

Abstract
Introduction
Systemic lupus erythematosus (SLE), rheumatoid arthritis (RA) and Sjögren’s syndrome (SjS) are inflammatory systemic autoimmune diseases (SADs) that share several clinical and pathological features. The shared biological mechanisms are not yet fully characterized. The objective of this study was to perform a meta-analysis using publicly available gene expression data about the three diseases to identify shared gene expression signatures and overlapping biological processes.
Methods
Previously reported gene expression datasets were selected and downloaded from the Gene Expression Omnibus database. Normalization and initial preprocessing were performed using the statistical programming language R and random effects model–based meta-analysis was carried out using INMEX software. Functional analysis of over- and underexpressed genes was done using the GeneCodis tool.
Results
The gene expression meta-analysis revealed a SAD signature composed of 371 differentially expressed genes in patients and healthy controls, 187 of which were underexpressed and 184 overexpressed. Many of these genes have previously been reported as significant biomarkers for individual diseases, but others provide new clues to the shared pathological state. Functional analysis showed that overexpressed genes were involved mainly in immune and inflammatory responses, mitotic cell cycles, cytokine-mediated signaling pathways, apoptotic processes, type I interferon–mediated signaling pathways and responses to viruses. Underexpressed genes were involved primarily in inhibition of protein synthesis.
Conclusions
We define a common gene expression signature for SLE, RA and SjS. The analysis of this signature revealed relevant biological processes that may play important roles in the shared development of these pathologies.
Electronic supplementary material
The online version of this article (doi:10.1186/s13075-014-0489-x) contains supplementary material, which is available to authorized users.
Introduction
Autoimmunity refers to the failure of the immune system to recognize its own constituent parts, eliciting an immune response against the tissues themselves. Currently, there are more than 80 clinically distinct autoimmune diseases [1], and the biological mechanisms that cause them are not clearly understood. It has been suggested that both genetic and environmental factors influence the development of autoimmune diseases [2]. Three of these inflammatory, autoimmune diseases are systemic lupus erythematosus (SLE), rheumatoid arthritis (RA) and Sjögren’s syndrome (SjS).
Although at first appearance these disorders have different phenotypes, they all are heterogeneous, multifactorial disorders that share molecular mechanisms which elicit similar clinical and pathogenic features. In fact, a differential diagnosis between these immune disorders at an early stage is not always reliable, and treatments are similar for all three, except when organ damage ensues or features of one dominate over another. Therefore, one of the major aims in this field is to discover similarities and differences at the molecular level between these diseases and between groups of patients across diseases. This will lead to a better understanding of the specific biological mechanisms and the development of more efficient and personalized treatments.
In this context, the analysis of gene expression patterns can provide useful information for understanding the molecular mechanisms by defining specific gene expression signatures that underlie these disorders. These studies are becoming more plentiful as a result of the development of high-throughput technologies such as microarrays and next-generation sequencing. These methods allow us to measure gene expression on a genome-wide scale, including for autoimmune diseases, and have been widely used during the past decade (see, for example, [3-5]). In this field, meta-analysis techniques offer the potential to integrate and jointly analyze data from different sources. In previous meta-analyses, investigators have combined data related to the same disease from different studies to get more consistent and reliable results. For example, Song et al. [6] performed a meta-analysis integrating three public SjS datasets. Arasappan et al. [7] conducted a pathway-based meta-analysis of four SLE datasets and identified a 37-gene signature associated with this disease. Olsen et al. [8,9] analyzed different RA datasets and found several genes related to pathways such as type I interferon (IFN), apoptotic processes and cell cycles.
Moreover, meta-analytic techniques have also been used to integrate data from different diseases to uncover similar patterns. In this context, Tuller et al. [4] analyzed public data on six different autoimmune diseases (multiple sclerosis, SLE, juvenile RA, Crohn’s disease, ulcerative colitis and type 1 diabetes) from peripheral blood mononuclear cells (PBMCs). Silva et al. [10] combined SLE and RA data to uncover coexpression patterns, and Higgs et al. [11,12] integrated data on SLE, myositis, RA and scleroderma and defined a common type I IFN-related signature.
In this study, we performed a gene expression meta-analysis using publicly available gene expression data from PBMC samples of SLE, RA and SjS patients and controls. To the best of our knowledge, this is the first study in which gene expression data from these three diseases have been integrated, together with analysis of the common gene expression signatures with respect to healthy controls.
Methods
Search and selection of datasets
We mined the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database [13] to find all publicly available gene expression datasets related to SLE, RA and SjS. From among all published studies, we selected for our analysis those that fit the following criteria: (1) They had to include control and case samples in separate arrays (one-channel arrays); (2) they had to have been performed with human PBMC samples; and (3) the samples had to have been obtained without any type of treatment.
Processing of the datasets and meta-analysis
Initial processing of the data was carried out using the R statistical programming language. Each dataset was downloaded from the NCBI GEO database using the GEOquery R package [14], and probes were annotated with the Entrez Gene identifiers, which were used to merge data from different platforms for further analysis. In each dataset, gene expression profiles were averaged for duplicate genes by computing the median values. Genes with missing values in more than 10% of samples were filtered out, and the remaining missing values were imputed using the average expression values within the group (case or control). The integration of different datasets and gene expression meta-analysis was performed using the INMEX software package [15]. Gene expression values were log-transformed and normalized by applying quantile normalization. The dataset for identification of genes specifically overexpressed in lupus CD4 T and B cells [GEO:GSE4588] contains samples from SLE and RA patients; therefore, these two subpopulations were treated as two different datasets.
Differential expression meta-analysis across diseases and healthy controls was carried out by using a random effects model (REM) [16,17], which is based on combining the effect sizes (ESs) or changes of gene expression from different studies and obtaining an overall mean.
Functional analysis
In order to obtain biological information from the list of differentially expressed genes, we performed a functional analysis using the GeneCodis tool [18-21]. This software allows evaluation of which annotations are significantly enriched in a gene list, which can be used as functional descriptors of the biological processes that are acting in experimental conditions [19,20]. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes pathway annotations were evaluated in the analysis.
Results
Studies selected for the meta-analysis
After conducting a thorough search, we identified 19 datasets related to SLE, RA and SjS. From among this initial set, we dismissed three datasets that included treated samples, four datasets without control samples, five with too many missing values (more than 50% of missing values) and three generated with two-channel arrays. Thus, our final meta-analysis included four datasets (Table 1). The selected datasets comprise a total of 371 samples with a breakdown of 94 controls, 190 SjS samples, 54 SLE samples and 33 RA samples.
Table 1.
Datasets used in the study a
| GEO ID | Platform | Disease | Cases/controls | Description | Reference | Key findings |
|---|---|---|---|---|---|---|
| [GEO:GSE10325] | Affymetrix Human Genome U133A Array | SLE | 39/28 | Expression data from human peripheral blood subsets | [22] | Some apoptotic genes are related to SLE phenotype |
| [GEO:GSE15573] | Affymetrix Human Genome U133 Plus 2.0 Array | RA | 18/15 | Immunity and defense genes in PBMCs of RA patients | [23] | 283 underexpressed genes (involved in metabolic processes), 101 overexpressed (immunity, calcium transport) |
| [GEO:GSE4588] | Illumina Human 6 v2.0 expression BeadChip | SLE, RA | 15 SLE, 15 RA/19 | Identification of genes specifically overexpressed in SLE CD4 T and B cells | – | – |
| [GEO:GSE51092] | Illumina Human WG-6 v3.0 expression BeadChip | SjS | 190/32 | Variants at multiple loci implicated in both innate and adaptive immune responses are associated with SjS | [24] | Risk loci for SjS (related to IFN pathway, STATs, chemokine receptors, interleukins proteins or a BLK |
aNCBI GEO ID: Unique gene expression series identifier for each dataset in the National Center for Biotechnology Information Gene Expression Omnibus database; Platform: Microarray platform; Disease: Type of disease and number of cases/controls in each dataset; Description: Brief description of the study; Reference: Publication; Key findings: Main findings in the original studies. BLK, B Lymphocyte Kinase Protein; IFN, Interferon; PBMCs, Peripheral blood mononuclear cells; RA, Rheumatoid arthritis; SLE, Systemic lupus erythematosus; SjS, Sjögren’s syndrome; STAT, Signal transducers and activators of transcription.
Identifying a common gene expression signature among systemic lupus erythematosus, rheumatoid arthritis and Sjögren’s syndrome
For meta-analysis, the processed data were loaded into the INMEX web tool, and ES statistical analysis was used to find genes that were differentially expressed among diseases and healthy controls across different studies. Rather than requiring us to merge the original datasets, this method allowed us to combine them through high-level summary statistics, thus avoiding the problem of interstudy variation.
We identified 412 genes that were consistently differentially expressed (P < 0.05). Among the different studies (see Additional file 1), 210 genes were overexpressed and 202 were underexpressed.
Among this initial set, we found 187 overexpressed genes in all diseases with respect to healthy controls and 184 that were underexpressed. Additional file 1 gives the average fold changes of each gene in all datasets. This set comprises the common gene expression signature—that is, genes that are significantly differentially expressed in all diseases with respect to healthy controls. Figure 1 shows the top 50 over- and underexpressed genes.
Figure 1.
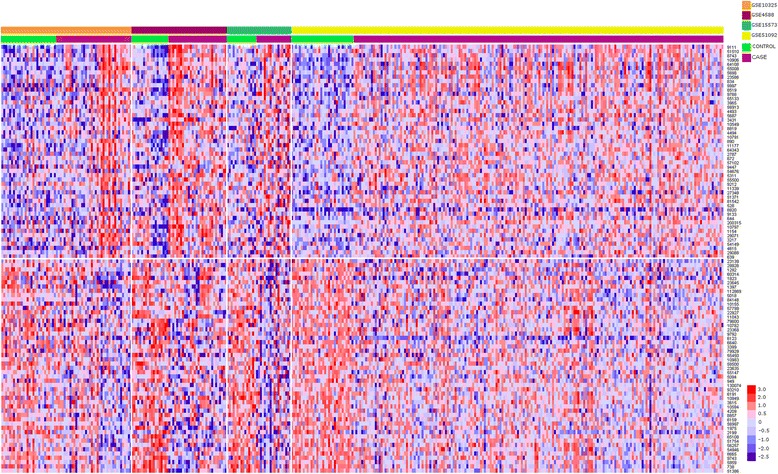
Heatmap of top differentially expressed genes. The heatmap represents the log2- transformed expression values with the top 50 overexpressed (top) and top 50 underexpressed (below) genes.
Some of the most differentially expressed genes were HERC6, which belongs to the HERC family of E3 ubiquitin ligases, and RTP4, which encodes a receptor (chemosensory) transporter protein related to “chemotaxis” and was previously described as an IFN-inducible gene [25]. In addition, the most overexpressed genes with the largest ESs were RSAD2 and IFI44L. IFI44L encodes the IFN-induced protein 44-like, which has been described in several autoimmune diseases in conjunction with other genes involved in the type I IFN signaling pathway, such as IFIT1, IFI27 and IFITM1. We also found overexpression of these genes in our results.
The gene with the lowest ES (ES = −1.2545) was eukaryotic translation elongation factor 2 (EEF2), a biomarker protein of some types of cancer [26] that plays an important role in protein synthesis. This was, in fact, the most relevant pathway that was associated with underexpressed genes.
Meta-analytic techniques have been also used to evaluate reproducibility and bias across microarray studies. This is especially important when comparing replicated samples or samples of the same condition or phenotype. In this sense, there are different methods that can be used for this purpose [27]. In this context, we also evaluated the meta-analysis results with those obtained from individual analyses of studies and/or diseases. We found 132 gained genes and 2,168 lost genes in our meta-analysis (see Additional file 1). Gained genes are the differentially expressed genes identified only in the meta-analysis and not in the individual analysis, because they show weak signals but consistent expression patterns across the different datasets. Lost genes are genes identified as differentially expressed genes in any individual analysis, but not in the meta-analysis. These genes show either conflicting changes in expression profiles or very large variations across different studies [6,15]. Additional file 2 contains a detailed study of the different datasets and the analysis used in our study.
Functional and pathway analysis
For the analysis of biological processes associated with the differentially expressed genes, we evaluated the enrichment of functional annotations using the GeneCodis tool [18]. GO annotations for biological processes were significantly overrepresented in the gene list if they showed a P-value <0.05. Results for biological pathways of overexpressed and underexpressed genes are shown in Figure 2 and Additional file 1. Functions such as “mitotic cell cycle,” “cytokine-mediated signaling pathway,” “response to virus” or “type I IFN-mediated signaling pathway” or “immune response” were significantly associated with this set of genes. This is in agreement with previous work that has associated these pathways with each of the diseases [12,28-31].
Figure 2.
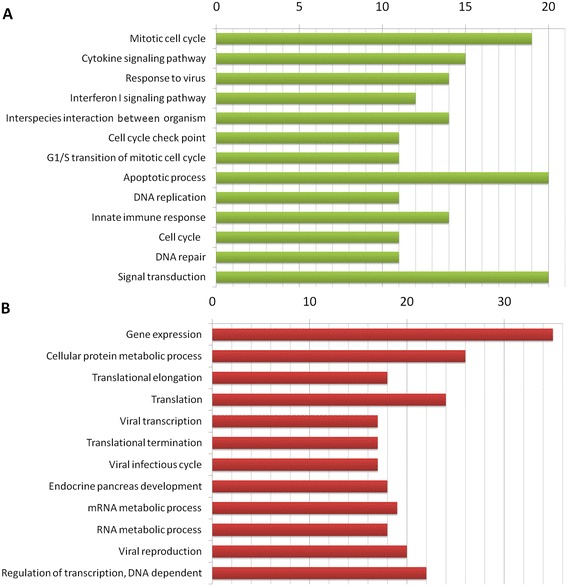
Biological function related to the differentially expressed genes. This graphic shows the main Gene Ontology (GO) biological functions identified and related to overexpressed genes (A) and underexpressed genes (B). GO annotations were considered significantly enriched in the list of genes if they had a P-value <0.01 and were associated with at least ten genes. The x-axis represents the number of genes, and the y-axis shows the names of the significant GO categories sorted by decreasing P-values.
Similarly, the most significant GO categories or pathways in the analysis of underexpressed genes were “gene expression” and others related to protein biosynthesis mechanisms previously reported [32].
Discussion
In this study, we define a signature of differentially expressed genes for SLE, RA and SjS using a gene expression meta-analytic strategy showing common biological mechanisms across three otherwise clinically separate entities. The combined ES and REM meta-analytic method was chosen because it allowed us to integrate microarray datasets from different platforms consistently, without the obstacle of the batch effects that we clearly observed when we began our analyses. We performed the meta-analysis using four publicly available datasets and defined a common signature composed of 187 overexpressed genes and 184 underexpressed genes in all diseases compared to healthy controls. We found significant pathways related to overexpressed genes, such as “immune response,” “type I IFN-mediated signaling pathway,” “cytokine-mediated signaling pathway,” “mitotic cycle” and “response to virus,” as well as pathways related to underexpressed genes that highlight gene expression and metabolic processes.
In this context, independent studies of SLE, RA and/or SjS have shown that overexpression of type I IFN-related genes is very consistent [28-31,33,34], and it appears that these genes roughly form a common pattern in all inflammatory autoimmune diseases [4,6,7,9-12]. The cytokine signaling pathway-related genes behave similarly. In fact, both routes are tightly related. IFN proteins regulate several signaling pathways normally in response to pathogens, such as apoptosis or immune stimulation. In the gene expression signature, we identified several type I IFN-related genes as the most significantly overexpressed genes in all diseases, such as IFI44L, IFI44, IFI27 and IFIT1. We also found other genes, such as JAK2, involved in the Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway and previously related to immune disorders [35]. JAK/STAT signaling promotes IFN-stimulated gene transcription. OASL is and IFN-inducible protein related to antiviral activity [36]. In addition, we found genes related to apoptosis, with genes such as FAS, which has been described as a risk allele in some autoimmune diseases [37,38]; TNFSF10; and CASP1. We also identified several proteasome subunits, such as PSM2, PSM6 and PSMC2. Apoptotic processes have been related to autoimmune diseases in different studies [39]. Apoptotic cells are not immunologically neutral, and the accumulation of apoptotic material not properly phagocytosed can be an important source of autoimmune antigens, enabling the development of autoimmune disorders [40]. In this context, there are some hypotheses focused on the increase in apoptosis, lazy phagocytes or the interaction between apoptotic material and antigen-presenting cells as potential triggers for these diseases.
In addition, some of these genes have been described previously as biomarkers of one or a variety of autoimmune diseases, such as IFN-induced protein 44-like, chemokine receptor 1 and FAS. Therefore, our results are consistent with previously published data for each of the three disorders, but show, for the first time to our knowledge, and formally, their shared genetic signatures.
Moreover, we found interesting results, such as the overexpression of EIF2AK2 gene (or PKR) (see Additional file 1). This gene is initially related to the response to virus and the innate immune response and encodes a serine/threonine protein kinase that is activated by autophosphorylation after binding to double-stranded RNA [41]. The activated form can phosphorylate multiple substrates, including several translation initiation factors, such as eIF2A, eIF3F eIF2S1 and eEF2, impairing the recycling of these factors between successive rounds of initiation and leading to inhibition of translation, which eventually results in shutdown of cellular protein synthesis and a reduction in cell proliferation [42,43]. This is in agreement with our finding of biological processes related to inhibition of gene expression and protein synthesis in the list of underexpressed genes.
In a previous study of SLE, Groulleau et al. [32] described the relationship between PKR and the phosphorylation of the eIF2A translation initiation factor, but this action was attributed only to SLE, whereas researchers in other studies independently related PKR with RA [44,45]. In addition, PKR phosphorylates p38, JNK and nuclear factor κB (NF-κB), which are proteins of the mitogen-activated protein kinase signaling pathway related to the production of cytokines and tumor necrosis factor [46]. These in turn intervene in apoptotic processes, regulation of signal transduction or cell proliferation and differentiation. PKR has direct influences on the production of IFNs [47,48]. We also found other genes related to the immune system, such as MYD88, which is an adaptor protein of Toll-like receptors that activates NF-κB and translocates to the nucleus to stimulate the expression of certain genes for the production of cytokines and IFN proteins [49].
Regarding the pathways related to underexpressed genes, the two most relevant processes were “gene expression” and “cellular protein metabolic process”. Analysis of genes associated with these annotations revealed that many were genes involved in translation, such as ribosomal protein-encoding genes, and different eukaryotic translation initiation factor subunits, such as eEF2 or eIF3F mentioned above. The relationship between the underexpression of these genes and autoimmune disease is largely undefined. We also found genes involved in translation and cell growth, which are underexpressed, as mentioned above.
Conclusions
We performed a gene expression meta-analysis using previously published datasets obtained from PBMCs of SLE, RA and SjS patients. A common gene expression signature was defined, comprising many genes that have been previously related to one, two or each of the three diseases. Although there are previous gene expression meta-analyses of immune-related diseases, our present study is the first one, to our knowledge, in which data on these three specific disorders have been integrated, which allowed us to define common biological processes. We found that pathways in our results, such as “type I IFN-mediated signaling pathway,” apoptotic processes, “immune response,” reduction in translation processes and “response to virus.” This suggests that a majority of these pathways are related to the action of the IFN proteins. However, we found other pathways, such as mitotic cell cycles, whose relationship to the IFN pathway, or even to the diseases themselves, has not been described. Future functional and specific studies of the genes we identified are needed to define the roles of these genes in the pathogenesis of SADs.
Acknowledgements
This study was possible thanks to grants from the Instituto de Salud Carlos III (IP12/02558), supported in part by European Regional Development Fund (to MAR), and from Innovative Medicines Initiative (grant GA-115565, coordinated by MAR).
Abbreviations
- ES
Effect size
- GEO
Gene Expression Omnibus
- GO
Gene ontology
- IFN
Interferon
- JAK/STAT
Janus kinase/signal transducer and activator of transcription
- NCBI
National Center for Biotechnology Information
- NF-κB
Nuclear factor κB
- PBMC
Peripheral blood mononuclear cell
- RA
Rheumatoid arthritis
- REM
Random effects model
- SAD
Inflammatory and systemic autoimmune disease
- SjS
Sjögren’s syndrome
- SLE
Systemic lupus erythematosus
Additional files
Four Excel spreadsheets with results from the meta-analysis. Spreadsheet 1: Differential gene expression analysis results with list of the names of the genes and their Entrez ID, the value of the combined ESs among the different experiments for each gene and the associated P-values. The left column shows the upexpressed genes, and the right column shows the underexpressed genes. Spreadsheet 2: The fold changes of each gene in all datasets. Genes marked with an asterisk were removed from the gene expression signature, as they did not show a consistent pattern of overexpression or underexpression in all diseases. Spreadsheet 3: Gain and loss gene lists. Spreadsheet 4: All GO categories enriched in the gene signature are listed, with the minimum number of genes set to three. The table contains the number of genes in each pathway, the corrected P-values (Hyp*), the GO annotation numbers and the names of the pathways and the genes related to each pathway.
Detailed study of the different datasets used and their influence in the final results of the meta-analysis.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
PCS and MAR conceived of and supervised the project. PCS and DTD designed the study and the data analysis pipeline. DTD implemented the meta-analysis workflow and performed the analysis. PCS, MAR and DTD contributed to the interpretation of data. All authors wrote and revised the manuscript critically. All authors read and approved the final manuscript.
Contributor Information
Daniel Toro-Domínguez, Email: daniel.toro@genyo.es.
Pedro Carmona-Sáez, Email: pedro.carmona@genyo.es.
Marta E Alarcón-Riquelme, Email: marta.alarcon@genyo.es.
References
- 1.Karopka T, Fluck J, Mevissen HT, Glass Ä. The Autoimmune Disease Database: a dynamically compiled literature-derived database. BMC Bioinformatics. 2006;7:325. doi: 10.1186/1471-2105-7-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salaman MR. A two-step hypothesis for the appearance of autoimmune disease. Autoimmunity. 2003;36:57–61. doi: 10.1080/0891693031000068637. [DOI] [PubMed] [Google Scholar]
- 3.Burska AN, Roget K, Blits M, Soto Gomez L, van de Loo F, Hazelwood LD, Verweij CL, Rowe A, Goulielmos GN, van Baarsen LGM, Ponchel F. Gene expression analysis in RA: towards personalized medicine. Pharmacogenomics J. 2014;14:93–106. doi: 10.1038/tpj.2013.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tuller T, Atar S, Ruppin E, Gurevich M, Achiron A. Common and specific signatures of gene expression and protein–protein interactions in autoimmune diseases. Genes Immun. 2013;14:67–82. doi: 10.1038/gene.2012.55. [DOI] [PubMed] [Google Scholar]
- 5.van ’t Veer LJ, Bernards R: Enabling personalized cancer medicine through analysis of gene-expression patterns.Nature 2008, 452:564–570. [DOI] [PubMed]
- 6.Song GG, Kim JH, Seo YH, Choi SJ, Ji JD, Lee YH. Meta-analysis of differentially expressed genes in primary Sjogren’s syndrome by using microarray. Hum Immunol. 2014;75:98–104. doi: 10.1016/j.humimm.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 7.Arasappan D, Tong W, Mummaneni P, Fang H, Amur S. Meta-analysis of microarray data using a pathway-based approach identifies a 37-gene expression signature for systemic lupus erythematosus in human peripheral blood mononuclear cells. BMC Med. 2011;9:65. doi: 10.1186/1741-7015-9-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olsen N, Sokka T, Seehorn CL, Kraft B, Maas K, Moore J, Aune TM. A gene expression signature for recent onset rheumatoid arthritis in peripheral blood mononuclear cells. Ann Rheum Dis. 2004;63:1387–1392. doi: 10.1136/ard.2003.017194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olsen NJ, Moore JH, Aune TM. Gene expression signatures for autoimmune disease in peripheral blood mononuclear cells. Arthritis Res Ther. 2004;6:120–128. doi: 10.1186/ar1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Silva GL, Junta CM, Mello SS, Garcia PS, Rassi DM, Sakamoto-Hojo ET, Donadi EA, Passos GAS. Profiling meta-analysis reveals primarily gene coexpression concordance between systemic lupus erythematosus and rheumatoid arthritis. Ann N Y Acad Sci. 2007;1110:33–46. doi: 10.1196/annals.1423.005. [DOI] [PubMed] [Google Scholar]
- 11.Higgs BW, Liu Z, White B, Zhu W, White WI, Morehouse C, Brohawn P, Kiener PA, Richman L, Fiorentino D, Greenberg SA, Jallal B, Yao Y. Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Ann Rheum Dis. 2011;70:2029–2036. doi: 10.1136/ard.2011.150326. [DOI] [PubMed] [Google Scholar]
- 12.Higgs BW, Zhu W, Richman L, Fiorentino DF, Greenberg SA, Jallal B, Yao Y. Identification of activated cytokine pathways in the blood of systemic lupus erythematosus, myositis, rheumatoid arthritis, and scleroderma patients. Int J Rheum Dis. 2012;15:25–35. doi: 10.1111/j.1756-185X.2011.01654.x. [DOI] [PubMed] [Google Scholar]
- 13.National Center for Biotechnology Information: Gene Expression Omnibus. [http://www.ncbi.nlm.nih.gov/geo/] (accessed 17 December 2014).
- 14.Davis S, Meltzer PS. GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics. 2007;23:1846–1847. doi: 10.1093/bioinformatics/btm254. [DOI] [PubMed] [Google Scholar]
- 15.Xia J, Fjell CD, Mayer ML, Pena OM, Wishart DS, Hancock REW: INMEX—a web-based tool for integrative meta-analysis of expression data.Nucleic Acids Res 2013, 41(Web server issue):W63–W70. [DOI] [PMC free article] [PubMed]
- 16.Choi JK, Yu U, Kim S, Yoo OJ. Combining multiple microarray studies and modeling interstudy variation. Bioinformatics. 2003;19:i84–i90. doi: 10.1093/bioinformatics/btg1010. [DOI] [PubMed] [Google Scholar]
- 17.Marot G, Foulley JL, Mayer CD, Jaffrézic F. Moderated effect size and P-value combinations for microarray meta-analyses. Bioinformatics. 2009;25:2692–2699. doi: 10.1093/bioinformatics/btp444. [DOI] [PubMed] [Google Scholar]
- 18.GeneCodis: [http://genecodis.cnb.csic.es/] (accessed 17 December 2014).
- 19.Carmona-Saez P, Chagoyen M, Tirado F, Carazo JM, Pascual-Montano A. GENECODIS: a web-based tool for finding significant concurrent annotations in gene lists. Genome Biol. 2007;8:R3. doi: 10.1186/gb-2007-8-1-r3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nogales-Cadenas R, Carmona-Saez P, Vazquez M, Vicente C, Yang X, Tirado F, Carazo JM, Pascual-Montano A: GeneCodis: interpreting gene lists through enrichment analysis and integration of diverse biological information.Nucleic Acids Res 2009, 37(Web server issue):W317–W322. [DOI] [PMC free article] [PubMed]
- 21.Tabas-Madrid D, Nogales-Cadenas R, Pascual-Montano A: GeneCodis3: a non-redundant and modular enrichment analysis tool for functional genomics.Nucleic Acids Res 2012, 40(Webserver issue):W478–W483. [DOI] [PMC free article] [PubMed]
- 22.Hutcheson J, Scatizzi JC, Siddiqui AM, Haines GK, 3rd, Wu T, Li QZ, Davis LS, Mohan C, Perlman H. Combined deficiency of proapoptotic regulators Bim and Fas results in the early onset of systemic autoimmunity. Immunity. 2008;28:206–217. doi: 10.1016/j.immuni.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 23.Teixeira VH, Olaso R, Martin-Magniette ML, Lasbleiz S, Jacq L, Oliveira CR, Hilliquin P, Gut I, Cornelis F, Petit-Teixeira E. Transcriptome analysis describing new immunity and defense genes in peripheral blood mononuclear cells of rheumatoid arthritis patients. PLoS One. 2009;4:e6803. doi: 10.1371/journal.pone.0006803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lessard CJ, Li H, Adrianto I, Ice JA, Rasmussen A, Grundahl KM, Kelly JA, Dozmorov MG, Miceli-Richard C, Bowman S, Lester S, Eriksson P, Eloranta ML, Brun JG, Gøransson LG, Harboe E, Guthridge JM, Kaufman KM, Kvarnström M, Jazebi H, Graham DSC, Grandits ME, Nazmul-Hossain ANM, Patel K, Adler AJ, Maier-Moore JS, Farris AD, Brennan MT, Lessard JA, Chodosh J, et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjögren’s syndrome. Nat Genet. 2013;45:1284–1292. doi: 10.1038/ng.2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yao Y, Higgs BW, Morehouse C, de los Reyes M, Trigona W, Brohawn P, White W, Zhang J, White B, Coyle AJ, Kiener PA, Jallal B. Development of potential pharmacodynamic and diagnostic markers for anti-IFN-α monoclonal antibody trials in systemic lupus erythematosus. Hum Genomics Proteomics. 2009;1:374312. doi: 10.4061/2009/374312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun HG, Dong XJ, Lu T, Yang MF, Wang XM. Clinical value of eukaryotic elongation factor 2 (eEF2) in non-small cell lung cancer patients. Asian Pac J Cancer Prev. 2014;14:6533–6535. doi: 10.7314/APJCP.2013.14.11.6533. [DOI] [PubMed] [Google Scholar]
- 27.Tseng GC, Ghosh D, Feingold E. Comprehensive literature review and statistical considerations for microarray meta-analysis. Nucleic Acids Res. 2012;40:3785–3799. doi: 10.1093/nar/gkr1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, Behrens TW. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A. 2003;100:2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bezalel S, Guri KM, Elbirt D, Asher I, Sthoeger ZM. Type I interferon signature in systemic lupus erythematosus. Isr Med Assoc J. 2014;16:246–249. [PubMed] [Google Scholar]
- 31.Salloum R, Niewold TB. Interferon regulatory factors in human lupus pathogenesis. Transl Res. 2011;157:326–331. doi: 10.1016/j.trsl.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grolleau A, Kaplan MJ, Hanash SM, Beretta L, Richardson B. Impaired translational response and increased protein kinase PKR expression in T cells from lupus patients. J Clin Invest. 2000;106:1561–1568. doi: 10.1172/JCI9352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brkic Z, Versnel MA. Type I IFN signature in primary Sjögren’s syndrome patients. Expert Rev Clin Immunol. 2014;10:457–467. doi: 10.1586/1744666X.2014.876364. [DOI] [PubMed] [Google Scholar]
- 34.Le Page C, Génin P, Baines MG, Hiscott J. Interferon activation and innate immunity. Rev Immunogenet. 2000;2:374–386. [PubMed] [Google Scholar]
- 35.Malemud CJ. Intracellular signaling pathways in rheumatoid arthritis. J Clin Cell Immunol. 2013;4:160. doi: 10.4172/2155-9899.1000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu J, Zhang Y, Ghosh A, Cuevas RA, Forero A, Dhar J, Ibsen MS, Schmid-Burgk JL, Schmidt T, Ganapathiraju MK, Fujita T, Hartmann R, Barik S, Hornung V, Coyne CB, Sarkar SN. Antiviral activity of human OASL protein is mediated by enhancing signaling of the RIG-I RNA sensor. Immunity. 2014;40:936–948. doi: 10.1016/j.immuni.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu MM, Ye QL, Feng CC, Yang J, Zhang T, Li J, Leng RX, Pan HF, Yuan H, Ye DQ. Association of FAS gene polymorphisms with systemic lupus erythematosus: a case–control study and meta-analysis. Exp Ther Med. 2012;4:497–502. doi: 10.3892/etm.2012.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Treviño-Talavera BA, Palafox-Sánchez CA, Muñoz-Valle JF, Orozco-Barocio G, Navarro-Hernández RE, Vázquez-Del Mercado M, García de la Torre I, Oregon-Romero E. FAS −670A>G promoter polymorphism is associated with soluble Fas levels in primary Sjögren’s syndrome. Genet Mol Res. 2014;13:4831–4838. doi: 10.4238/2014.July.2.12. [DOI] [PubMed] [Google Scholar]
- 39.Colonna L, Lood C, Elkon KB. Beyond apoptosis in lupus. Curr Opin Rheumatol. 2014;26:459–466. doi: 10.1097/BOR.0000000000000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Biermann MH, Veissi S, Maueröder C, Chaurio R, Berens C, Herrmann M, Munoz LE. The role of dead cell clearance in the etiology and pathogenesis of systemic lupus erythematosus: dendritic cells as potential targets. Expert Rev Clin Immunol. 2014;10:1151–1164. doi: 10.1586/1744666X.2014.944162. [DOI] [PubMed] [Google Scholar]
- 41.Zhang S, Sun Y, Chen H, Dai Y, Zhan Y, Yu S, Qiu X, Tan L, Song C, Ding C. Activation of the PKR/eIF2α signaling cascade inhibits replication of Newcastle disease virus. Virol J. 2014;11:62. doi: 10.1186/1743-422X-11-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bullido MJ, Martínez-García A, Tenorio R, Sastre I, Muñoz DG, Frank A, Valdivieso F. Double stranded RNA activated EIF2α kinase (EIF2AK2; PKR) is associated with Alzheimer’s disease. Neurobiol Aging. 2008;29:1160–1166. doi: 10.1016/j.neurobiolaging.2007.02.023. [DOI] [PubMed] [Google Scholar]
- 43.Sadler AJ, Williams BRG. Structure and function of the protein kinase R. Curr Top Microbiol Immunol. 2007;316:253–292. doi: 10.1007/978-3-540-71329-6_13. [DOI] [PubMed] [Google Scholar]
- 44.Gilbert SJ, Duance VC, Mason DJ. Does protein kinase R mediate TNF-α- and ceramide-induced increases in expression and activation of matrix metalloproteinases in articular cartilage by a novel mechanism? Arthritis Res Ther. 2003;6:R46. doi: 10.1186/ar1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gilbert SJ, Duance VC, Mason DJ. Protein kinase R: a novel mediator of articular cartilage degradation in arthritis. Curr Rheumatol Rev. 2006;2:9–21. doi: 10.2174/157339706775697026. [DOI] [Google Scholar]
- 46.Yeung MC, Liu J, Lau AS. An essential role for the interferon-inducible, double-stranded RNA-activated protein kinase PKR in the tumor necrosis factor-induced apoptosis in U937 cells. Proc Natl Acad Sci U S A. 1996;93:12451–12455. doi: 10.1073/pnas.93.22.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ward SV, Samuel CE. The PKR kinase promoter binds both Sp1 and Sp3, but only Sp3 functions as part of the interferon-inducible complex with ISGF-3 proteins. Virology. 2003;313:553–566. doi: 10.1016/S0042-6822(03)00347-7. [DOI] [PubMed] [Google Scholar]
- 48.McAllister CS, Taghavi N, Samuel CE. Protein kinase PKR amplification of interferon β induction occurs through initiation factor eIF-2α-mediated translational control. J Biol Chem. 2012;287:36384–36392. doi: 10.1074/jbc.M112.390039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun. 2009;388:621–625. doi: 10.1016/j.bbrc.2009.08.062. [DOI] [PubMed] [Google Scholar]