

Abstract
RASSF1A may be the most frequently inactivated tumor suppressor identified in human cancer so far. It is a proapoptotic Ras effector and plays an important role in the apoptotic DNA damage response (DDR). We now show that in addition to DDR regulation, RASSF1A also plays a key role in the DNA repair process itself. We show that RASSF1A forms a DNA damage-regulated complex with the key DNA repair protein xeroderma pigmentosum A (XPA). XPA requires RASSF1A to exert full repair activity, and RASSF1A-deficient cells exhibit an impaired ability to repair DNA. Moreover, a cancer-associated RASSF1A single-nucleotide polymorphism (SNP) variant exhibits differential XPA binding and inhibits DNA repair. The interaction of XPA with other components of the repair complex, such as replication protein A (RPA), is controlled in part by a dynamic acetylation/deacetylation cycle. We found that RASSF1A and its SNP variant differentially regulate XPA protein acetylation, and the SNP variant hyperstabilizes the XPA-RPA70 complex. Thus, we identify two novel functions for RASSF1A in the control of DNA repair and protein acetylation. As RASSF1A modulates both apoptotic DDR and DNA repair, it may play an important and unanticipated role in coordinating the balance between repair and death after DNA damage.
INTRODUCTION
The human genome is under constant assault from environmental factors such as UV light, mutagenic chemicals, and oxidative metabolic by-products. The ability to repair DNA is essential to maintain genome integrity and to minimize the acquisition of oncogenic lesions (1, 2). Accumulated mutation loads beyond the capacity of the DNA repair systems to correct provoke a DNA damage response (DDR) leading to apoptotic or senescent cell death (3–5).
RASSF1A is a tumor suppressor that is frequently inactivated by epigenetic mechanisms in human tumor cells (6–8). Knockout of RASSF1A in mice promotes an enhanced rate of cancer development (9). RASSF1A appears to act as a scaffolding protein that complexes with and modulates the activity of other apoptotic tumor suppressors such as MST/LATS1 (10). It has been shown to play an important role in the apoptotic DNA damage response by activating the Hippo pathway (11). RASSF1A also contains a Ras association (RA) domain (12) and may form an endogenous complex with the activated Ras oncoprotein (13). Thus, it has the potential to serve as a Ras effector, integrating progrowth pathways with progrowth arrest/death pathways.
Nucleotide excision repair (NER) is a major DNA repair pathway in eukaryotic cells that removes aberrant nucleotides from one strand and replaces them using the undamaged strand as the template. The repair process involves the formation of a dynamic, multicomponent repair complex at the site of damage whose members cycle in and out as the repair proceeds. The xeroderma pigmentosum A protein (XPA) is an important member of the complex and is essential for effective NER. It directly binds the damaged single strand of DNA (14) and scaffolds multiple DNA repair components as they cycle in and out of the dynamic NER complex (15). The regulation of XPA protein/protein interactions after DNA damage involves both phosphorylation of XPA by ATR and deacetylation by SIRT1 (16, 17). Hereditary defects in XPA in humans cause an impaired ability to repair UV-induced DNA damage and a predisposition to cancer (18). Thus, XPA is one of an elite group of proteins that is confirmed as a bona fide human tumor suppressor.
The RASSF1 gene was originally identified from a yeast two-hybrid screen using XPA as the bait (19). This suggested that RASSF1A might play a role in DNA repair as well as modulating the DDR. However, the interaction has never been confirmed in mammalian cells, and the effects of RASSF1A on NER have not been reported. We have now determined that RASSF1A forms an endogenous complex with XPA after DNA damage and that this interaction is essential for full XPA repair activity. A single-nucleotide polymorphism (SNP) variant of RASSF1A has been identified in approximately 22% of the Caucasian population of the United States (20). This variant has repeatedly been associated with an enhanced risk of cancer (20), but the mechanism responsible for such an effect is unclear. When we examined the SNP variant, we found that it differentially binds XPA and exerts a marked inhibitory effect on DNA damage repair.
XPA undergoes a cycle of acetylation/deacetylation which regulates how it interacts with other members of the dynamic DNA repair complex, such as replication protein A (RPA), over the course of the lesion repair (16). We show that both wild-type RASSF1A and its SNP variant promote the formation of deacetylated XPA in the absence of UV. However, in the presence of UV, the two RASSF1A isoforms have opposite effects. The wild-type RASSF1A enhances the deacetylation of XPA, while the SNP variant inhibits it. In RASSF1A SNP variant cells, this results in a hyperstabilized XPA-RPA complex and prevents normal XPA cycling in and out of the nucleus. Thus, we identify new roles for RASSF1A in the regulation of DNA repair and protein acetylation. We also provide a mechanistic explanation for the association of the RASSF1A SNP with cancer.
MATERIALS AND METHODS
Plasmids and DNA.
RASSF1A expression plasmids have been described previously (20, 21). Wild-type XPA cDNA was a gift from K. Kraemer (NCI, Bethesda, MD) and was cloned into pEGFP (EGFP stands for enhanced green fluorescent protein) and pHC-Red1 (Clontech, Palo Alto, CA) or pcDNA3.1 modified to add a 5′ hemagglutinin (HA) epitope tag. The acetylation mimicking mutant of XPA (XPA-2KQ) was kindly provided by Jianyuan Luo (University of Maryland) and was cloned into pHC-Red1 or pcDNA3.1 modified to add a 5′ HA epitope tag. pCMV-Luc (CMV stands for cytomegalovirus, and Luc stands for luciferase) and TK-Renilla-Luc (TK stands for thymidine kinase, and Renilla-Luc stands for renilla luciferase) were obtained commercially (Promega, Madison, WI). RASSF1A short hairpin RNA (shRNA) plasmids were obtained from Origene, Rockville, MD (catalog no. TR307696).
Tissue culture and cell lines.
NCI-H1792, Calu-6, NCI-H1299, and HEK-293 cells were obtained from the ATCC (Manassas, VA). They were cultured in RPMI 1640 or Dulbecco modified Eagle medium (DMEM), respectively (Invitrogen, Carlsbad, CA), each with 10% fetal bovine serum (FBS) (Valley Biologicals, VA). The cells were treated with cisplatin at 10 μM for 12 to 16 h prior to analysis. HEK-293T cells were cultured in DMEM supplemented with 10% FBS. XPA−/− human fibroblasts were a gift from K. Kraemer (NCI, Bethesda, MD). Stable transfectants were generated by transfecting cells with 1 μg of plasmid DNA using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) following the manufacturer's instructions. RASSF1A transfectants were selected in 500 μg/ml G418 and used as an early passage pooled population. shRNA transfectants were selected in 1 μg/ml puromycin. Transient transfections were performed using Lipofectamine 2000 and 1 μg of each plasmid DNA. UVC (UV C [100- to 280-nm wavelength]) irradiation was performed in a Stratalinker (Stratagene, La Jolla, CA).
Western blot analysis and immunoprecipitation.
Cells were lysed in modified radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 50 mM Tris [pH 7.5], 1% NP-40), and the lysate was Western blotted using a Novex NuPage gel system (Invitrogen). XPA, green fluorescent protein (GFP), and RASSF1A antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). FLAG and HA antibodies were obtained from Sigma (Newark, NJ). Immunoprecipitations were performed using HA-conjugated Sepharose beads (Sigma), antiacetyllysine-conjugated beads (Immunechem, Burnaby, BC, Canada) and anti-XPA antibody (Santa Cruz) as appropriate. Horseradish peroxidase (HRP)-conjugated Trueblot secondary antibodies were purchased from eBioscience Inc. (San Diego, CA). Western blots were developed using a Pierce enhanced chemiluminescence (ECL) detection system (Thermo Scientific, Rockford, IL) and Kodak X-ray film. Images were scanned and quantified using a Pharos FX plus molecular imager (Bio-Rad, Hercules, CA) and Quantity One software (Bio-Rad).
Luciferase-based DNA repair assay.
CMV-Luc plasmid was diluted to a concentration of 200 ng/μl and irradiated in a clear Eppendorf tube with 900 J/m2 of UV in a Stratalinker (Stratagene, La Jolla, CA). After checking that the irradiated plasmid had no activity in fibroblasts derived from an XPA patient, target cells were transfected with 100 ng irradiated CMV-Luc and 5 ng nonirradiated TK-Renilla-Luc per well in a six-well plate. Each transfection also included 1 μg of vector or XPA expression plasmid. After 4 h, cells were lysed and assayed with a dual luciferase kit (Promega). Samples were read in a Berthold luminometer.
Comet assay.
Comet assays were performed using a Trevigen (Boston, MA) kit per the manufacturer's instructions. Results were visualized on an Olympus fluorescence inverted microscope and quantified using CASPLab software (22).
CPD assay.
UV-induced cyclobutane pyrimidine dimers (CPD) were quantitated using the OxiSelect UV-induced DNA damage enzyme-linked immunosorbent assay (ELISA) kit (Cell Biolabs, Inc., San Diego, CA) per the manufacturer's instructions.
Fluorescence quantitation.
Fluorescence intensity of the XPA-deficient fibroblasts transiently expressing either GFP-tagged vector (GFP-vector) or RASSF1A was quantitated using ImageJ software. Images were captured at a magnification of ×10, and the fluorescence intensity of at least 50 cells was determined after subtracting the surrounding background.
Transgenic animals.
RASSF1A knockout mice (generous gift from Gerd Pfeifer, Beckman Research Institute, City of Hope, Duarte, CA) (9) were crossed with XPA knockout mice (23) or wild-type parental mice. UV treatments were performed as described in reference 24. All animal experiments were performed with the permission of the Institutional IACUC committee.
Statistical analysis.
Data are reported as means ± standard deviations. Statistical significance of differences between the mean values was assessed by Student's t test or analysis of variance (ANOVA), as appropriate. Data were considered significant at a P of <0.05.
RESULTS
RASSF1A forms a DNA damage-regulated complex with XPA.
Previous studies have implicated RASSF1A as a potential binding partner for the DNA repair protein XPA in yeast two-hybrid systems (19), but these results have never been confirmed in mammalian cells. To determine whether RASSF1A and XPA can form a physiological complex in mammalian cells, we treated NCI-H1792 cells with carrier or the DNA damaging agent cisplatin, immunoprecipitated cell lysates with XPA, and immunoblotted for RASSF1A. In the absence of DNA damage, we observed little evidence of coimmunoprecipitation between endogenous RASSF1A and XPA proteins. However, when the cells were challenged with cisplatin, the association of endogenous RASSF1A with endogenous XPA could be detected (Fig. 1A). Exogenous expression of RASSF1A and XPA in HEK-293T cells challenged with either cisplatin or UV irradiation gave similar results (Fig. 1B).
FIG 1.

RASSF1A forms an endogenous complex with XPA that is enhanced by DNA damaging agents. (A) Equal amounts of protein lysates from H1792 lung cancer cells that had been treated with 10 μM cisplatin (+) or vehicle for 16 h were immunoprecipitated with an anti-XPA antibody, fractionated on an SDS-polyacrylamide gel, and then immunoblotted for the presence of RASSF1A in the complex. (B) HEK-293T cells were transiently transfected with GFP-tagged RASSF1A and HA-tagged XPA expression constructs. The cells were left untreated (−) or treated with 40 J/m2 UVC or 10 μM cisplatin (CP). One hour after UV exposure or 16 h after adding cisplatin, the cells were lysed, and equal amounts of protein were immunoprecipitated (IP) for GFP. The immunoprecipitate was fractionated on an SDS-polyacrylamide gel and then Western blotted (or immunoblotted [IB]) with anti-HA and anti-GFP antibodies. (C) HEK-293T cells were cotransfected with expression constructs for HA-tagged RASSF1A and Flag-tagged XPA, in the absence (−) or presence (+) of GFP-tagged ATR. Equal amounts of protein were immunoprecipitated with an anti-HA antibody, and the immunoprecipitates fractionated on an SDS-polyacrylamide gel and Western blotted with anti-HA and anti-Flag antibodies. (D) HEK-293T cells were cotransfected with expression constructs for HA-tagged XPA and GFP-tagged wild-type RASSF1A or a mutant that is defective for phosphorylation at serine 131 (S131A). Cells were left untreated or exposed to 40 J/m2 UV. One hour after exposure to UV, the cells were lysed, and equal amounts of protein were immunoprecipitated with an anti-GFP antibody. The immunoprecipitates were fractionated on an SDS-polyacrylamide gel and analyzed by Western blotting using anti-HA and anti-GFP antibodies.
RASSF1A contains a consensus ATM/ATR phosphorylation site (7) and is phosphorylated at serine 131 by ATM in response to DNA damaging agents (11). ATM and ATR are the principal checkpoint kinases activated in response to DNA damage (25), and ATR is the primary sensor of single-stranded (ssDNA) breaks (SSB) caused by UV damage and replication stress (26). We therefore sought to determine whether phosphorylation of RASSF1A by ATM/ATR at serine 131 is required for its binding to XPA. Cotransfection experiments of XPA and RASSF1A in the presence and absence of exogenous ATR showed that ATR enhanced the interaction between RASSF1A and XPA (Fig. 1C). A mutant of RASSF1A that is unable to be phosphorylated at serine 131, RASSF1A S(131)A, did not exhibit enhanced binding to XPA after UV exposure (Fig. 1D).
RASSF1A is required for full XPA activity.
To examine the functional consequences of the interaction between RASSF1A and XPA, we generated a series of stable RASSF1A knockdown cell lines from XPA-deficient patient-derived fibroblasts using two different RASSF1A Hush shRNAs. The ability of XPA to stimulate the repair of a UV-irradiated CMV-Luc plasmid was then assayed in the cells. Increased relative luciferase activity is an indirect measure of XPA-mediated DNA repair. Figure 2A shows that after irradiation, the CMV-Luc reporter lost most of its ability to express luciferase in XPA-deficient fibroblasts. Figure 2B shows that cotransfection of XPA into the XPA-deficient cells acts to restore the activity, but this is impaired when RASSF1A is suppressed by shRNA. The blots at the bottom of Fig. 2B show the degree of RASSF1A protein knockdown in the cells.
FIG 2.

Suppression of RASSF1A impairs XPA-mediated DNA repair. (A) The CMV-Luc plasmid was irradiated with 900 J/m2 UVC and transfected into XPA-deficient human fibroblasts, and 4 h later, luciferase activity was assayed. The UV-irradiated plasmid showed severely reduced luciferase activity compared to the unirradiated control. (B) XPA-deficient human fibroblasts were stably transfected with 2 independent RASSF1A shRNA constructs and a scrambled control. Cell lines were transiently cotransfected with the irradiated CMV-Luc reporter plasmid together with an expression plasmid for XPA and TK-Renilla as an internal control. DNA repair was assessed by an increase in luciferase activity compared to the renilla luciferase activity (relative luciferase units [RLU]). Data are derived from three independent experiments performed in duplicate, and error bars show standard errors. Data were considered significant at a P of <0.05. Effective knockdown of RASSF1A expression was confirmed by Western blot analysis. Actin was used as a loading control.
The cancer-associated A(133)S SNP variant of RASSF1A differentially binds XPA.
A common SNP of the RASSF1A gene has been identified and associated with a predisposition for cancer (20, 27–29). The SNP variant produces a protein with an A(133)S substitution, which interferes with the consensus phosphorylation site for the DNA damage-activated ATM/ATR kinases (11). This suggests that the SNP variant protein might mediate a differential response to DNA damage (29). When we examined the interaction of the SNP variant with XPA before and after UV damage, we were surprised to find that it demonstrated the interaction profile opposite to that of the wild-type RASSF1A, forming a complex with XPA that was readily detectable in the absence of DNA damage that dissociated upon UV exposure (Fig. 3A). Similar results were obtained when the cells were damaged with cisplatin (Fig. 3B). This suggested that the SNP variant might differentially regulate XPA and DNA repair.
FIG 3.
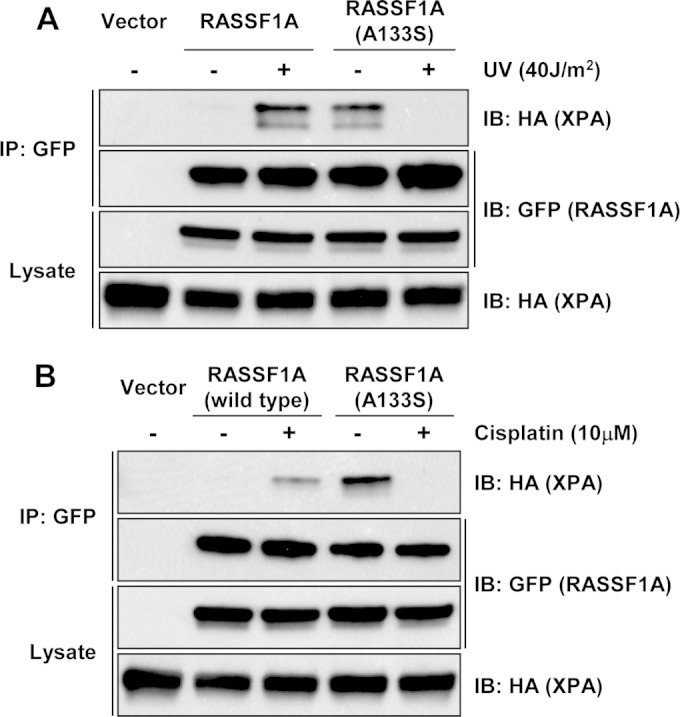
The A(133)S RASSF1A SNP variant differentially binds XPA. (A) HEK-293T cells were transiently transfected with GFP-tagged RASSF1A or the A(133)S SNP variant and HA-tagged XPA expression constructs and left untreated or exposed to 40 J/m2 UV. One hour after exposure to UV, the cells were lysed, and equal amounts of protein were immunoprecipitated with an anti-GFP antibody. The immunoprecipitates were fractionated on an SDS-polyacrylamide gel and Western blotted with anti-HA and anti-GFP antibodies. (B) Similar experiments were performed using cisplatin as a DNA damaging stimulus.
Differential regulation of UV-induced DNA repair by RASSF1A and its A(133)S SNP variant.
To confirm the DNA repair results on chromosomal DNA and to investigate the effects of the RASSF1A SNP variation, we generated a RASSF1A+/− matched set of NCI-H1299 human lung tumor cells (which are RASSF1A negative [30].) Cells were transfected with vector, wild-type RASSF1A, or A(133)S SNP variant RASSF1A expression plasmids, and a stable pool of cells was obtained after selection in G418 that expressed the relevant proteins within approximately 5-fold of the endogenous levels observed in the RASSF1A-positive tumor cell line Calu-6 (Fig. 4A). We then subjected the cells to UV-mediated DNA damage and assayed the DNA repair capacity of the cells using an alkaline Comet assay kit (Fig. 4B). The average size of the DNA tail moment from lysed cells is a measure of unrepaired DNA present. Cells with restored wild-type RASSF1A expression exhibited smaller tails, indicating enhanced ability to repair DNA after damage relative to the vector-transfected cells. However, the SNP variant expressing cells demonstrated much larger tails, evidencing a marked suppression of DNA repair. Figure 4C shows a representative cell comet tail after UV damage from each group.
FIG 4.
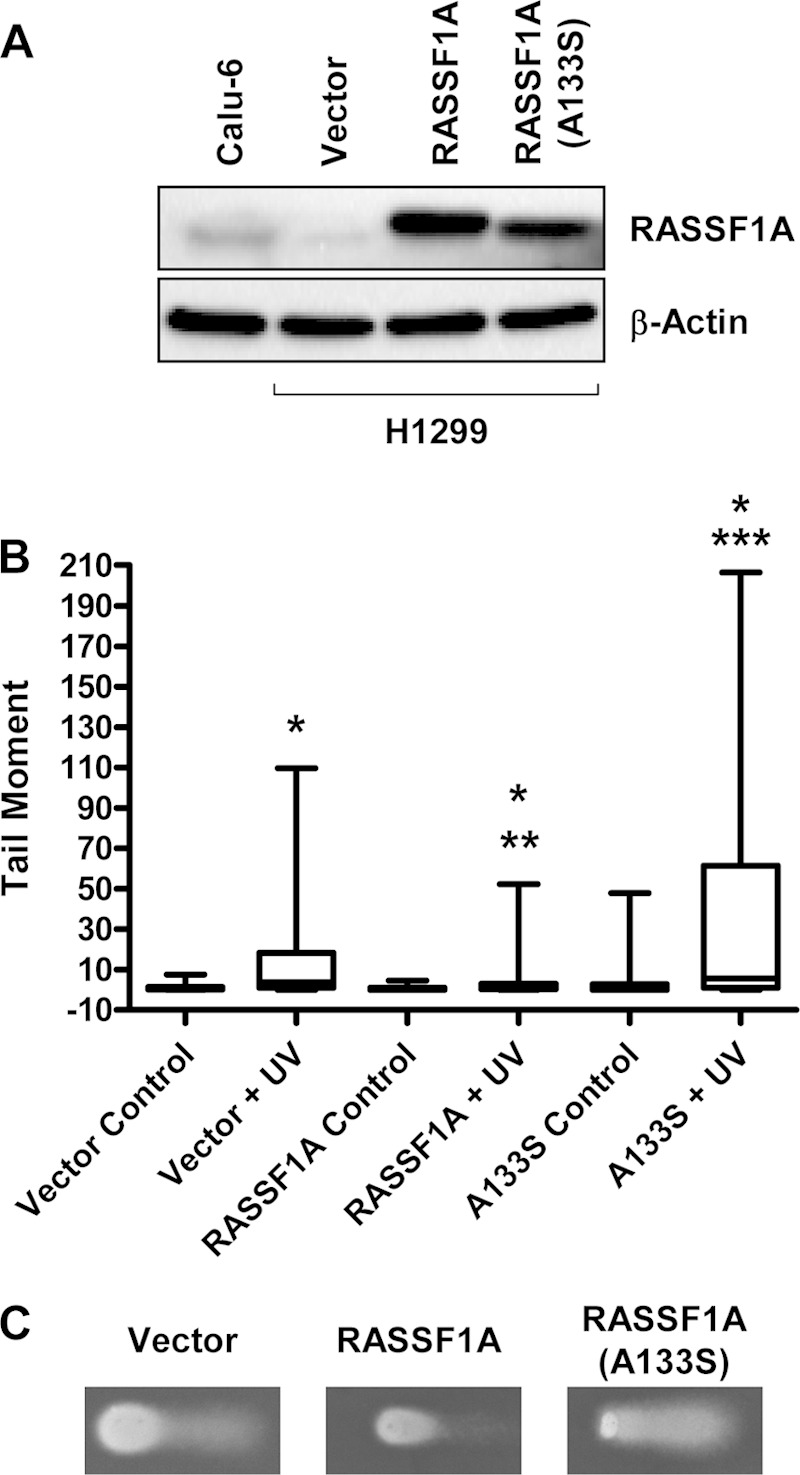
Wild-type RASSF1A and the A(133)S SNP variant of RASSF1A differentially modulate DNA repair. (A) NCI-H1299 cells were stably transfected with vectors expressing wild-type RASSF1A or the A(133)S SNP variant form of RASSF1A. Expression of the exogenous protein was measured by immunoblotting with anti-RASSF1A antibodies and found to be within 5-fold of that observed for the endogenous protein in Calu-6 cells. (B) The cells were irradiated with UV and assayed for changes in DNA repair activity by Comet assay. Box-and-whisker plots show data from 100 cells from two independent experiments. Values that are significantly different are indicated by asterisks as follows: *, P < 0.001 compared to the value for untreated control cells; **, P < 0.01 compared to the value for UV-treated control cells; ***, P < 0.0001 compared to the value for UV-treated RASSF1A-expressing cells. (C) Representative comet tails after irradiation.
Colocalization of RASSF1A and XPA in live cells.
As the wild-type RASSF1A and the SNP variant of RASSF1A exhibit different effects on binding XPA and modulating DNA repair activity, we sought to compare their colocalization with XPA in live cells. We transfected COS-7 cells with GFP-tagged RASSF1A (GFP-RASSF1A) and red fluorescent protein (RFP)-tagged XPA. After 24 h, cells were examined by fluorescence microscopy. In the absence of RASSF1A, XPA exhibited uniform distribution in the nucleus (Fig. 5A). RASSF1A is most prominently localized to the cytoplasm on microtubules (31–33). When XPA and RASSF1A or the A(133)S SNP variant of RASSF1A were coexpressed in the cells, although the RASSF1A proteins were still prominently localized in the cytoplasm on microtubules, they also colocalized in foci in the nucleus with XPA, as evidenced by the yellow color in the merged image (Fig. 5A). The wild-type RASSF1A appeared to give stronger general nuclear staining, and most of the nuclear RASSF1A SNP variant appeared to be associated with the foci. The exact nature of the XPA foci remains to be determined. XPA has been found in conjunction with other DNA repair proteins, such as RPA and PCNA, in a variety of nuclear foci linked to sites of DNA repair, including sites of single-strand DNA damage (34), replication foci, where XPA plays a key role in mediating DNA repair (35), and even in sites of DNA double-strand breaks (36). There was little apparent difference in the numbers of foci/cell between the wild type and SNP variant of RASSF1A in unstimulated cells. After irradiation with UV, the number of focus-positive cells increased after 10 min. After 2 h, the numbers had returned to background level for the wild-type RASSF1A cells, but the levels of SNP variant cells with foci remained elevated (Fig. 5B).
FIG 5.
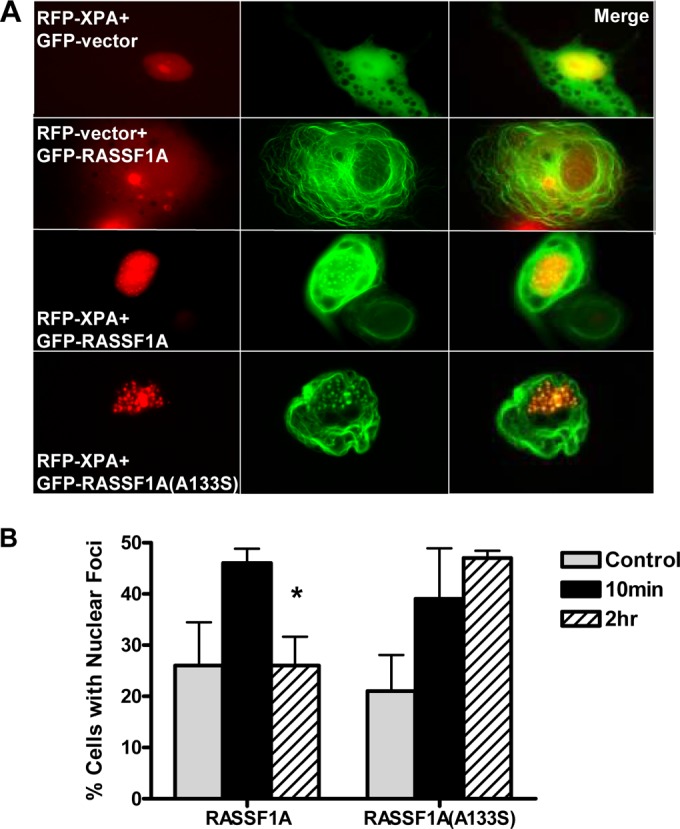
RASSF1A and XPA colocalize in nuclear foci. (A) COS-7 cells were cotransfected with GFP vector or GFP-tagged RASSF1A (GFP-RASSF1A) or the A(133)S SNP variant of RASSF1A and RFP-XPA, and images were captured 24 h later using a fluorescence microscope. Magnification, ×100. (B) The cells were exposed to UV irradiation, and the percentage of cells with XPA and RASSF1A colocalization was quantitated 10 min and 2 h after exposure by randomly selecting 50 cells expressing both GFP-RASSF1A and RFP-tagged XPA (RFP-XPA). Results are expressed as means plus standard deviations (SD) (error bars) from triplicate experiments. The value that is significantly different (P < 0.05) from the value for control cells is indicated by an asterisk.
Differential subcellular localization of RASSF1A variants and XPA.
The fluorescence experimental results hinted that there were significant differences in the subcellular localization of the RASSF1A proteins and that they might differentially affect the localization of XPA. To quantify such effects, we UV treated the stable H1299 cell system, fractionated lysates into cytoplasmic and nuclear fractions, and Western blotted the fractions for the relative levels of RASSF1A and XPA. Figure 6A shows a representative blot. Figure 6B shows quantification of two experiments. In H1299 cells expressing wild-type RASSF1A, the nuclear levels of XPA essentially doubled after 5 min (Fig. 6A and B) and fell back to almost normal after 1 h (by which time most repair has been completed [Fig. 4B]). In contrast, although XPA in the cells expressing RASSF1A A(133)S SNP also increased after UV irradiation, the nuclear levels remained elevated after 1 h (Fig. 6A and B). When we examined the status of RASSF1A in the H1299 cell system, we found that wild-type RASSF1A appeared to be almost equally cytoplasmic and nuclear and that there was no statistically significant change in relative localization 1 h after treatment (Fig. 6A and C). In contrast, the SNP variant RASSF1A protein was less nuclear without treatment and became even less nuclear after UV irradiation (Fig. 6A and C). Thus, the wild-type RASSF1A and SNP variant exhibit differential subcellular localization.
FIG 6.

Wild-type RASSF1A and the A(133)S SNP variant differentially regulate DNA repair complex stability. (A) The NCI-H1299 cells stably expressing RASSF1A or the A(133)S SNP were exposed to UV, and nuclear (Nuc) and cytoplasmic (Cyt) lysates were prepared 5 min (5′) and 1 h after exposure. Equal amounts of lysates were analyzed by Western blotting for XPA expression in each fraction using anti-XPA and anti-HA antibodies. p38 and transcription factor IIH (TFIIH) were used as controls for cytoplasmic and nuclear fractions, respectively. The experiment was repeated twice, and a representative blot is shown. (B and C) The blots were quantified densitometrically to generate a ratio of nuclear versus cytoplasmic XPA (B) or RASSF1A (C). (D) The NCI-H1299 RASSF1A/A133S+/− cells were exposed to UV, and 1 h later, the cells were lysed, and equal amounts of protein were immunoprecipitated with an anti-XPA antibody. Immunoprecipitates were resolved on SDS-polyacrylamide gels and immunoblotted with anti-XPA and anti-RPA70 antibodies.
The RASSF1A A(133)S SNP variant hyperstabilizes the XPA-RPA70 complex after DNA damage.
After DNA damage has occurred, a repair complex assembles at the site of damage. The complex is dynamic, and XPA enters the complex to help scaffold the cycle of repair proteins that enter and leave during the DNA repair process (15, 37, 38). One of the components of the complex that helps anchor XPA to DNA is the RPA trimer (39, 40). As the RASSF1A SNP variant appeared to promote the retention of XPA in the nucleus after UV treatment, we wondered whether the SNP-expressing cells might demonstrate changes in the association of XPA with RPA.
When we examined the association between endogenous XPA and endogenous RPA70 in the NCI-H1299 matched set of cells, we found little stable association between the proteins in the absence of UV irradiation in any of the cells. When we treated vector-transfected cells or cells expressing wild-type RASSF1A with UV, we still observed no coprecipitation of XPA and RPA. However, in the irradiated cells expressing the SNP variant of RASSF1A, we detected a strong, stable, endogenous complex between XPA and RPA70 (Fig. 6D). This suggests that the XPA is being trapped on the DNA in the nucleus in a frozen repair complex.
Differential regulation of XPA deacetylation by RASSF1A and the A(133)S SNP variant.
Having established that RASSF1A can complex with XPA and modulate NER, we sought to determine the mechanism. XPA is regulated by posttranslational modifications, including phosphorylation (41) and acetylation (16). Acetylation/deacetylation is essential for the correct dynamic assembly/disassembly of XPA repair complexes, and in particular is essential for the interaction with RPA (16).
To determine whether RASSF1A might be modulating XPA acetylation, we cotransfected HEK-293 cells with XPA and wild-type RASSF1A or RASSF1A SNP variant expression constructs. Immunoprecipitation and immunoblotting with an acetyllysine antibody was used to measure the degree of XPA acetylation. Figure 7A shows the results of a representative experiment, with a quantification of the average of three experiments shown below (Fig. 7B). In the absence of RASSF1A, the intrinsic XPA acetylation was relatively high and showed little decrease upon UV treatment. However, in the presence of wild-type RASSF1A, XPA acetylation was reduced, and UV irradiation provoked a further, marked decrease in the levels of acetylated XPA. Cells expressing the SNP variant of RASSF1A also showed a reduced basal XPA acetylation. However, upon UV treatment, the acetylation increased, in striking contrast to the cells expressing wild-type RASSF1A. Thus, RASSF1A and its SNP variant not only differentially bind XPA after UV damage but also differentially modulate its acetylation status.
FIG 7.
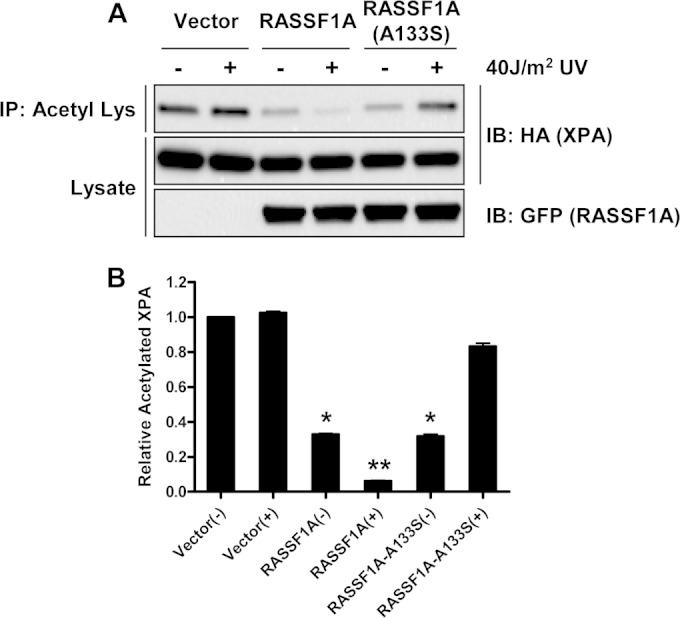
RASSF1A and the A(133)S SNP variant differentially modulate XPA acetylation. (A) HEK-293 cells were transiently cotransfected with expression constructs for HA-tagged XPA and GFP-tagged RASSF1A or A(133)S SNP and left untreated (−) or exposed to 40 J/m2 UV (+). One hour after UV exposure, the cells were lysed, and equal amounts of protein were immunoprecipitated with anti-acetyl-Lys beads. The immunoprecipitates were fractionated on an SDS-polyacrylamide gel and Western blotted with anti-HA and anti-GFP antibodies. A representative Western blot is shown in panel A. (B) Data from 3 independent experiments are quantitated in the bar graph. Data are the means plus SD of three independent experiments. Values that are significantly different are indicated by asterisks as follows: *, P < 0.05 compared to the value for control cells exposed to UV; **, P < 0.05 compared to the value for untreated RASSF1A-expressing cells.
In Fig. 1 we showed that inactivating the ATR phosphorylation site of RASSF1A blocked the increase in RASSF1A/XPA binding. When we examined this S(131)A RASSF1A mutant in the acetylating assays, we found that it was rendered unresponsive to UV (Fig. 8A). Thus, the action of wild-type RASSF1A appears to be dependent upon ATR phosphorylation, but the action of the SNP variant does not. As further confirmation of the role of ATR, we performed experiments in the presence of the ATR/ATM inhibitor caffeine. The inhibitor blocked both the basal effects of RASSF1A on XPA deacetylation and the effects after UV treatment (Fig. 8B).
FIG 8.
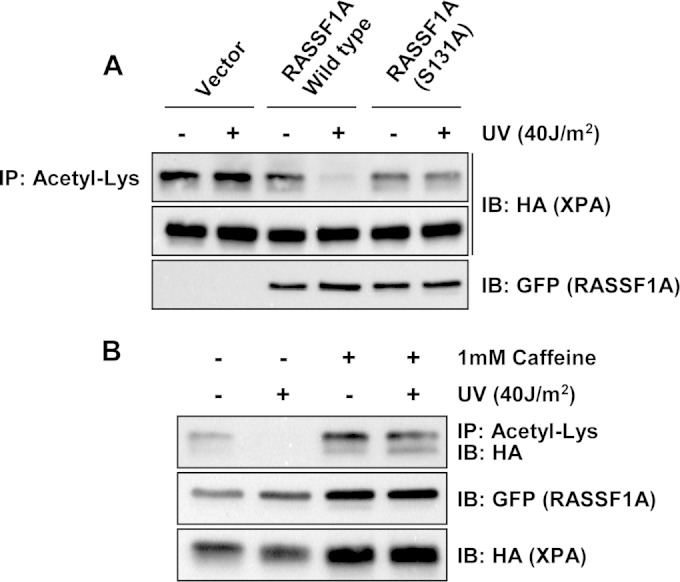
Phosphorylation at Ser131 by ATR is critical for RASSF1A-mediated XPA deacetylation. (A) HEK-293 cells were transiently cotransfected with expression constructs for HA-tagged XPA and GFP-tagged RASSF1A or the S(131)A nonphosphorylatable mutant of RASSF1A and left untreated (−) or exposed to 40 J/m2 UV (+). One hour after UV exposure, the cells were lysed, and equal amounts of protein were immunoprecipitated with anti-acetyl-Lys beads. The immunoprecipitates were fractionated on an SDS-polyacrylamide gel and immunoblotted with anti-HA and anti-GFP antibodies. (B) Similar experiments were performed with wild-type RASSF1A except the cells were treated with 1 mM caffeine or vehicle 16 h prior to exposure to UV. One hour after UV exposure, the cells were processed as described above.
To confirm the effects of RASSF1A on XPA acetylation under conditions that are closer to physiological conditions, we examined the status of endogenous XPA in the H1299 cell system. We could detect low levels of XPA acetylation in the RASSF1A-negative cells but not in the cells expressing wild-type RASSF1A or the SNP variant in the absence of irradiation (Fig. 9). We observed a reduction in XPA acetylation in the absence of RASSF1A after irradiation. This contrasts with the results in the HEK-293 overexpression studies, perhaps because of the lower levels of the endogenous protein making the assay more sensitive. The most striking result obtained was with the SNP variant-expressing cells. These repair defective cells demonstrated a massive failure to deacetylate XPA after irradiation (Fig. 9). This result was similar but much more marked than that observed in the overexpression studies, emphasizing the importance of measuring the status of the endogenous protein.
FIG 9.
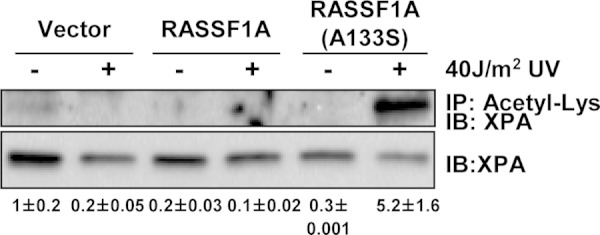
RASSF1A and the A(133)S SNP variant differentially modulate XPA acetylation in stable cell lines. The NCI-H1299 cells stably expressing wild-type RASSF1A or the A(133)S SNP variant were assayed in a manner similar to that described in the legend to Fig. 7. Endogenous XPA present in the antiacetyl-lysine IP was detected using anti-XPA antibodies. The experiment was repeated twice, and densitometric quantitation of the blots is shown below the blots.
RASSF1A-induced deacetylation of XPA is critical for XPA-mediated repair of UV-induced DNA damage.
We have shown that RASSF1A is required for XPA-mediated DNA repair and can modulate the acetylation status of XPA. Deacetylation of XPA has been shown to be essential for the repair of cyclobutane pyrimidine dimers (CPD), one of the predominant types of DNA damage induced by UV. To determine whether RASSF1A was acting via modulating XPA acetylation, we generated a matched set of cell lines in which we stably reexpressed either wild-type XPA or a mutant of XPA that mimics the hyperacetylated form (XPA-2KQ) (7) in XPA-deficient human fibroblasts. We then transiently transfected the cells with expression constructs for either the GFP-tagged vector or RASSF1A, exposed the cells to UV irradiation, and then quantitated the levels of cyclobutane pyrimidine dimers. We found that there was a sharp increase in CPD formation in the XPA-deficient cells stably expressing a control vector after exposure to UV (Fig. 10A), and this was decreased, although not significantly in the cells overexpressing RASSF1A. There was a significant decrease in CPD levels in the cells reexpressing wild-type XPA, which was further decreased in the presence of RASSF1A (Fig. 10A). In contrast, the XPA-2KQ mutant of XPA abolished the ability of RASSF1A to repair CPD adducts in the XPA-deficient cells, suggesting that RASSF1A-induced XPA deacetylation is critical for XPA-mediated repair of UV-induced DNA damage. Although the level of the exogenous GFP-tagged RASSF1A (GFP-RASSF1A) was slightly lower than for that of the cells overexpressing the GFP vector, as quantitated using ImageJ software, the levels of RASSF1A expression were consistent among the three different XPA-deficient cell lines (Fig. 10B).
FIG 10.
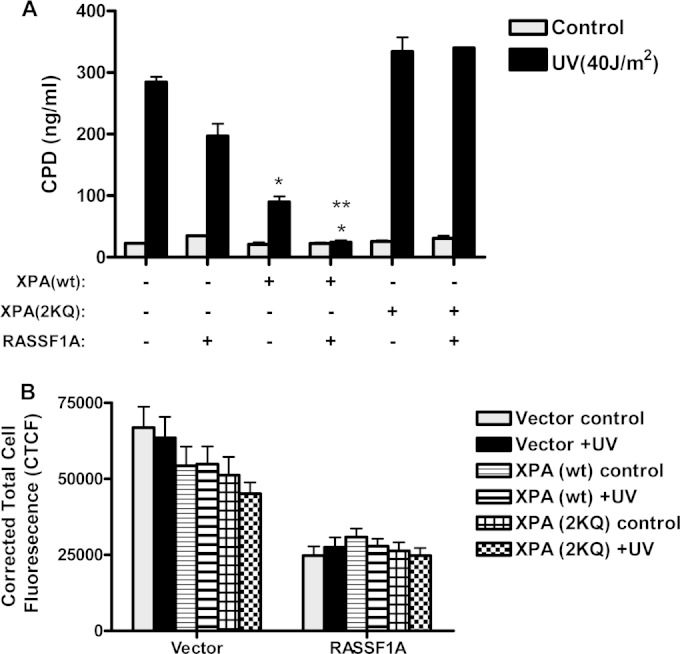
RASSF1A-mediated deacetylation of XPA is essential for repair of UV-induced cyclobutane pyrimidine dimers (CPD). (A) XPA-deficient fibroblasts stably expressing either wild-type (wt) XPA or an XPA acetylation mimicking mutant (XPA-2KQ) or transfected with the empty vector were transiently transfected with either GFP-vector or RASSF1A. Twenty-four hours after transfection, the cells were left untreated or exposed to 40 J/m2 UV. One hour later, DNA was isolated from the cells, and equal amounts were added to a high-binding 96-well plate. CPD levels were quantitated by ELISA using an anti-CPD antibody. Values are means plus SD for two independent experiments performed in duplicate. Values that are significantly different are indicated by asterisks as follows: *, P < 0.05 compared to the value for vector control cells exposed to UV; **, P < 0.05 compared to the value for XPA-deficient cells expressing wild-type XPA without RASSF1A expression. (B) The fluorescence intensity of the cells was quantitated using ImageJ software after subtracting the background fluorescence and is represented as corrected total cell fluorescence.
RASSF1A modulation of XPA deacetylation is Hippo independent and SIRT1 dependent.
RASSF1A activates the MST1/Hippo pathway (10). To determine whether the effects of RASSF1A were via Hippo signaling, we examined the ability of a RASSF1A point mutant that is defective for MST1 binding (42) to modulate XPA deacetylation. Figure 11A shows that the point mutant retains the ability to promote XPA deacetylation, suggesting that the effect is Hippo independent.
FIG 11.
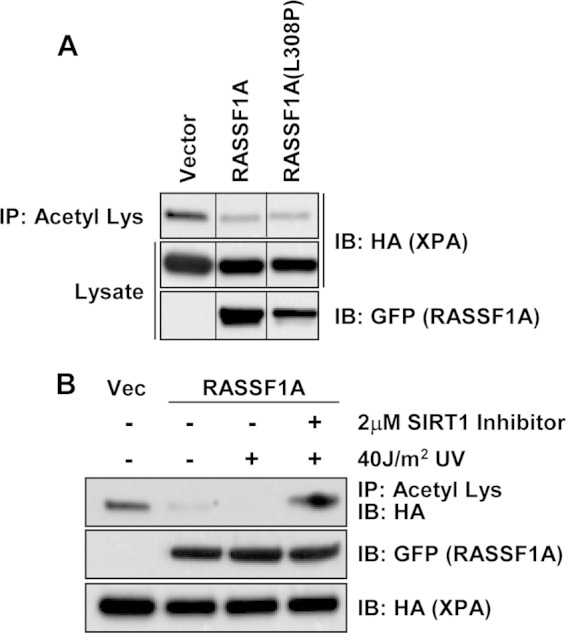
RASSF1A-mediated XPA deacetylation is Hippo pathway independent and is mediated by SIRT1. HEK-293 cells were transiently cotransfected with expression constructs for HA-tagged XPA and GFP-tagged RASSF1A or a RASSF1A mutant that is defective for binding to Mst1 (42) [RASSF1A L(308)P]. Equal amounts of protein were immunoprecipitated with anti-acetyl-Lys beads, and the immunoprecipitates were fractionated on an SDS-polyacrylamide gel and Western blotted with anti-HA and anti-GFP antibodies. (B) HEK-293 cells were transiently cotransfected with expression constructs for HA-tagged XPA and GFP-tagged RASSF1A. Thirty-six hours after transfection, a 2 μM concentration of the SIRT1 inhibitor inauhzin was added to the cells. Sixteen hours later, the cells were exposed to 40 J/m2 UV, and 1 h after exposure to UV, the cells were lysed, and lysates were immunoprecipitated with anti-acetyl-Lys beads. Immunoprecipitates were resolved on SDS-polyacrylamide gels, transferred to nitrocellulose membranes, and blotted with anti-HA and anti-GFP antibodies. Vec, vector.
The deacetylase SIRT1 is known to play major role in controlling the acetylation of XPA (16). As RASSF1A appears to be modulating XPA acetylation, we wondered whether RASSF1A might be acting via SIRT1. To determine the role of SIRT1 in the action of RASSF1A, we used a specific SIRT1 inhibitor and showed that the ability of RASSF1A to promote XPA deacetylation after UV treatment is abolished when SIRT1 is inhibited (Fig. 11B).
RASSF1A and XPA cooperate in vivo.
XPA patients are highly susceptible to UV-induced skin tumors (43). Previous work has shown that mice heterozygous for XPA exhibit a tumor response to UV that is identical to that of wild-type controls (24). Both wild-type and heterozygous XPA animals begin to develop tumors approximately 200 days after the start of UV treatment with 50% of the animals scoring tumor positive by 300 days. In contrast, animals with defects in both XPA alleles show more rapid tumor development, with the animals scoring positive for tumors 70 days after the start of the UV irradiation protocol (24).
The in vitro data suggest that XPA+/− mice might be sensitized to UV if RASSF1A function were also impaired. We performed a small-scale in vivo experiment where we crossed RASSF1A and XPA knockout mice to generate a population of double heterozygous animals. The animals were subjected to the standard UV exposure procedure (24) and monitored to determine the rate of skin tumor induction. One hundred days after exposure to UV, we found that three of six or 50% of mice that were heterozygous for both RASSF1A and XPA (RASSF1A−/+ XPA−/+) exhibited single or multiple skin tumors, whereas the three wild-type heterozygous control mice (XPA+/+ RASSF1A−/+) remained completely tumor free. We hope to expand these experiments to include a larger sample size and additional controls in the future.
DISCUSSION
Loss of RASSF1A function due to epigenetic inactivation or point mutation is a common event in human cancer. Indeed, some tumor types appear to undergo almost 100% inactivation (7), suggesting that it may be one of the most important genetic changes involved in the development of cancer. RASSF1A knockout mice exhibit an enhanced rate of spontaneous tumor formation, but the role of RASSF1A in suppressing tumor development is not well understood. It is proapoptotic and can mediate the apoptotic response to Ras activation (21). It is also a key player in the apoptotic response to DNA damage (11). Here we show that it also plays a vital role in the regulation of DNA repair itself by forming a direct endogenous complex with the nucleotide excision repair (NER) protein XPA. The interaction of wild-type RASSF1A with XPA after UV damage appears to be regulated by ATR phosphorylation. Suppression of RASSF1A inhibits DNA repair, and overexpression of RASSF1A enhances it.
Although XPA is best characterized as an NER protein that facilitates the repair of single-strand DNA breaks, it has now also been implicated in the repair of double-strand breaks (44), and it may be present at stalled replication forks where it can participate in postreplication repair (35). Therefore, RASSF1A may exert a more general effect on DNA repair than just NER. As RASSF1A mediates the DDR as well, it may be an integral part of the complex machinery that determines the cellular response to DNA damage: repair or death. Cells with inactivated RASSF1A should both resist the induction of apoptosis due to DNA damage and exhibit enhanced genomic instability after DNA damage due to defective NER. This would suggest that defects in RASSF1A and XPA might act together to enhance tumorigenesis. Our small-scale in vivo study supports this concept, as RASSF1A XPA double heterozygous mice showed a synergistic increase in tumorigenesis after UV irradiation.
The RASSF1A A(133)S SNP variant has been linked to an enhanced risk of cancer (28, 29). It is partially defective for the ability to activate the MST kinases and therefore, the apoptotic DNA damage response (29). We have found that the RASSF1A SNP variant exhibits differential binding to XPA and suppresses DNA repair. Therefore, cells expressing the SNP are more likely to both survive and accumulate more oncogenic lesions after DNA damage.
The mechanism underlying the differential association with XPA of the wild-type RASSF1A and SNP variants of RASSF1A remains unclear. The failure of an S(131)A mutant of RASSF1A to behave like the A(133)S SNP variant after UV treatment shows that the differential binding of the SNP variant is not due to loss of phosphorylation at serine 131 by ATR. Perhaps the serine introduced at residue 133 in the SNP variant is subject to an as yet unknown posttranslational modification that can be regulated by DNA damage and modulates its interaction with XPA.
Despite its potentially deleterious effects, the RASSF1A A(133)S SNP variant is present in approximately ∼22% of the Caucasian population in the United States (20). It seems curious that such an apparently deleterious SNP variant would be so evolutionarily successful. Even more intriguing is the finding that the SNP is relatively uncommon in populations of African descent (4 to 6%) (20). We hypothesize that the lower levels of ambient UV in the ancestral Caucasian environment may have permitted the evolution of a tempered DNA damage response by the SNP. This could provide an evolutionary advantage in a low-UV environment because of the recently discovered cardiovascular role of RASSF1A (45). We have found that a mutant of RASSF1A that is defective for MST/Hippo activation suppresses cardiomyocyte apoptosis and fibrosis in a mouse model for heart disease (45). As the RASSF1A SNP variant is partially defective for MST activation (29), carriers may gain an enhanced resistance to cardiac disease. The SNP could make a major contribution to reported racial disparities in heart disease (46).
RASSF1A has typically been observed associated with the microtubule network (32, 33). The interaction of RASSF1A with XPA and its effects on DNA repair suggested that RASSF1A might be present in the nucleus. Using fluorescence microscopy allowed us to detect colocalization of RASSF1A and XPA in nuclear foci in live cells. The precise identity of the foci remains unclear. The fluorescence suggested that the RASSF1A SNP variant might be less nuclear than the wild-type RASSF1A is. Fractionation experiments confirmed this. Whether this is a cause or effect of reduced XPA/SNP binding after UV treatment is not yet known.
The XPA protein is thought to act as a scaffold for the cycling in and out of various components of the DNA repair complex as the repair process proceeds (15). The binding of XPA to other components of the complex is regulated, in part, by the acetylation/deacetylation of XPA. Mutants of XPA which are defective for acetylation exhibit a defective interaction with other repair complex components, such as the RPA trimer, and are defective for DNA repair (16). We found that the wild-type form of RASSF1A shows enhanced binding to XPA after DNA damage and promotes the formation of deacetylated XPA, and this RASSF1A-mediated deacetylation of XPA is crucial for repair of UV-induced DNA damage. However, the A(133)S SNP variant of RASSF1A exhibits the binding profile opposite to that of XPA as the wild-type protein and blocks the deacetylation cycle of XPA after DNA damage. Moreover, the RASSF1A SNP variant had a powerful effect on the stabilization of the endogenous XPA-RPA70 complex. Such deregulation of the XPA-RPA complex cycling inhibits DNA repair (39). Thus, we have identified a new mechanism of action for RASSF1A in the control of its binding partners by modulating their acetylation.
An acetylation cycle is essential for XPA regulation (16). One of the key enzymes regulating the acetylation status of XPA is the SIRT1 deacetylase (16). When we used an inhibitor of SIRT1, RASSF1A was no longer able to promote XPA deacetylation. This shows that RASSF1A can modulate SIRT1 activity. MST1 has been shown to phosphorylate and inhibit the SIRT1 deacetylase (47), and RASSF1A can bind and activate MST1. Since RASSF1A-mediated activation of MST kinases can be induced by DNA damage (11), RASSF1A could be modulating XPA acetylation via MST1/SIRT1. However, when we used a point mutant form of RASSF1A that cannot bind or activate MST1 (42), we found that it still induced XPA deacetylation. Therefore, while the control of XPA acetylation by RASSF1A is mediated by SIRT1, it appears largely MST independent. How the A(133)S SNP variant can inhibit XPA deacetylation after UV irradiation when it does not bind XPA remains to be determined. It seems likely that the SNP variant is acting indirectly on XPA, via SIRT1.
Dynamic protein acetylation is a major component of posttranslational control of protein activity. Many proteins known to serve a critical function in growth, development, and cancer are regulated by protein acetylation, for example the p53 tumor suppressor (48). There are at least 9 families of deacetylase/acetyltransferase proteins that mediate the process in humans (49). Our data suggest that SIRT1 is one of these components that are modulated by RASSF1A. Exactly which substrates in addition to XPA can be modulated by RASSF1A and its SNP variant may prove an interesting avenue for future studies. One of the most pertinent could be the tumor suppressor p53, as RASSF1A has been linked to p53 regulation, and the p53 status of a cell can affect XPA nuclear localization dynamics (50).
Thus, DNA repair and regulation of protein acetylation are two more properties that can be assigned to the RASSF1A tumor suppressor. RASSF1A can form an endogenous complex with the K-Ras oncoprotein (13, 51). Moreover, activated Ras has been reported to enhance DNA repair (52) but also to promote DNA damage (53) under different conditions. Perhaps the interaction of RASSF1A with XPA may be one more connection that allows Ras to influence DNA repair, and RASSF1A levels could affect whether the effect is positive or negative.
ACKNOWLEDGMENTS
The work was supported by awards 1P20 RR18733, NIH R01 CA133171, and NCI intramural funds (G.J.C.).
We thank Lindsey J. Stallons, Caleb Greenwell, and Glenn McGregor for technical assistance and thoughtful comments.
H.D. and G.J.C. designed research. H.D., J.C., F.R., N.N., T.B., K.R.H., M.D.V., B.S., and G.J.C performed research. H.D. and G.J.C. analyzed data. G.J.C. wrote the paper.
REFERENCES
- 1.Friedberg EC, Wood RD, Walker GC, Schultz RA, Siede W, Ellenberger T. 2006. DNA repair and mutagenesis. ASM Press, Washington, DC. [Google Scholar]
- 2.Nouspikel T. 2009. DNA repair in mammalian cells: so DNA repair really is that important? Cell Mol Life Sci 66:965–967. doi: 10.1007/s00018-009-8734-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Larsson LG. 2011. Oncogene- and tumor suppressor gene-mediated suppression of cellular senescence. Semin Cancer Biol 21:367–376. doi: 10.1016/j.semcancer.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 4.Reinhardt HC, Schumacher B. 2012. The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet 28:128–136. doi: 10.1016/j.tig.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roos WP, Kaina B. 2013. DNA damage-induced apoptosis: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett 332:237–248. doi: 10.1016/j.canlet.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 6.Dammann R, Schagdarsurengin U, Seidel C, Strunnikova M, Rastetter M, Baier K, Pfeifer GP. 2005. The tumor suppressor RASSF1A in human carcinogenesis: an update. Histol Histopathol 20:645–663. [DOI] [PubMed] [Google Scholar]
- 7.Donninger H, Vos MD, Clark GJ. 2007. The RASSF1A tumor suppressor. J Cell Sci 120:3163–3172. doi: 10.1242/jcs.010389. [DOI] [PubMed] [Google Scholar]
- 8.van der Weyden L, Adams DJ. 2007. The Ras-association domain family (RASSF) members and their role in human tumourigenesis. Biochim Biophys Acta 1776:58–85. doi: 10.1016/j.bbcan.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tommasi S, Dammann R, Zhang Z, Wang Y, Liu L, Tsark WM, Wilczynski SP, Li J, You M, Pfeifer GP. 2005. Tumor susceptibility of Rassf1a knockout mice. Cancer Res 65:92–98. [PubMed] [Google Scholar]
- 10.Guo C, Tommasi S, Liu L, Yee JK, Dammann R, Pfeifer GP. 2007. RASSF1A is part of a complex similar to the Drosophila Hippo/Salvador/Lats tumor-suppressor network. Curr Biol 17:700–705. doi: 10.1016/j.cub.2007.02.055. [DOI] [PubMed] [Google Scholar]
- 11.Hamilton G, Yee KS, Scrace S, O'Neill E. 2009. ATM regulates a RASSF1A-dependent DNA damage response. Curr Biol 19:2020–2025. doi: 10.1016/j.cub.2009.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vos MD, Ellis CA, Bell A, Birrer MJ, Clark GJ. 2000. Ras uses the novel tumor suppressor RASSF1 as an effector to mediate apoptosis. J Biol Chem 275:35669–35672. doi: 10.1074/jbc.C000463200. [DOI] [PubMed] [Google Scholar]
- 13.Matallanas D, Romano D, Al-Mulla F, O'Neill E, Al-Ali W, Crespo P, Doyle B, Nixon C, Sansom O, Drosten M, Barbacid M, Kolch W. 2011. Mutant K-Ras activation of the proapoptotic MST2 pathway is antagonized by wild-type K-Ras. Mol Cell 44:893–906. doi: 10.1016/j.molcel.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 14.Jones CJ, Wood RD. 1993. Preferential binding of the xeroderma pigmentosum group A complementing protein to damaged DNA. Biochemistry 32:12096–12104. doi: 10.1021/bi00096a021. [DOI] [PubMed] [Google Scholar]
- 15.Camenisch U, Nageli H. 2008. XPA gene, its product and biological roles. Adv Exp Med Biol 637:28–38. doi: 10.1007/978-0-387-09599-8_4. [DOI] [PubMed] [Google Scholar]
- 16.Fan W, Luo J. 2010. SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Mol Cell 39:247–258. doi: 10.1016/j.molcel.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 17.Wu X, Shell SM, Yang Z, Zou Y. 2006. Phosphorylation of nucleotide excision repair factor xeroderma pigmentosum group A by ataxia telangiectasia mutated and Rad3-related-dependent checkpoint pathway promotes cell survival in response to UV irradiation. Cancer Res 66:2997–3005. doi: 10.1158/0008-5472.CAN-05-3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cleaver JE. 2005. Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nat Rev Cancer 5:564–573. doi: 10.1038/nrc1652. [DOI] [PubMed] [Google Scholar]
- 19.Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP. 2000. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet 25:315–319. doi: 10.1038/77083. [DOI] [PubMed] [Google Scholar]
- 20.Donninger H, Barnoud T, Nelson N, Kassler S, Clark J, Cummins TD, Powell DW, Nyante SJ, Millikan RC, Clark GJ. 2011. RASSF1A and the rs2073498 cancer associated SNP. Front Oncol 1:54. doi: 10.3389/fonc.2011.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vos MD, Dallol A, Eckfeld K, Allen NP, Donninger H, Hesson LB, Calvisi D, Latif F, Clark GJ. 2006. The RASSF1A tumor suppressor activates Bax via MOAP-1. J Biol Chem 281:4557–4563. doi: 10.1074/jbc.M512128200. [DOI] [PubMed] [Google Scholar]
- 22.Konca K, Lankoff A, Banasik A, Lisowska H, Kuszewski T, Gozdz S, Koza Z, Wojcik A. 2003. A cross-platform public domain PC image-analysis program for the comet assay. Mutat Res 534:15–20. doi: 10.1016/S1383-5718(02)00251-6. [DOI] [PubMed] [Google Scholar]
- 23.de Vries A, van Steeg H. 1996. Xpa knockout mice. Semin Cancer Biol 7:229–240. doi: 10.1006/scbi.1996.0031. [DOI] [PubMed] [Google Scholar]
- 24.Berg RJ, de Vries A, van Steeg H, de Gruijl FR. 1997. Relative susceptibilities of XPA knockout mice and their heterozygous and wild-type littermates to UVB-induced skin cancer. Cancer Res 57:581–584. [PubMed] [Google Scholar]
- 25.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. 2004. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 26.Zou L, Elledge SJ. 2003. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 27.El-Kalla M, Onyskiw C, Baksh S. 2010. Functional importance of RASSF1A microtubule localization and polymorphisms. Oncogene 29:5729–5740. doi: 10.1038/onc.2010.316. [DOI] [PubMed] [Google Scholar]
- 28.Gao B, Xie XJ, Huang C, Shames DS, Chen TT, Lewis CM, Bian A, Zhang B, Olopade OI, Garber JE, Euhus DM, Tomlinson GE, Minna JD. 2008. RASSF1A polymorphism A133S is associated with early onset breast cancer in BRCA1/2 mutation carriers. Cancer Res 68:22–25. doi: 10.1158/0008-5472.CAN-07-5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yee KS, Grochola L, Hamilton G, Grawenda A, Bond EE, Taubert H, Wurl P, Bond GL, O'Neill E. 2012. A RASSF1A polymorphism restricts p53/p73 activation and associates with poor survival and accelerated age of onset of soft tissue sarcoma. Cancer Res 72:2206–2217. doi: 10.1158/0008-5472.CAN-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shivakumar L, Minna J, Sakamaki T, Pestell R, White MA. 2002. The RASSF1A tumor suppressor blocks cell cycle progression and inhibits cyclin D1 accumulation. Mol Cell Biol 22:4309–4318. doi: 10.1128/MCB.22.12.4309-4318.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dallol A, Agathanggelou A, Fenton SL, Ahmed-Choudhury J, Hesson L, Vos MD, Clark GJ, Downward J, Maher ER, Latif F. 2004. RASSF1A interacts with microtubule-associated proteins and modulates microtubule dynamics. Cancer Res 64:4112–4116. doi: 10.1158/0008-5472.CAN-04-0267. [DOI] [PubMed] [Google Scholar]
- 32.Liu L, Tommasi S, Lee DH, Dammann R, Pfeifer GP. 2003. Control of microtubule stability by the RASSF1A tumor suppressor. Oncogene 22:8125–8136. doi: 10.1038/sj.onc.1206984. [DOI] [PubMed] [Google Scholar]
- 33.Vos MD, Martinez A, Elam C, Dallol A, Taylor BJ, Latif F, Clark GJ. 2004. A role for the RASSF1A tumor suppressor in the regulation of tubulin polymerization and genomic stability. Cancer Res 64:4244–4250. doi: 10.1158/0008-5472.CAN-04-0339. [DOI] [PubMed] [Google Scholar]
- 34.Hass CS, Lam K, Wold MS. 2012. Repair-specific functions of replication protein A. J Biol Chem 287:3908–3918. doi: 10.1074/jbc.M111.287441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gilljam KM, Muller R, Liabakk NB, Otterlei M. 2012. Nucleotide excision repair is associated with the replisome and its efficiency depends on a direct interaction between XPA and PCNA. PLoS One 7:e49199. doi: 10.1371/journal.pone.0049199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y, Wang Y, Rusinol AE, Sinensky MS, Liu J, Shell SM, Zou Y. 2008. Involvement of xeroderma pigmentosum group A (XPA) in progeria arising from defective maturation of prelamin A. FASEB J 22:603–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coin F, Oksenych V, Mocquet V, Groh S, Blattner C, Egly JM. 2008. Nucleotide excision repair driven by the dissociation of CAK from TFIIH. Mol Cell 31:9–20. doi: 10.1016/j.molcel.2008.04.024. [DOI] [PubMed] [Google Scholar]
- 38.de Laat WL, Jaspers NG, Hoeijmakers JH. 1999. Molecular mechanism of nucleotide excision repair. Genes Dev 13:768–785. doi: 10.1101/gad.13.7.768. [DOI] [PubMed] [Google Scholar]
- 39.Li L, Lu X, Peterson CA, Legerski RJ. 1995. An interaction between the DNA repair factor XPA and replication protein A appears essential for nucleotide excision repair. Mol Cell Biol 15:5396–5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saijo M, Takedachi A, Tanaka K. 2011. Nucleotide excision repair by mutant xeroderma pigmentosum group A (XPA) proteins with deficiency in interaction with RPA. J Biol Chem 286:5476–5483. doi: 10.1074/jbc.M110.172916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shell SM, Li Z, Shkriabai N, Kvaratskhelia M, Brosey C, Serrano MA, Chazin WJ, Musich PR, Zou Y. 2009. Checkpoint kinase ATR promotes nucleotide excision repair of UV-induced DNA damage via physical interaction with xeroderma pigmentosum group A. J Biol Chem 284:24213–24222. doi: 10.1074/jbc.M109.000745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Donninger H, Allen N, Henson A, Pogue J, Williams A, Gordon L, Kassler S, Dunwell T, Latif F, Clark GJ. 2011. Salvador protein is a tumor suppressor effector of RASSF1A with hippo pathway-independent functions. J Biol Chem 286:18483–18491. doi: 10.1074/jbc.M110.214874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rapin I. 2013. Disorders of nucleotide excision repair. Handb Clin Neurol 113:1637–1650. doi: 10.1016/B978-0-444-59565-2.00032-0. [DOI] [PubMed] [Google Scholar]
- 44.Ricceri F, Porcedda P, Allione A, Turinetto V, Polidoro S, Guarrera S, Rosa F, Voglino F, Pezzotti A, Minieri V, Accomasso L, Cibrario RE, Orlando L, Giachino C, Matullo G. 2011. Involvement of MRE11A and XPA gene polymorphisms in the modulation of DNA double-strand break repair activity: a genotype-phenotype correlation study. DNA Repair (Amst) 10:1044–1050. doi: 10.1016/j.dnarep.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 45.Del Re DP, Matsuda T, Zhai P, Gao S, Clark GJ, Van Der Weyden L, Sadoshima J. 2010. Proapoptotic Rassf1A/Mst1 signaling in cardiac fibroblasts is protective against pressure overload in mice. J Clin Invest 120:3555–3567. doi: 10.1172/JCI43569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mensah GA, Brown DW. 2007. An overview of cardiovascular disease burden in the United States. Health Aff (Millwood) 26:38–48. doi: 10.1377/hlthaff.26.1.38. [DOI] [PubMed] [Google Scholar]
- 47.Yuan F, Xie Q, Wu J, Bai Y, Mao B, Dong Y, Bi W, Ji G, Tao W, Wang Y, Yuan Z. 2011. MST1 promotes apoptosis through regulating Sirt1-dependent p53 deacetylation. J Biol Chem 286:6940–6945. doi: 10.1074/jbc.M110.182543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marouco D, Garabadgiu AV, Melino G, Barlev NA. 2013. Lysine-specific modifications of p53: a matter of life and death? Oncotarget 4:1556–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arif M, Senapati P, Shandilya J, Kundu TK. 2010. Protein lysine acetylation in cellular function and its role in cancer manifestation. Biochim Biophys Acta 1799:702–716. doi: 10.1016/j.bbagrm.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 50.Li Z, Musich PR, Zou Y. 2011. Differential DNA damage responses in p53 proficient and deficient cells: cisplatin-induced nuclear import of XPA is independent of ATR checkpoint in p53-deficient lung cancer cells. Int J Biochem Mol Biol 2:138–145. [PMC free article] [PubMed] [Google Scholar]
- 51.Del Re DP, Matsuda T, Zhai P, Maejima Y, Jain MR, Liu T, Li H, Hsu CP, Sadoshima J. 2014. Mst1 promotes cardiac myocyte apoptosis through phosphorylation and inhibition of Bcl-xL. Mol Cell 54:639–650. doi: 10.1016/j.molcel.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cho HJ, Jeong HG, Lee JS, Woo ER, Hyun JW, Chung MH, You HJ. 2002. Oncogenic H-Ras enhances DNA repair through the Ras/phosphatidylinositol 3-kinase/Rac1 pathway in NIH3T3 cells. Evidence for association with reactive oxygen species. J Biol Chem 277:19358–19366. doi: 10.1074/jbc.M200933200. [DOI] [PubMed] [Google Scholar]
- 53.Tu Z, Aird KM, Bitler BG, Nicodemus JP, Beeharry N, Xia B, Yen TJ, Zhang R. 2011. Oncogenic RAS regulates BRIP1 expression to induce dissociation of BRCA1 from chromatin, inhibit DNA repair, and promote senescence. Dev Cell 21:1077–1091. doi: 10.1016/j.devcel.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]