

Abstract
Myelin membranes are sheet-like extensions of oligodendrocytes that can be considered membrane domains distinct from the cell's plasma membrane. Consistent with the polarized nature of oligodendrocytes, we demonstrate that transcytotic transport of the major myelin-resident protein proteolipid protein (PLP) is a key element in the mechanism of myelin assembly. Upon biosynthesis, PLP traffics to myelin membranes via syntaxin 3-mediated docking at the apical-surface-like cell body plasma membrane, which is followed by subsequent internalization and transport to the basolateral-surface-like myelin sheet. Pulse-chase experiments, in conjunction with surface biotinylation and organelle fractionation, reveal that following biosynthesis, PLP is transported to the cell body surface in Triton X-100 (TX-100)-resistant microdomains. At the plasma membrane, PLP transiently resides within these microdomains and its lateral dissipation is followed by segregation into 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS)-resistant domains, internalization, and subsequent transport toward the myelin membrane. Sulfatide triggers PLP's reallocation from TX-100- into CHAPS-resistant membrane domains, while inhibition of sulfatide biosynthesis inhibits transcytotic PLP transport. Taking these findings together, we propose a model in which PLP transport to the myelin membrane proceeds via a transcytotic mechanism mediated by sulfatide and characterized by a conformational alteration and dynamic, i.e., transient, partitioning of PLP into distinct membrane microdomains involved in biosynthetic and transcytotic transport.
INTRODUCTION
Oligodendrocytes (OLGs) synthesize a multilamellar membrane structure known as the myelin sheath (in vitro: myelin sheet), which extends from their plasma membrane and wraps around axons, thereby facilitating rapid saltatory conduction and axonal survival in the central nervous system. Myelin is enriched in galactolipids and cholesterol and contains myelin-specific proteins, the major ones of which are myelin basic protein (MBP) and proteolipid protein (PLP) (1–3). In generating a myelin membrane, OLGs acquire a polarized phenotype. The sphingolipid-enriched myelin membrane is served by a basolateral-surface-like mechanism, while the cell body plasma membrane is served by an apical-surface-like transport mechanism (4–8). MBP and PLP stabilize apposed myelin membrane surfaces in compact myelin, and their transport routes are segregated. Thus, following its biosynthesis at the rough endoplasmic reticulum and processing through the Golgi apparatus (9, 10), the transmembrane protein PLP is transported via vesicles to the myelin membrane (11, 12). In marked contrast, MBP, a peripheral membrane protein facing the cytoplasm, is synthesized at and associates with the myelin membrane on site, involving myelin membrane-directed transport of its mRNA rather than the native protein (13, 14). In addition, to limit ectopic assembly and premature compaction of myelin membranes, the protein-mediated biogenesis of myelin membranes is tightly regulated in both time and space (reviewed in reference 6). In fact, MBP associates with myelin membranes prior to PLP (2). Regulation of the trafficking of either protein is under tight neuronal control, with MBP trafficking being regulated mainly by adhesive interactions (15, 16) while the flow of PLP appears to be governed by secreted factors (17). Clearly, defining the processes of cellular sorting and transport is essential for an understanding of both normal OLG development and the process of remyelination in demyelinating diseases such as multiple sclerosis (MS), in which myelin membrane organization is perturbed. For example, the molecular composition of the myelin galactolipid sulfatide and MBP in MS differs from their composition under control conditions (18, 19), while paranodal structures are disrupted (20, 21).
The underlying sorting machinery by which PLP reaches its final destination is still unclear, but insight into the overall transport pathway is gradually emerging and favors indirect transport of PLP to myelin. Thus, prior to reaching the myelin membrane, PLP is sorted to and stored in a late LAMP1-positive endosomal compartment (11, 17, 22), from where the protein, along with cholesterol and in galactosylceramide (GalC)-enriched microdomains, is subsequently transported to the myelin membrane (17). To reach this late endosomal compartment, PLP is internalized from the plasma membrane via clathrin-independent but cholesterol-dependent endocytosis (17, 22, 23). Recently, delivery of PLP to the surface has been claimed to occur by two independent pathways, involving the vesicle-soluble N-ethylmaleimide-sensitive factor attachment protein receptors (v-SNAREs) VAMP3 and VAMP7, respectively (24). Therefore, in line with the presence of polarity-based transport mechanisms in OLGs, newly synthesized PLP might reach its final destination via an indirect pathway. Consistent with this hypothesis is the observation that PLP, stably expressed in polarized Madin-Darby canine kidney cells, is transported to the apical rather than the basolateral surface (25), i.e., membrane domains reminiscent of the cell body plasma membrane and the myelin sheet in cultured OLGs, respectively (4, 5).
Thus far, knowledge concerning the docking of PLP and its final insertion into the myelin membrane is scanty. A variety of syntaxins, i.e., membrane-localized target SNAREs (t-SNAREs) involved in the docking and fusion of transport vesicles at target membranes, have been detected in OLGs (6, 26, 27), including syntaxins 3 and 4, the respective putative binding partners of VAMP7 and VAMP3 (24, 27). Specifically, in previous work, we have shown a polarized distribution of these syntaxins, with syntaxin 3 localizing largely at the plasma membrane of the OLG cell body whereas syntaxin 4 is enriched at the myelin sheet (6). To obtain further insight into the molecular mechanism underlying PLP trafficking and its assembly into myelin membranes, the present study was undertaken. Our data demonstrate that PLP is transported to the myelin membrane via a transcytotic route, with first localization at the OLG cell body plasma membrane involving syntaxin 3 and then a sulfatide-facilitated shift into a different lateral plasma membrane pool, accompanied by a conformational alteration and/or oligomerization, prior to the protein's delivery to the myelin sheet.
MATERIALS AND METHODS
Cell cultures. (i) OLGs.
Primary cultures of OLGs were prepared from forebrains of 1- to 2-day-old Wistar rats as described previously (28, 29). OLG progenitor cells (OPCs) were plated in SATO medium (28) supplemented with the growth factors fibroblast growth factor 2 (FGF-2) (10 ng/ml; Peprotech, London, United Kingdom) and platelet-derived growth factor-AA (10 ng/ml; Peprotech) on poly-l-lysine (PLL, 5 μg/ml; Sigma, St. Louis, MO)-coated 10-cm dishes (Nunc, Naperville, IL) at 1.0 × 106 cells/dish or eight-well Permanox chamber slides (Nunc) at 20,000 cells/well for biochemical and immunocytochemical analyses, respectively. After 2 days, differentiation was initiated by switching to SATO supplemented with 0.5% fetal calf serum (FCS; Bodinco, Alkmaar, The Netherlands) and cells were grown for 3 days (immature OLGs [imOLGs]) or 7 to 10 days (mature OLGs [mOLGs]). To inhibit sulfatide biosynthesis, cells were cultured in the presence of 30 μM sodium chlorate for at least 4 to 7 days. All experimental procedures were approved by the Animal Ethical Committee of the University Medical Center Groningen (UMCG).
(ii) OLN-PLP cells.
OLN-PLP cells were made in the laboratory of Zeger Debyser (University of Leuven, Leuven, Belgium) in collaboration with Rik Gijsbers and Veerle Baekelandt (University of Leuven, Leuven, Belgium) and were a kind gift of Ellen Gielen (Hasselt University, Hasselt, Belgium). Briefly, PLP was cloned into the lentiviral plasmid pCOMBI-eGFP-IRES-puro and PLP-enhanced green fluorescent protein (eGFP)-containing lentiviral particles were produced by using HEK-293T cells. Subsequently, the rat-derived oligodendroglia-derived cell line OLN-93, a kind gift of Christiane Richter Landsberg (University of Oldenburg, Oldenburg, Germany) (30) was lentivirally transduced with PLP-eGFP-containing lentiviral particles. OLN-PLP cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% heat-inactivated FCS and 2 μg/ml puromycin (Sigma). Cells were plated on eight-well Permanox chamber slides for immunocytochemistry analysis (5,000 cells/well) or on 10-cm tissue culture dishes for biochemical analysis (500,000 cells/well). Cells were grown in DMEM supplemented with 10% FCS for 3 days, unless otherwise indicated.
(iii) HepG2 cells.
HepG2 cells were cultured in DMEM containing 10% FCS. Transfection of HepG2 cells with plasmid pLEGFP-N1-PLP was performed with Lipofectamine 2000 (Invitrogen, Breda, The Netherlands) according to the manufacturer's instructions. After transfection, the cells were cultured in the presence of 1.0 mg/ml Geneticin (G418; Invitrogen) and colonies were isolated 14 days after transfection. Stably transfected cells were plated on eight-well Permanox chamber slides for immunocytochemistry analysis (5,000 cells/well) or on 10-cm tissue culture dishes for biochemical analysis (500,000 cells/well).
Constructs.
The cDNAs encoding syntaxins 3 and 4 were a kind gift of Thomas Weimbs (University of California, Santa Barbara, CA) (31), and PLP-eGFP was a kind gift of Niels Hellings (Hasselt University, Hasselt, Belgium). For cloning of the syntaxin 3 and 4 genes into retroviral vector pLXIN (Clontech Biosciences, Mountain View, CA), an XhoI restriction site at the ATG start codon and an XhoI restriction site after the stop codon were introduced by PCR. The primers used were 5′-CATGTATTCGAAGAGCTCTTCGCACATG-3′ (forward syntaxin 3), 5′-CTAGGTGATCAAGAGCTCCTAGGGCCCACG-3′ (reverse syntaxin 3), 5′-CGAATAGCTATGAGCTCCATGGTCTAG-3′ (forward syntaxin 4), and 5′-GATCTCCTAGAGCTCACGTAGGGAC-3′ (reverse syntaxin 4). The cDNAs encoding ceramide sulfatide transferase (CST) and ceramide galactosyltransferase (CGT) were kind gifts of Matthias Eckhardt (University of Bonn, Bonn, Germany) and Brian Popko (University of Chicago, Chicago, IL), respectively. For cloning, the genes for CST and CGT were inserted into the EcoRI site of the retroviral vector pLXIN (Clontech Biosciences, Mountain View, CA). The orientation and integrity of the pLXIN constructs obtained were confirmed by DNA sequencing.
Retroviral transduction.
The production of retroviral particles and the subsequent infection of OPCs and OLN-PLP cells were performed according to Klunder et al. (5) and Maier et al. (32), respectively, Transductions were carried out by exposing cells to retroviral particles and 8 μg/ml hexadimethrine bromide (Polybrene; Sigma) for 16 to 18 h. The cells were cultured for 24 h and then cultured with selection under proliferating conditions in the presence of 400 μg/ml (OPCs) or 2 mg/ml (OLN-PLP cells) G418 (Roche, Mannheim, Germany) for 5 days. OLN-PLP cells were first infected with CGT (OLN-PLP-G cells) and then selected; this was followed by a second infection with CST (OLN-PLP-GS cells). An OLN-mock cell line was obtained by retroviral infection with the vector only (pLXIN). The transduction efficiency was nearly 100%. OLN-PLP-G and OLN-PLP-GS cells were cultured in the presence of 2 μg/ml puromycin and 2 mg/ml G418.
PLP antibodies.
PLP consists of four hydrophobic α-helices that span the lipid bilayer with two extracytoplasmic domains and three cytoplasmic domains, while both the C and N termini face the cytoplasm (33, 34). Anti-PLP antibodies 4C2, directed against a nonconformational epitope in the first extracellular loop (PLP amino acids 50 to 69), and 2D2, directed against an intracellular region absent from DM-20 (PLP amino acids 100 to 123), were kind gifts of Vijay Kuchroo (Harvard Medical School, Boston, MA) (35). The epitope recognized by anti-PLP antibody O10 is acquired posttranslationally and is directed against a conformation-dependent epitope in the second extracellular loop (kind gift of Evi Albers-Krämer, Mainz, Germany) (36). The anti-PLP antibody ET3 was raised in rabbits against the second extracellular loop of PLP. To this end, a synthetic peptide, PLP181-230, with the two disulfide bridges was generated (37). Two rabbits were injected subcutaneously on days 0, 21, and 35 with an emulsion containing 300 μg of this synthetic peptide in Freund's complete (day 0) or incomplete (days 21 and 35) adjuvant. At day 49, 600 μg of the peptide in incomplete adjuvant was injected. The rabbits were bled on day 56, and the serum was kept frozen at −20°C. The ET3 antibody reacts with the peptide in an enzyme-linked immunosorbent assay and recognizes a conformational epitope, given the absence of a band in reducing Western blot assays (data not shown).
Pulse-chase experiments.
ImOLGs were preincubated in DMEM without methionine (Invitrogen, Breda, The Netherlands) for 1 h and then labeled for 10 min with Tran35S-label (GE Healthcare, Biosciences, Buckinghamshire, United Kingdom) at 200 μCi/dish. The cells were then chased with medium supplemented with 10 mM methionine (Merck, Darmstadt, Germany) for 0, 15, 30, and 60 min. Cells were scraped into phosphate-buffered saline (PBS), extracted with TNE lysis buffer (20 mM Tris-HCl [pH 7.4], 150 mM NaCl, and 1 mM EDTA supplemented with 1% Triton X-100 [TX-100] and a cocktail of protease inhibitors [Complete Mini; Roche]) for 30 min at 4°C, and separated by centrifugation (15 min at 13,300 × g) into TX-100-soluble supernatants and TX-100-insoluble pellets. PLP was immunoprecipitated from both fractions with protein G-Sepharose (GE Healthcare) beads overnight at 4°C. Protein G-Sepharose beads were washed four times with TNE lysis buffer supplemented with 0.2% SDS and once with PBS. Washed protein G-Sepharose beads were resuspended in SDS sample buffer and counted for 2 min in a microplate scintillation and luminescence counter (Packard Instrument Company, Meriden, CT).
Preparation of detergent extracts.
Cells were washed with PBS and harvested by scraping the cells in TNE lysis buffer containing either 1% TX-100 or 20 mM 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS). The solution was passed 10 times through a 21-gauge needle and incubated on ice for at least 30 min. The protein content was determined by a DC protein assay (Bio-Rad Laboratories, Hercules, CA) using bovine serum albumin (BSA) as a standard.
OptiPrep density gradient centrifugation.
For density gradient centrifugation, a discontinuous OptiPrep gradient was prepared. To this end, 250 μl of total cell detergent extracts (equal protein) was added to 500 μl of 60% OptiPrep. This 40% OptiPrep solution was overlaid with 30% and 10% OptiPrep. Gradients were centrifuged overnight at 152,000 × g (4°C, Beckman SW55 rotor), and seven gradient fractions were collected from the top (fraction 1) to the bottom (fraction 7). To concentrate proteins, equal fraction volumes were adjusted to a final volume of 1 ml with TNE buffer and treated with deoxycholate (125 μg/ml) for 5 min at 4°C; this was followed by precipitation with 6.5% trichloric acid (TCA) for 15 min at 4°C. Precipitates were centrifuged for 20 min at 9,200 × g and 4°C. The pellets were dried and resuspended in SDS reducing sample buffer. After the pH was adjusted to 6.8 by exposure to ammonia, the samples were heated for 30 min at 37°C and subjected to SDS-PAGE and Western blotting. The lateral distribution of PLP-eGFP was calculated from the protein's (infrared) intensity in either fractions 3 and 4 (membrane microdomains) or fractions 6 and 7 (nonmembrane microdomains), relative to the total intensity, i.e., measured collectively in all of the fractions.
Surface biotinylation.
Cells were washed twice with ice-cold PBS and incubated for 1 h with sulfo-NHS-L-C-biotin (0.1 mg/ml in PBS; Pierce, Rockford, IL) at 4°C. The cells were washed three times for 5 min each with cell wash buffer (CWB; 65 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2) to remove excess biotin and twice with PBS. The cells were harvested by being scraped into 350 μl of TNE lysis buffer and pressed 18 times through a 21-gauge needle. Lysis occurred on ice for 30 min, and the protein content was determined by the Bio-Rad DC protein assay. Equal amounts of protein were centrifuged for 20 min at 15,600 × g to obtain soluble and insoluble fractions or subjected to OptiPrep density gradient centrifugation. Biotinylated proteins were immunoprecipitated from equal volumes of the fractions with streptavidin (SA)-agarose for 16 to 18 h at 4°C. After centrifugation, the SA-agarose beads (biotinylated proteins) were washed four times with CWB supplemented with 1% NP-40 and 0.35 M NaCl and once with PBS. Nonbiotinylated proteins (supernatants) were concentrated by TCA precipitation as described above. Samples from SA-agarose beads (surface) and supernatant (intracellular) fractions were mixed with SDS reducing sample buffer, heated for 2 min at 95°C or 30 min at 37°C, and subjected to SDS-PAGE and Western blotting.
Isolation of endosomes and lysosomes.
Endosome- and lysosome-enriched fractions were isolated from cells by the flotation gradient fractionation method (38, 39). Cells were harvested by being scraped into a mixture of 250 mM sucrose, 20 mM HEPES, and 0.5 mM EGTA at pH 7.0 (homogenization buffer [HB]) and immediately subjected to the isolation procedure. Cells were washed twice with HB by centrifugation at 800 × g for 5 min at 4°C. The pellet was resuspended in 1 ml of HB supplemented with protease inhibitors and homogenized with a grinding glass cell Dounce homogenizer (15× loose and 10× tight). The homogenate was centrifuged at 800 × g for 10 min at 4°C. The postnuclear supernatant obtained was centrifuged at 15,000 × g for 15 min at 4°C to remove mitochondria. Subsequent centrifugation of the supernatant at 128,000 × g for 1 h at 4°C removed the microsomal fraction. The remaining endosome- and lysosome-enriched fractions were separated from each other on a discontinuous sucrose density gradient. To this end, the pellet was resuspended in 1 ml of a 40.6% sucrose solution and passed 10 times through a 25-gauge needle. The 40.6% sucrose–protein mixture was overlaid sequentially with sucrose solutions of 35% (1.5 ml), 30% (1.5 ml), 25% (2 ml), and HB (6 ml). The gradient was centrifuged at 125,000 × g for 2 h at 4°C (SW41-Ti rotor). Fractions of 1 ml were collected from the top (fraction 1) to the bottom (fraction 12). The fractions were diluted with 2 ml of 20 mM HEPES and 0.5 mM EGTA at pH 7.0 and centrifuged at 153,000 × g for 30 min at 4°C (TLA 100.3 rotor). Pellets were resuspended in 160 μl of TNE, passed five times through a 25-gauge needle, and stored at −20°C. Of note, the pellets of fractions 1 to 4 were pooled.
Analysis of cellular glycosphingolipids.
Cells were washed three times with PBS, harvested by scraping in PBS, and centrifuged at 9,200 × g at room temperature (RT); this was followed by lipid extraction of the cell pellet according to Bligh and Dyer (40). Lipids were separated on thin-layer chromatography (TLC) plates by using C3H6O2-CH3CH(OH)CH3-CHCl3-CH3OH–25% KCl (25:25:25:10:9, vol/vol/vol/vol/vol) as the running solvent. To visualize the glycosphingolipids, the plates were dried and sprayed with 10% H2SO4 and 5% CH3OH and heated to 120°C.
Immunocytochemistry analysis and in situ extraction.
Antibody staining of cell surface components was performed on living cells at 4°C. After blocking of nonspecific binding with 4% BSA in PBS, cells were incubated with anti-surface PLP antibodies 4C2 (1:5), ET3 (1:50), and O10 (1:3) or with antisulfatide antibody O4 or anti-GalC antibody O1 (1:1 and 1:10, respectively; kind gifts of Guus Wolswijk) (41) for 30 min; washed three times; and incubated for 25 min with appropriate fluorescein isothiocyanate (FITC)- or tetramethyl rhodamine isocyanate (TRITC)-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA). The cells were fixed with 4% paraformaldehyde (PFA; Merck) in PBS for 20 min at RT. For double or single staining of intracellular antigens, PFA-fixed cells were permeabilized and blocked with 0.1% TX-100 and 4% BSA, respectively, in PBS for 30 min at RT. The cells were incubated for 1 to 2 h with anti-PLP (1:5, 4C2 or 2D2), anti-MRP2 (1:300, Axxora, Lörrach, Germany), or anti-GFP antibodies (1:100; Invitrogen, Molecular Probes, Eugene, OR) at RT. The cells were washed with PBS and incubated with appropriate FITC- or TRITC-conjugated secondary antibodies and 4′,6-diamidino-2-phenylindole (DAPI, 1 μg/ml; Sigma) for 25 min at RT. After being washed with PBS, the cells were covered with 2.5% 1,4-diazobicyclo[2.2.2]octane (DABCO) in 90% glycerol–10% PBS to prevent image fading. For in situ extraction, cells were exposed to cold 0.5% TX-100 or 10 mM CHAPS in PBS for 2 min at 4°C; this was followed by PFA fixation at 4°C. The samples were analyzed with a conventional immunofluorescence microscope (Olympus AX70) equipped with analySIS software or by confocal microscopy with a TCS SP2 or SP8 AOBS confocal laser scanner microscope (Leica Heidelberg, Germany) in combination with Leica Confocal software. Data were processed using Adobe Photoshop software.
Dot blotting.
Equal volumes (10 μl) of gradient fractions were applied to nitrocellulose membranes. When dried, the membranes were incubated for 1 h at RT in blocking solution (5% nonfat dry milk in PBS); this was followed by overnight incubation with the antisulfatide O4 antibody (ammonium sulfated precipitated, O4 at 1:750). After washing with PBS containing 0.1% Tween 20 (PBS-T), the membranes were incubated for 2 h with horseradish peroxidase-conjugated anti-IgM antibodies in 1% nonfat dry milk in PBS-T. Signals were detected by enhanced chemiluminescence (GE Healthcare).
Antibody trafficking assay.
The basolateral surface of HepG2 cells that were stably transfected with PLP-eGFP was labeled with primary antibodies diluted in 0.2% BSA in PBS for 30 min on ice; this was followed by a 90-min chase in culture medium at 37°C. The primary antibodies used were anti-extracellular PLP ET3 (1:5) and anti-dipeptidylpeptidase IV (DPPIV; R&D Systems) antibodies. The tight junctions prevent access of the antibodies to apical membrane (42). Cells were fixed in 4% PFA for 30 min on ice, permeabilized with ice-cold methanol for 10 min, blocked with 4% BSA in PBS, and incubated with the appropriate TRITC-labeled secondary antibodies. Transcytosis of the antibody-antigen complex to the apical surface was analyzed with a conventional immunofluorescence microscope (Olympus AX70) equipped with analySIS software.
SDS-PAGE and Western blotting.
Equal volumes (gradient fractions) or protein amounts (cell lysates) were mixed with SDS reducing sample buffer, heated for 5 min at 95°C (syntaxin 3, syntaxin 4, GFP, caveolin, LAMP1, EEA1) or 30 min at 37°C (PLP) and subjected to SDS-PAGE and Western blotting. Samples were loaded onto 10 or 12.5% SDS-polyacrylamide gels, transferred to Immobilon-FL (Millipore, Bedford, MA) by semidry blotting, and subjected to immunoblot detection as described previously (29). The primary antibodies used were anti-syntaxin 3 (1:1,000; Synaptic Systems, Göttingen, Germany), anti-syntaxin 4 (1:500; Synaptic Systems), anti-PLP (1:100; 4C2 or 2D2), anti-GFP (1:500), anti-caveolin-1 (BD Transduction Laboratories, Lexington, KY), anti-LAMP1 (1:250; GeneTex Inc., Irvine, CA), and anti-EEA1 (1:250; Abcam, Cambridge, United Kingdom) antibodies. The signals were detected with the Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE) and analyzed with Odyssey V3.0 analysis software.
Statistics.
Data are expressed as the mean values of at least three independent experiments ± the standard deviations (SDs). Statistical analysis was performed with the Student t test when two means were compared and with a one-sample t test when a value was compared to the control, which was set to 100% in each independent experiment. When absolute values of more than two means were compared, statistical significance was calculated by one-way analysis of variance (ANOVA), followed by Tukey's posttest. In all cases, P < 0.05 was considered significant.
RESULTS
PLP undergoes a conformational alteration during transport to the myelin membrane.
The multimembrane-spanning protein PLP is integrated into the myelin membrane via vesicular transport. To accurately define its intracellular trafficking pathway following biosynthesis in primary OLGs, we first determined the localization of PLP at the onset of myelination (imOLGs) and at a more advanced stage (mOLGs). In imOLGs, when processes develop, PLP is abundantly localized in vesicular structures in the perinuclear region of the cell (Fig. 1A), often showing a punctate appearance when associated with primary processes. Furthermore, in a subset of cells, prominent labeling of the plasma membrane was observed (Fig. 1A, arrow), suggesting an association of PLP with the plasma membrane of the cell body. Next to its presence in the cell body, in mature, well-differentiated OLGs, PLP localizes in particular to vesicular structures in the processes and myelin sheets (Fig. 1A). Staining of live cells with anti-PLP antibody ET3 (directed against a synthetic second extracellular loop) (37) and O10 (directed against a conformational epitope) (36) confirmed the presence of PLP at the cell body plasma membrane and myelin sheet (Fig. 1B). Remarkably, although both antibodies are directed against a conformational epitope in the second extracellular loop, ET3 most prominently binds to PLP present at the cell body plasma membrane, whereas O10 staining is more intense when PLP is localized in the processes and myelin sheets. This staining pattern was unaltered upon double labeling, implying that the antibodies detect different epitopes in the protein when present at either the cell body plasma membrane or the myelin membrane (Fig. 1C). Since the secondary structure of the extracellular loop changes as a function of the environment (37), the data might thus indicate that PLP undergoes conformational alterations during transport and/or oligomerizes in a molecular-environment-dependent manner.
FIG 1.
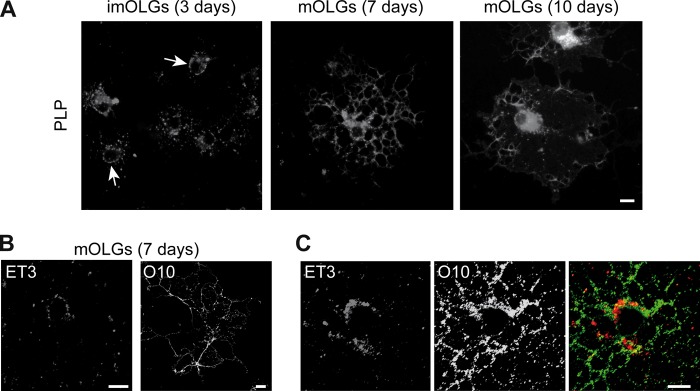
PLP undergoes a conformational alteration during transport to myelin membranes. (A) Localization of PLP in developing OLGs analyzed by immunostaining (4C2). Note that in imOLGs, PLP is localized at the cell body plasma membrane (arrows), whereas in mOLGs, PLP-containing vesicular structures in the perinuclear region and myelin sheet were observed. (B, C) Surface staining of mOLGs with anti-PLP antibodies that recognize an extracellular epitope (ET3, O10). Single labeling is shown in panel B, and double labeling is shown in panel C. Note that ET3 binds primarily to the cell body plasma membrane, whereas O10 staining is more prominent in the myelin sheet. Representative pictures of four independent experiments are shown. The scale bars are 10 μm.
Overexpression of syntaxin 3 inhibits cell body plasma membrane-directed transport of PLP.
In previous work, we have shown that the t-SNARE syntaxin 3, which localizes predominantly at the apical plasma membrane in epithelial cells (43, 44), largely localizes at or near the plasma membrane of the OLG cell body (6), which is served by an apical-surface-like vesicular transport route (4, 5). In contrast, syntaxin 4, which localizes in the basolateral domain of epithelial cells (43, 44), localizes at the myelin membrane in cultured OLGs (6). To assess whether PLP transport toward the myelin sheet relies on transcytosis, i.e., involves prior transport to and delivery at the cell body plasma membrane, we examined the effect of syntaxin 3 and 4 overexpression on PLP localization. Overexpression of syntaxins leads to nonfunctional SNARE complexes (43), thus mimicking a dominant negative approach. Double labeling with anti-PLP antibodies recognizing extracellular (O10) and intracellular (2D2) PLP epitopes (35, 36) revealed that in OLGs that overexpress syntaxin 3, transport of PLP to myelin sheets was apparently blocked, while in cells that overexpress syntaxin 4, PLP is present at the surface (Fig. 2A). Thus, at approximately 5-fold overexpression of syntaxin 3, PLP prominently accumulated in intracellular vesicles in the cell body, with no significant appearance at the cell body plasma membrane or in the myelin sheet (Fig. 2A, middle panels). Importantly, upon syntaxin 3 and 4 overexpression, no difference from mock (vector-only)-transduced cells in the number of cells expressing PLP was observed (data not shown). Hence, our data suggest that malfunctioning of plasma membrane-localized syntaxin 3, as a consequence of its overexpression, precludes docking of PLP-containing transport vesicles, which remain trapped in the cell body cytoplasm.
FIG 2.

Overexpression of syntaxin 3 inhibits surface transport of PLP in OLGs concomitant with TX-100 resistance. (A, B) OPCs were transduced with the vector only (mock), syntaxin 3 (S3↑), or syntaxin 4 (S4↑). (A) Surface and intracellular localization of PLP in mOLGs as determined by immunostaining with anti-PLP antibodies directed against extracellular (O10) and intracellular PLP epitopes (2D2). Syntaxin 3 and 4 overexpression was confirmed by Western blotting. Note the lack of PLP surface expression upon the overexpression of syntaxin 3. (B) TX-100 and CHAPS membrane microdomain association of PLP in mOLGs. PLP (4C2) and caveolin were visualized by Western blotting. Detergent-resistant membrane microdomains are present in fractions 3 and 4. Note that PLP is TX-100 resistant in syntaxin 3-overexpressing OLGs but largely TX-100 soluble in mock-transduced OLGs. (C) ImOLGs were pulse-labeled with Tran35S-label and chased for the times indicated. PLP was immunoprecipitated from the TX-100-soluble (black bars) and -insoluble (white bars) fractions, and the amounts were analyzed by radioactivity counting. The statistical significance of differences between 0-min values and values obtained at the time points indicated is shown (n = 3; **, P < 0.01; ***, P < 0.001; one-way ANOVA with Tukey's posttest). Note that following biosynthesis, PLP is transiently TX-100 resistant. (D) Cell surface proteins of mOLGs were biotinylated, lysed, and subjected to TX-100 extraction and OptiPrep density gradient centrifugation. Biotinylated, surface-localized proteins and nonbiotinylated, i.e., intracellularly localized, proteins were separated by SA immunoprecipitation. PLP was visualized by Western blotting (4C2 antibody). Note that TX-100-resistant PLP is present at the surface. (E) imOLGs were subjected to in situ detergent extraction with either TX-100 or CHAPS prior to fixation. Note that TX-100-resistant PLP localized to the cell body plasma membrane (arrow), whereas CHAPS-resistant PLP resided mainly intracellularly. Pictures and blots representative of at least three independent experiments are shown. The scale bars are 10 μm.
Following biosynthesis, PLP transiently associates with TX-100-resistant microdomains.
To obtain further support and mechanistic insight into the potential involvement of a transcytotic mechanism underlying myelin-directed trafficking of PLP, we next examined PLP dynamics by characterizing its lateral membrane organization with detergent insolubility as a criterion. It has been well documented that PLP ultimately integrates into CHAPS-resistant membrane microdomains (11, 45). Given the apical-surface-like nature of trafficking directed toward the cell body plasma membrane in OLGs (4, 5), the observed inhibition of PLP transport to the surface upon overexpression of syntaxin 3 therefore suggests inhibition of an apical-surface-directed PLP transport step. In general, apical protein transport is associated with the protein's integration into TX-100-resistant membrane microdomains (4, 46). To verify this in the case of PLP transport, syntaxin 3-overexpressing OPCs were allowed to differentiate into myelin sheet-forming mOLGs, extracted with either CHAPS or TX-100, loaded onto an OptiPrep density gradient, and analyzed by Western blotting. Whereas PLP essentially localized to TX-100-soluble (Fig. 2B, fractions 6 and 7, approximately 60%) but CHAPS-insoluble fractions (Fig. 2B, fractions 3 and 4, approximately 75%) in mock-transduced cells, in syntaxin 3-overexpressing cells, PLP was largely recovered in the TX-100-insoluble fraction (Fig. 2B, fractions 3 and 4, approximately 39%) and much less so in the CHAPS-insoluble fraction (Fig. 2B, fractions 3 and 4, approximately 46%). Caveolin, a control protein present in membrane microdomains, remained insoluble under the conditions used (Fig. 2B), indicating that, rather than a general phenomenon, the effect of syntaxin 3 on lateral distribution is specific for PLP transport. Thus, these data suggest that PLP, following its biosynthesis, transiently integrates into TX-100-resistant microdomains, reflecting the apical-surface-like plasma membrane-directed transport mechanism. To verify this potential scenario further, a pulse-chase experiment was carried out to better define the initial steps in PLP trafficking. OLGs were metabolically labeled with Tran35S-label for 10 min at 37°C. Subsequently, excess nonradioactive methionine was added and the cells were chased for 0, 15, 30, and 60 min, followed by extraction with TX-100 and analysis of radiolabeled PLP in the pellet (insoluble) and supernatant (soluble). As shown in Fig. 2C, during the first 15 min following de novo biosynthesis of PLP, essentially the entire PLP fraction was present within a TX-100-resistant fraction (white bars), while over the next 45 min, PLP gradually partitioned into a TX-100-soluble fraction (black bars). To determine whether TX-100-resistant PLP-containing membrane fractions are, in fact, present at the cell surface, mOLGs were surface biotinylated, followed by TX-100 extraction and OptiPrep density gradient fractionation. To separate surface-localized (i.e., biotinylated) TX-100-resistant PLP from intracellular (i.e., nonbiotinylated) TX-100-resistant PLP, the gradient fractions were subjected to SA precipitation and analyzed by Western blotting. As shown in Fig. 2D, PLP was present at the surface and this surface pool partitioned roughly equally between the TX-100-resistant (fractions 3 and 4, approximately 39%) and TX-100-soluble fractions (fractions 6 and 7, approximately 42%). Of the intracellular fraction, less than 15% was resistant to solubilization by TX-100. As this biochemical characterization does not provide information about the actual (surface) membrane localization of PLP, i.e., cell body plasma membrane versus myelin sheet, we next visualized PLP in TX-100-resistant membrane domains by in situ extraction with TX-100 prior to fixation. TX-100-resistant PLP was observed mainly in the cell body plasma membrane (Fig. 2E, arrow). In contrast, CHAPS-resistant PLP resides mainly intracellularly. Hence, these findings are consistent with the notion that de novo-synthesized PLP is initially assembled into TX-100-insoluble membrane microdomains localized within transport vesicles that presumably dock by a syntaxin 3-dependent mechanism at the plasma membrane. During this early processing, the protein appears to reside transiently in membrane microdomains at the cell body plasma membrane that are TX-100 resistant. The prominent intracellular CHAPS resistance of PLP then presumably reflects the ensuing transport vesicle-mediated processing of the protein from the plasma membrane toward the myelin sheet. To obtain further support for the ability of PLP to engage in a transcytotic transport route, PLP transport was analyzed in the well-polarized hepatic cell line HepG2.
PLP is present in apical and basolateral domains of polarized HepG2 cells.
Polarized HepG2 cells display two structurally and functionally different plasma membrane domains (Fig. 3A). The two membrane domains are separated by tight junctions and comprise the apical membrane, which faces the bile canaliculus (BC), and the basolateral domain, which faces adjacent cells. In these cells, direct routes for basolateral and apical delivery exist, as well as indirect transcytotic routes (47). More specifically, our previous work showed that in these cells, resident apical proteins may employ different pathways to reach the apical surface following biosynthesis (42). Thus, whereas single transmembrane proteins traffic to the apical membrane via the basolateral membrane, multimembrane-spanning proteins may arrive at the apical surface along a direct pathway. To define the polarized transport properties of multimembrane-spanning PLP, HepG2 cells were transfected with PLP-eGFP. In stably transfected cells, PLP-eGFP was present prominently at the apical surface (Fig. 3B, arrow) whereas a fraction was also detectable at the basolateral surface (Fig. 3B, arrowhead). Surprisingly, the PLP-eGFP at the basolateral membrane was not recognized by O10 (data not shown), implying inaccessibility of PLP's conformational O10 epitope in HepG2 cells. However, antibody ET3 does recognize extracellular PLP-eGFP at the basolateral surface (Fig. 3C). Unfortunately, given the relative inaccessibility of the apical surface in HepG2 cells for exogenous labeling (Fig. 3A and C) (42), determination of antibody ET3 recognition at this surface is essentially precluded. Under steady-state conditions, PLP-eGFP associated primarily with CHAPS-insoluble membrane microdomains (Fig. 3D, fractions 3 and 4, approximately 56%) whereas only a minor fraction was TX-100 insoluble (Fig. 3D, fractions 3 and 4, approximately 21%). Hence, under these conditions, PLP-eGFP partitioned in microdomains, displaying detergent resistance properties similar to those seen in primary OLGs (cf. Fig. 2B, mock). In situ extraction revealed that CHAPS-resistant PLP is present at both the apical and basolateral membrane domains, whereas the PLP fraction that resisted TX-100 extraction was associated exclusively with the apical domain (Fig. 3E). Quantitative analysis based on the use of MRP2 as a detergent-resistant apical marker revealed that upon in situ extraction with TX-100, only 5 to 10% of the MRP2-positive apical membranes are positive for PLP-eGFP, whereas upon in situ extraction with CHAPS, 60 to 70% of the apical membranes are positive for PLP (Fig. 3F). In untreated cells, approximately 85% of the MRP2-positive apical membranes are positive for PLP-eGFP. Since PLP localized at both surfaces, it was of obvious interest to better define its transport itinerary. To verify whether PLP-eGFP is transported in a direct manner to the apical surface or, alternatively, may employ an indirect transcytotic pathway from the basolateral to the apical surface, an antibody-trafficking assay was performed (42). HepG2-PLP-eGFP cells were incubated for 30 min (on ice) with ET3 and then incubated at 37°C for 90 min to activate trafficking. Subsequent immunocytochemical analysis of the localization of the antibody-antigen complex revealed that the antibody-stained PLP-eGFP became internalized but did not reach the apical membrane (Fig. 3G). In contrast, DPPIV, a single-pass resident apical membrane protein and a marker for basolateral-to-apical transcytosis in HepG2 cells (42), was transported to the apical domain upon antibody internalization over the same incubation period (Fig. 3G), indicating that basolateral-to-apical transcytosis is functional in HepG2 cells that express PLP-eGFP. These findings suggest that PLP is directly and effectively transported to the apical membrane in polarized HepG2 cells following biosynthesis. Subsequently, PLP may engage in transcytotic transport to the basolateral surface, where reinternalization may occur without ensuing effective redelivery of PLP to the apical surface.
FIG 3.

PLP-eGFP is present in the apical and basolateral membrane domains of polarized HepG2 cells. (A) Schematic model of polarized HepG2 cells. The apical (BC, red) and basolateral (blue) membrane domains are separated by tight junctions (green). (B) Localization of PLP-eGFP upon stable expression in HepG2 cells. Note that PLP-eGFP is present in the apical (arrow) and basolateral (arrowhead) membrane domains. (C) Localization of extracellular PLP (ET3) and total PLP-eGFP (inset) at the basolateral surface of HepG2 cells. Nuclei were visualized with DAPI. (D) TX-100 and CHAPS membrane microdomain association of PLP-eGFP in HepG2 cells. PLP-eGFP was visualized by Western blotting (anti-GFP antibody). Detergent-resistant membrane microdomains are present in fractions 3 and 4. (E, F) HepG2-PLP-eGFP cells were left untreated (ctrl) or subjected to in situ detergent extraction with either TX-100 or CHAPS prior to fixation. Apical membranes were visualized by immunostaining for MRP2 (E, insets, red). Note that TX-100-resistant PLP-eGFP localized to apical membranes, whereas CHAPS-resistant PLP-eGFP resided at both membrane domains. The number of PLP-positive BCs among the total MRP2-positive BCs was determined under the conditions indicated (F). Each bar represents the mean result + the SD of four independent experiments. (G) Basolateral-to-apical transcytosis of PLP-eGFP and DPPIV as analyzed by an antibody (Ab) trafficking assay. Antibodies (red) were visualized by immunostaining, and nuclei were stained with DAPI. Note that DPPIV, but not PLP-eGFP (ET3), was transcytosed to the apical membrane domain. The localization of total PLP-eGFP is shown in the insets (green) (H) TLC analysis of cellular glycosphingolipids in HepG2 and HepG2-PLP-eGFP cells. Pictures and blots representative of at least three independent experiments are shown. Scale bars are 10 μm.
To obtain further insight into the underlying mechanism that drives polarized PLP transport, we focused on the potential role of the galactolipids GalC and sulfatide. These galactolipids are highly enriched in myelin; their expression precedes the appearance of PLP in OLGs (2). Since GalC and sulfatide are not enriched in HepG2 cells, we analyzed the expression of GalC and sulfatide in PLP-eGFP-expressing HepG2 cells. Upon stable expression of PLP-eGFP, the expression of sulfatide was dramatically greater than that in parental cells (Fig. 3H, approximately 2-fold), suggesting that HepG2 cells likely adapt to PLP expression by increasing the levels of sulfatide. This finding is rather surprising, given a reported role for GalC-enriched microdomains but not sulfatide in PLP transport to the myelin sheet (11). However, although it is controversial, sulfatide has been implicated in PLP transport (48, 49). The apparently contradictory findings likely originate from the different experimental conditions, including, among others, the polarized status of the cells. Therefore, we next revisited the role of sulfatide in PLP transport in OLGs.
Sulfatide is not essential for the transport of PLP to the cell body plasma membrane.
Previously, we have shown that GalC and sulfatide are differently distributed in mOLGs, with GalC preferentially localizing to myelin sheets and compact myelin whereas sulfatide is largely excluded from myelin sheets and enriched in paranodes (50). To assess whether sulfatide is a priori involved in mediating the transport of PLP, we next examined the cellular localization of PLP in primary imOLGs, i.e., conditions under which PLP transport to the cell body plasma membrane is already apparent (cf. Fig. 1A), upon inhibition of sulfatide biosynthesis with sodium chlorate from OPCs onward. Sodium chlorate is a competitive inhibitor of sulfation that inhibits sulfatide biosynthesis but not GalC biosynthesis (51–53). Furthermore, this compound does not affect OLG viability and differentiation when the cells are cultured on a PLL substrate (53). Labeling of cell surface PLP (ET3), followed by fixation, permeabilization, and staining of PLP with 4C2, revealed that in cells in which sulfatide biosynthesis was blocked, PLP localized predominantly at the plasma membrane of the cell body, whereas in untreated cells, PLP was localized mainly intracellularly (Fig. 4A, left panel). A quantitative analysis of the cells showing cell surface staining revealed a 3-fold increase in PLP surface staining upon inhibition of sulfatide biosynthesis, compared to that in untreated cells (Fig. 4B). In conjunction with the reduced intracellular staining of PLP (Fig. 4A), the accumulated surface fraction of PLP presumably reflects a decrease in PLP internalization by the cells. Remarkably, irrespective of sulfatide inhibition, PLP expressed on the surface of imOLGs does not yet expose the O10 epitope (Fig. 4A and B). Nevertheless, sulfatide is not essential for PLP transport to the membrane surface of the cell body, given the presence of PLP at the surface upon sulfatide depletion (Fig. 4A and B). To examine the lateral membrane distribution of PLP at the cell surface in the absence of sulfatide, untreated and sodium chlorate-treated mOLGs were surface biotinylated; this was followed by detergent extraction, density gradient centrifugation, and SA precipitation. The fraction of PLP present at the surface of untreated OLGs was partly TX-100 resistant and CHAPS resistant, whereas surface PLP was still partly TX-100 resistant but CHAPS soluble in sodium chlorate-treated, i.e., sulfatide-depleted, cells (Fig. 4C). Thus, sulfatide depletion results in the elimination of PLP's localization in CHAPS-resistant microdomains at the cell surface, indicating that in cultured OLGs, the presence of sulfatide mediates a microdomain reallocation of cell surface-localized PLP, as reflected by a detergent (in)solubility transition of PLP from TX-100 to CHAPS resistance. To further assess the role of sulfatide in PLP transport and its lateral membrane distribution, we next made use of the oligodendroglia-derived cell line OLN-93, which is deficient in both PLP (30) and galactosphingolipids (Fig. 5A, mock).
FIG 4.

Sulfatide is not essential for the transport of PLP to the cell body plasma membrane. OPCs were differentiated for 3 (A, B) or 7 (C) days in the absence (ctrl) or presence of sodium chlorate, an inhibitor of sulfatide synthesis. (A) The surface localization of PLP was determined by immunostaining with anti-PLP antibodies directed against extracellular epitopes (ET3, O10); this was followed by fixation, permeabilization, and staining with the 4C2 anti-PLP antibody. The percentage of PLP surface-positive cells is shown in panel B. Statistically significant differences from the results obtained with untreated cells (ctrl) were assessed with the Student t test (n = 4; **, P < 0.001). Note the prominent PLP surface expression (ET3 antibody) at the cell body plasma membrane upon inhibition of sulfatide biosynthesis. (C) TX-100 and CHAPS membrane microdomain association of surface PLP. Cell surface proteins were biotinylated and subjected to TX1-100 or CHAPS extraction, density gradient fractionation, and immunoprecipitation with SA-agarose. PLP was visualized by Western blotting (4C2 antibody). Note that surface PLP is TX-100 and CHAPS resistant in untreated cells but CHAPS soluble upon inhibition of sulfatide synthesis (chlorate). Pictures and blots representative of at least three independent experiments are shown. Scale bars are 10 μm.
FIG 5.

Sulfatide is essential for segregation to TX-100-soluble membrane microdomains. (A) TLC analysis of cellular glycosphingolipids in OLN-PLP (mock), OLN-PLP-G (GalC), and OLN-PLP-GS (GalC and sulfatide) cells. (B) Western blot analysis of PLP-eGFP expression in OLN-PLP cells (mock), OLN-PLP-G cells, OLN-PLP-GS cells, OPCs, and OLGs (10 days of differentiation). PLP was visualized with anti-PLP antibody 2D2. (C) Cell surface proteins were biotinylated, lysed, and immunoprecipitated with SA-agarose to separate surface-localized proteins (lanes S) and nonbiotinylated, i.e., intracellularly localized proteins (lanes I). PLP-eGFP was visualized by Western blotting (anti-GFP antibody). Note that PLP-eGFP is present at the surface of OLN-PLP cells (mock), i.e., in the absence of GalC (G) and sulfatide (S). (D) The surface localization of PLP-eGFP (ET3) was determined in mock-transduced (vector only) and syntaxin 3-overexpressing cells (S3↑). Note the decreased PLP-eGFP surface expression upon the overexpression of syntaxin 3 (arrow). (E) TX-100 and CHAPS membrane microdomain association of PLP-eGFP in OLN-PLP, OLN-PLP-G, and OLN-PLP-GS cells. PLP-eGFP was visualized by Western blotting (anti-GFP antibody). Detergent-resistant membrane microdomains are present in fractions 3 and 4. Note that PLP-eGFP is TX-100 resistant in the absence of sulfatide. PLP (4C2) and caveolin were visualized by Western blotting. Pictures and blots representative of at least three independent experiments are shown. The scale bar is 10 μm.
Sulfatide is necessary for PLP's reallocation to TX-100-soluble membrane microdomains.
Since OLN-93 cells do not endogenously express PLP (30), we first stably expressed PLP-eGFP in OLN-93 cells (OLN-PLP). Notably, the introduction of PLP-eGFP did not affect the viability of OLN-93 cells (data not shown), while the expression level did not exceed the PLP expression levels in primary OLGs (Fig. 5B, mock versus OLG). PLP-eGFP was expressed prominently at the surface of galactolipid-deficient OLN-PLP cells (Fig. 5D, mock), confirming that GalC and sulfatide are not required for transport to the plasma membrane. Surface biotinylation experiments confirmed the plasma membrane localization of PLP-eGFP in OLN-PLP cells (Fig. 5C, mock, surface [S] versus intracellular [I]). Notably, a fraction of the PLP-eGFP remained in the endoplasmic reticulum. Importantly, as in primary OLGs, overexpression of syntaxin 3 dramatically reduced the levels of PLP on the OLN-PLP cell surface (Fig. 5D, S3↑, arrow), suggesting a similarity in the underlying mechanisms of plasma membrane-directed transport in both cell types. Therefore, these findings are consistent with the previous finding that sulfatide is not essential for the transport of PLP to the OLG plasma membrane (Fig. 4) (51, 52). To further clarify the role of GalC and sulfatide in the lateral membrane distribution of PLP, OLN-PLP cells were transduced with constructs that express CGT, the enzyme catalyzing the final step in GalC biosynthesis and/or CST, which catalyzes the biosynthesis of sulfatide. The GalC-overexpressing and GalC- and sulfatide-overexpressing OLN-PLP cells are referred to as OLN-PLP-G and OLN-PLP-GS, respectively. TLC analysis revealed that, compared to mock-transduced cells (OLN-PLP, i.e., vector only), OLN-PLP-G cells express significant levels of GalC, while both GalC and sulfatide are present in OLN-PLP-GS cells (Fig. 5A). Of interest, the presence of both galactolipids and, to a lesser extent, the presence of GalC alone resulted in an increase in total PLP expression (Fig. 5B) and an approximately 2-fold increase in the surface expression of PLP-eGFP (Fig. 5C). Given that PLP partitions in either TX-100- or CHAPS-resistant membrane domains, the lateral distribution of PLP-eGFP in OLN-PLP, OLN-PLP-G, and OLN-PLP-GS cells was analyzed. In OLN-PLP-GS cells, PLP-eGFP was CHAPS resistant (Fig. 5E, fractions 3 and 4, approximately 77%) and TX-100 soluble (Fig. 5E, fractions 6 and 7, approximately 52%), in line with PLP's resistance in primary OLGs (Fig. 2B, mock) and HepG2 cells (Fig. 3D). Strikingly, in galactolipid-deficient OLN-PLP-mock cells, PLP-eGFP is TX-100 resistant (Fig. 5E, fractions 3 and 4, approximately 86%) and partly CHAPS resistant (Fig. 5E, fractions 3 and 4, approximately 42%), while in sulfatide-deficient OLN-PLP-G cells, PLP-eGFP is CHAPS resistant (Fig. 5E, fractions 3 and 4, approximately 70%) and only partly TX-100 soluble (Fig. 5E, fractions 6 and 7, approximately 32%). The observed redistribution, reflected by the changes in detergent solubility, appeared to be specific for PLP, since the localization of caveolin in membrane microdomains remained unaltered, irrespective of the presence of galactolipids. In all of the OLN lines, PLP acquired CHAPS resistance, indicating that in these cells, the presence of sulfatide is not a prerequisite for PLP's sequestration in CHAPS-resistant domains. Hence, the presence of sulfatide caused PLP-eGFP to redistribute into membrane microdomains that are soluble in TX-100. Since PLP transiently associates with TX-100-resistant membrane domains, i.e., upon its biosynthesis, while the protein acquires CHAPS resistance in subsequent transcytotic transport, we next determined the location and underlying driving force of this shift in lateral membrane distribution.
Sulfatide-dependent redistribution of PLP to different microdomains at the plasma membrane.
To determine whether PLP, after arrival at the plasma membrane, and sulfatide may partition within the same membrane domains at the cell surface, we first determined the potential colocalization of PLP with either anti-GalC (O1) or antisulfatide (O4) antibodies in live OLN-PLP-GS cells. PLP-eGFP colocalized with sulfatide but not GalC at the cell surface (Fig. 6A, inset). Of note, surface labeling with anti-PLP antibody O10 showed colocalization with PLP-eGFP (data not shown). In fixed and permeabilized OLN-PLP-GS cells (Fig. 6A), intracellular PLP-eGFP colocalized with GalC but not sulfatide (Fig. 6A). The intracellular sulfatide patches colocalized with LAMP1 but not EEA1 (data not shown, Fig. 6B), and this might be a reflection of a reduced ability to degrade sulfatide in OLN-PLP-GS cells. As shown, in the presence of sulfatide, an increasing PLP fraction redistributed from TX-100-insoluble to TX-100-soluble membrane domains (Fig. 5E and 4C). To determine the cellular location of PLP's redistribution to TX-100-soluble membrane microdomains, i.e., at the plasma membrane or upon internalization, subcellular fractions enriched in endosomes and lysosomes were isolated and separated on a discontinuous sucrose gradient (38, 39) (Fig. 6B); this was followed by either TX-100 or CHAPS extraction. In OLN-PLP-GS cells, PLP-eGFP is TX-100 soluble but CHAPS resistant in both endosomes and lysosomes (Fig. 6C), indicating that the detergent (in)solubility shift, as in HepG2 cells (Fig. 3, data not shown), likely occurred at the cell surface or immediately following internalization. In OLN-PLP cells, i.e., cells that do not express galactolipids, PLP-eGFP is CHAPS soluble in the enriched endosomal fraction but CHAPS resistant in the enriched lysosomal fraction (Fig. 6C). Of note, in OLN-PLP cells, a larger fraction of PLP-eGFP is present in the enriched endosomal fraction than in the enriched lysosomal fraction, compared to its distribution in OLN-PLP-GS cells (Fig. 6B). Hence, the detergent analysis suggests that sulfatide potentially induces a lateral redistribution of PLP in the plasma membrane of OLGs, which was examined next in live cells.
FIG 6.

PLP is conveyed from TX-100- to CHAPS-resistant membrane microdomains at the plasma membrane. (A) Colocalization of PLP-eGFP with GalC and/or sulfatide in OLN-PLP-GS cells. Immunostaining with anti-GalC (O1) and antisulfatide (O4) antibodies was performed with live or fixed and permeabilized cells. Note that PLP-eGFP colocalized with sulfatide but not GalC at the surface of OLN-PLP-GS cells, whereas intracellular PLP-eGFP partially colocalized with GalC but not sulfatide (insets). (B, C) Endosomes and lysosomes were separated by organelle fractionation and centrifugation on a discontinuous sucrose gradient. The distribution of EEA1, LAMP1, and PLP-eGFP (anti-GFP antibody) was visualized by Western blotting. The distribution of sulfatide was visualized with a dot blotting assay using antisulfatide antibody O4 for detection. The boxes mark the pooled fractions of the gradient used for TX-100 and CHAPS extraction of the enriched endosomal and lysosomal fractions shown in panel C. Detergent-resistant membrane microdomains are present in fractions 3 and 4. Note that PLP-eGFP is TX-100 soluble and CHAPS insoluble in both endosomes and lysosomes in OLN-PLP-GS cells but still CHAPS soluble in endosomes in galactolipid-deficient OLN-PLP cells. The scale bar is 10 μm.
Sulfatide modulates the conformation of PLP.
Presumably, a change in the lateral microenvironment of a membrane protein may be related to changes in the protein's structure, including its oligomerization. By employing the three available anti-extracellular PLP antibodies in the OLN-PLP cell lines, we investigated the possibility that PLP could undergo such changes when sensing the presence of sulfatide at the level of the plasma membrane. Immunocytochemical analysis of live cells revealed that antibody 4C2, which is directed against a nonconformational epitope in the first extracellular loop (35), recognizes PLP-eGFP at the surface of virtually all OLN-PLP, OLN-PLP-G, and OLN-PLP-GS cells (Fig. 7A), confirming the biochemical analysis (cf. Fig. 5C) that PLP reaches the surface independently of the presence of sulfatide. Similarly, when applying antibody ET3, which recognizes a conformational epitope in the second extracellular loop, prominent surface staining was observed on all three OLN-PLP lines (Fig. 7A). Remarkably, in line with the observation in primary OLGs, in OLN-PLP-GS cells, the ET3 epitope is exposed mainly at the cell body, while in OLN-PLP and OLN-PLP-G cells, ET3 binds in addition to PLP present at the processes. In contrast, the O10 epitope emerged in OLN-PLP-GS cells (Fig. 7A and B) but was hardly detectable in OLN-PLP and OLN-PLP-G cells, suggesting that the presence of sulfatide is sensed by PLP, which apparently adjusted its structure in terms of conformation. Importantly, the O10 epitope emerges only when PLP reaches a functional tertiary structure and/or oligomerizes (36), while the secondary structure of the second extracellular loop changes as a function of its environment (37). Given that, in OLGs and OLN-PLP-GS cells, ET3 recognizes PLP mainly at the cell body plasma membrane, where sulfatide is enriched (50), whereas the O10 epitope is more prominently exposed at the myelin sheet (Fig. 1B and C), it is tempting to suggest that sulfatide likely initiates structural changes in PLP upon its internalization. In addition, given that O10 does recognize PLP-eGFP at the surface of OLN-PLP-GS cells but not that of HepG2 cells (see above), this finding indicates that a component that is necessary for exposure of the O10 epitope is apparently lacking in the basolateral membrane of HepG2 cells.
FIG 7.

Sulfatide modulates the conformation of PLP at the plasma membrane. Surface expression of PLP-eGFP in differentiated OLN-PLP, OLN-PLP-G, and OLN-PLP-GS cells with three different anti-PLP antibodies against extracellular epitopes, i.e., the first (4C2) and second (O10, ET3) extracellular loops. Note that O10 recognizes surface PLP-eGFP only when sulfatide is present (A). Quantification of the number of O10-positive cells among the total number of cells is shown in panel B. Each bar represents the mean value plus the SD. Statistically significant differences from the results obtained with OLN-PLP cells were assessed by one-way ANOVA with Tukey's posttest (n = 3; ***, P < 0.001). The scale bar is 10 μm.
DISCUSSION
In the present work, we have shown that myelin biogenesis in OLGs at least partly relies on the involvement of a transcytotic transport mechanism, as reflected by myelin-directed transport of PLP. Thus, our data support a model in which PLP, early after biosynthesis, integrates into distinct membrane domains characterized by TX-100 insolubility for transport to the plasma membrane of the OLG cell body, which relies on syntaxin 3-mediated docking (Fig. 8). At the plasma membrane, at least part of the PLP fraction is redistributed into membrane domains that are characterized by TX-100 solubility but CHAPS resistance, consistent with previously reported observations (11). Interestingly, inhibition of sulfatide biosynthesis in primary OLGs and parallel experiments with OLN-93 cells expressing GalC or GalC and sulfatide indicate that sulfatide is instrumental in this shift in membrane domain localization, likely related to a sulfatide-induced conformational change in and/or oligomerization of the protein. Given that PLP is transported to the plasma membrane in the absence of sulfatide, while it remains TX-100 resistant for 30 to 60 min following its biosynthesis, PLP likely encounters sulfatide after reaching the cell body plasma membrane. As shown previously, as part of the apical-to-basolateral transport process, PLP becomes integrated into cholesterol- and GalC-enriched microdomains in myelin membranes, suggesting its eventual dissociation from sulfatide (11). Evidently, a transcytotic mechanism would be entirely in line with our previous contentions that OLGs can be envisioned as polarized cells, displaying properties similar to those reported for typical polarized cells like epithelial cells. In fact, in the present work, we demonstrate that in such cells, i.e., hepatocytes, the major epithelial cells in the liver, PLP transcytotic transport may occur between the apical and basolateral surfaces.
FIG 8.
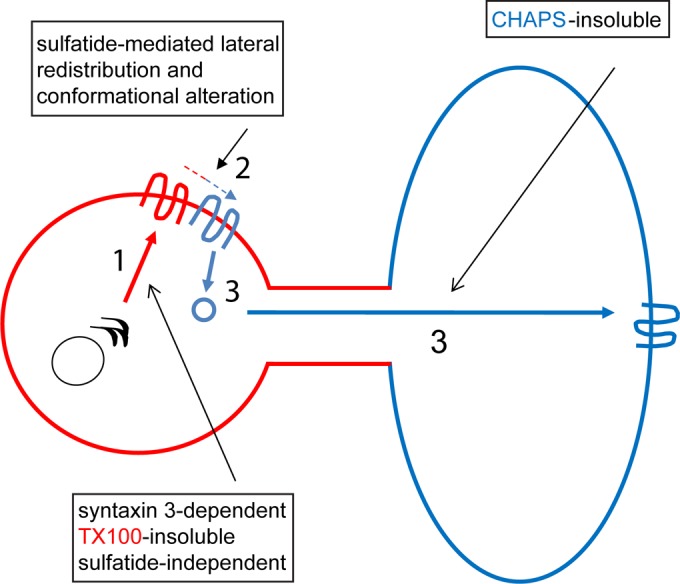
Schematic overview of the trafficking of de novo-synthesized PLP to myelin sheets via transcytosis. (Part 1) Transport of PLP from the trans-Golgi network to the apical-surface-like (red) cell body plasma membrane is syntaxin 3 dependent and sulfatide independent and occurs as part of a TX-100-insoluble, CHAPS-soluble membrane microdomain (part 2). At the plasma membrane, PLP is redistributed to TX-100-soluble, CHAPS-insoluble microdomains, a process that is sulfatide mediated and likely involves a conformational alteration and/or oligomerization (part 3). Transport from the plasma membrane of the cell body to the basolateral-surface-like (blue) myelin-like membranes occurs by means of TX-100-soluble, CHAPS-insoluble microdomains. Along this route, PLP likely accumulates temporally in a late endosomal compartment (11, 12). See the text for further details.
Our observations indicate that transport of PLP to the myelin membrane relies on a syntaxin 3- but not syntaxin 4-dependent mechanism. Thus, following biosynthesis, PLP is transported to the apical-surface-like plasma membrane of the cell body in a syntaxin 3-dependent manner, after which the protein is laterally reallocated, internalized, and directed toward the myelin sheet. A transient localization of PLP at the cell body plasma membrane, followed by its subsequent internalization via endocytosis, would, as a matter of mechanistic principle, also be consistent with observations reported by others (17, 23). Although syntaxin 3 is clearly involved in the delivery of PLP at the cell body plasma membrane following biosynthesis, the t-SNARE involved in docking of PLP at the myelin membrane remains to be determined. A possible candidate is syntaxin 2, which localizes to myelin sheets (our unpublished observations; 27).
At first glance, our data are inconsistent with the studies of Feldmann et al. (24). They proposed that vesicular delivery of PLP from endosomal compartments to the myelin membrane relies on VAMP7, the cognate v-SNARE partner of syntaxin 3, whereas our findings indicate an earlier role for syntaxin 3, i.e., in the biosynthetic pathway to the plasma membrane, rather than the transcytotic pathway. However, the conclusion of Feldmann et al. (24) on the localization of VAMP7 was based primarily on studies with GFP-VAMP7 constructs in mouse-derived Oli-neu cells that express sulfatide (54), whereas our data are derived from mature rat OLGs. On the basis of the present findings, it would be of interest to determine the nature of the detergent sensitivity of the PLP-containing transport vesicles and sulfatide dependence upon VAMP7 dysfunctioning, either by downregulation or overexpression of the v-SNARE, analogously as reported in the present study for a syntaxin 3 dependence of PLP transport. Nevertheless, myelin defects arise when interfering with VAMP7-mediated but not VAMP3-mediated vesicular transport, which emphasizes that the cognate VAMP7-syntaxin 3 pair but not the VAMP3-syntaxin 4 pair, is essential for PLP transport to myelin membranes (24), which is entirely consistent with our observation in primary OLGs.
A hallmark of the transcytotic transport route of PLP is its transient partitioning into distinct membrane domains at different stages of its processing after de novo biosynthesis. Thus, following its biosynthesis, PLP is transported from the trans-Golgi network in a TX-100-insoluble membrane microdomain fraction to the cell body plasma membrane, where the protein is redistributed to TX-100-soluble membrane domains that, in time, become resistant to CHAPS solubilization. A similar sequential shift between distinct membrane domains, reflected by differences in detergent (in)solubility, was observed in polarized HepG2 cells. In further support of such a sequence of events and its functional consequences are observations reported by Simons et al. (11, 55) that the kinetics of PLP's acquirement of TX-100 solubility appear to be commensurate with those of acquiring CHAPS insolubility. More specifically, PLP is largely CHAPS soluble at the Golgi apparatus, and only after 30 to 60 min, i.e., likely the time required to reach the plasma membrane, PLP is redistributed from a TX-100-insoluble to a CHAPS-insoluble domain (Fig. 2) (11). As noted above, this detergent-sensitive shift likely takes place at the apical-surface-like cell body plasma membrane rather than within endocytic compartments. Presumably, this lateral sorting step might be involved in the subsequent internalization and transport of PLP from the plasma membrane to endosomal compartments, which are instrumental in transcytosis (56) and subsequent delivery to the myelin membrane.
Our data indicate that sulfatide is presumably responsible for the lateral reallocation to distinct membrane microdomains. Thus, in primary OLGs, sulfatide, rather than GalC, conveys TX-100 solubility to PLP, prior to its integration into CHAPS-resistant domains, a finding that is consistent with observations in mice that are unable to synthesize GalC and sulfatide. In these mice, a major fraction of PLP resides in TX-100-resistant domains and fails to segregate into CHAPS-resistant domains (11). The underlying mechanism by which sulfatide sequesters PLP from TX-100-insoluble domains remains to be determined, although a sulfatide-induced conformational alteration and/or oligomerization of PLP, as stated in the present report, may be of relevance in this regard. However, of interest in this context is also the reported cholesterol dependence of PLP's internalization before the acquirement of CHAPS insolubility and exposure of the O10 epitope (11, 22). Given that PLP and sulfatide may both associate with cholesterol (45, 57), such interactions can affect the physicochemical properties of microdomains and hence their content (58). As a consequence, sulfatide- and cholesterol-dependent invagination may be triggered, allowing subsequent transcytotic transport to the myelin sheet. Moreover, a preferential role for sulfatide in endocytosis in OLGs has been reported (59).
Taking the results together, we show here that, along with being a key compound of the regulation of the timing of terminal OLG differentiation (53, 60, 61), sulfatide is also a key regulator of transcytotic PLP transport, presumably relying in part on a sulfatide-induced conformational change. This may allow for the regulated timing of PLP's appearance in myelin membranes, thereby preventing premature and ectopic compaction. Remarkably, in vivo, PLP transport to myelin membranes is uninterrupted in the absence of sulfatide (51, 52, 62). It is likely, under these conditions, that PLP trafficking is not temporally regulated and is transported to myelin membranes by bulk flow or by an unknown compensatory, likely nontranscytotic, mechanism that allows CHAPS-soluble and possibly TX-100-insoluble PLP to insert into myelin membranes. Of interest in this context is that in the absence of sulfatide, a nonselective association with myelin-directed vesicles has been noted (63). In addition, sulfatide interacts with several key adhesion and ECM proteins, including laminin-2 (64, 65), which is secreted by developing axons (66) and promotes myelin biogenesis (reviewed in reference 67). Hence, it is tempting to suggest that the recently reported interaction between laminin-2 and sulfatide in OLGs (53) might regulate (transcytotic) PLP transport in vivo, a process that may be disrupted in MS. These issues are currently being investigated in our laboratory.
ACKNOWLEDGMENTS
We gratefully acknowledge Joao Relvas for expert help in applying the retroviral expression system in OLGs and Sven van Ijzendoorn for expert support in the HepG2 cell experiments.
This work was supported by grants from the Dutch MS Research Foundation (Stichting MS Research) (W.B., B.K., and D.H.), The Netherlands Organization of Scientific Research (NWO, VIDI, Aspasia) (W.B.), and the Foundation Jan Kornelis de Cock. Parts of this work were performed at the UMCG Microscopy and Imaging Center, sponsored by NWO grant 175-010-2009-023.
REFERENCES
- 1.Baumann N, Pham-Dinh D. 2001. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev 81:871–927. [DOI] [PubMed] [Google Scholar]
- 2.Pfeiffer SE, Warrington AE, Bansal R. 1993. The oligodendrocyte and its many cellular processes. Trends Cell Biol 3:191–197. doi: 10.1016/0962-8924(93)90213-K. [DOI] [PubMed] [Google Scholar]
- 3.Jahn O, Tenzer S, Werner HB. 2009. Myelin proteomics: molecular anatomy of an insulating sheath. Mol Neurobiol 40:55–72. doi: 10.1007/s12035-009-8071-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Vries H, Schrage C, Hoekstra D. 1998. An apical trafficking pathway is present in cultured oligodendrocytes but the sphingolipid-enriched myelin membrane is target of a basolateral-type pathway. Mol Biol Cell 9:599–609. doi: 10.1091/mbc.9.3.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klunder B, Baron W, Schrage C, de Jonge J, de Vries H, Hoekstra D. 2008. Sorting signals and regulation of cognate basolateral trafficking in myelin biogenesis. J Neurosci Res 86:1007–1016. doi: 10.1002/jnr.21556. [DOI] [PubMed] [Google Scholar]
- 6.Baron W, Hoekstra D. 2010. On the biogenesis of myelin membranes: sorting, trafficking and cell polarity. FEBS Lett 584:1760–1770. doi: 10.1016/j.febslet.2009.10.085. [DOI] [PubMed] [Google Scholar]
- 7.Masaki T. 2012. Polarization and myelination in myelinating glia. ISRN Neurol 2012:769412. doi: 10.5402/2012/769412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Snaidero N, Möbius W, Czopka T, Hekking LHP, Mathisen C, Verkleij D, Goebbels S, Edgar J, Merkler D, Lyons DA, Nave K-A, Simons M. 2014. Myelin membrane wrapping of CNS axons by PI(3,4,5)P3-dependent polarized growth at the inner tongue. Cell 156:277–290. doi: 10.1016/j.cell.2013.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwob VS, Clark HB, Agrawal D, Agrawal HC. 1985. Electron microscopic immunocytochemical localization of myelin proteolipid protein and myelin basic protein to oligodendrocytes in rat brain during myelination. J Neurochem 45:559–571. doi: 10.1111/j.1471-4159.1985.tb04024.x. [DOI] [PubMed] [Google Scholar]
- 10.Gow A, Friedrich VL Jr, Lazzarini RA. 1994. Intracellular transport and sorting of the oligodendrocyte transmembrane proteolipid protein. J Neurosci Res 37:563–573. doi: 10.1002/jnr.490370503. [DOI] [PubMed] [Google Scholar]
- 11.Simons M, Krämer EM, Thiele C, Stoffel W, Trotter J. 2000. Assembly of myelin by association of proteolipid protein with cholesterol- and galactosylceramide-rich membrane domains. J Cell Biol 151:143–154. doi: 10.1083/jcb.151.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krämer EM, Schardt A, Nave KA. 2001. Membrane traffic in myelinating oligodendrocytes. Microsc Res Tech 52:656–671. doi: 10.1002/jemt.1050. [DOI] [PubMed] [Google Scholar]
- 13.Ainger K, Avossa D, Morgan F, Hill SJ, Barry C, Barbarese E, Carson JH. 1993. Transport and localization of exogenous myelin basic protein mRNA microinjected into oligodendrocytes. J Cell Biol 123:431–441. doi: 10.1083/jcb.123.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Müller C, Bauer NM, Schäfer I, White R. 2013. Making myelin basic protein—from mRNA transport to localized translation. Front Cell Neurosci 7:169. doi: 10.3389/fncel.2013.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laursen LS, Chan CW, ffrench-Constant C. 2011. Translation of myelin basic protein mRNA in oligodendrocytes is regulated by integrin activation and hnRNP-K. J Cell Biol 192:797–811. doi: 10.1083/jcb.201007014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.White R, Gonsior C, Krämer-Albers E-M, Stöhr N, Hüttelmaier S, Trotter J. 2008. Activation of oligodendroglial Fyn kinase enhances translation of mRNAs transported in hnRNP A2-dependent RNA granules. J Cell Biol 181:579–586. doi: 10.1083/jcb.200706164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trajkovic K, Dhaunchak AS, Goncalves JT, Wenzel D, Schneider A, Bunt G, Nave K-A, Simons M. 2006. Neuron to glia signaling triggers myelin membrane exocytosis from endosomal storage sites. J Cell Biol 172:937–948. doi: 10.1083/jcb.200509022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marbois BN, Faull KF, Fluharty AL, Raval-Fernandes S, Rome LH. 2000. Analysis of sulfatide from rat cerebellum and multiple sclerosis white matter by negative ion electrospray mass spectrometry. Biochim Biophys Acta 1484:59–70. doi: 10.1016/S1388-1981(99)00201-2. [DOI] [PubMed] [Google Scholar]
- 19.Harauz G, Boggs JM. 2013. Myelin management by the 18.5-kDa and 21.5-kDa classic myelin basic protein isoforms. J Neurochem. 125:334–361. doi: 10.1111/jnc.12195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coman I, Aigrot MS, Seilhean D, Reynols R, Girault JA, Zalc B, Lubetzki C. 2006. Nodal, paranodal and juxtaparanodal axonal proteins during demyelination and remyelination in multiple sclerosis. Brain 129:3186–3195. doi: 10.1093/brain/awl144. [DOI] [PubMed] [Google Scholar]
- 21.Maier O, Baron W, Hoekstra D. 2007. Reduced raft-association of NF155 in active MS lesions is accompanied by the disruption of the paranodal junction. Glia 55:885–895. doi: 10.1002/glia.20510. [DOI] [PubMed] [Google Scholar]
- 22.Krämer-Albers E-M, Gehrig-Burger K, Thiele C, Trotter J, Nave K-A. 2006. Perturbed interactions of mutant proteolipid protein/DM20 with cholesterol and lipid rafts in oligodendroglia: implications for dysmyelination in spastic paraplegia. J Neurosci 26:11743–11752. doi: 10.1523/JNEUROSCI.3581-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winterstein C, Trotter J, Krämer-Albers E-M. 2008. Distinct endocytic recycling of myelin proteins promotes oligodendroglial membrane remodeling. J Cell Sci 121:834–842. doi: 10.1242/jcs.022731. [DOI] [PubMed] [Google Scholar]
- 24.Feldmann A, Amphornrat J, Schönherr M, Winterstein C, Möbius W, Ruhwedel T, Danglot L, Nave K-A, Galli T, Bruns D, Trotter J, Krämer-Albers E-M. 2011. Transport of the major myelin proteolipid protein is directed by VAMP3 and VAMP7. J Neurosci 31:5659–5672. doi: 10.1523/JNEUROSCI.6638-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kroepfl JF, Gardinier MV. 2001. Mutually exclusive apicobasolateral sorting of two oligodendroglial membrane proteins, proteolipid protein and myelin/oligodendrocyte glycoprotein, in Madin-Darby canine kidney cells. J Neurosci Res 66:1140–1148. doi: 10.1002/jnr.10035. [DOI] [PubMed] [Google Scholar]
- 26.Madison DL, Krueger WH, Cheng D, Trapp BD, Pfeiffer SE. 1999. SNARE complex proteins, including the cognate pair VAMP-2 and syntaxin-4, are expressed in cultured oligodendrocytes. J Neurochem 72:988–998. [DOI] [PubMed] [Google Scholar]
- 27.Feldmann A, Winterstein C, White R, Trotter J, Krämer-Albers E-M. 2009. Comprehensive analysis of expression, subcellular localization, and cognate pairing of SNARE proteins in oligodendrocytes. J Neurosci Res 87:1760–1772. doi: 10.1002/jnr.22020. [DOI] [PubMed] [Google Scholar]
- 28.Maier O, van der Heide T, van Dam A-M, Baron W, de Vries H, Hoekstra D. 2005. Alteration of the extracellular matrix interferes with raft association of neurofascin in oligodendrocytes. Potential significance for multiple sclerosis? Mol Cell Neurosci 28:390–401. doi: 10.1016/j.mcn.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 29.Bsibsi M, Nomden A, van Noort JM, Baron W. 2012. Toll-like receptors 2 and 3 agonists differentially affect oligodendrocyte survival, differentiation, and myelin membrane formation. J Neurosci Res 90:388–398. doi: 10.1002/jnr.22767. [DOI] [PubMed] [Google Scholar]
- 30.Richter-Landsberg C, Heinrich M. 1996. OLN-93: a new permanent oligodendroglia cell line derived from primary rat brain glial cultures. J Neurosci Res 45:161–173. [DOI] [PubMed] [Google Scholar]
- 31.Low SH, Chapin SJ, Weimbs T, Kömüves LG, Bennett MK, Mostov KE. 1996. Differential localization of syntaxin isoforms in polarized Madin-Darby canine kidney cells. Mol Biol Cell 7:2007–2018. doi: 10.1091/mbc.7.12.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maier O, van der Heide T, Johnson R, de Vries H, Baron W, Hoekstra D. 2006. The function of neurofascin155 in oligodendrocytes is regulated by metalloprotease-mediated cleavage and ectodomain shedding. Exp Cell Res 312:500–511. doi: 10.1016/j.yexcr.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 33.Weimbs T, Stoffel W. 1992. Proteolipid protein (PLP) of CNS myelin: positions of free, disulfide-bonded, and fatty acid thioester-linked cysteine residues and implications for the membrane topology of PLP. Biochemistry 31:12289–12296. doi: 10.1021/bi00164a002. [DOI] [PubMed] [Google Scholar]
- 34.Gow A, Gragerov A, Gard A, Colman DR, Lazzarini RA. 1997. Conservation of topology, but not conformation, of the proteolipid proteins of the myelin sheath. J Neurosci 17:181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greenfield EA, Reddy J, Lees A, Dyer CA, Koul O, Nguyen K, Bell S, Kassam N, Hinojoza J, Eaton MJ, Lees MB, Kuchroo VK, Sobel RA. 2006. Monoclonal antibodies to distinct regions of human myelin proteolipid protein simultaneously recognize central nervous system myelin and neurons of many vertebrate species. J Neurosci Res 83:415–431. doi: 10.1002/jnr.20748. [DOI] [PubMed] [Google Scholar]
- 36.Jung M, Sommer I, Schachner M, Nave KA. 1996. Monoclonal antibody O10 defines a conformationally sensitive cell-surface epitope of proteolipid protein (PLP): evidence that PLP misfolding underlies dysmyelination in mutant mice. J Neurosci 16:7920–7929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trifilieff E. 2005. Synthesis and secondary structure of loop 4 of myelin proteolipid protein: effect of a point mutation found in Pelizaeus-Merzbacher disease. J Pept Res 66:101–110. doi: 10.1111/j.1399-3011.2005.00278.x. [DOI] [PubMed] [Google Scholar]
- 38.Zeng Y, Cheng H, Jiang X, Han X. 2008. Endosomes and lysosomes play distinct roles in sulfatide-induced neuroblastoma apoptosis: potential mechanisms contributing to abnormal sulfatide metabolism in related neuronal diseases. Biochem J 410:81–92. doi: 10.1042/BJ20070976. [DOI] [PubMed] [Google Scholar]
- 39.Wainszelbaum MJ, Proctor BM, Pontow SE, Stahl PD, Barbieri MA. 2006. IL4/PGE2 induction of an enlarged early endosomal compartment in mouse macrophages is Rab5-dependent. Exp Cell Res 312:2238–2251. doi: 10.1016/j.yexcr.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 40.Bligh EG, Dye WJ. 1959. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 41.Sommer I, Schachner M. 1981. Monoclonal antibodies (O1 to O4) to oligodendrocyte cell surfaces: an immunocytological study in the central nervous system. Dev Biol 83:311–327. doi: 10.1016/0012-1606(81)90477-2. [DOI] [PubMed] [Google Scholar]
- 42.Slimane TA, Trugnan G, Van IJzendoorn SCD, Hoekstra D. 2003. Raft-mediated trafficking of apical resident proteins occurs in both direct and transcytotic pathways in polarized hepatic cells: role of distinct lipid microdomains. Mol Biol Cell 14:611–624. doi: 10.1091/mbc.E02-08-0528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Low SH, Chapin SJ, Wimmer C, Whiteheart SW, Kömüves LG, Mostov KE, Weimbs T. 1998. The SNARE machinery is involved in apical plasma membrane trafficking in MDCK cells. J Cell Biol 141:1503–1513. doi: 10.1083/jcb.141.7.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fujita H, Tuma PL, Finnegan CM, Locco L, Hubbard AL. 1998. Endogenous syntaxins 2, 3 and 4 exhibit distinct but overlapping patterns of expression at the hepatocyte plasma membrane. Biochem J 329(Pt 3):527–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Werner HB, Krämer-Albers E-M, Strenzke N, Saher G, Tenzer S, Ohno-Iwashita Y, De Monasterio-Schrader P, Möbius W, Moser T, Griffiths IR, Nave K-A. 2013. A critical role for the cholesterol-associated proteolipids PLP and M6B in myelination of the central nervous system. Glia 61:567–586. doi: 10.1002/glia.22456. [DOI] [PubMed] [Google Scholar]
- 46.Coskun U, Simons K. 2010. Membrane rafting: from apical sorting to phase segregation. FEBS Lett 584:1685–1693. doi: 10.1016/j.febslet.2009.12.043. [DOI] [PubMed] [Google Scholar]
- 47.Aït Slimane T, Hoekstra D. 2002. Sphingolipid trafficking and protein sorting in epithelial cells. FEBS Lett 529:54–59. doi: 10.1016/S0014-5793(02)03183-6. [DOI] [PubMed] [Google Scholar]
- 48.Pasquini JM, Guarna MM, Besio-Moreno MA, Iturregui MT, Oteiza PI, Soto EF. 1989. Inhibition of the synthesis of glycosphingolipids affects the translocation of proteolipid protein to the myelin membrane. J Neurosci Res 22:289–296. doi: 10.1002/jnr.490220309. [DOI] [PubMed] [Google Scholar]
- 49.Brown MC, Moreno MB, Bongarzone ER, Cohen PD, Soto EF, Pasquini JM. 1993. Vesicular transport of myelin proteolipid and cerebroside sulfates to the myelin membrane. J Neurosci Res 35:402–408. doi: 10.1002/jnr.490350407. [DOI] [PubMed] [Google Scholar]
- 50.Maier O, Hoekstra D, Baron W. 2008. Polarity development in oligodendrocytes: sorting and trafficking of myelin components. J Mol Neurosci 35:35–53. doi: 10.1007/s12031-007-9024-8. [DOI] [PubMed] [Google Scholar]
- 51.Bansal R, Pfeiffer SE. 1994. Inhibition of protein and lipid sulfation in oligodendrocytes blocks biological responses to FGF-2 and retards cytoarchitectural maturation, but not developmental lineage progression. Dev Biol 162:511–524. doi: 10.1006/dbio.1994.1105. [DOI] [PubMed] [Google Scholar]
- 52.van der Haar ME, Visser HW, de Vries H, Hoekstra D. 1998. Transport of proteolipid protein to the plasma membrane does not depend on glycosphingolipid cotransport in oligodendrocyte cultures. J Neurosci Res 51:371–381. doi:. [DOI] [PubMed] [Google Scholar]
- 53.Baron W, Bijlard M, Nomden A, de Jonge JC, Teunissen CE, Hoekstra D. 2014. Sulfatide-mediated control of extracellular matrix-dependent oligodendrocyte maturation. Glia 62:927–942. doi: 10.1002/glia.22650. [DOI] [PubMed] [Google Scholar]
- 54.Jung M, Krämer E, Grzenkowski M, Tang K, Blakemore W, Aguzzi A, Khazaie K, Chlichlia K, von Blankenfeld G, Kettenmann H, Trotter J. 1995. Lines of murine oligodendroglial precursor cells immortalized by an activated neu tyrosine kinase show distinct degrees of interaction with axons in vitro and in vivo. Eur J Neurosci 7:1245–1265. doi: 10.1111/j.1460-9568.1995.tb01115.x. [DOI] [PubMed] [Google Scholar]
- 55.Simons M, Kramer E-M, Macchi P, Rathke-Hartlieb S, Trotter J, Nave K-A, Schulz JB. 2002. Overexpression of the myelin proteolipid protein leads to accumulation of cholesterol and proteolipid protein in endosomes/lysosomes: implications for Pelizaeus-Merzbacher disease. J Cell Biol 157:327–336. doi: 10.1083/jcb.200110138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoekstra D, Tyteca D, van IJzendoorn SCD. 2004. The subapical compartment: a traffic center in membrane polarity development. J Cell Sci 117:2183–2192. doi: 10.1242/jcs.01217. [DOI] [PubMed] [Google Scholar]
- 57.Pernber Z, Richter K, Mansson J-E, Nygren H. 2007. Sulfatide with different fatty acids has unique distributions in cerebellum as imaged by time-of-flight secondary ion mass spectrometry (TOF-SIMS). Biochim Biophys Acta 1771:202–209. doi: 10.1016/j.bbalip.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 58.Hao C, Sun R, Zhang J, Chang Y, Niu C. 2009. Behavior of sulfatide/cholesterol mixed monolayers at the air/water interface. Colloids Surf B Biointerfaces 69:201–206. doi: 10.1016/j.colsurfb.2008.11.013. [DOI] [PubMed] [Google Scholar]
- 59.Watanabe R, Asakura K, Rodriguez M, Pagano RE. 1999. Internalization and sorting of plasma membrane sphingolipid analogues in differentiating oligodendrocytes. J Neurochem 73:1375–1383. [DOI] [PubMed] [Google Scholar]
- 60.Hirahara Y, Bansal R, Honke K, Ikenaka K, Wada Y. 2004. Sulfatide is a negative regulator of oligodendrocyte differentiation: development in sulfatide-null mice. Glia 45:269–277. doi: 10.1002/glia.10327. [DOI] [PubMed] [Google Scholar]
- 61.Bansal R, Winkler S, Bheddah S. 1999. Negative regulation of oligodendrocyte differentiation by galactosphingolipids. J Neurosci 19:7913–7924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Coetzee T, Fujita N, Dupree J, Shi R, Blight A, Suzuki K, Suzuki K, Popko B. 1996. Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability. Cell 86:209–219. doi: 10.1016/S0092-8674(00)80093-8. [DOI] [PubMed] [Google Scholar]
- 63.Fewou SN, Fernandes A, Stockdale K, Francone VP, Dupree JL, Rosenbluth J, Pfeiffer SE, Bansal R. 2010. Myelin protein composition is altered in mice lacking either sulfated or both sulfated and non-sulfated galactolipids. J Neurochem 112:599–610. doi: 10.1111/j.1471-4159.2009.06464.x. [DOI] [PubMed] [Google Scholar]
- 64.Colognato H, Yurchenco PD. 2000. Form and function: the laminin family of heterotrimers. Dev Dyn 218:213–234. doi:. [DOI] [PubMed] [Google Scholar]
- 65.Timpl R, Tisi D, Talts JF, Andac Z, Sasaki T, Hohenester E. 2000. Structure and function of laminin LG modules. Matrix Biol 19:309–317. doi: 10.1016/S0945-053X(00)00072-X. [DOI] [PubMed] [Google Scholar]
- 66.Colognato H, Baron W, Avellana-Adalid V, Relvas JB, Baron-Van Evercooren A, Georges-Labouesse E, ffrench Constant-C. 2002. CNS integrins switch growth factor signalling to promote target-dependent survival. Nat Cell Biol 4:833–841. doi: 10.1038/ncb865. [DOI] [PubMed] [Google Scholar]
- 67.Colognato H, Tzvetanova ID. 2011. Glia unglued: how signals from the extracellular matrix regulate the development of myelinating glia. Dev Neurobiol 71:924–955. doi: 10.1002/dneu.20966. [DOI] [PMC free article] [PubMed] [Google Scholar]