

Abstract
Background
Eye development in vertebrates relies on the critical regulation of SOX2 expression. Humans with mutations in SOX2 often suffer from eye defects including anophthalmia (no eye) and microphthalmia (small eye). In mice, deletion of Sox2 in optic cup progenitor cells results in loss of neural competence and cell fate conversion of the neural retina to a non-neurogenic fate, specifically the acquisition of fate associated with progenitors of the ciliary epithelium. This fate is also promoted with constitutive expression of stabilized β-Catenin in the optic cup, where the WNT pathway is up-regulated. We addressed whether SOX2 co-ordinates the neurogenic boundary of the retina through modulating the WNT/β-Catenin pathway by using a genetic approach in the mouse.
Results
Upon deletion of Sox2 in the optic cup, response to WNT signaling was expanded, correlating with loss of neural competence, cell fate conversion of the neural retina to ciliary epithelium primordium and, in addition, increased cell cycle time of optic cup progenitors. Removal of Ctnnb1 rescued the cell fate conversion; however, the loss of neural competence and the proliferation defect resulting from lack of SOX2 were not overcome. Lastly, central Sox2-deficient optic cup progenitor cells exhibited WNT-independent up-regulation of D-type Cyclins.
Conclusion
We propose two distinct roles for SOX2 in the developing retina. Our findings suggest that SOX2 antagonizes the WNT pathway to maintain a neurogenic fate and, in contrast, regulates cycling of optic cup progenitors in a WNT-independent manner. Given that WNT signaling acting upstream of SOX2 has been implicated in the tumorigenicity of embryonic stem cell-derived retinal progenitor cells, our results distinguish the endogenous role of WNT signaling in early optic cup patterning and support a WNT-independent role for SOX2 in maintaining retinal progenitor cell proliferation.
Electronic supplementary material
The online version of this article (doi:10.1186/1749-8104-9-27) contains supplementary material, which is available to authorized users.
Keywords: β-Catenin, Canonical WNT signaling, Cell cycle, Eye development, Neural progenitor cells, Proliferation, Retina, SOX2
Background
How neural progenitor cells maintain the ability to generate neurons over time is a major question in developmental neurobiology. The eyecup is an ideal tool to address this question, because it consists of neurogenic and non-neurogenic structures derived from a common progenitor pool originating in the eyefield of the medial anterior neural plate [1, 2]. The eyefield gives rise to the optic vesicle, which invaginates to form the optic cup (OC) around embryonic day (E) 10.0 of mouse development [3]. The OC is regionalized along the central-peripheral axis: the central OC consists of neurogenic retinal progenitor cells (RPCs) that give rise to the six neuronal and one glial cell type that make up the neural retina (NR). The peripheral OC rim, also known as the ciliary margin (CM), gives rise to the non-neurogenic epithelium of the iris and ciliary body (CB), herein referred to as ciliary epithelium (CE) (reviewed in [4]). Multipotent progenitor cells at the central boundary of the CM make a binary cell fate decision to become NR or CE [5].
Relatively little is known about the molecular mechanisms that specify neurogenic versus non-neurogenic fate in the OC. Canonical WNT signaling, which functions through its downstream transcriptional effector β-Catenin, has been identified as a major regulator of CE fate specification in many species [6–12]. In the chick eye, WNT/β-Catenin signaling was found to inhibit NR fate and promote CE fate [7]. In the mouse, a genetic reporter under the control of β-Catenin/TCF/LEF response elements showed WNT activity to be concentrated to the CM [6], and specific ablation of Ctnnb1 in OC progenitor cells (OCPCs) reduced the size of the CE progenitor cell pool [8, 13]. Conversely, stabilized expression of Ctnnb1 in mouse OCPCs induced ectopic expression of CE-specific genes [8]. However, these ectopic CE-like cells did not express Pax6 or Chx10, two well-known transcriptional regulators of CE fate, suggesting that there was only a partial transformation of NR-to-CE upon activation of Ctnnb1.
Unsurprisingly, in light of the above, removing the inhibition of WNT signaling also induced CE fate; Foxg1-null embryos showed expanded WNT activity into the central OC and ectopic formation of CE positive for both Pax6 and Chx10, suggesting that near physiological levels of β-Catenin may be required to generate all the hallmarks of CE [14]. Thus, precise regulation of the level of WNT activity is crucial for establishing the proper boundary between CE and NR, where factors mediating this function are not known.
SRY (sex determining region Y)-box (SOX) proteins are known regulators of WNT signaling in several developmental systems and disease states. SOX2, a member of the SOXB1 family of transcription factors, is a major regulator of neural competence in vertebrates [15–17]. Heterozygous mutations in human SOX2 are associated with anophthalmia (absent eye) and account for 10 to 20% of cases of severe bilateral ocular malformation, including microphthalmia (small eye) [18–20] indicating a defect in OCPC proliferation or survival. In the mouse OC, SOX2 expression is restricted to the presumptive NR, and ablation of Sox2 in OCPCs resulted in loss of neural competence and cell fate conversion of the NR to CE primordium, accompanied by an increase in WNT signaling [5]. The genetic relationship between SOX2 and WNT signaling in this context was not investigated.
In addition to eye defects, human patients with SOX2 mutations often have pituitary abnormalities, and WNT signaling is known to be involved in hypothalamic and pituitary development. Human SOX2 protein can inhibit β-Catenin-driven reporter expression in vitro, but several SOX2 proteins with the identified human mutations cannot. Therefore, it has been suggested that an inability to repress WNT/β-Catenin signaling may contribute to the pathogenesis of SOX2 loss-of-function (LOF) mutations in human patients [21, 22]. In support of this hypothesis, a SOX2 binding site was identified in the Lef1 promoter and was found to function as a repressor of β-Catenin-dependent Lef1 expression in primary airway epithelial cells [23]. Additionally, in osteoblasts, SOX2 was shown to physically associate with β-Catenin to down-regulate the expression of many WNT target genes, but the HMG domain was not required, suggesting that SOX2 may antagonize WNT signaling via β-Catenin sequestration [24].
The complementary eye phenotypes associated with Sox2 and Ctnnb1 LOF suggest antagonism between these two pathways in mammalian OC development. In lower vertebrates and in RPCs differentiated from induced pluripotent stem cells, these two pathways have been found to work somewhat synergistically to promote retinal neural progenitor proliferation [25, 26]. These findings may reflect species-specific differences in the role of WNT signaling in OC development. Alternatively, WNT signaling may play different roles over developmental time: constitutive activation of WNT signaling later in development, in a subset of committed neural precursors, may have different effects than that of widespread WNT activation at earlier time points, in uncommitted OCPCs. Given the evidence that SOX2 and WNT signaling play complex and crucial roles in the eye development of many species, we chose to dissect the relationship between these two factors using a genetic approach in the mouse.
In this study, we investigated the hypothesis that SOX2 antagonizes canonical WNT signaling to maintain neurogenic fate in the mouse OC. We present whole-genome expression arrays comparing wild-type and Sox2-mutant OCs demonstrating the deregulation of the WNT pathway and serving as a resource for identifying genes involved in CE fate and function, with direct relevance to understanding the pathogenesis of diseases associated with the anterior segment. We show that removal of Ctnnb1 from the Sox2-mutant OC partially rescued the Sox2-mutant phenotype. Conversely, ectopic activation of the WNT pathway in Sox2-expressing cells resulted in acquisition of CE fate following loss of Sox2 expression. Our data provide evidence that SOX2 antagonizes CE fate via modulation of WNT signaling and highlight a β-Catenin-independent role for SOX2 to promote proliferation and prevent aberrant expression of cell cycle regulators in OCPCs.
Results
Canonical WNT signaling is ectopically activated in Sox2-mutant optic cups
Ablation of Sox2 in the mouse OC from E10.5 leads to eventual loss of NR fate and expansion of the non-neurogenic CE [5]. To determine the molecular mechanisms underlying this phenotype, we performed a whole genome expression screen of Sox2cond/+;αP0CREiresGFP ('control') and Sox2cond/cond;αP0CREiresGFP ('mutant') eyes at E16.5, when the loss of neural fate is taking place (Figure 1A). We ran one microarray for each of six pairs of eyes per genotype, for a total of twelve microarrays. The complete results from this screen have been deposited in NCBI’s Gene Expression Omnibus [27] and can be accessed through the GEO series accession number GSE46796 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE46796). We identified 880 significantly up-regulated genes and 951 significantly down-regulated genes in mutant OCs compared with controls (see Methods). To confirm the efficacy of this screen, we first verified that transcripts found to change by in situ hybridization (ISH) [5] behaved as expected by microarray (Figure 1C). As anticipated, in mutants compared with controls, Sox2 was decreased over 4-fold, Hes5, an NR marker, which was found to be absent in Sox2-ablated cells, was decreased almost 5-fold, and Chx10, a definitive marker of OC progenitors at this stage, and which was shown to be maintained, was not significantly changed.
Figure 1.
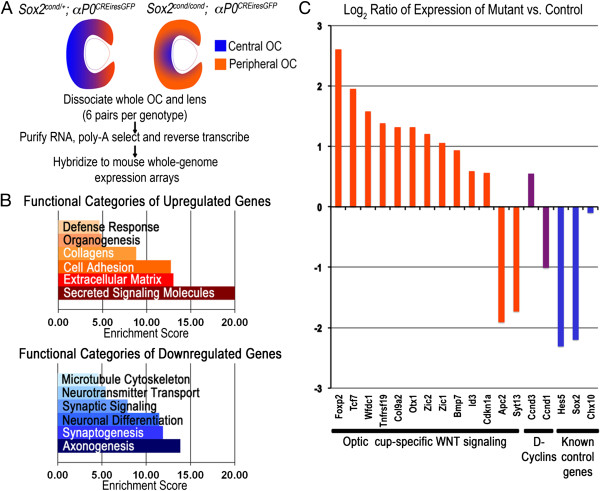
Genome- wide expression analysis of E16.5 Sox2-mutant optic cups (OCs) confirms loss of neural retina (NR) fate, activation of ciliary epithelium (CE) primordium genes, and suggests activated WNT signaling. Global changes in gene expression in Sox2-ablated OCs undergoing NR-to-CE primordium cell fate conversion were analyzed by microarray. (A) Diagram of expansion of the peripheral OC/prospective CE (orange) at the expense of the central OC/prospective NR (blue) in mutants compared with controls. (B) Ontologies of increased transcripts (orange) are consistent with CE function, while those of decreased transcripts (blue) are consistent with NR function. (C) Subsets of aberrantly expressed genes suggest activated WNT signaling and perturbed cell cycle. Decreased Hes5 and Sox2, and unchanged Chx10, confirm the validity of the screen.
We then used DAVID analysis software [28, 29] to search for enriched functional terms in each group of genes and determine if they were consistent with a change in cell fate. The full list of genes for each enriched functional term for each group is provided in Additional file 1. Up-regulated genes were enriched for terms associated with secreted signaling, extracellular matrix, cell adhesion, organogenesis and the defense response, all of which are consistent with the function of the CE to maintain the intraocular pressure through secretion [30, 31] and to produce proteins of the inner limiting membrane and the vitreous body [32, 33] (Figure 1B). By contrast, down-regulated genes were enriched for terms associated with axonogenesis, synaptic development, neuronal differentiation and microtubule cytoskeleton, consistent with the loss of neurogenic fate observed in mutants. Collectively, these data confirm that our screen provides a reliable indication of functional pathways disrupted by Sox2 ablation.
We next queried which signaling pathways were disrupted in Sox2-mutant eyes. Two of the most significantly affected pathways were canonical WNT signaling and transforming growth factor beta (TGFβ) signaling, both known for their involvement in CE fate specification [7, 8, 34, 35]. We focused further on WNT signaling, given that SOX2 was shown to antagonize the pathway in the Xenopus OC [25], and we have observed ectopic expression of WNT pathway genes, including Lef1, Sfrp2 and Axin2, in the Sox2-mutant OC at E14.5 [5]. We scanned our set of significantly changed transcripts for known OC-specific WNT target genes and identified 13 for which expression was changed at least 1.47-fold (Figure 1C, orange bars; Additional file 2) [9, 11, 12, 36, 37]. We also confirmed that Axin2, a well-characterized read-out of WNT signaling, was increased (1.29-fold, P = 0.0155). Collectively, these data suggest that response to the WNT pathway was activated upon Sox2 ablation. We thus hypothesized that SOX2 normally antagonizes canonical WNT signaling to maintain neurogenic fate in the mouse OC.
Genetic removal of Ctnnb1does not rescue the neurogenesis defect of Sox2-mutant retinas
If increased WNT signaling causes loss of neural competence upon ablation of Sox2, then reduction of WNT activity should rescue neurogenesis in Sox2-mutant eyes. SOX2 protein has been shown to interact directly with β-Catenin/Ctnnb1, the main transcriptional mediator of canonical WNT signaling, to inhibit the activation of target genes in many biological systems [24, 38–40]. So to address this hypothesis, we performed epistasis analysis using conditional alleles of both Sox2 and Ctnnb1 an OC-specific Cre. Ctnnb1lox(ex2–6)/+ mice, in which exons 2 to 6 of the Ctnnb1 locus are flanked by loxP sites [41], were crossed with transgenic mice carrying Chx10CreGFP, a BAC transgene expressing Cre recombinase in OCPCs as early as E10.5 [42]. For this experiment, we chose to use Chx10CreGFP in place of αP0CreiresGFP in order to avoid differential regulation of the αP0Cre transgene in the absence of SOX2 and β-Catenin, as previously observed [5, 8]. Distinctions between our uses of αP0CreiresGFP and Chx10CreGFP are summarized in Table 1.
Table 1.
Distinctions between the two optic cup (OC)-specific Cre lines used in this study
| CRE line | Onset | Expression pattern | Applications in this study |
|---|---|---|---|
| αP0 CREiresGFP | E10.5 | Prospective CE and NR (PeripheralHI - CentralLO) | Whole genome expression screen; Proliferation analysis |
| Chx10 CRE-GFP | E10.5 | Prospective CE and NR (Mosaic) | Genetic epistasis (Cre expression is not affected by SOX2 or β-Catenin) |
Abbreviations: CE ciliary epithelium, NE neural retina.
Ctnnb1lox(ex2–6)/+;Chx10CreGFP mice were crossed with Sox2cond/+ mice to produce Sox2/Ctnnb1 double-mutant OCPCs. We compared the eyes of Sox2 single-mutants (Sox2cond/cond;Chx10CreGFP) and Ctnnb1 single-mutants (Ctnnb1lox(ex2–6)/lox(ex2–6);Chx10CreGFP) with Sox2/Ctnnb1 double-mutants (Sox2cond/cond;Ctnnb1lox(ex2–6)/lox(ex2–6);Chx10CreGFP) and stage-matched controls (Sox2cond/+;Ctnnb1lox(ex2–6)/+;Chx10CreGFP).
We first verified that Sox2 and Ctnnb1 were efficiently ablated with Chx10CreGFP and that co-ablation did not increase cell death (Additional file 3). In double-mutant OCs, SOX2 and β-Catenin proteins were absent from GFP-positive cells, demonstrating that Chx10CreGFP was able to recombine all floxed alleles in the same nuclei (Additional file 3D, E versus A, B). Moreover, deletion of both genes did not increase cell death as assayed by activated-Caspase 3 staining (Additional file 3C, F).
To test the hypothesis that removal of Ctnnb1 from Sox2-mutant cells would rescue the loss of neural competence, we examined expression of prospective NR markers in single-mutant and double-mutant OCs at E13.5 (Figure 2; Table 2). We first examined expression of β-Tubulin III, a neuronal marker, to determine whether neurogenesis was restored in double- mutant OCs. β-Tubulin III was expressed in the central OC of controls, marking retinal ganglion cells (RGCs), but was broadly lost in Sox2 single-mutants and in Sox2/Ctnnb1 double-mutants (Figure 2B and E versus H and K). RGCs express Sonic Hedgehog (Shh), so to confirm the observed loss of neurogenesis, we examined Shh expression in control and mutant eyes. Like β-Tubulin III, Shh was expressed in the RGC layer of controls (Figure 2C, F, area between closed arrowheads), but expression was essentially lost in Sox2 single-mutant and double-mutant eyes (Figure 2I, L, closed arrowhead indicating residual Shh in a double-mutant eye correlating with residual β-Tubulin III expression). Shh expression in the presumptive eyelids remained unchanged in all genotypes, confirming the efficacy of the staining (Figure 2C, F, I, L, white arrowheads).
Figure 2.

Deletion of Ctnnb1 does not restore neurogenesis to Sox2- mutant cells. Transverse sections of E13.5 control and mutant eyes were analyzed for restoration of neurogenic fate to Sox2-ablated regions. (A, D, G, J) Ccnd1 mRNA localizes to the central optic cup (OC) of wild-type controls (A) and Ctnnb1 lox(ex2–6)/lox(ex2–6) single-mutants (D) and is maintained in the central OC of Sox2 single-mutants (G) and Sox2/Ctnnb1 double-mutants (J). (B, E, H, K) β-Tubulin III, marking neurons, is expressed in the retinal ganglion cell (RGC) layer of controls (B) and Ctnnb1 single-mutants (E), but its expression is markedly reduced in Sox2 single-mutant (H) and double-mutant (K) cells. (C, F, I, L) Sonic Hedgehog (Shh) mRNA expression correlates with the pattern of β-Tubulin III staining, and thus is significantly reduced in mutants (I, L) compared with controls (F, C). Black arrowheads in (C) and (F) indicate the limits of Shh expression. White arrowheads in (C, F, I and L) point to an unaffected region of Shh expression in the presumptive eyelid. Scale bars: 200 μm.
Table 2.
Differences in the expression patterns of central optic cup progenitor cells (OCPCs) of wild-type, single-mutant and double-mutant eyes
| NR markers | Ciliary epithelium markers | Optic cup progenitor cell TFs | D-Cyclins (protein) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | Hes5 | β-Tubulin/ Shh | Msx1 | Otx1 | BMP4 | Zic1 | Chx10 | Rax | Pax6 | CyclinD3 | CyclinD1 |
| Wild-type | expressed | expressed | absent | absent | absent | absent | expressed | expressed | expressed | absent | expressed |
| Sox2-/-; Ctnnb1-/+ | absent | absent | expressed | expressed | expressed | expressed | expressed | expressed | increased | expressed | increased |
| Sox2-/+; Ctnnb1-/- | expressed | expressed | absent | absent | absent | absent | expressed | expressed | expressed | absent | expressed |
| Sox2-/-; Ctnnb1-/- | absent | absent | absent | absent | absent | absent | expressed | expressed | increased | expressed | increased |
Abbreviation: TFs transcription factors.
CyclinD1/Ccnd1 is normally restricted to the central OC and is often used to mark NR progenitor cells [8, 14]. In wild-type controls and Ctnnb1 single-mutant eyes, Ccnd1 mRNA was expressed in central neurogenic progenitor cells. Ccnd1 expression persisted in Sox2 single-mutant OCs despite the loss of NR fate (Figure 2A and D versus G). Overall, Ccnd1 staining appeared decreased in Sox2-ablated cells consistent with the gene expression results of the microarray analysis, which showed a 2-fold decrease (P = 0.001). Moreover, removal of Ctnnb1 from Sox2-mutant OCPCs did not further diminish Ccnd1 expression (Figure 2J). In summary, genetic reduction of WNT activation did not restore neural competence to Sox2-ablated eyes at E13.5, and expression of Ccnd1 was sustained despite loss of either pathway.
Genetic ablation of β-Catenin prevents cell fate conversion of Sox2-mutant retinas
We next examined single- and double-mutant neonatal eyes to determine whether removal of Ctnnb1 could rescue the NR-to-CE cell fate conversion previously observed in Sox2-mutant eyes [5]. At P0, Sox2 single-mutant and Sox2/Ctnnb1 double-mutant eyes were small, and the optic neuroepithelium of both was hypoplastic compared with controls (Figure 3A, B and E, F versus I, J and M, N). To clearly identify mutant cells in each background, we applied two methods: first we used an antibody against β-Catenin to distinguish positive and negative cells (Figure 3B”, E”, H”, K”). Secondly, we backcrossed all genotypes to reporter mice expressing β-galactosidase (β-gal) from the ROSA26 locus [43] to mark recombined cells (Figure 3C”, F”, I”, L”).
Figure 3.
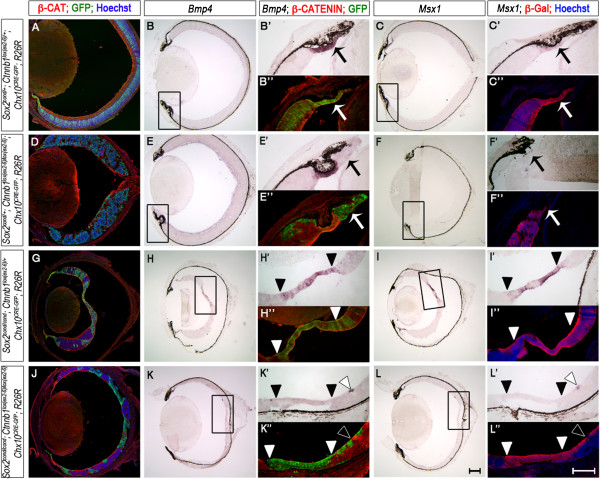
Deletion of Ctnnb1 prevents the ectopic expression of ciliary epithelium (CE) genes in Sox2- mutant cells. Transverse sections of P0 eyes were analyzed for CE fate. (A, D, G, J) Low magnification view of β-Catenin/GFP-stained eyes indicates hypoplasia of Sox2 single-mutants (G) and Sox2/Ctnnb1 double-mutants (J) compared with Ctnnb1 lox(ex2–6)/lox(ex2–6) single-mutants (D) and wild-type littermates (A). (B - C ” ) In situ hybridization of Bmp4 (C) and Msx1 (D) shows specific expression of these genes in GFP-positive (B”) and β-galactosidase (β-gal)-positive (C”) CE of wild-type controls. (E-F”) Bmp4 (E) and Msx1 (F) are not expressed in the GFP-positive (E”, arrow) and β-gal-positive (F”, arrow) CE of Ctnnb1 single-mutants. (H-I”) Conversely, Bmp4 (H) and Msx1 (I) are ectopically expressed in the GFP-positive (H”, closed arrowheads) and β-gal-positive (I”, closed arrowheads) central optic cup (OC) of Sox2 single-mutants. (K-L”) Ectopic expression of Bmp4 (K) and Msx1 (L) is rescued in the GFP-positive (K”, closed arrowheads) and β-gal-positive (L”, closed arrowheads) central OC of Sox2/Ctnnb1 double-mutants. Open arrowheads indicate faint expression of CE genes at the periphery of the NR of double-mutants. Boxes in ( B-C), (E-F), (H-I) and (K- L) are magnified in ’ and ”. Scale bars: 100 μm.
Bmp4 and Msx1 were expressed in the presumptive CE of wild-type controls as expected (Figure 3B, C) [8, 44]. By contrast, Bmp4 and Msx1 were absent from presumptive CE cells of Ctnnb1 single-mutants, supporting the hypothesis that canonical WNT signaling directly regulates the expression of CE primordium genes (Figure 3E, F, E’ and F’ arrows). As seen in our previous study, Bmp4 and Msx1 were ectopically expressed in Sox2-ablated regions (Figure 3H, I, H’ and I’, area between closed arrowheads) [5]. This ectopic up-regulation of Bmp4 and Msx1 occurred in approximately 30 to 60% of recombined cells based on GFP or β-gal expression. Strikingly, genetic deletion of Ctnnb1 from these cells prevented the ectopic expression of Bmp4 and Msx1 (Figure 3K, L, K’, L’ area between closed arrowheads) in all retinas analyzed (n = 3 per genotype, per stage). We noted marginal CE gene expression at the periphery of β-Catenin-positive/Cre-negative regions in double-mutants (Figure 3 K’, L’ open arrowheads), possibly indicating a signaling center for CE specification at the edges of NR, as has been reported in the chick OC [45].
We next examined expression of additional NR and CE markers in control, single-mutant and double-mutant OCs at P0, and these results are summarized in Table 2. Similar to Bmp4 and Msx1, ectopic expression of the CE genes Otx1 and Zic1 was rescued in double-mutants. The pan-OCPC transcription factors Chx10 [42] and Rax [46] were unchanged in all genotypes. Notably, Pax6, which was increased in Sox2 single-mutants [5], remained so in double-mutants, suggesting that SOX2 antagonizes Pax6 independently of WNT signaling. Together, these data suggest that SOX2 antagonizes CE identity, in part through mediation of WNT/β-Catenin activity.
Ectopic WNT pathway activation in SOX2-expressing cells results in loss of NR fate
To confirm the notion that up-regulation of the WNT pathway is sufficient to cause the NR-to-CE fate transformation observed in Sox2 mutants, we used a complementary approach, expressing a stable mutant form of Ctnnb1 specifically in NR-committed Sox2-expressing cells. Sox2CreERT2/+ males [47] were crossed with Ctnnb1lox(ex3)/lox(ex3) females, which were injected with tamoxifen at E11.5 to activate Cre expression. Analysis of Sox2CreERT2/+;Ctnnb1lox(ex3)/+ mutant eyes at E15.5 revealed abnormal NR morphology showing signs of detachment (Figure 4). We observed ectopic areas of activation of the CM genes Bmp4 and Msx1 (Figure 4D, E), which corresponded to regions accumulating β-Catenin, as seen on serial sections (arrowheads, Figure 4G), and lacking the bipolar cell and photoreceptor marker Otx2 (Figure 4F) [48]. These areas also showed gross reduction in SOX2 protein (Figure 4I) and Ki67 (Figure 4H), further suggestive of loss of NR fate and consistent with a reduction in proliferative capacity in the CE. Of note, not all regions accumulating β-Catenin activated expression of CE genes; however, upon close examination, it was evident that SOX2 and Ki67 were excluded from β-Catenin-accumulating cells (arrows, Figure 4J-O). Taken together, these data suggest that WNT pathway activation can override establishment of NR fate even in cells initially expressing Sox2. Despite WNT activation being a consequence of SOX2 loss, stable expression of Ctnnb1 resulted in a subsequent reduction in SOX2 expression.
Figure 4.

Expression of an activated mutant form of Ctnnb1 in the neural retina (NR) induces ciliary epithelium (CE)- like cell fate transformation. Transverse sections of mutant eyes induced with tamoxifen to express Ctnnb1 lox(ex3) in Sox2-expressing cells at E11.5 were analyzed at E15.5. (A-I) In situ hybridization for Bmp4 (A, D) and Msx1 (B, E) shows specific expression of these CE genes in ectopic regions, which accumulate β-Catenin in serial sections (G). Loss of NR fate is evident through in situ hybridization for Otx2 (F) normally marking the NR (C). Yellow and white arrowheads indicate two regions of β-Catenin accumulation across the different markers in serial sections (D-I). The cell fate transformation is accompanied by loss of proliferative capacity as seen by anti-Ki67 immunofluorescence (H) and loss of SOX2 (I). (J-O) Double immunofluorescence for β-Catenin and Ki67 (J-L) and β-Catenin together with SOX2 (M-O) reveal that areas of β-Catenin accumulation in induced Sox2 CreERT2/+;Ctnnb1 lox(ex3)/+ retinas are accompanied by the loss of proliferative capacity and the specific reduction of SOX2 indicating loss of NR fate. Two representative regions of β-Catenin accumulation are indicated across the three markers with yellow arrows (panels J- O). Scale bars: 200 μm.
Attenuation of WNT signaling does not alter RPC proliferation
Down-regulation of WNT signaling was not sufficient to improve Sox2-mutant microphthalmia (Figure 3). Moreover, activation of WNT signaling led to a simultaneous reduction in both SOX2 and Ki67 expression (Figure 4). We therefore hypothesized that SOX2 regulation of RPC proliferation occurs independently of WNT activation. This contrasts the finding that WNT signaling promotes proliferation of embryonic stem cell-derived RPCs upstream of Sox2 [49]. Alternatively, widespread WNT activation in vivo at an early developmental time point (E11.5) may have a different effect than that of WNT activation in vitro in differentiating neurons. To analyze the roles of SOX2 and WNT signaling in RPC proliferation in vivo, we quantified BrdU incorporation in single-mutant, double-mutant and control eyes over two hours following a single injection at E14.5 (Figure 5).
Figure 5.

Sox2- ablated optic cup progenitor cells (OCPCs) exhibit Ctnnb1- independent proliferation defects. BrdU incorporation of cycling cells was quantified in single-mutants, double-mutants and wild-type controls after a single pulse at E14.5, and OCPC proliferation in wild-type controls and Sox2 single-mutants was assessed at 3 developmental time-points. (A) No significant difference in the BrdU/Ki67 index between wild-types and Ctnnb1 lox(ex2–6)/lox(ex2–6) single-mutants at E14.5 is detected, but Sox2 single-mutants show significantly reduced BrdU incorporation. There is no significant difference in BrdU incorporation between Sox2 single-mutants and Sox2/Ctnnb1 double-mutants. (B-G) Incorporation of BrdU and concomitant staining for phospho-Histone-H3 between E14.5 and E18.5. Normal ciliary epithelium (CE) development from E14.5-E18.5 is characterized by a gradual reduction in BrdU incorporation over time (B-D, white brackets in C and D). In Sox2 single-mutants, the peripheral BrdU-negative region expands over time (E-G, white brackets in F and G). (H-J) Ki67 is maintained in the peripheral optic cup (OC) of controls at E14.5 (G) but is increasingly reduced at E16.5 (H) and E18.5 (I). (K-M) Ki67 expression in the peripheral OC of Sox2-mutants is similar to that of controls at E14.5 (J) but is increasingly reduced in the periphery at E16.5 (K) and E18.5 (L). Insets in (A -F) show GFP staining to indicate where Cre was expressed. Scale bars: 50 μm.
By two-factor analysis of variance (ANOVA), Sox2 deletion showed a significant effect on percent BrdU incorporation (51.3% and 56.8% for wild-type controls and Ctnnb1 single-mutants versus 20.3% and 26.9% for Sox2 single-mutants and double-mutants, respectively), F(1,15) = 114.95, P < 0.001 (Figure 5A). Genetic deletion of Ctnnb1 alone had no significant effect, F(1,15) = 0.52, P > 0.05. Sox2/Ctnnb1 double-mutants did not show a significant difference in BrdU incorporation compared with Sox2 single-mutants, suggesting that β-Catenin-mediated WNT signaling may not act in synergy with SOX2 in the regulation of RPC proliferation, F(1,15) = 4.41,P > 0.05. Together, these data demonstrate that removing Ctnnb1 has little effect on RPC proliferation in both wild-type and Sox2-mutant contexts.
Ablation of SOX2 leads to increased cell cycle time
Cell cycle exit and the onset of neuronal differentiation are tightly coupled. Given the proliferation and neurogenesis defects of the Sox2-mutant OC, we further characterized cell cycle dynamics of control and mutant eyes by examining the patterns of BrdU incorporation, phospho-Histone-H3 and Ki67 expression in wild-type and Sox2-mutant eyes over time (E14.5-E18.5; Figure 5B-M). BrdU incorporation was observed in RPCs from the center to the periphery of control OCs; however, at E14.5, fewer cells in the periphery of Sox2-mutant OCs incorporated BrdU compared with controls (Figure 5E versus B). This peripheral region of decreased BrdU incorporation expanded centrally at E16.5 and E18.5 (Figure 5F, G versus C, D). By contrast, at E14.5, there was no difference in the pattern of Ki67 expression between control and mutant OCs (Figure 5K versus H). However, at E16.5, the Ki67-depleted peripheral region was expanded in mutants compared with controls (Figure 5L versus I), and this expansion further increased by E18.5 (Figure 5M versus J). By P0, the Ki67-negative region was extended far into the center of the Sox2-mutant OC (Additional file 4A-C versus D-F, area between arrowheads). Notably, some central Sox2-ablated cells still expressed Ki67 at P0, as did wild-type CE cells just adjacent to the NR of control eyes (Additional file 4B, C versus E, F arrows).
These observations raise two non-mutually exclusive possibilities: 1) Sox2-mutant cells prematurely exit the cell cycle and/or 2) increase total cell cycle length in a peripheral-to-central gradient. To more precisely characterize the cell cycle dynamics of Sox2-ablated cells, we quantified the lengths of S-phase (TS) and cell cycle (TC) at E14.5 and E16.5 by pulse-chase labeling with thymidine analogs (Figure 6A-J) [50]. At E14.5, there was a marked difference in the TS of central mutant RPCs compared with controls (24.08 versus 16.30 hours, respectively, P = 0.0002; Figure 6K). TC was also increased in mutants compared with controls (51.84 versus 25.51 hours, respectively, P = 0.00002). At E16.5, there was an additional difference in cell cycle dynamics within Sox2-mutant RPCs but not within controls. More peripheral RPCs of mutants appeared to cycle more slowly than those toward the center of the OC. We therefore divided the E16.5 mutant OC into 'central' and 'peripheral' bins to quantify the difference in cell cycle rate between the two regions (Figure 6L). Notably, for central RPCs, the difference in TS between control and GFP+ mutant cells was not significant (15.08 versus 15.16 hours, respectively, P = 0.9), but the difference in TC was significant (23.68 versus 27.22 hours, respectively, P = 0.007). Moreover, both TS and TC were significantly longer in peripheral RPCs of mutants compared with controls (TS: 19.65 versus 15.08 hours, respectively, P = 0.0002 and TC: 51.44 versus 23.68 hours, respectively, P = 0.002).
Figure 6.
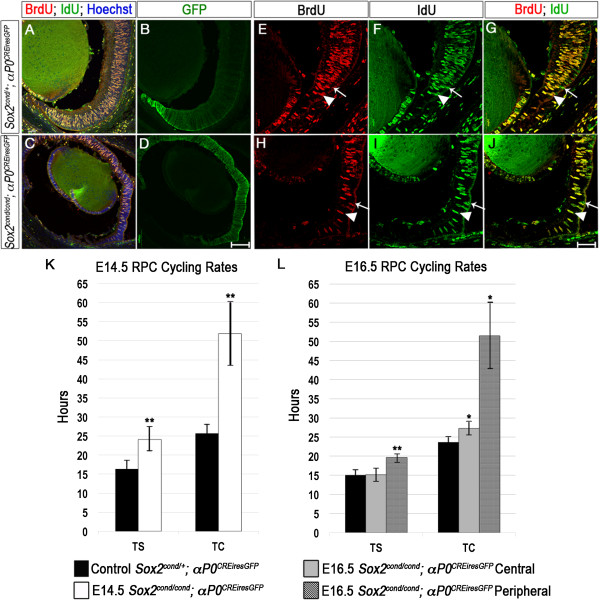
Sox2- mutant retinal progenitor cells (RPCs) take longer to complete the cell cycle. TS and TC were quantified in control and Sox2 single-mutant RPCs at E14.5 and E16.5. (A-J) Co-labeling with BrdU and IdU indicates which cells have finished S-phase during the protocol (green only, arrows) and which cells remain in S-phase (yellow, arrowheads) in wild-type (A-G) and mutant (C-J) optic cups (OCs). (K, L) Both TS and TC are significantly longer in mutant RPCs compared with controls at E14.5 (K). AT E16.5, TS and TC vary between central and peripheral mutant RPCs (L). TS is significantly longer in peripheral RPCs of mutants compared with controls. TC is significantly longer in both central and peripheral RPCs of mutants compared with controls. (E-J) are high power magnifications of (A-D). Scale bars: D (for A-D): 100 μm; J (for E-J): 50 μm.
Taken together, these findings suggest that Sox2-mutant RPCs increase cell cycle time and exit the cell cycle in a graded manner from the periphery to the center. At E16.5, central mutant RPCs appear to be unaffected by, or perhaps compensate for, loss of SOX2 and progress through S-phase at the proper rate.
D-type Cyclins are increased in Sox2-mutant cells
Despite losing neural competence, Sox2-mutant RPCs continue to cycle well after deletion of Sox2. We next asked how RPC proliferation is supported in the absence of SOX2. D-type Cyclins are expressed in RPCs and promote progression through the cell cycle [51–53]. We examined expression of CyclinD1 and CyclinD3 protein by immunofluorescence in control and mutant OCs at E16.5 and P0. In control embryos, CyclinD1 was localized to the prospective NR, with higher levels at the boundary of the NR and CE and extending into the presumptive CE (Figure 7A, B arrowheads) as observed in zebrafish [11]. Notably, CyclinD1 protein was increased in some Sox2-negative (GFP-positive) RPCs (Figure 7D, E arrowheads). This increase appeared cell-autonomous in regions where Cre expression was mosaic (Figure 7C, F arrowheads). CyclinD3 protein was confined to the prospective CE of controls but expanded into the central OC of mutants (Figure 7G, H versus J, K). Co-labeling with CyclinD1 and CyclinD3 indicated that these two proteins, which are normally mutually exclusive, were aberrantly co-expressed in some Sox2-depleted cells (Figure 7M-P; arrows) [53].
Figure 7.
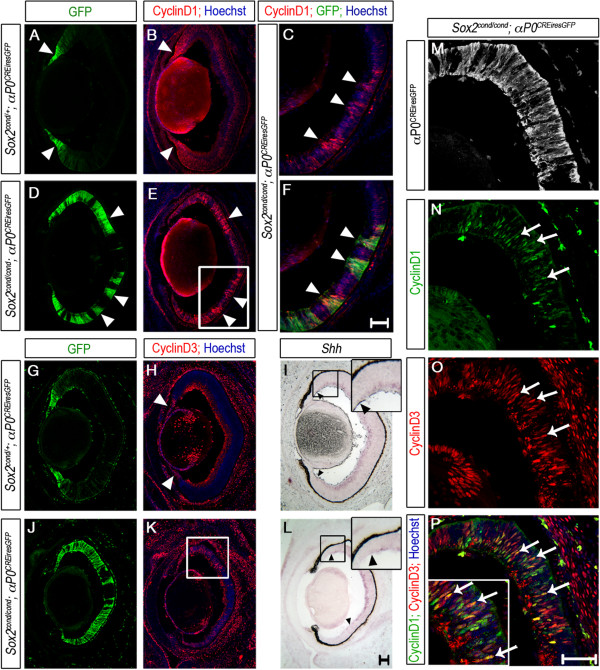
D- type Cyclins are aberrantly expressed in Sox2- ablated optic cup progenitor cells (OCPCs) at E16.5. Expression of D-Cyclins and Shh was assayed on transverse sections through E16.5 eyes. (A-F) CyclinD1 protein is increased in GFP-positive/Sox2-ablated cells (C-F arrowheads) compared with controls (A, B). High power magnification of the boxed area in (E) shows cell-autonomous up-regulation of CyclinD1 (C, F). CyclinD1 protein is also elevated in the peripheral NR of controls (arrowheads in A, B). (G- H, J- K) CyclinD3 is restricted to the peripheral OC/prospective ciliary epithelium (CE) of controls (G, H, arrowhead) and is ectopically expressed in central mutant OCPCs (box in K), supporting NR-to-CE cell fate conversion. Note that speckled staining in the inner region of the optic cup visible in (H, K, O, P) is non-specific background. (I, L) Shh mRNA expression is decreased in central OCPCs of mutants (L) compared with controls (I). (M-P) High power magnification of the boxed area in (K) shows that CyclinD1 and CyclinD3 are aberrantly co-expressed in some Sox2-mutant central OCPCs (arrows in P). Black arrowheads in (I) and (L) indicate the extent of Shh expression. The boxed regions in (I) and (L) are magnified twice in the inserts in each panel. Scale bars: 50 μm.
Sonic Hedgehog (SHH) regulates RPC proliferation in part through CyclinD1 [54–57]. To address whether an increase in SHH at E16.5 could explain the increase in CyclinD1 protein, we analyzed Shh mRNA levels by microarray and ISH. Shh expression was decreased 4.5-fold in mutant retinas compared with controls (P = 0.0014), and the decrease was confirmed by ISH (Figure 7I versus L). The primary source of SHH in the developing retina is thought to be retinal ganglion cells (RGCs), as we have also shown by ISH at E13.5 [58] (Figure 2). Given that Sox2-ablated OCs generate far fewer RGCs and thus have decreased Shh expression, elevated CyclinD1 protein in Sox2-ablated cells is most likely driven by an additional mitogenic pathway not involving SHH.
CyclinD1 expression in the retina is independent of WNT and PAX6 signaling
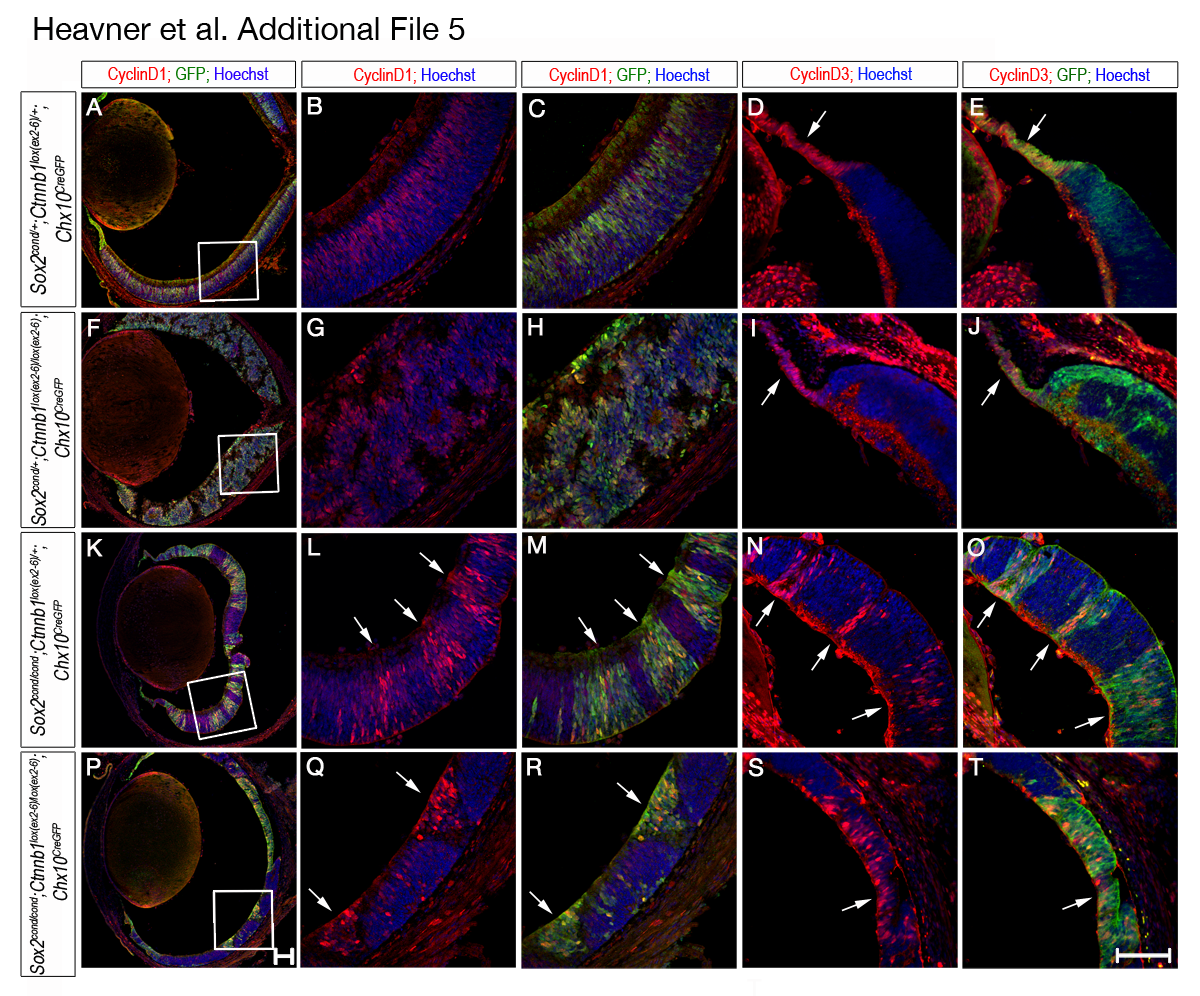
We next queried if aberrant CyclinD1 expression is due to WNT activation given that Ccnd1 is a major transcriptional target of β-Catenin [59]. To address whether ectopic WNT activity drove the increase in D-Cyclins observed in Sox2-mutant OCs, we examined expression of D-Cyclins in single-mutant, double-mutant and wild-type control eyes at P0 (Additional file 5). CyclinD1 was expressed in GFP-positive central OCPCs in wild-type and Ctnnb1 single-mutant eyes (Additional file 5A-B, E-F, I- J). As expected, CyclinD1 protein was increased in central RPCs of P0 Sox2 single-mutants (Additional file 5C, G, K; arrows). Moreover, CyclinD1 remained increased even after deletion of Ctnnb1 (Additional file 5D, H, L; arrows), suggesting that retinal CyclinD1 levels are completely independent of Ctnnb1. CyclinD3 protein was restricted to the CE of both wild-type and Ctnnb1 single-mutant eyes at P0, and it was ectopically expressed in the central OC of Sox2 single-mutants and Sox2/Ctnnb1 double-mutants (Additional file 5M-T). Collectively, these results suggest that SOX2 antagonizes D-Cyclins in the OC independently of WNT/β-Catenin signaling.
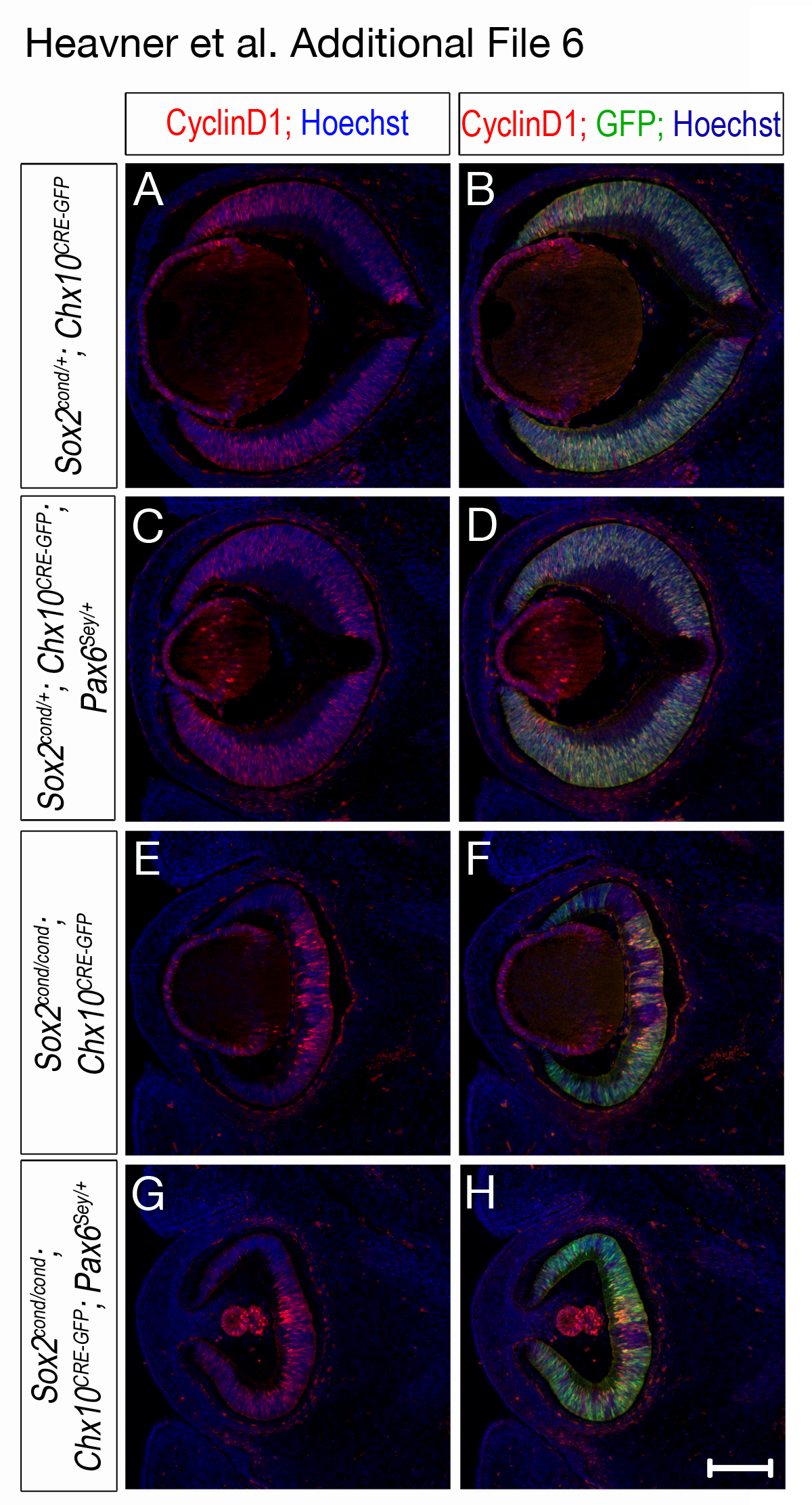
The paired-box transcription factor PAX6 is crucial for maintaining RPC proliferation and multipotency [60, 61]. Despite increased cell cycle exit and decreased Ccnd1 transcription in Pax6-depleted cells, CyclinD1 protein remained elevated, suggesting that PAX6 regulates CyclinD1 expression post-transcriptionally or post-translationally. This study thus revealed a complex role for PAX6 in the regulation of RPC proliferation and CyclinD1 expression [62]. To test the alternative hypothesis, that increased PAX6 protein in the Sox2-ablated OC was responsible for the elevated CyclinD1, we genetically decreased Pax6 in Sox2-ablated RPCs using Pax6Sey/+ (small eye) mice as previously described [5]. The Pax6Sey/+ allele contains an early nonsense mutation in the Pax6 locus, resulting in a truncated protein widely used as a Pax6-null [63, 64]. We compared CyclinD1 expression in Sox2cond/cond;Chx10CreGFP single-mutant OCs with Sox2cond/cond;Pax6Sey/+;Chx10CreGFP double-mutant OCs and Pax6Sey/+ single-mutant and wild-type controls at E14.5 (Additional file 6). As expected, CyclinD1 protein was elevated in central Sox2 single-mutant cells when compared with wild-type and Pax6 single-mutant cells (Additional file 6E, F versus A-D). Moreover, CyclinD1 protein remained up-regulated in Pax6/Sox2 double-mutant OCPCs in the central OC. Elevated CyclinD1 protein was therefore independent of PAX6 expression in the Sox2-mutant OC (Additional file 6G, H).
Collectively, our data indicate that loss of Sox2 in the developing OC resulted in a prolonged cell cycle and eventual cell cycle exit. During mid-retinogenesis (E16.5), centrally located Sox2-mutant OCPCs appeared to cycle at the normal rate and exhibited WNT- and PAX6-independent increased CyclinD1. By contrast, the NR-to-CE primordium cell fate conversion observed upon ablation of Sox2 was regulated by WNT/β-Catenin signaling. The prevention of ectopic CE/CM gene expression by removal of Ctnnb1 from Sox2-mutant cells, but not of increased D-Cyclin proteins, supports the conclusion that β-Catenin is a major regulator of CE fate but not of OCPC proliferation.
Discussion
SOX2 regulates the neurogenic/non-neurogenic boundary of the retina through modulation of WNT signaling
SOX proteins have been shown to physically interact with β-Catenin to modulate the expression of WNT target genes [24, 38–40]. The SOX2 HMG domain is not required to inhibit β-Catenin-induced gene expression in osteoblasts, suggesting that SOX2 antagonizes WNT signaling through its C-terminus, most likely via β-Catenin sequestration [24]. Alternatively, SOX2, distantly related to the TCF/LEF family of transcription factors, which also bind DNA via an HMG domain, could compete with TCF/LEF for direct DNA binding [65].
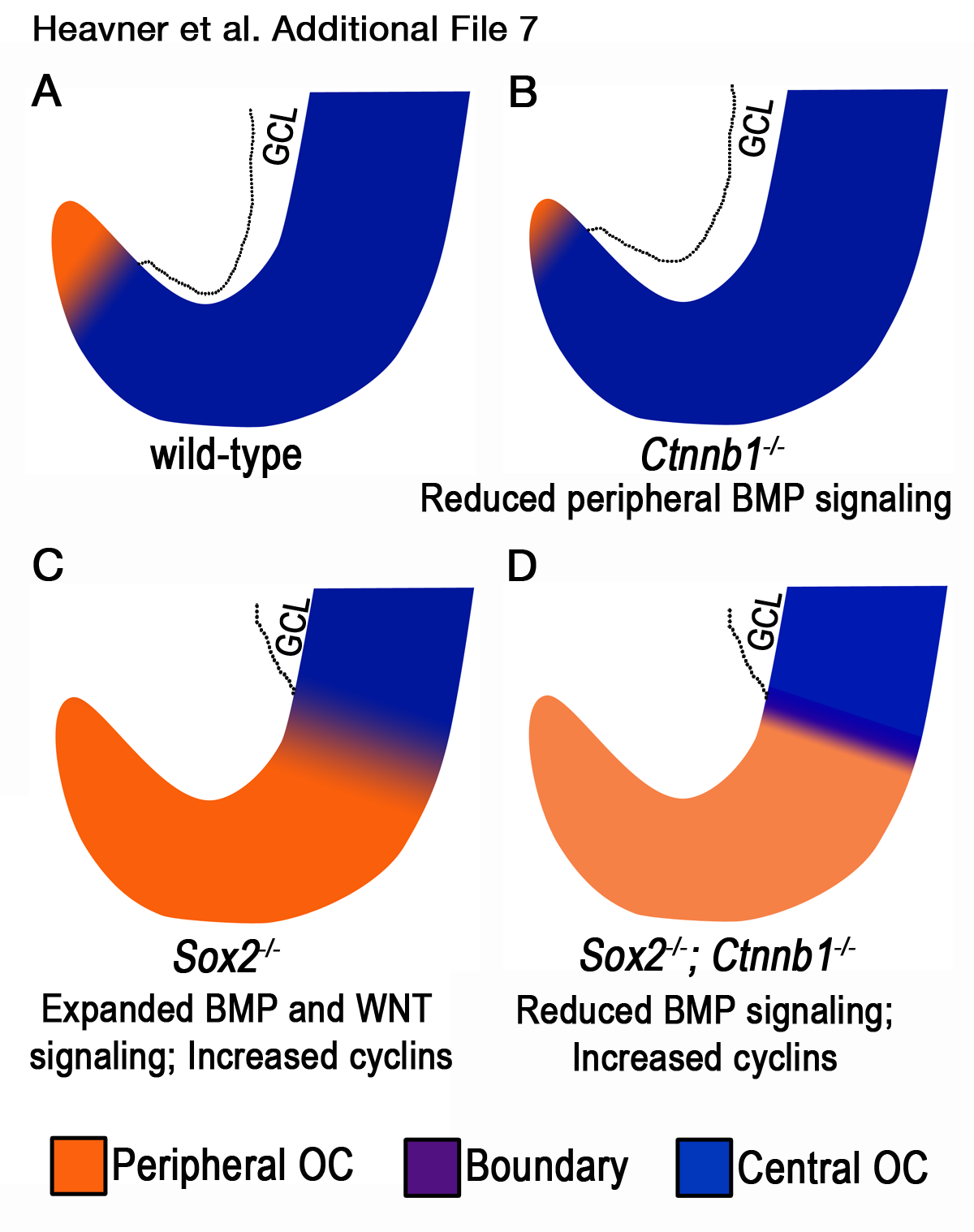
Our data support a model in which SOX2 and WNT signaling intersect at the level of target gene expression. It is still unclear, however, how SOX2 antagonizes CE fate. Using genetic epistasis analysis, we demonstrated that SOX2 and canonical WNT signaling coordinate opposing OC fates (modeled in Additional file 7). Both Sox2 LOF and Ctnnb1 GOF resulted in expansion of CE at the expense of NR, which is a direct consequence of elevated WNT signaling, as we find an up-regulation of WNT response in Sox2 LOF mutants. In line with this, Ctnnb1 LOF resulted in partial restoration of NR fates in Sox2 LOF eyes, where CE-specific genes failed to become up-regulated, demonstrating that regulation of CE identity by SOX2 is mediated by WNT signaling. However, the neurogenesis defect observed in Sox2 LOF mutants was not restored by limiting activation of the WNT pathway, suggesting a WNT-independent role of SOX2 in the regulation of neurogenic fates. Our data reveal that precise coordination of SOX2 and WNT signaling is necessary to ensure proper development of the neurogenic retina and circumjacent CB.
SOX2 regulates the cell cycle independently of the WNT pathway
Genetic ablation of Sox2 in the OC resulted in prolonged RPC cell cycle times. This increase is in line with a fate transition of the NR to CE, where cells have a longer cell cycle [4, 7]. In the Xenopus OC, SOX2 was found to be necessary for both proliferation and neural competence, agreeing with the findings of our study [17]. SOX2 was shown to perform this function downstream of WNT while also feeding back on the WNT pathway to inhibit it, thus painting a more complex picture of WNT-SOX2 interactions in OC development [17, 25].
Our findings indicate that even a modest increase in WNT activity upon SOX2 inactivation is associated with reduced proliferation. Reduced proliferation following WNT activation has been seen previously [8, 11, 66] and aberrant WNT activity was associated with restricted regenerative capacity in the planarian Procotyla fluviatilis [67]. Despite WNT activation in Sox2-mutant OCs, the cell cycle defect of Sox2-mutant cells was not rescued through deletion of Ctnnb1 and successful down-regulation of the WNT pathway. Conversely, expression of activated Ctnnb1 in Sox2-expressing RPCs promoted CE fate and reduced Ki67 only after a dramatic loss of SOX2 in cells accumulating β-Catenin, which was not investigated in previous reports [8, 11, 25]. Our experimental evidence thereby supports a model in which SOX2 regulates RPC proliferation independently of WNT signaling in the mouse.
The Sox2-mutant phenotype suggests complex regulation of CyclinD1 by SOX2
It was somewhat surprising that RPCs continued to proliferate well after loss of SOX2. One possible explanation for this sustained proliferation is that stabilized D-Cyclins permitted OCPCs to progress through the cell cycle. These data raise an additional contradiction: increased Cyclins would not be expected to correlate with the observed increase in cell cycle time and decreased BrdU incorporation. Nonetheless, in support of our finding, at least one other instance of increased CyclinD1 protein and decreased proliferation has been observed in the OC: Pax6-ablated OCPCs showed elevated CyclinD1 protein and increased cell cycle exit. In this context, CyclinD1 was not sufficient to maintain the proliferation of Pax6-ablated cells, since the CyclinD1-positive cells did not incorporate BrdU [62]. Thus, CyclinD1 expression may not indicate sustained proliferation. Moreover, Sox2-ablated cells undergo a cell fate transformation from a more proliferative to a less proliferative cell type (NR-to-CE). The presence of high CyclinD1 protein at the CM at E16.5 (Figure 7b, arrowhead) raises the possibility that cells normally undergoing the NR/CE cell fate decision have elevated CyclinD1 levels [68].
The decrease in Ccnd1 mRNA suggests that CyclinD1 transcription is positively regulated by SOX2, as SOX2 has been shown to be capable of transactivating the Ccnd1 promoter in vitro [39]. The unexpected negative regulation of CyclinD1 protein may take place through an alternative cascade influenced by SOX2. In human embryonic stem cells (ESCs), SOX2 and OCT4 promote the expression of miR-302, a cluster of 8 microRNAs expressed in pluripotent cells, and miR-302a post-transcriptionally represses Ccnd1 [69]. Moreover, OC-specific ablation of Dicer1, a major mediator of microRNA biosynthesis, resulted in increased CyclinD1 protein in mouse OCPCs, creating a persistent CM-like region [70]. Therefore, even with decreased Ccnd1 transcription, CyclinD1 protein could be stabilized upon the loss of microRNA biosynthesis influenced by the lack of SOX2.
Lastly, SOX2 could antagonize CyclinD1 via direct regulation of the cell cycle inhibitor p27Kip1, as has been observed in inner pillar cells of the mouse auditory sensory epithelium [71]. Indeed, p27Kip1 protein was absent in Sox2-null RPCs (WEH, unpublished observations). In agreement with this notion, genetic ablation of Sox2 specifically in the developing pituitary gland led to a severe reduction in proliferative capacity of pituitary embryonic precursors, although p27Kip1 expression was elevated rather than repressed [72].
Canonical WNT signaling acts upstream of BMPs to specify CE fate
A role for BMP signaling in CE specification has been demonstrated, although the specific sources of ocular BMPs are currently unclear [34, 35, 45, 73]. Results from various studies suggest that the developing lens may instruct the peripheral OC margin to become CE [35, 73]. However, the CE is correctly specified in the absence of a lens, suggesting that the lens may be more important for maintaining CE fate [10, 74]. Our data suggest that WNT signaling within the OC acts upstream of BMP signaling such that expression of Bmp4/7 and the BMP target gene Msx1 are lost in Ctnnb1-mutant cells and are up-regulated with stabilization of the pathway.
The Sox2-deficient OC provides a resource to identify genes important for CE development
Downstream of BMP and WNT signaling, much of CE development remains a mystery. Many of the significantly up-regulated genes in this study were associated with ion exchange and secretion (Figure 1). Up-regulation of these groups of genes is consistent with the function of the CB to maintain constant intraocular pressure (IOP) through the active transport of fluid via Na+-K+ exchange pumps and Cl- channels [30]. Given that the presumptive CB epithelium significantly expands upon Sox2 ablation, our genome-wide screen may reveal previously unidentified genes important for the function of the CB.
Conclusions
Our study demonstrates that SOX2 regulates the neurogenic boundary of the retina in part through mediation of canonical WNT signaling, such that genetic removal of Ctnnb1 from Sox2-mutant eyes partially restores neural retina cell fate. By contrast, Sox2, but not Ctnnb1, is required to maintain RPC proliferation. The increase in total cell cycle time of Sox2-mutant RPCs may explain how mutations in human SOX2 contribute to microphthalmia.
Methods
Animals
All animal work was carried out in compliance with the University of North Carolina Institutional Animal Care and use committee (IACUC) policies and ethical approval by the Division of Laboratory Animal Medicine (DLAM) at The University of North Carolina at Chapel Hill. Generation of the Sox2Cond/+ mouse line was described previously [16]. Sox2cond/+ and Sox2cond/+;αP0CREiresGFP mice were maintained on a C57BL/6 J background, and all others described in this study were maintained on a mixed background containing 129/Sv and C57BL/6 J. αP0CREiresGFP [60], Chx10CreGFP [42], Ctnnb1lox(ex2–6)/+ [41], ROSA26Reporter [43], Pax6Sey/+ [64], Ctnnb1lox(ex3)/+ [75] and Sox2CreERT2/+ [47] mice have been previously described. For CreERT2 activation, pregnant dams received a single injection of tamoxifen totaling 1.5 mg, and simultaneous injection with 0.75 mg progesterone (both Sigma-Aldrich, St Louis, MO, USA) to reduce the risk of spontaneous abortion.
Tissue preparation, immunostaining and in situhybridization
Immunostaining was performed as previously described [5]. Additional antibodies used in this study include CyclinD1 (1:400, Thermo Scientific, Waltham, MA, USA), CyclinD3 (1:100, Cell Signaling, Beverly, MA, USA), β-Catenin (1:250, Abcam, Cambridge, MA, USA) and β-galactosidase (1:10,000 Molecular Probes, Eugene, OR, USA). Antigen retrieval was used for antibodies against BrdU, CyclinD3 and phospho-Histone-H3 as previously described [76]. In situ hybridization was performed on 14-μm frozen sections as described [5]. The following probes were used: Bmp4, Msx1, Ccnd1 3’UTR, Shh, Otx2 and Hes5 (kind gifts from Dr A LaMantia, Dr Y Liu, Dr C Cepko, Dr E Tucker, Dr JP Martinez-Barbera and Dr E Anton, respectively). Images were captured on a Leica inverted microscope (Leica DMIRB, Leica Microsystems GmbH, Germany) equipped with a Retiga (SRV-1394, QImaging, BC, Canada) camera or on an Olympus laser scanning confocal microscope (Olympus Fluoview FV1000, Olympus America, PA, USA) and processed using Adobe Photoshop software. Unless otherwise stated, a minimum of three samples per genotype were analyzed for each assay.
BrdU and IdU labeling
Pregnant dams (E14.5 or E16.5) were injected intraperitoneally with 120 mg IdU per kg body weight at time (T) 0. At T = 1.5 hours, dams were injected with 100 mg BrdU (Sigma B5002, Sigma-Aldrich, St Louis, MO, USA) per kg body weight. At T = 2 hours, dams were culled and embryos were fixed in cold 4% PFA. A mouse antibody against IdU and BrdU (BD) detected both thymidine analogs, and a rat antibody specific to BrdU (Serotec, Raleigh, NC, USA) detected BrdU alone [50].
Cell counting, S-phase and cell cycle measurements
For each eye, a section through the middle of the eyecup was imaged using an Olympus Fluoview FV1000 confocal microscope (Olympus America, PA, USA). Four eyes from separate animals were collected for each genotype, for each stage. For E16.5 eyes, the prospective NR was divided into 2 bins (central and peripheral) using Olympus Fluoview 2.1c software, totaling to 2 regions per section, covering the entire prospective retina. Note, for all proliferation experiments in this study (Figures 5 and 6), we quantified data for RPCs (central OCPCs) in controls and mutants. Data for prospective CE progenitor cells (peripheral OCPCs) were too variable to support any reliable conclusions. In wild-type eyes, all proliferating cells were counted. In mutant eyes, only GFP-positive proliferating cells were counted (ranges E14.5 1,200 to 2,400 cells per section, E16.5 1,500 to 3,200 cells per section). At least 3 samples from each genotype were used in the final calculations at E16.5. For studies at E14.5, we analyzed 3 wild-type controls, 4 each from single mutant genotypes and 6 double-mutants. To determine the effect of Sox2 and Ctnnb1 on BrdU index, a two-factor ANOVA was performed. To determine the significance of differences in cell cycle times between controls and Sox2 single-mutants, a Student’s t-test was carried out. P-values less than 0.05 were considered significant, and those less than 0.001 were considered highly significant. The following formula was used to calculate BrdU index:
S-phase and cell cycle times were calculated as described [50]. The ratio of 'S' cells (BrdU/IdU double-positive) to 'L' cells (IdU-only) was measured, and the following formulae were used:
Whole-genome expression analysis
Pairs of eyes were enucleated from 6 wild-type (Sox2cond/+;αP0CREresGFP) and 6 mutant (Sox2cond/cond;αP0CREresGFP) embryos at E16.5 (for a total of 12 eyes per genotype). Each pair of eyes was immediately placed in RNA-Later RNA stabilization solution and stored at 4°C for no longer than 30 days. RNA was purified using an Ambion (Foster City, CA, USA) RNaqueous kit following the manufacturer’s instructions. Five hundred nanograms of RNA from each pair of eyes were amplified and biotin-labeled using an Ambion Illumina (San Diego, CA, USA) TotalPrep RNA Amplification kit. One and a half micrograms of labeled cRNA were hybridized to Illumina (San Diego, CA, USA) mouse WG-6 expression bead chips (six replicate arrays per genotype, for twelve arrays total). Microarray data were generated by Expression Analysis (Durham, NC, USA). Briefly, to detect significant changes in gene expression between controls and mutants, a permutation analysis for differential expression (PADE) was performed. PADE repeatedly randomly reassigns each sample to one of the two groups to determine if the calculated difference in levels of a transcript is non-random. PADE was also used to estimate the FDR for sets of differentially expressed transcripts. A P-value for each transcript was calculated using a two-sample t-test. Transcripts with an accumulated false discovery rate (FDR) of less than 5%, a P-value of less than 0.05, and a fold change of greater than ±1.6, totaling 2,194 transcripts, were chosen for further analysis. Functional gene annotation was identified using DAVID [28, 29] and potentially affected pathways were determined using Ingenuity Pathway Analysis (Qiagen, Redwood City, CA, USA).
Electronic supplementary material
Additional file 1: Significantly changed optic cup (OC) WNT target genes. Thirteen known OC WNT target genes were found to be changed at least 1.47-fold (log ratio of at least ±0.55) in mutant compared with control eyes. (XLSX 640 KB)
Additional file 2: Functional Annotation of the Sox2-ablated optic cup (OC) reveals cell fate change. Functional terms significantly enriched for up-regulated genes (first tab) and down-regulated genes (second tab) resulting from optic cup-specific ablation of Sox2. (DOCX 51 KB)
{kind=link}
Additional file 3: Chx10 CreGFP efficiently ablates Sox2 and Ctnnb1 from the optic cup (OC) without causing increased cell death. The efficiency of Chx10 CreGFP and cell death were assessed in double-mutants at E14.5. (A-B, D-E) SOX2 and β-Catenin are absent from CreGFP-positive cells in double-mutants (C,D) compared with controls (A,B). (C,F) Cleaved Caspase 3 is similar between controls and mutants. Scale bar: 200 μm. (PNG 1 MB)
{kind=link}
Additional file 4: Sox2-deficient OCPCs prematurely exit the cell cycle. Cycling cells in neonatal controls and mutants were identified by Ki67 staining. (A-F) In controls, Ki67 is expressed in RPCs (red in A) and in some GFP-positive CE cells adjacent to the NR (B-C; arrows) but not in the most peripheral CE cells (B-C; arrowheads). In mutants, Ki67 is expressed in centrally located Sox2-ablated RPCs (E-F; arrows) but not in peripheral Sox2-ablated RPCs (E-F; arrowheads). Boxed areas in (A) and (D) are magnified in (B-C) and (E-F), respectively. Scale bars: 100 μm. (PNG 776 KB)
{kind=link}
Additional file 5: Deletion of Ctnnb1 in Sox2-ablated optic cup progenitor cells (OCPCs) does not rescue increased CyclinD1 or ectopic expression of CyclinD3. CyclinD1 and CyclinD3 expression were analyzed in single-mutants, double-mutants and controls at P0. CyclinD1 is restricted to retinal progenitor cells (RPCs) in controls (A-C) and Ctnnb1 single-mutants (F-H). CyclinD1 is increased in GFP-positive cells in the central OC of Sox2 single-mutants (K-M; arrows) and in Sox2/Ctnnb1 double-mutants (P-R; arrows). CyclinD3 is restricted to the CE of controls (D, E; arrows) and Ctnnb1 single-mutants (I, J; arrows) and ectopically expressed in GFP-positive cells in the central OC of Sox2 single-mutants (N, O; arrows) and Sox2/Ctnnb1 double-mutants (S, T; arrows). Boxed areas in (A, F, K and P) are magnified in (B, C; G, H; L, M; and Q, R), respectively. Scale bars: 100 μm. (PNG 1 MB)
{kind=link}
Additional file 6: Reduction of Pax6 in Sox2-ablated optic cup progenitor cells (OCPCs) does not rescue increased CyclinD1. CyclinD1 was assayed on transverse sections through the eyes of Sox2 single-mutants, Pax6 Sey/+ single-mutants, double-mutants and wild-type controls at E14.5. (A-D) CyclinD1 is expressed in retinal progenitor cells (RPCs) of controls (A, B) and Pax6 Sey/+ single-mutants (C, D). (E-H) CyclinD1 is increased in central OCPCs of Sox2 single-mutants (E, F) and Sox2/Pax6 Sey/+double-mutants (G, H). Scale bar: 200 μm. (PNG 3 MB)
{kind=link}
Additional file 7: Model of how SOX2 and canonical WNT signaling regulate the neurogenic boundary of the optic cup (OC). OC-specific genetic ablation of Sox2 or Ctnnb1 results in complementary phenotypes. (A-D) The boundary between neural retina (NR) (blue) and ciliary epithelium (CE) (orange) is shifted peripherally in Ctnnb1 single-mutants (B) compared with controls (A). Conversely, the boundary between NR and CE is shifted centrally in Sox2 single-mutants such that WNT and BMP signaling are expanded (C) compared with controls (A). The boundary between the NR and CE remains centrally shifted in Sox2/Ctnnb1 double-mutants (D). However, BMP signaling and other classical CE markers fail to be expressed in this expanded CE-like region. D-type cyclins are increased in both Sox2 single-mutants and Sox2/Ctnnb1 double-mutants. (PNG 167 KB)
Acknowledgments
Work in the LHP laboratory is supported by NIH (NIMH - R01MH071822 and NEI - R01EY018261), in the UNC Neuroscience in situ Core by NIH (NINDS - P30-S045892-02) and in the UNC Neuroscience Multiphoton and Confocal Imaging Core by NIH (5P30NS045892). The authors would like to thank the UNC Neuroscience Center, particularly Dr Vladimir Ghukasyan for help with confocal microscopy, Dr Megumi Aita (UNC) and Gabriela Carreno (UCL) for help with in situ hybridizations. We would also like to thank Dr Scott Magness, Dr Natalia Surzenko and Dr Constance Cepko for graciously sharing reagents. For critical reading of the manuscript, we extend deep gratitude to Dr Juan Pedro Martinez-Barbera, Dr Dino Leone and Dr Susan McConnell. For continued scientific discussions, camaraderie and support, we thank past and present members of the Pevny lab.
Dedication
This manuscript is dedicated to our beloved advisor, mentor and friend, Dr Larysa Pevny. Her devotion will be forever in our hearts and rigor forever in our minds.
Abbreviations
- ΑΝΟVΑ
analysis of variance
- β-gal
β-galactosidase
- BAC
bacterial artificial chromosome
- BrdU
bromodeoxyuridine
- CB
ciliary body
- Ccnd1
CyclinD1
- Ccnd3
CyclinD3
- CE
ciliary epithelium
- CM
ciliary margin
- Ctnnb1
β-Catenin
- E
embryonic day
- ESCs
embryonic stem cells
- FDR
false discovery rate
- GFP
green fluorescent protein
- GOF
gain-of-function
- IdU
iododeoxyuridine
- IHC
immunohistochemistry
- ISH
in situ hybridization
- IOP
intraocular pressure
- LOF
loss-of-function
- NR
neural retina
- OC
optic cup
- OCPC
optic cup progenitor cell
- P
postnatal day
- PADE
permutation analysis for differential expression
- PCR
polymerase chain reaction
- PFA
paraformaldehyde
- RGC
retinal ganglion cell
- RPC
retinal progenitor cell
- Sey
small-eye mutation
- Shh
Sonic Hedgehog
- TC
cell cycle time
- TGFβ
transforming growth factor beta
- TS
S-phase time.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LHP and WEH conceived the study and designed the experiments. WEH conducted all Sox2 and Pax6 mutant analyses, and CLA conducted the β-Catenin gain-of-function studies. WEH and CLA interpreted the data and wrote the manuscript. WEH and CLA read and approved the final manuscript.
Contributor Information
Whitney E Heavner, Email: heavner@stanford.edu.
Cynthia L Andoniadou, Email: cynthia.andoniadou@kcl.ac.uk.
References
- 1.Li H, Tierney C, Wen L, Wu JY, Rao Y. A single morphogenetic field gives rise to two retina primordia under the influence of the prechordal plate. Development. 1997;124:603–615. doi: 10.1242/dev.124.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zaghloul NA, Yan B, Moody SA. Step-wise specification of retinal stem cells during normal embryogenesis. Biol Cell Auspices Eur Cell Biol Organ. 2005;97:321–337. doi: 10.1042/BC20040521. [DOI] [PubMed] [Google Scholar]
- 3.Heavner W, Pevny L. Eye development and retinogenesis. Cold Spring Harb Perspect Biol. 2012;4:a008391. doi: 10.1101/cshperspect.a008391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beebe DC. Development of the ciliary body: a brief review. Trans Ophthalmol Soc U K. 1986;105(Pt 2):123–130. [PubMed] [Google Scholar]
- 5.Matsushima D, Heavner W, Pevny LH. Combinatorial regulation of optic cup progenitor cell fate by SOX2 and PAX6. Development. 2011;138:443–454. doi: 10.1242/dev.055178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu H, Mohamed O, Dufort D, Wallace VA. Characterization of Wnt signaling components and activation of the Wnt canonical pathway in the murine retina. Dev Dyn. 2003;227:323–334. doi: 10.1002/dvdy.10315. [DOI] [PubMed] [Google Scholar]
- 7.Cho S-H, Cepko CL. Wnt2b/beta-catenin-mediated canonical Wnt signaling determines the peripheral fates of the chick eye. Development. 2006;133:3167–3177. doi: 10.1242/dev.02474. [DOI] [PubMed] [Google Scholar]
- 8.Liu H, Xu S, Wang Y, Mazerolle C, Thurig S, Coles BLK, Ren J-C, Taketo MM, van der Kooy D, Wallace VA. Ciliary margin transdifferentiation from neural retina is controlled by canonical Wnt signaling. Dev Biol. 2007;308:54–67. doi: 10.1016/j.ydbio.2007.04.052. [DOI] [PubMed] [Google Scholar]
- 9.Trimarchi JM, Cho S-H, Cepko CL. Identification of genes expressed preferentially in the developing peripheral margin of the optic cup. Dev Dyn. 2009;238:2327–2329. doi: 10.1002/dvdy.21973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kitamoto J, Hyer J. The expression of Wnt2b in the optic cup lip requires a border between the pigmented and nonpigmented epithelium. Mol Vis. 2010;16:2701–2717. [PMC free article] [PubMed] [Google Scholar]
- 11.Stephens WZ, Senecal M, Nguyen M, Piotrowski T. Loss of adenomatous polyposis coli (apc) results in an expanded ciliary marginal zone in the zebrafish eye. Dev Dyn. 2010;239:2066–2077. doi: 10.1002/dvdy.22325. [DOI] [PubMed] [Google Scholar]
- 12.Ha A, Perez-Iratxeta C, Liu H, Mears AJ, Wallace VA. Identification of Wnt/β-catenin modulated genes in the developing retina. Mol Vis. 2012;18:645–656. [PMC free article] [PubMed] [Google Scholar]
- 13.Fu X, Sun H, Klein WH, Mu X. Beta-catenin is essential for lamination but not neurogenesis in mouse retinal development. Dev Biol. 2006;299:424–437. doi: 10.1016/j.ydbio.2006.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fotaki V, Smith R, Pratt T, Price DJ. Foxg1 is required to limit the formation of ciliary margin tissue and Wnt/β-catenin signalling in the developing nasal retina of the mouse. Dev Biol. 2013;380:299–313. doi: 10.1016/j.ydbio.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pevny L, Placzek M. SOX genes and neural progenitor identity. Curr Opin Neurobiol. 2005;15:7–13. doi: 10.1016/j.conb.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 16.Taranova OV, Magness ST, Fagan BM, Wu Y, Surzenko N, Hutton SR, Pevny LH. SOX2 is a dose-dependent regulator of retinal neural progenitor competence. Genes Dev. 2006;20:1187–1202. doi: 10.1101/gad.1407906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Raay TJ, Moore KB, Iordanova I, Steele M, Jamrich M, Harris WA, Vetter ML. Frizzled 5 signaling governs the neural potential of progenitors in the developing Xenopus retina. Neuron. 2005;46:23–36. doi: 10.1016/j.neuron.2005.02.023. [DOI] [PubMed] [Google Scholar]
- 18.Fantes J, Ragge NK, Lynch S-A, McGill NI, Collin JRO, Howard-Peebles PN, Hayward C, Vivian AJ, Williamson K, van Heyningen V, FitzPatrick DR. Mutations in SOX2 cause anophthalmia. Nat Genet. 2003;33:461–463. doi: 10.1038/ng1120. [DOI] [PubMed] [Google Scholar]
- 19.Fitzpatrick DR, van Heyningen V. Developmental eye disorders. Curr Opin Genet Dev. 2005;15:348–353. doi: 10.1016/j.gde.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 20.Ragge NK, Lorenz B, Schneider A, Bushby K, de Sanctis L, de Sanctis U, Salt A, Collin JRO, Vivian AJ, Free SL, Thompson P, Williamson KA, Sisodiya SM, van Heyningen V, Fitzpatrick DR. SOX2 anophthalmia syndrome. Am J Med Genet A. 2005;135:1–7. doi: 10.1002/ajmg.a.30642. [DOI] [PubMed] [Google Scholar]
- 21.Kelberman D, Rizzoti K, Avilion A, Bitner-Glindzicz M, Cianfarani S, Collins J, Chong WK, Kirk JMW, Achermann JC, Ross R, Carmignac D, Lovell-Badge R, Robinson ICAF, Dattani MT. Mutations within Sox2/SOX2 are associated with abnormalities in the hypothalamo-pituitary-gonadal axis in mice and humans. J Clin Invest. 2006;116:2442–2455. doi: 10.1172/JCI28658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alatzoglou KS, Andoniadou CL, Kelberman D, Buchanan CR, Crolla J, Arriazu MC, Roubicek M, Moncet D, Martinez-Barbera JP, Dattani MT. SOX2 haploinsufficiency is associated with slow progressing hypothalamo-pituitary tumours. Hum Mutat. 2011;32:1376–1380. doi: 10.1002/humu.21606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie W, Lynch TJ, Liu X, Tyler SR, Yu S, Zhou X, Luo M, Kusner DM, Sun X, Yi Y, Zhang Y, Goodheart MJ, Parekh KR, Wells JM, Xue H-H, Pevny LH, Engelhardt JF. Sox2 modulates Lef-1 expression during airway submucosal gland development. Am J Physiol Lung Cell Mol Physiol. 2014;306:L645–L660. doi: 10.1152/ajplung.00157.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mansukhani A, Ambrosetti D, Holmes G, Cornivelli L, Basilico C. Sox2 induction by FGF and FGFR2 activating mutations inhibits Wnt signaling and osteoblast differentiation. J Cell Biol. 2005;168:1065–1076. doi: 10.1083/jcb.200409182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Agathocleous M, Iordanova I, Willardsen MI, Xue XY, Vetter ML, Harris WA, Moore KB. A directional Wnt/beta-catenin-Sox2-proneural pathway regulates the transition from proliferation to differentiation in the Xenopus retina. Development. 2009;136:3289–3299. doi: 10.1242/dev.040451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meyers JR, Hu L, Moses A, Kaboli K, Papandrea A, Raymond PA. β-catenin/Wnt signaling controls progenitor fate in the developing and regenerating zebrafish retina. Neural Develop. 2012;7:30. doi: 10.1186/1749-8104-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 30.Civan MM, Macknight ADC. The ins and outs of aqueous humour secretion. Exp Eye Res. 2004;78:625–631. doi: 10.1016/j.exer.2003.09.021. [DOI] [PubMed] [Google Scholar]
- 31.Rainger J, Keighren M, Keene DR, Charbonneau NL, Rainger JK, Fisher M, Mella S, Huang JT-J, Rose L, Van’t Hof R, Sakai LY, Jackson IJ, Fitzpatrick DR. A trans-acting protein effect causes severe eye malformation in the Mp mouse. PLoS Genet. 2013;9:e1003998. doi: 10.1371/journal.pgen.1003998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coulombre AJ. The role of intraocular pressure in the development of the chick eye. II. Control of corneal size. AMA Arch Ophthalmol. 1957;57:250–253. doi: 10.1001/archopht.1957.00930050260015. [DOI] [PubMed] [Google Scholar]
- 33.Coulombre AJ, Coulombre JL. The role of intraocular pressure in the development of the chick eye: III. Ciliary body. Am J Ophthalmol. 1957;44(4 Pt 2):85–93. doi: 10.1016/0002-9394(57)90435-X. [DOI] [PubMed] [Google Scholar]
- 34.Flügel-Koch C, Ohlmann A, Piatigorsky J, Tamm ER. Disruption of anterior segment development by TGF-beta1 overexpression in the eyes of transgenic mice. Dev Dyn. 2002;225:111–125. doi: 10.1002/dvdy.10144. [DOI] [PubMed] [Google Scholar]
- 35.Zhao S, Chen Q, Hung F-C, Overbeek PA. BMP signaling is required for development of the ciliary body. Development. 2002;129:4435–4442. doi: 10.1242/dev.129.19.4435. [DOI] [PubMed] [Google Scholar]
- 36.Thut CJ, Rountree RB, Hwa M, Kingsley DM. A large-scale in situ screen provides molecular evidence for the induction of eye anterior segment structures by the developing lens. Dev Biol. 2001;231:63–76. doi: 10.1006/dbio.2000.0140. [DOI] [PubMed] [Google Scholar]
- 37.Kubota R, McGuire C, Dierks B, Reh TA. Identification of ciliary epithelial-specific genes using subtractive libraries and cDNA arrays in the avian eye. Dev Dyn. 2004;229:529–540. doi: 10.1002/dvdy.20000. [DOI] [PubMed] [Google Scholar]
- 38.Zorn AM, Barish GD, Williams BO, Lavender P, Klymkowsky MW, Varmus HE. Regulation of Wnt signaling by Sox proteins: XSox17 alpha/beta and XSox3 physically interact with beta-catenin. Mol Cell. 1999;4:487–498. doi: 10.1016/S1097-2765(00)80200-2. [DOI] [PubMed] [Google Scholar]
- 39.Chen Y, Shi L, Zhang L, Li R, Liang J, Yu W, Sun L, Yang X, Wang Y, Zhang Y, Shang Y. The molecular mechanism governing the oncogenic potential of SOX2 in breast cancer. J Biol Chem. 2008;283:17969–17978. doi: 10.1074/jbc.M802917200. [DOI] [PubMed] [Google Scholar]
- 40.Kormish JD, Sinner D, Zorn AM. Interactions between SOX factors and Wnt/beta-catenin signaling in development and disease. Dev Dyn. 2010;239:56–68. doi: 10.1002/dvdy.22046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- 42.Rowan S, Cepko CL. Genetic analysis of the homeodomain transcription factor Chx10 in the retina using a novel multifunctional BAC transgenic mouse reporter. Dev Biol. 2004;271:388–402. doi: 10.1016/j.ydbio.2004.03.039. [DOI] [PubMed] [Google Scholar]
- 43.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 44.Monaghan AP, Davidson DR, Sime C, Graham E, Baldock R, Bhattacharya SS, Hill RE. The Msh-like homeobox genes define domains in the developing vertebrate eye. Development. 1991;112:1053–1061. doi: 10.1242/dev.112.4.1053. [DOI] [PubMed] [Google Scholar]
- 45.da Silva MRD, Tiffin N, Mima T, Mikawa T, Hyer J. FGF-mediated induction of ciliary body tissue in the chick eye. Dev Biol. 2007;304:272–285. doi: 10.1016/j.ydbio.2006.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mathers PH, Grinberg A, Mahon KA, Jamrich M. The Rx homeobox gene is essential for vertebrate eye development. Nature. 1997;387:603–607. doi: 10.1038/42475. [DOI] [PubMed] [Google Scholar]
- 47.Andoniadou CL, Matsushima D, Mousavy Gharavy SN, Signore M, Mackintosh AI, Schaeffer M, Gaston-Massuet C, Mollard P, Jacques TS, Le Tissier P, Dattani MT, Pevny LH, Martinez-Barbera JP. Sox2(+) stem/progenitor cells in the adult mouse pituitary support organ homeostasis and have tumor-inducing potential. Cell Stem Cell. 2013;13:433–445. doi: 10.1016/j.stem.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 48.Nishida A, Furukawa A, Koike C, Tano Y, Aizawa S, Matsuo I, Furukawa T. Otx2 homeobox gene controls retinal photoreceptor cell fate and pineal gland development. Nat Neurosci. 2003;6:1255–1263. doi: 10.1038/nn1155. [DOI] [PubMed] [Google Scholar]
- 49.Cui L, Guan Y, Qu Z, Zhang J, Liao B, Ma B, Qian J, Li D, Li W, Xu G-T, Jin Y. WNT signaling determines tumorigenicity and function of ESC-derived retinal progenitors. J Clin Invest. 2013;123:1647–1661. doi: 10.1172/JCI65048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martynoga B, Morrison H, Price DJ, Mason JO. Foxg1 is required for specification of ventral telencephalon and region-specific regulation of dorsal telencephalic precursor proliferation and apoptosis. Dev Biol. 2005;283:113–127. doi: 10.1016/j.ydbio.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 51.Tong W, Pollard JW. Genetic evidence for the interactions of cyclin D1 and p27(Kip1) in mice. Mol Cell Biol. 2001;21:1319–1328. doi: 10.1128/MCB.21.4.1319-1328.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Das G, Choi Y, Sicinski P, Levine EM. Cyclin D1 fine-tunes the neurogenic output of embryonic retinal progenitor cells. Neural Develop. 2009;4:15. doi: 10.1186/1749-8104-4-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Das G, Clark AM, Levine EM. Cyclin D1 inactivation extends proliferation and alters histogenesis in the postnatal mouse retina. Dev Dyn. 2012;241:941–952. doi: 10.1002/dvdy.23782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moshiri A, Reh TA. Persistent progenitors at the retinal margin of ptc+/- mice. J Neurosci. 2004;24:229–237. doi: 10.1523/JNEUROSCI.2980-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Y, Dakubo GD, Thurig S, Mazerolle CJ, Wallace VA. Retinal ganglion cell-derived sonic hedgehog locally controls proliferation and the timing of RGC development in the embryonic mouse retina. Development. 2005;132:5103–5113. doi: 10.1242/dev.02096. [DOI] [PubMed] [Google Scholar]
- 56.Sakagami K, Gan L, Yang X-J. Distinct effects of Hedgehog signaling on neuronal fate specification and cell cycle progression in the embryonic mouse retina. J Neurosci. 2009;29:6932–6944. doi: 10.1523/JNEUROSCI.0289-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wall DS, Mears AJ, McNeill B, Mazerolle C, Thurig S, Wang Y, Kageyama R, Wallace VA. Progenitor cell proliferation in the retina is dependent on Notch-independent Sonic hedgehog/Hes1 activity. J Cell Biol. 2009;184:101–112. doi: 10.1083/jcb.200805155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang YP, Dakubo G, Howley P, Campsall KD, Mazarolle CJ, Shiga SA, Lewis PM, McMahon AP, Wallace VA. Development of normal retinal organization depends on Sonic hedgehog signaling from ganglion cells. Nat Neurosci. 2002;5:831–832. doi: 10.1038/nn918. [DOI] [PubMed] [Google Scholar]
- 59.Megason SG, McMahon AP. A mitogen gradient of dorsal midline Wnts organizes growth in the CNS. Development. 2002;129:2087–2098. doi: 10.1242/dev.129.9.2087. [DOI] [PubMed] [Google Scholar]
- 60.Marquardt T, Ashery-Padan R, Andrejewski N, Scardigli R, Guillemot F, Gruss P. Pax6 is required for the multipotent state of retinal progenitor cells. Cell. 2001;105:43–55. doi: 10.1016/S0092-8674(01)00295-1. [DOI] [PubMed] [Google Scholar]
- 61.Davis-Silberman N, Kalich T, Oron-Karni V, Marquardt T, Kroeber M, Tamm ER, Ashery-Padan R. Genetic dissection of Pax6 dosage requirements in the developing mouse eye. Hum Mol Genet. 2005;14:2265–2276. doi: 10.1093/hmg/ddi231. [DOI] [PubMed] [Google Scholar]
- 62.Farhy C, Elgart M, Shapira Z, Oron-Karni V, Yaron O, Menuchin Y, Rechavi G, Ashery-Padan R. Pax6 is required for normal cell-cycle exit and the differentiation kinetics of retinal progenitor cells. PLoS One. 2013;8:e76489. doi: 10.1371/journal.pone.0076489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hogan BL, Hirst EM, Horsburgh G, Hetherington CM. Small eye (Sey): a mouse model for the genetic analysis of craniofacial abnormalities. Development. 1988;103(Suppl):115–119. doi: 10.1242/dev.103.Supplement.115. [DOI] [PubMed] [Google Scholar]
- 64.Hill RE, Favor J, Hogan BL, Ton CC, Saunders GF, Hanson IM, Prosser J, Jordan T, Hastie ND, van Heyningen V. Mouse small eye results from mutations in a paired-like homeobox-containing gene. Nature. 1991;354:522–525. doi: 10.1038/354522a0. [DOI] [PubMed] [Google Scholar]
- 65.Haremaki T, Tanaka Y, Hongo I, Yuge M, Okamoto H. Integration of multiple signal transducing pathways on Fgf response elements of the Xenopus caudal homologue Xcad3. Development. 2003;130:4907–4917. doi: 10.1242/dev.00718. [DOI] [PubMed] [Google Scholar]
- 66.Kubo F, Takeichi M, Nakagawa S. Wnt2b controls retinal cell differentiation at the ciliary marginal zone. Development. 2003;130:587–598. doi: 10.1242/dev.00244. [DOI] [PubMed] [Google Scholar]
- 67.Sikes JM, Newmark PA. Restoration of anterior regeneration in a planarian with limited regenerative ability. Nature. 2013;500:77–80. doi: 10.1038/nature12403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Simpson PJ, Moon C, Kleman AM, Connolly E, Ronnett GV. Progressive and inhibitory cell cycle proteins act simultaneously to regulate neurotrophin-mediated proliferation and maturation of neuronal precursors. Cell Cycle. 2007;6:1077–1089. doi: 10.4161/cc.6.9.4132. [DOI] [PubMed] [Google Scholar]
- 69.Card DAG, Hebbar PB, Li L, Trotter KW, Komatsu Y, Mishina Y, Archer TK. Oct4/Sox2-regulated miR-302 targets cyclin D1 in human embryonic stem cells. Mol Cell Biol. 2008;28:6426–6438. doi: 10.1128/MCB.00359-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Davis N, Mor E, Ashery-Padan R. Roles for Dicer1 in the patterning and differentiation of the optic cup neuroepithelium. Development. 2011;138:127–138. doi: 10.1242/dev.053637. [DOI] [PubMed] [Google Scholar]
- 71.Liu Z, Walters BJ, Owen T, Brimble MA, Steigelman KA, Zhang L, Mellado Lagarde MM, Valentine MB, Yu Y, Cox BC, Zuo J. Regulation of p27Kip1 by Sox2 maintains quiescence of inner pillar cells in the murine auditory sensory epithelium. J Neurosci. 2012;32:10530–10540. doi: 10.1523/JNEUROSCI.0686-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jayakody SA, Andoniadou CL, Gaston-Massuet C, Signore M, Cariboni A, Bouloux PM, Le Tissier P, Pevny LH, Dattani MT, Martinez-Barbera JP. SOX2 regulates the hypothalamic-pituitary axis at multiple levels. J Clin Invest. 2012;122:3635–3646. doi: 10.1172/JCI64311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hung FC, Zhao S, Chen Q, Overbeek PA. Retinal ablation and altered lens differentiation induced by ocular overexpression of BMP7. Vision Res. 2002;42:427–438. doi: 10.1016/S0042-6989(01)00242-5. [DOI] [PubMed] [Google Scholar]
- 74.Zhang Y, Overbeek PA, Govindarajan V. Perinatal ablation of the mouse lens causes multiple anterior chamber defects. Mol Vis. 2007;13:2289–2300. [PubMed] [Google Scholar]
- 75.Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, Taketo MM. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J. 1999;18:5931–5942. doi: 10.1093/emboj/18.21.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Surzenko N, Crowl T, Bachleda A, Langer L, Pevny L. SOX2 maintains the quiescent progenitor cell state of postnatal retinal Muller glia. Development. 2013;140:1445–1456. doi: 10.1242/dev.071878. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Significantly changed optic cup (OC) WNT target genes. Thirteen known OC WNT target genes were found to be changed at least 1.47-fold (log ratio of at least ±0.55) in mutant compared with control eyes. (XLSX 640 KB)
Additional file 2: Functional Annotation of the Sox2-ablated optic cup (OC) reveals cell fate change. Functional terms significantly enriched for up-regulated genes (first tab) and down-regulated genes (second tab) resulting from optic cup-specific ablation of Sox2. (DOCX 51 KB)
Additional file 3: Chx10 CreGFP efficiently ablates Sox2 and Ctnnb1 from the optic cup (OC) without causing increased cell death. The efficiency of Chx10 CreGFP and cell death were assessed in double-mutants at E14.5. (A-B, D-E) SOX2 and β-Catenin are absent from CreGFP-positive cells in double-mutants (C,D) compared with controls (A,B). (C,F) Cleaved Caspase 3 is similar between controls and mutants. Scale bar: 200 μm. (PNG 1 MB)
Additional file 4: Sox2-deficient OCPCs prematurely exit the cell cycle. Cycling cells in neonatal controls and mutants were identified by Ki67 staining. (A-F) In controls, Ki67 is expressed in RPCs (red in A) and in some GFP-positive CE cells adjacent to the NR (B-C; arrows) but not in the most peripheral CE cells (B-C; arrowheads). In mutants, Ki67 is expressed in centrally located Sox2-ablated RPCs (E-F; arrows) but not in peripheral Sox2-ablated RPCs (E-F; arrowheads). Boxed areas in (A) and (D) are magnified in (B-C) and (E-F), respectively. Scale bars: 100 μm. (PNG 776 KB)
Additional file 5: Deletion of Ctnnb1 in Sox2-ablated optic cup progenitor cells (OCPCs) does not rescue increased CyclinD1 or ectopic expression of CyclinD3. CyclinD1 and CyclinD3 expression were analyzed in single-mutants, double-mutants and controls at P0. CyclinD1 is restricted to retinal progenitor cells (RPCs) in controls (A-C) and Ctnnb1 single-mutants (F-H). CyclinD1 is increased in GFP-positive cells in the central OC of Sox2 single-mutants (K-M; arrows) and in Sox2/Ctnnb1 double-mutants (P-R; arrows). CyclinD3 is restricted to the CE of controls (D, E; arrows) and Ctnnb1 single-mutants (I, J; arrows) and ectopically expressed in GFP-positive cells in the central OC of Sox2 single-mutants (N, O; arrows) and Sox2/Ctnnb1 double-mutants (S, T; arrows). Boxed areas in (A, F, K and P) are magnified in (B, C; G, H; L, M; and Q, R), respectively. Scale bars: 100 μm. (PNG 1 MB)
Additional file 6: Reduction of Pax6 in Sox2-ablated optic cup progenitor cells (OCPCs) does not rescue increased CyclinD1. CyclinD1 was assayed on transverse sections through the eyes of Sox2 single-mutants, Pax6 Sey/+ single-mutants, double-mutants and wild-type controls at E14.5. (A-D) CyclinD1 is expressed in retinal progenitor cells (RPCs) of controls (A, B) and Pax6 Sey/+ single-mutants (C, D). (E-H) CyclinD1 is increased in central OCPCs of Sox2 single-mutants (E, F) and Sox2/Pax6 Sey/+double-mutants (G, H). Scale bar: 200 μm. (PNG 3 MB)
Additional file 7: Model of how SOX2 and canonical WNT signaling regulate the neurogenic boundary of the optic cup (OC). OC-specific genetic ablation of Sox2 or Ctnnb1 results in complementary phenotypes. (A-D) The boundary between neural retina (NR) (blue) and ciliary epithelium (CE) (orange) is shifted peripherally in Ctnnb1 single-mutants (B) compared with controls (A). Conversely, the boundary between NR and CE is shifted centrally in Sox2 single-mutants such that WNT and BMP signaling are expanded (C) compared with controls (A). The boundary between the NR and CE remains centrally shifted in Sox2/Ctnnb1 double-mutants (D). However, BMP signaling and other classical CE markers fail to be expressed in this expanded CE-like region. D-type cyclins are increased in both Sox2 single-mutants and Sox2/Ctnnb1 double-mutants. (PNG 167 KB)