 |
| (C1653T + G1896A) | 27.3% (6/20) | 2.7143 | 2.7143-0.7495 | 0.1680 |
| (T1753V + G1896A) | 66.7% (14/21) | 12.6667 | 12.6667-3.6262 | 0.0000 |
| (A1846T + G1896A) | 46.2% (6/13) | 5.4286 | 5.4286-1.3530 | 0.0200 |
| Dominant combinations in sextuplet mutations: | ||||
| (G1613A + C1653T + A1846T + G1896A) | 71.4% (5/7) | 14.5142 | 1.8869-185.1359 | 0.0033 |
| (G1613A + C1653T + A1846T + G1899A) | 83.3% (5/6) | 28.2555 | 2.5885-1517.9673 | 0.0012 |
| Dominant combinations in septuplet mutations: | ||||
| (G1613A + C1653T + T1753V + A1846T + G1896A) | 100% (6/6) | Infinity | 5.4236-infinity | 0.0001 |
| (G1613A + C1653T + A1846T + G1896A + G1899A) | 85.7% (6/7) | 39.3553 | 4.0487-2018.0433 | 0.0001 |
Hepatocellular carcinoma;
Basal core promoter;
Reference of odds ratio analyses; P-value was calculated by Fisher’s exact test for count data. BCP: Basal core promoter; HCC: Hepatocellular carcinoma.
Table 2.
Comparison of hepatocellular carcinoma rate between low and high titers of hepatitis B e antigen in each mutation number group
| Mutation |
HBeAg-positive rate |
HCC rate |
HBeAg < 35 or |
P-value1 | |||
| number |
Total |
HBeAg < 35 |
HBeAg-positive |
HBeAg-negative | HBeAg-negative rate | ||
| % (No./total cases) | % (No./total cases) | HBeAg > 35 | HBeAg < 35 | % (No./total cases) | among HCCs | ||
| % (No./total cases) | % (No./total cases) | % (No./total cases) | |||||
| 0 | 96.6% (28/29) | 7.1% (2/28) | 0% (0/26) | 50% (1/2) | 0% (0/1) | 100% (1/1) | - |
| 1 | 65.2% (15/23) | 13.3% (2/15) | 0% (0/13) | 0% (0/2) | 25.0% (2/8) | 100% (2/2) | - |
| 2-12 | 45.5% (5/11) | 60.0% (3/5) | 0% (0/2) | 33.3% (1/3) | 16.7% (1/6) | 100% (2/2) | - |
| 2-23 | 84.1% (37/44) | 16.2% (6/37) | 6.5% (2/31) | 33.3% (2/ 6) | 28..6% (2/7) | 66.7% (4/6) | 0.0530 |
| 3 | 71.4% (60/84) | 25.0% (15/60) | 10.4% (5/48) | 31.3% (5/16) | 31.4% (11/35) | 76.2% (16/21) | 0.0137 |
| 4 | 48.5% (47/97) | 42.6% (20/47) | 22.2% (6/27) | 45.0% (9/20) | 38.9% (21/54) | 83.3% (30/36) | 0.1047 |
| 5 | 40.0% (26/65) | 46.2% (12/26) | 33.3% (5/15) | 41.7% (5/12) | 28.2% (11/39) | 76.2% (16/21) | 1.0000 |
| 6 | 46.2% (18/39) | 66.7% (12/18) | 57.1% (4/7) | 53.8% (7/13) | 50.0% (11/22) | 81.8% (18/22) | 1.0000 |
| 7 | 47.4% (9/19) | 100% (9/9) | 0% (0/9) | 100% (9/9) | 60.0% (6/10) | 100% (15/15) | - |
| 8 | 12.5% (1/8) | 100% (1/1) | 0% (0/1) | 100% (1/1) | 71.4% (5/7) | 100% (6/6) | - |
Fisher’s exact test for count data was carried out to compare the significance of HBeAg status in the prediction of HCC risk between HCC patients with HBeAg > 35 and with HBeAg < 35 or -negative. Low titer HBeAg or HBeAg-negativity is significantly predominant among HCC patients;
Only one of two mutations is A1762T or G1764A;
Basal core promoter double mutations (A1762T/G1764A). HCC: Hepatocellular carcinoma; HBeAg: Hepatitis B e antigen.
Footnotes
P- Reviewer: Bellanti F, Vespasiani-Gentilucci U, Wang SK
S- Editor: Tian YL L- Editor: Webster JR E- Editor: Liu SQ
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: August 28, 2014
First decision: September 28, 2014
Article in press: October 29, 2014
References
- 1.Fattovich G. Natural history and prognosis of hepatitis B. Semin Liver Dis. 2003;23:47–58. doi: 10.1055/s-2003-37590. [DOI] [PubMed] [Google Scholar]
- 2.McMahon BJ. The natural history of chronic hepatitis B virus infection. Semin Liver Dis. 2004;24 Suppl 1:17–21. doi: 10.1055/s-2004-828674. [DOI] [PubMed] [Google Scholar]
- 3.McMahon BJ. The natural history of chronic hepatitis B virus infection. Hepatology. 2009;49:S45–S55. doi: 10.1002/hep.22898. [DOI] [PubMed] [Google Scholar]
- 4.Shi YH, Shi CH. Molecular characteristics and stages of chronic hepatitis B virus infection. World J Gastroenterol. 2009;15:3099–3105. doi: 10.3748/wjg.15.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chung WK, Moon SK, Popper H. Anicteric hepatitis in korea: comparative studies of asymptomatic and symptomatic series. Gastroenterology. 1965;48:1–11. [PubMed] [Google Scholar]
- 6.Chung WK. Chronic hepatitis in Korea. Prog Liver Dis. 1986;8:469–484. [PubMed] [Google Scholar]
- 7.McMahon BJ. Epidemiology and natural history of hepatitis B. Semin Liver Dis. 2005;25 Suppl 1:3–8. doi: 10.1055/s-2005-915644. [DOI] [PubMed] [Google Scholar]
- 8.Fattovich G, Brollo L, Giustina G, Noventa F, Pontisso P, Alberti A, Realdi G, Ruol A. Natural history and prognostic factors for chronic hepatitis type B. Gut. 1991;32:294–298. doi: 10.1136/gut.32.3.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen TJ, Liaw YF. The prognostic significance of bridging hepatic necrosis in chronic type B hepatitis: a histopathologic study. Liver. 1988;8:10–16. doi: 10.1111/j.1600-0676.1988.tb00960.x. [DOI] [PubMed] [Google Scholar]
- 10.Lok AS. Hepatitis B: liver fibrosis and hepatocellular carcinoma. Gastroenterol Clin Biol. 2009;33:911–915. doi: 10.1016/j.gcb.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 11.Ohkubo K, Kato Y, Ichikawa T, Kajiya Y, Takeda Y, Higashi S, Hamasaki K, Nakao K, Nakata K, Eguchi K. Viral load is a significant prognostic factor for hepatitis B virus-associated hepatocellular carcinoma. Cancer. 2002;94:2663–2668. doi: 10.1002/cncr.10557. [DOI] [PubMed] [Google Scholar]
- 12.Lin CL, Kao JH. Hepatitis B viral factors and clinical outcomes of chronic hepatitis B. J Biomed Sci. 2008;15:137–145. doi: 10.1007/s11373-007-9225-8. [DOI] [PubMed] [Google Scholar]
- 13.Yeh CT, So M, Ng J, Yang HW, Chang ML, Lai MW, Chen TC, Lin CY, Yeh TS, Lee WC. Hepatitis B virus-DNA level and basal core promoter A1762T/G1764A mutation in liver tissue independently predict postoperative survival in hepatocellular carcinoma. Hepatology. 2010;52:1922–1933. doi: 10.1002/hep.23898. [DOI] [PubMed] [Google Scholar]
- 14.Chu CM, Lin CC, Lin SM, Lin DY, Liaw YF. Viral load, genotypes, and mutants in hepatitis B virus-related hepatocellular carcinoma: special emphasis on patients with early hepatocellular carcinoma. Dig Dis Sci. 2012;57:232–238. doi: 10.1007/s10620-011-1844-2. [DOI] [PubMed] [Google Scholar]
- 15.Huang YH, Wu JC, Chang TT, Sheen IJ, Huo TI, Lee PC, Su CW, Lee SD. Association of core promoter/precore mutations and viral load in e antigen-negative chronic hepatitis B patients. J Viral Hepat. 2006;13:336–342. doi: 10.1111/j.1365-2893.2005.00688.x. [DOI] [PubMed] [Google Scholar]
- 16.Liu CJ, Chen PJ, Lai MY, Lin FY, Wang T, Kao JH, Chen DS. Viral factors correlate with hepatitis B e antigen seroconverson in patients with chronic hepatitis B. Liver Int. 2006;26:949–955. doi: 10.1111/j.1478-3231.2006.01319.x. [DOI] [PubMed] [Google Scholar]
- 17.Tai DI, Lin SM, Sheen IS, Chu CM, Lin DY, Liaw YF. Long-term outcome of hepatitis B e antigen-negative hepatitis B surface antigen carriers in relation to changes of alanine aminotransferase levels over time. Hepatology. 2009;49:1859–1867. doi: 10.1002/hep.22878. [DOI] [PubMed] [Google Scholar]
- 18.Fang ZL, Sabin CA, Dong BQ, Wei SC, Chen QY, Fang KX, Yang JY, Wang XY, Harrison TJ. The association of HBV core promoter double mutations (A1762T and G1764A) with viral load differs between HBeAg positive and anti-HBe positive individuals: a longitudinal analysis. J Hepatol. 2009;50:273–280. doi: 10.1016/j.jhep.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tacke F, Gehrke C, Luedde T, Heim A, Manns MP, Trautwein C. Basal core promoter and precore mutations in the hepatitis B virus genome enhance replication efficacy of Lamivudine-resistant mutants. J Virol. 2004;78:8524–8535. doi: 10.1128/JVI.78.16.8524-8535.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yuen MF, Tanaka Y, Shinkai N, Poon RT, But DY, Fong DY, Fung J, Wong DK, Yuen JC, Mizokami M, et al. Risk for hepatocellular carcinoma with respect to hepatitis B virus genotypes B/C, specific mutations of enhancer II/core promoter/precore regions and HBV DNA levels. Gut. 2008;57:98–102. doi: 10.1136/gut.2007.119859. [DOI] [PubMed] [Google Scholar]
- 21.Herbers U, Amini-Bavil-Olyaee S, Mueller A, Luedde T, Trautwein C, Tacke F. Hepatitis B e antigen-suppressing mutations enhance the replication efficiency of adefovir-resistant hepatitis B virus strains. J Viral Hepat. 2013;20:141–148. doi: 10.1111/j.1365-2893.2012.01639.x. [DOI] [PubMed] [Google Scholar]
- 22.Lin CL, Liao LY, Liu CJ, Chen PJ, Lai MY, Kao JH, Chen DS. Hepatitis B genotypes and precore/basal core promoter mutants in HBeAg-negative chronic hepatitis B. J Gastroenterol. 2002;37:283–287. doi: 10.1007/s005350200036. [DOI] [PubMed] [Google Scholar]
- 23.Chan HL, Hui AY, Wong ML, Tse AM, Hung LC, Wong VW, Sung JJ. Genotype C hepatitis B virus infection is associated with an increased risk of hepatocellular carcinoma. Gut. 2004;53:1494–1498. doi: 10.1136/gut.2003.033324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chu CM, Lin CC, Chen YC, Jeng WJ, Lin SM, Liaw YF. Basal core promoter mutation is associated with progression to cirrhosis rather than hepatocellular carcinoma in chronic hepatitis B virus infection. Br J Cancer. 2012;107:2010–2015. doi: 10.1038/bjc.2012.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pujol FH, Navas MC, Hainaut P, Chemin I. Worldwide genetic diversity of HBV genotypes and risk of hepatocellular carcinoma. Cancer Lett. 2009;286:80–88. doi: 10.1016/j.canlet.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 26.McMahon BJ. The influence of hepatitis B virus genotype and subgenotype on the natural history of chronic hepatitis B. Hepatol Int. 2009;3:334–342. doi: 10.1007/s12072-008-9112-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang HI, Yeh SH, Chen PJ, Iloeje UH, Jen CL, Su J, Wang LY, Lu SN, You SL, Chen DS, et al. Associations between hepatitis B virus genotype and mutants and the risk of hepatocellular carcinoma. J Natl Cancer Inst. 2008;100:1134–1143. doi: 10.1093/jnci/djn243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JM, Ahn SH, Chang HY, Shin JE, Kim DY, Sim MK, Hong SP, Chung HJ, Kim SO, Han KH, et al. [Reappraisal of HBV genotypes and clinical significance in Koreans using MALDI-TOF mass spectrometry] Korean J Hepatol. 2004;10:260–270. [PubMed] [Google Scholar]
- 29.Song BC, Cui XJ, Kim H. Hepatitis B virus genotypes in Korea: an endemic area of hepatitis B virus infection. Intervirology. 2005;48:133–137. doi: 10.1159/000081740. [DOI] [PubMed] [Google Scholar]
- 30.Kim H, Jee YM, Song BC, Shin JW, Yang SH, Mun HS, Kim HJ, Oh EJ, Yoon JH, Kim YJ, et al. Molecular epidemiology of hepatitis B virus (HBV) genotypes and serotypes in patients with chronic HBV infection in Korea. Intervirology. 2007;50:52–57. doi: 10.1159/000096313. [DOI] [PubMed] [Google Scholar]
- 31.Cho JH, Yoon KH, Lee KE, Park DS, Lee YJ, Moon HB, Lee KR, Choi CS, Cho EY, Kim HC. [Distribution of hepatitis B virus genotypes in Korea] Korean J Hepatol. 2009;15:140–147. doi: 10.3350/kjhep.2009.15.2.140. [DOI] [PubMed] [Google Scholar]
- 32.Kim JK, Chang HY, Lee JM, Baatarkhuu O, Yoon YJ, Park JY, Kim do Y, Han KH, Chon CY, Ahn SH. Specific mutations in the enhancer II/core promoter/precore regions of hepatitis B virus subgenotype C2 in Korean patients with hepatocellular carcinoma. J Med Virol. 2009;81:1002–1008. doi: 10.1002/jmv.21501. [DOI] [PubMed] [Google Scholar]
- 33.Jang JW, Chun JY, Park YM, Shin SK, Yoo W, Kim SO, Hong SP. Mutational complex genotype of the hepatitis B virus X /precore regions as a novel predictive marker for hepatocellular carcinoma. Cancer Sci. 2012;103:296–304. doi: 10.1111/j.1349-7006.2011.02170.x. [DOI] [PubMed] [Google Scholar]
- 34.Park YM, Kim BS, Tabor E. Precore codon 28 stop mutation in hepatitis B virus from patients with hepatocellular carcinoma. Korean J Intern Med. 1997;12:201–207. doi: 10.3904/kjim.1997.12.2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cho SW, Shin YJ, Hahm KB, Jin JH, Kim YS, Kim JH, Kim HJ. Analysis of the precore and core promoter DNA sequence in liver tissues from patients with hepatocellular carcinoma. J Korean Med Sci. 1999;14:424–430. doi: 10.3346/jkms.1999.14.4.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lok AS, Akarca U, Greene S. Mutations in the pre-core region of hepatitis B virus serve to enhance the stability of the secondary structure of the pre-genome encapsidation signal. Proc Natl Acad Sci USA. 1994;91:4077–4081. doi: 10.1073/pnas.91.9.4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park YM, Jang JW, Yoo SH, Kim SH, Oh IM, Park SJ, Jang YS, Lee SJ. Combinations of eight key mutations in the X/preC region and genomic activity of hepatitis B virus are associated with hepatocellular carcinoma. J Viral Hepat. 2014;21:171–177. doi: 10.1111/jvh.12134. [DOI] [PubMed] [Google Scholar]
- 38.Tong SP, Li JS, Vitvitski L, Trépo C. Replication capacities of natural and artificial precore stop codon mutants of hepatitis B virus: relevance of pregenome encapsidation signal. Virology. 1992;191:237–245. doi: 10.1016/0042-6822(92)90185-r. [DOI] [PubMed] [Google Scholar]
- 39.Liu CJ, Jeng YM, Chen CL, Cheng HR, Chen PJ, Chen TC, Liu CH, Lai MY, Chen DS, Kao JH. Hepatitis B virus basal core promoter mutation and DNA load correlate with expression of hepatitis B core antigen in patients with chronic hepatitis B. J Infect Dis. 2009;199:742–749. doi: 10.1086/596655. [DOI] [PubMed] [Google Scholar]
- 40.Lee JH, Han KH, Lee JM, Park JH, Kim HS. Impact of hepatitis B virus (HBV) x gene mutations on hepatocellular carcinoma development in chronic HBV infection. Clin Vaccine Immunol. 2011;18:914–921. doi: 10.1128/CVI.00474-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu X, Mertz JE. Promoters for synthesis of the pre-C and pregenomic mRNAs of human hepatitis B virus are genetically distinct and differentially regulated. J Virol. 1996;70:8719–8726. doi: 10.1128/jvi.70.12.8719-8726.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cui XJ, Cho YK, Song HJ, Choi EK, Kim HU, Song BC. Molecular characteristics and functional analysis of full-length hepatitis B virus quasispecies from a patient with chronic hepatitis B virus infection. Virus Res. 2010;150:43–48. doi: 10.1016/j.virusres.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 43.Liu S, Zhang H, Gu C, Yin J, He Y, Xie J, Cao G. Associations between hepatitis B virus mutations and the risk of hepatocellular carcinoma: a meta-analysis. J Natl Cancer Inst. 2009;101:1066–1082. doi: 10.1093/jnci/djp180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takahashi K, Akahane Y, Hino K, Ohta Y, Mishiro S. Hepatitis B virus genomic sequence in the circulation of hepatocellular carcinoma patients: comparative analysis of 40 full-length isolates. Arch Virol. 1998;143:2313–2326. doi: 10.1007/s007050050463. [DOI] [PubMed] [Google Scholar]
- 45.Tangkijvanich P, Sa-Nguanmoo P, Mahachai V, Theamboonlers A, Poovorawan Y. A case-control study on sequence variations in the enhancer II/core promoter/precore and X genes of hepatitis B virus in patients with hepatocellular carcinoma. Hepatol Int. 2010;4:577–584. doi: 10.1007/s12072-010-9197-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yin J, Xie J, Liu S, Zhang H, Han L, Lu W, Shen Q, Xu G, Dong H, Shen J, et al. Association between the various mutations in viral core promoter region to different stages of hepatitis B, ranging of asymptomatic carrier state to hepatocellular carcinoma. Am J Gastroenterol. 2011;106:81–92. doi: 10.1038/ajg.2010.399. [DOI] [PubMed] [Google Scholar]
- 47.Cho EY, Choi CS, Cho JH, Kim HC. Association between Hepatitis B Virus X Gene Mutations and Clinical Status in Patients with Chronic Hepatitis B Infection. Gut Liver. 2011;5:70–76. doi: 10.5009/gnl.2011.5.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ouneissa R, Bahri O, Alaya-Bouafif NB, Chouaieb S, Ben Yahia A, Sadraoui A, Hammami W, Filali N, Azzouz MM, Mami NB, et al. Frequency and clinical significance of core promoter and precore region mutations in Tunisian patients infected chronically with hepatitis B. J Med Virol. 2012;84:1719–1726. doi: 10.1002/jmv.23394. [DOI] [PubMed] [Google Scholar]
- 49.Tatsukawa M, Takaki A, Shiraha H, Koike K, Iwasaki Y, Kobashi H, Fujioka S, Sakaguchi K, Yamamoto K. Hepatitis B virus core promoter mutations G1613A and C1653T are significantly associated with hepatocellular carcinoma in genotype C HBV-infected patients. BMC Cancer. 2011;11:458. doi: 10.1186/1471-2407-11-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kitab B, Essaid El Feydi A, Afifi R, Trepo C, Benazzouz M, Essamri W, Zoulim F, Chemin I, Alj HS, Ezzikouri S, et al. Variability in the precore and core promoter regions of HBV strains in Morocco: characterization and impact on liver disease progression. PLoS One. 2012;7:e42891. doi: 10.1371/journal.pone.0042891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen CH, Hung CH, Lee CM, Hu TH, Wang JH, Wang JC, Lu SN, Changchien CS. Pre-S deletion and complex mutations of hepatitis B virus related to advanced liver disease in HBeAg-negative patients. Gastroenterology. 2007;133:1466–1474. doi: 10.1053/j.gastro.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 52.Thompson AJ, Nguyen T, Iser D, Ayres A, Jackson K, Littlejohn M, Slavin J, Bowden S, Gane EJ, Abbott W, et al. Serum hepatitis B surface antigen and hepatitis B e antigen titers: disease phase influences correlation with viral load and intrahepatic hepatitis B virus markers. Hepatology. 2010;51:1933–1944. doi: 10.1002/hep.23571. [DOI] [PubMed] [Google Scholar]