

Abstract
Gene expression frequently requires chromatin-remodeling complexes, and it is assumed that these complexes have common gene targets across cell types. Contrary to this belief, we show by genome-wide expression profiling that Bptf, an essential and unique subunit of the nucleosome-remodeling factor (NURF), predominantly regulates the expression of a unique set of genes between diverse cell types. Coincident with its functions in gene expression, we observed that Bptf is also important for regulating nucleosome occupancy at nucleosome-free regions (NFRs), many of which are located at sites occupied by the multivalent factors Ctcf and cohesin. NURF function at Ctcf binding sites could be direct, because Bptf occupies Ctcf binding sites in vivo and has physical interactions with CTCF and the cohesin subunit SA2. Assays of several Ctcf binding sites using reporter assays showed that their regulatory activity requires Bptf in two different cell types. Focused studies at H2-K1 showed that Bptf regulates the ability of Klf4 to bind near an upstream Ctcf site, possibly influencing gene expression. In combination, these studies demonstrate that gene expression as regulated by NURF occurs partly through physical and functional interactions with the ubiquitous and multivalent factors Ctcf and cohesin.
INTRODUCTION
Cell differentiation requires the establishment and maintenance of specific gene expression profiles. Central to this process are transcription factors and their associated coregulatory complexes. The functions of these complexes are diverse and include ATP-dependent chromatin remodeling. ATP-dependent chromatin-remodeling complexes are frequently multiprotein enzymes which alter the position, composition, or presence of nucleosomes as a means to regulate chromatin structure. Changes in chromatin structure regulate the ability of trans-acting factors to access the underlying DNA, which in turn affects DNA-dependent processes like gene expression (1). Because of their importance for DNA-dependent processes, many subunits of chromatin-remodeling complexes are essential for mammalian development (2).
One chromatin-remodeling complex essential for mammalian development is the nucleosome-remodeling factor (NURF). In mammals, NURF is a three-subunit complex containing bromodomain PHD finger-containing transcription factor (BPTF), the ATPase SNF2L, and the Trp-Asp (WD) repeat protein pRBAP46/48 (3, 4). NURF slides nucleosomes in cis without eviction or the exchange of histones from the nucleosome. NURF is proposed to remodel chromatin through physical interactions with both cell-type-restricted (PR and Smad) and ubiquitous (AP-1, Srf, and USF1) transcription factors and ubiquitously utilized histone modifications (H3K4me2/3) (5). These observations are not exclusive to NURF; several studies have documented both cell-type-specific and ubiquitous functions for many chromatin-remodeling complexes through interactions with ubiquitous factors like the centrosome, RNA polymerases, nuclear lamins, and a variety of cell-type-specific transcription factors (5–9). The relative importance of cell-type-restricted and ubiquitous activities for these complexes during gene expression is unknown.
CCCTC-binding factor (Ctcf) is a ubiquitous multivalent chromatin regulator with essential functions at diverse regulatory elements, including promoters, enhancers, silencers, barrier, and enhancer blocking elements (10). In mammals, Ctcf frequently associates with cohesin, a ring-like complex that functions to facilitate distal interactions in chromatin, and regulates gene expression (11). The Ctcf/cohesin interaction is not obligate for either protein's function in vivo, as both Ctcf and cohesin can operate independently to regulate chromatin structure and gene expression (12–14). Ctcf correlates with a unique chromatin structure, which includes a nucleosome-free region (NFR) located at its binding site flanked by variant H2A.Z/H3.3 nucleosomes with high turnover (15). Once established, this specialized chromatin regulates access to DNA sequences by trans-acting factors which contribute to the diverse regulatory functions of Ctcf (16). Which chromatin-remodeling complexes establish the unique chromatin structures near Ctcf is not known.
To better understand in vivo functions for chromatin-remodeling complexes, we performed loss-of-function studies using a conditional knockout (KO) allele of the gene encoding Bptf, the largest and essential subunit of the NURF complex (17), in three primary cell types. From these studies, we show that Bptf regulates a distinct set of genes in each cell type studied. Subsequent studies uncovered functional and physical connections between NURF chromatin remodeling and the ubiquitous and multivalent regulators Ctcf and cohesin during gene expression. Detailed studies near H2-K1 showed that Bptf regulates the binding of a cell-type-specific transcription factor to sequences adjacent to Ctcf with consequences for cell-type-specific gene expression. In total, our results present a model of how chromatin-remodeling complexes can functionally interact with ubiquitous regulators of chromatin structure, like Ctcf and cohesin, to regulate gene expression.
MATERIALS AND METHODS
Animal husbandry and cell culture.
Abb/B2M double-knockout mice were purchased from Taconic Labs (stock number 4080). B6.129S1-Bptftm1.1Cwu/J, B6.Cg-Tg(tetO-cre)1Jaw/J, B6.Cg-Gt(ROSA)26Sortm1(rtTA*M2)Jae/J, and B6.Cg-Tg(Lck-cre)548Jxm/J mice were purchased from Jackson Laboratory (stock numbers 009367, 006234, 006965, and 003802, respectively). All mice were bred and maintained in a specific-pathogen-free (SPF) barrier facility located at Virginia Commonwealth University. All animal breeding and experiments were approved by the Virginia Commonwealth University Institutional Animal Care and Use Committee (IACUC).
Embryonic stem cell (ESC) lines, which were described previously (17), were maintained on gelatinized plates and grown in Dulbecco's modified Eagle medium (DMEM) as a base medium supplemented with 15% ESC-grade fetal bovine serum (FBS) (Life Technologies), 1× penicillin-streptomycin, 2 mM glutamine, 1× nonessential amino acids (NEAA), 0.1 mM 2-mercaptoethanol and 1,000 U/ml leukemia inhibitory factor (LIF) (ESGRO; Millipore). CD8+ CD4+ double-positive (DP) thymocytes were purified from the total thymocytes of Bptf KO (Lck-Cre Bptfflox/flox Abb/B2M−/−) and control (Bptfflox/flox Abb/B2M−/−) mice by anti-CD8 panning as described previously (18). DP thymocyte populations routinely exceeded 95% purity, as measured by flow cytometry (data not shown). Purified DP thymocytes were maintained in bacteriological-grade plasticware and grown in RPMI base medium supplemented with 10% FBS (Life Technologies), 1× penicillin-streptomycin, 2 mM glutamine, 1 mM sodium pyruvate, 1× NEAA, and 0.1 mM 2-mercaptoethanol. Mouse embryonic fibroblasts (MEFs) were prepared from Bptf KO (TetO-Cre rtTA Bptfflox/flox) and control (rtTA Bptfflox/flox) mice using standard methods. MEFs were grown in DMEM base medium supplemented with 10% FBS (Life Technologies), 1× penicillin-streptomycin, 2 mM glutamine, and 1× NEAA. Bptf KO was achieved by exposing MEFs to 10 μg/ml doxycycline for 2 days. This exposure resulted in complete deletion of the Bptfflox/flox alleles as measured by PCR (data not shown). MEFs were then grown in the absence of doxycycline for 8 additional days to deplete the Bptf protein, with passaging of the cells when necessary to maintain subconfluent growth. Bptf-depleted MEFs and corresponding controls were freshly prepared for all experiments. Forty-eight hours prior to being harvested, the cells were split onto separate plates for parallel RNA and nucleosome preparations. Total RNA was obtained from cells using Tri-Reagent (Sigma) according to the manufacturer's suggested protocol.
Short-hairpin RNA (shRNA) sequences were introduced into ESCs using the retrovirus Moloney murine leukemia virus (MMLV). Sequences were cloned into the pSIREN-RetroQ U6 expression construct and confirmed by sequencing. Viral particles were created by transient transfection of 293T cells using a three-plasmid system. Stable integration of retroviral sequences were selected with 5 μg/ml puromycin over 3 weeks. Successful Ctcf knockdown (KD) was confirmed by Western blotting. shRNA sequences are available in Table S5 in the supplemental material.
Gene expression.
Total RNA was prepared for microarray analysis using the Illumina gene expression kit according to the manufacturer's instructions. Subsequent hybridization to mouse WG-6v2 expression bead chips (Illumina), washing, scanning, and data analysis were performed using standard Illumina procedures.
RT-PCR analysis was performed by first reverse transcribing 5 μg of total RNA using Superscript II (Life Technologies) according to the manufacturer's instructions. All quantitative PCR (qPCR) measurements were performed using Absolute SYBR green ROX master mix (Fisher Scientific) on a 7900 HT fast real-time qPCR system (Applied Biosystems). Quantification of gene expression was determined using the delta-delta relative quantization method using GAPDH as a normalization control. Primer sequences are listed in Table S5 in the supplemental material.
Chromatin immunoprecipitation.
Fixed cells were thawed and suspended in lysis buffer (50 mM Tris [pH 8.0], 1% SDS, 10 mM EDTA) at a concentration of 10 × 106 cells/ml. The DNA was sheared to a peak distribution of ∼500 bp using a Bioruptor sonicator three times for 15 min of alternating 30 s on, 30 s off, on ice. Sonicated samples were centrifuged at high speed, and 100 to 200 μl of the supernatant was removed and diluted 1:10 in dilution buffer (16.7 mM Tris-HCl [pH 8.1], 167 mM NaCl, 1.1% Triton X-100, 1.2 mM EDTA). Twenty percent of the input sample was saved for quantification of immunoprecipitation efficiency. Protein G Dynal beads (Life Technologies) prebound with antibodies (10 μl Dynal beads with 5 μg antibodies per pulldown) were added to each immunoprecipitation mixture. The antibodies used for chromatin immunoprecipitation (ChIP) in this study included pan-histone H3 (Abcam), Ctcf (Cell Signaling Technologies), H2A.Z (Millipore), Smc1a (Abcam), Klf4 (Santa Cruz Biotech), and Bptf (Millipore). The immunoprecipitations were performed overnight at 4°C with mixing. Following mixing, the immunoprecipitation mixtures were concentrated by magnetic separation, washed sequentially with low-salt buffer (20 mM Tris [pH 8.0], 150 mM NaCl, 1% Triton X-100, and 2 mM EDTA), high-salt buffer (20 mM Tris [pH 8.0], 500 mM NaCl, 1% Triton X-100, and 2 mM EDTA), and LiCl buffer (10 mM Tris [pH 8.0], 0.25 M LiCl, 1% NP-40, 1% deoxycholic acid, and 1 mM EDTA) and then twice with TE (10 mM Tris [pH 8.0], 1 mM EDTA), for 5 min each time on ice. Bound DNA was eluted with two successive 250-μl washes with elution buffer (0.1 M NaHCO3, and 1% SDS) at room temperature. Cross-links were reversed (including those of the input) by adding 20 μl of 5 M NaCl and incubating overnight at 65°C. The proteins were treated with protease K for 4 h, extracted with phenol-chloroform–chloroform extractions, and then precipitated with ethanol. Precipitated DNA was dissolved in water, treated with RNase A, and subsequently used for qPCR. Primer sequenced used for ChIP are listed in Table S5 in the supplemental material.
Nucleosome occupancy measurements.
The cells used for mononucleosome preparations were fixed for 15 min at room temperature in freshly prepared 1% paraformaldehyde neutralized with phosphate-buffered saline (PBS). The fixed cells were washed three times with PBS and collected by scraping (ESCs or MEFs) or low-speed centrifugation (DP thymocytes). Fixed cells were frozen in aliquots of 30 × 106 at −80°C. Cells were thawed at a concentration of 60 × 106/ml in nucleus preparation buffer (10 mM Tris [pH 7.4], 10 mM NaCl, 0.5% NP-40, 3.5 mM MgCl2, 0.5 mM spermidine, and 0.15 mM spermine) on ice for 5 min followed by a 5-min spin in a refrigerated centrifuge at 1,000 × g. The washing step in nucleus preparation buffer was repeated 3 times. After the last wash, nuclei were resuspended at a concentration of 60 × 106 in micrococcal nuclease (MNase) digestion buffer (50 mM Tris [pH 8.0], 100 mM NaCl, 3 mM MgCl2, 2 mM CaCl2, 0.5 mM spermidine, and 0.15 mM spermine). The reaction mixture was prewarmed to 37°C prior to the addition of 1,500 U MNase/ml (Worthington), a concentration determined experimentally to completely digest fixed chromatin to mononucleosomes in 30 min. The reactions were stopped by adding 40 μl 0.5 M EDTA, 100 μl 10% SDS, and 100 μl 5 M NaCl per ml of reaction volume, and the products were incubated at 65°C overnight. The DNA was subsequently purified by phenol-chloroform-isoamyl alcohol, followed by a chloroform extraction and ethanol precipitation. The above procedure was performed in parallel with an identical aliquot of cells but without the addition of the MNase to provide a matching total-genomic-DNA control. Total genomic DNA was then sheared using a bath sonicator (Bioruptor) with conditions predetermined to yield a DNA fragment with a peak distribution of 100 to 200 bp.
Nucleosomal or sheared total control DNA was labeled for microarray analysis using a BioPrime array CGH genomic labeling system (Life Technologies) according to the manufacturer's instructions. Labeled DNA was then hybridized to custom high-density oligonucleotide arrays (Affymetrix), which were washed and scanned as recommended by the manufacturer. The custom high-density oligonucleotide arrays used in these experiments contained 60-bp probes tiled at a 10-bp resolution on both the Watson and Crick strands. The data were processed using an Affymetrix work station and associated software using default settings. Flagged data (saturated or nonuniform spots) and probes with low signal strength for the genomic DNA control were removed from the data set. Probes which were printed in duplicate on the array were averaged. Watson and Crick strand probes were subsequently averaged. Gaps from 15 to 25 bp were estimated using linear interpolation. Subsequent raw analysis included a log2 ratio of nuclear to genomic DNA signal intensities followed by smoothing using a sliding window program over a 50-bp window at 10-bp intervals. The smoothed data were Z-score normalized to allow comparison between biological replicates.
To identify probes with significant nucleosome occupancy changes in KO samples compared to controls, the probe nuclear/genomic ratio distribution was first determined among the three control samples. Ninety-five percent confidence intervals were then determined from the pairwise control-control comparisons. Probes with changes in nuclear/genomic ratio (control − KO) greater than the 95% confidence interval, as determined from control samples, were designated as having significant changes. A two-tailed t test was then preformed using a P value cutoff of <0.05 to identify probes with significant changes between 3 biological replicates.
Integrating reporter assays.
pNI-MCS was a kind gift from Rainer Renkawitz, Justus-Liebig-Universitaet, Germany. pNI-P-MCS was created by removing the human γ-globin promoter from pNI-MCS and replacing it with a multicloning site. Individual DNA fragments were amplified from mouse genomic DNA using Phusion polymerase (NEB) and subsequently cloned into either pNI-MCS or pNI-P-MCS. The resulting plasmids, and a pNI-MCS vector-only control, were linearized and then transfected into 1 × 106 to 2 × 106 ESCs or 8.0 × 104 NIH 3T3 fibroblasts using Lipofectamine 2000 (Life Technologies). Selection for neomycin-resistant ESC colonies started 24 to 48 h after transfection with 300 μg/ml Geneticin (Life Technologies). Selection lasted for 2 to 3 weeks depending on the cell type. Following colony formation, the colonies were stained with 0.1% methylene blue in 50% methanol, destained with water, and counted. Relative activity of DNA fragments is expressed as a ratio of the number of colonies obtained for the vector with the DNA fragment to the number obtained with pNI-MCS vector-only control. DNA fragments cloned into pNI-P-MCS were further normalized to the number of colonies obtained from the control cells.
Immunoprecipitations and glutathione S-transferase (GST) pulldowns.
ESCs were removed from the plate surface by scraping and frozen in aliquots of 50 × 106 cells. Then, 50 × 106 to 100 × 106 cells were suspended in 500 μl MNase digestion buffer (25 mM HEPES-KOH [pH 7.6], 100 mM NaCl, 5 mM MgCl2, 3 mM CaCl2, 10% glycerol, 0.2% NP-40, 2 μM pepstatin, 100 μM leupeptin, 5 μM phosphoramidon, 10 μM MG132, 1× phosphatase inhibitor cocktail [Sigma], and 1× EDTA-free protease inhibitor cocktail [Roche]). MNase (Worthington) was added to a concentration of 1,500 units/ml, and chromatin was digested on ice for 2 h. These conditions were sufficient to result in complete digestion of chromatin to mononucleosomes (data not shown). NaCl was added to a final concentration of 300 mM, the cells were lysed with 5 strokes from a Dounce homogenizer, ethidium bromide was added to 300 μg/ml, and then the cell extract was centrifuged at 10,000 × g at 4 C for 15 min to remove insoluble material. The soluble fraction was removed, and 50 to 100 μl was used for each immunoprecipitation reaction, with a portion saved as an input control. Protein G Dynal beads (Life Technologies) prebound with antibodies (10 μl Dynal beads with 5 μg antibodies per immunoprecipitation) were added to each pulldown. Antibodies used for in vivo pulldowns in this study were specific for Ctcf (Cell Signaling Technologies), Oct1 (Santa Cruz Biotech), SP1 (Santa Cruz Biotech), Nfatc1 (Santa Cruz Biotech), Smc1a (Abcam), Smc3 (Abcam), RAD21 (Abcam), SA2 (Abcam), and Bptf (Millipore). Antibody binding was performed overnight at 4°C with mixing. Following antibody binding, beads were washed 3 times for 5 min on ice with 1 ml of pulldown buffer (25 mM HEPES-KOH [pH 7.6], 300 mM NaCl, 5 mM MgCl2, 3 mM CaCl2, 10% glycerol, 0.2% NP-40, and 1× EDTA-free protease inhibitor cocktail [Roche]). Bound proteins were eluted with 50 μl of 1% SDS at 55 C for 30 min. Following elution, proteins were resolved by PAGE using standard methods. The proteins were transferred to polyvinylidene difluoride (PVDF) using standard transfer buffer (25 mM Tris, 190 mM glycine, and 20% methanol), except for Bptf, which was transferred using N-cyclohexyl-3-aminopropanesulfonic acid (CAPS) transfer buffer (10 mM CAPS-NaOH [pH 10.5], 15% methanol, and 2.5 mM dithiothreitol [DTT]). Incubations of the primary (4°C overnight) and secondary antibodies (2 h at room temperature) were performed in PBST (PBS, 0.1% Tween 20) with 5% NFDM. Enhanced chemiluminescence (ECL) with the Femto substrate (Fisher) was used to detect the signal of the horseradish peroxidase (HRP)-conjugated secondary antibody.
GST pulldowns were performed using purified proteins. Human Ctcf, Smc1a, and Smc3 cDNAs were cloned using standard methods from an MCF7 cDNA library into pGEX 4T-1 (GE Life Sciences). GST-RAD21 and SA2 were kind gifts from Chang-Woo Lee, Sungkyunkwan University, South Korea. The N-terminal, central (zinc finger), and C-terminal ends of Ctcf and SA2 were amplified from the full-length cDNA using Phusion high-fidelity polymerase (NEB) and subcloned into pGEX4T-1. GST fusion proteins were expressed in BL21(DE3) RIPL (Agilent) using 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside) at 37°C for 3 h. The expressed proteins were extracted from bacteria by sonication on ice in 1/10 culture volume of extraction buffer (25 mM triethanolamine, 1.5% l-laurylsarcosine, and 1 mM EDTA). Soluble extracted proteins were purified by glutathione-Sepharose affinity chromatography (GE Life Sciences). Bound GST fusion proteins were washed three times in 20 volumes of extraction buffer and then once in 20 volumes of phosphate-buffered saline. GST fusion protein-coupled resin was stored at −20°C after glycerol was added to 50%. Resin-bound full-length fusion protein was quantified by SDS-PAGE using bovine serum albumin (BSA) standards. Approximately 1 μg of resin-bound fusion protein (from 10 to 200 μl of 50% resin slurry) was used for each pulldown reaction. Resin was first washed and resuspended in 200 μl of binding buffer (25 mM HEPES-KOH [pH 7.6], 100 mM NaCl, 0.01% NP-40, 0.5 mM MgCl2, 10% glycerol, and 1× Roche EDTA-free protease inhibitor cocktail). Approximately 50 ng of recombinant human NURF complex was added to each pulldown reaction mixture, and reaction mixtures were incubated on ice for 1 h with occasional mixing. The resin was washed with three 1-ml volumes of binding buffer and then eluted with SDS sample buffer by heating at 50°C for 30 min. Eluted proteins were then resolved by 4% SDS-PAGE. Transfer of FLAG-BPTF onto PVDF was performed using CAPS transfer buffer (10 mM CAPS [pH 10.0], 15% methanol, 2.5 mM DTT) using a submerged transfer system (20 V, 20-mA limits, 17 h). Resin-bound human NURF complex was detected by Western blotting for the FLAG-BPTF subunit using FLAG M2 antibody (Sigma) as described above.
Statistical methods.
A two-tailed Student's t test was used to determine the significance of ChIP, gene expression, and integrating reporter assay results. McNemar's test was used to determine the significance of the differences in Bptf-dependent genes between cell types. Hypergeometric testing was used to determine the significance of overlap in nucleosome occupancy experiments. Significance of cooccupancy between Bptf-dependent changes in nucleosome occupancy and ChIP-Seq peaks was determined by Fisher's exact test followed by a Bonferroni testing correction.
Gene expression data accession numbers.
The gene expression data obtained here are available in the GEO database under accession number GSE48123, and data for probes and the custom array are available under accession number GSE47416.
RESULTS
Bptf is a regulator of gene expression and nucleosome occupancy in diverse cell types.
Bptf is the largest and essential subunit of the NURF chromatin-remodeling complex (3, 4). We created a conditional loss-of-function allele for the NURF complex in the mouse by flanking the second exon of the Bptf gene with loxP sites (17). Using this floxed allele, we created Bptf KO ESCs (embryonic stem cells), MEFs (mouse embryonic fibroblasts) and DP (double-positive) thymocytes by transient Cre expression. We chose these cell types because they can be isolated as >95% pure populations and can have the Bptf gene conditionally deleted by Cre-loxP (Fig. 1A). Western blot analysis shows that DP thymocytes have higher levels of the Bptf protein than ESCs or MEFs (Fig. 1B). Bptf-dependent changes in gene expression were subsequently determined genome-wide by microarray analysis in MEFs, as described previously for ESCs and DP thymocytes (see Tables S1 and S2 in the supplemental material) (17, 18). While the genes identified by this approach require Bptf for normal expression, the changes in gene expression could be the result of either direct or indirect effects of Bptf KO. A Venn diagram analysis of these data sets shows that Bptf regulates the expression of a largely nonoverlapping set of genes between cell types (Fig. 1C). Analysis of previously published RNA sequencing (RNA-Seq) data sets shows that Bptf regulates the expression of genes which change >5-fold in transcript abundance between cell types (Fig. 1D) (19). This is in contrast to genes with similar levels of expression between cell types (>0.95- to <1.05-fold), which are more often Bptf independent (Fig. 1E). This analysis also observed that Bptf-dependent genes have a wide distribution of expression, including those with high, moderate, and low expression levels (Fig. 1F). These results in combination show that Bptf regulates a distinct set of genes in each cell type and that many Bptf-dependent genes are differentially expressed between cell types.
FIG 1.

Bptf regulates a largely nonoverlapping set of genes between diverse cell types. (A) Efficiency of Bptf depletion was confirmed by Bptf Western blot analysis of control and Bptf KO total cell extracts derived from ESCs, MEFs, and DP thymocytes using Ponceau S as a loading control. (B) Bptf expression was determined by Bptf Western blot analysis of total cell extracts derived from control ESCs, MEFs, and DP thymocytes using Ponceau S as a loading control. (C) Venn diagram analysis of Bptf-dependent genes identified from Bptf KO ESCs, MEFs, and DP thymocytes (P values for differences using McNemar's test: for DP+MEF, <1.0E−6; for ESC+MEF, <1.0E−6; for ESC+DP, <1.0E−6). (D) Genes which change in expression >5-fold between cell types (either increase or decrease from one cell type to the other) were determined from quantile normalized RPKM values from RNA-Seq data sets (19). The genes were further divided into those which are Bptf dependent in either of the two cell types or Bptf independent, and the data are presented as fractions of total genes. Significance of enrichment of genes which are Bptf dependent relative to those which are Bptf independent was determined by Fisher's exact test (P values: for DP+MEF = 5.7E−13; ESC+MEF, 7.2E−37; ESC+DP, 7.8E−14). (E) Same data analysis as in panel D except that genes which change in expression >0.95- to <1.05-fold between cell types were analyzed (P values by Fisher's exact test: DP+MEF, 9.5E−4; ESC+MEF, 6.1E−4; ESC+DP, 3.8E−3) (19). (F) Transcript abundance of Bptf-dependent genes from ESCs, MEFs, and DP thymocytes was determined using quantile-normalized RPKM values from previously published RNA-Seq data sets (19). Transcript abundance for each gene was ranked as a percentage of the most abundant transcript from each cell type and binned into 5 groups (top 25% being most abundant) according to decreasing transcript abundance.
To explore how Bptf could regulate gene expression, we interrogated for changes in nucleosome occupancy between Bptf KO and control cells using an array of 60-bp oligonucleotides tiled at a 10-bp resolution across 3.3 Mbp of the mouse genome. We chose this approach because NURF is a well-documented nucleosome remodeler in several model organisms (5). For these studies, we digested chromatin with MNase to mononucleosomes, which has been shown by others to measure both nucleosome occupancy and nucleosome stability, each of which could be influenced by NURF (Fig. 2A) (20). As with our gene expression studies, any observed Bptf-dependent nucleosome occupancy changes could be either a direct or indirect effect of Bptf KO. The tiling arrays used for these experiments interrogated DNA sequences flanking 10 kb of 49 Bptf-dependent and 137 Bptf-independent genes, including a representative sample of genes expressed or silent in ESCs, MEFs, and DP thymocytes (see Table S3 in the supplemental material).
FIG 2.

Bptf regulates nucleosome occupancy at nucleosome-free regions (NFRs) in diverse cell types. (A) Cartoon showing work flow for the digestion, extraction, hybridization, and analysis and visualization of significant changes in nucleosome occupancy with Bptf KO in ESCs, MEFs, and DP thymocytes (see Materials and Methods for detailed procedures). (B) Venn diagram analysis of Bptf-dependent changes in nucleosome occupancy identified from ESCs, MEFs, and DP thymocytes (P values of overlap by Fisher's exact test: DP+MEF, 1.7E−13; ESC+MEF, 3.1E−24; ESC+DP, 8.9E−13). (C) Aggregate plots showing Bptf-dependent changes in nucleosome occupancy and their position at the NFR. Nucleosome occupancy is measured on the y axis as a Z-score-normalized log2 nucleosomal-DNA/genomic-DNA signal ratio from our tiling array (average from 3 biological replicates). Zero on the y axis represents the average nucleosome occupancy across the genome sampled by our tiling array. Individual traces were centered on the x axis at the probe with the greatest change in nucleosome occupancy with Bptf KO. Traces are aggregated based on an increase or decrease in nucleosome occupancy at the NFR with Bptf KO and by cell type. The number of traces used to make each aggregate is included inside each plot. (D) Bar graph showing average size of a Bptf-dependent change in the NFR from ESCs, MEFs, and DP thymocytes. The size of a change in NFR is measured by the number of consecutive tiled probes (60-bp probes tiled at 10-bp intervals) which significantly change in hybridization signal with Bptf KO. (E) Histogram plot showing the distance of a Bptf-dependent NFR to the closest transcription start site (TSS) or closest transcription termination site (TTS) from reference sequence (Ref Seq) genes. (F) Density of Bptf-dependent NFRs (NFRs per 100 kb) at the MHC and HOX genes (A, B, and C clusters) and all other genes analyzed. A higher density of Bptf-dependent NFRs is observed at the MHC and HOX genes than across the whole genome.
Venn diagram analysis comparing Bptf-dependent changes in nucleosome occupancy observed changes unique to each cell type but also those in common between cell types (Fig. 2B; also, see Table S4 in the supplemental material). An aggregation plot centered at the probe with the greatest Bptf-dependent change in nucleosome occupancy shows that it resides over an NFR in each of the cell types studied (Fig. 2C). The NFR is a conserved feature of regulatory elements in eukaryotes which is found at both promoters and distal regulatory elements (21). Further analysis shows that the average size of a Bptf-dependent change in nucleosome occupancy at the NFR is consistent between cell types at ∼80 to 100 bp (Fig. 2D). This size is measured by the number of consecutive probes at 10-bp intervals which change in nucleosome occupancy with Bptf KO. In each cell type there usually is an increase in nucleosome occupancy at the NFR with Bptf KO, suggesting that most often Bptf functions to keep the NFR depleted of nucleosomes (see numbers in the aggregation plots in Fig. 2C). Only a fraction (11 to 19%) of Bptf-dependent NFRs were found <1 kb from either a transcription start site (TSS) or a transcription termination site (TTS) (Fig. 2E). Interestingly, Bptf-dependent NFRs were overrepresented at the major histocompatibility complex (MHC) genes and HOX gene clusters, each of which contains many Bptf-dependent genes (Fig. 2F; also, see Table S1 in the supplemental material) (17, 18). These results suggest that Bptf has significance for maintaining unique chromatin structures at these regions, which are important for gene expression.
Bptf maintains the NFR located at Ctcf binding sites.
To identify factors which function with Bptf to regulate chromatin structure and possibly influence gene expression, we looked for overlap between Bptf-dependent NFRs and transcription factor occupancy. We began by comparing our data sets from ESCs to previously published ChIP-Seq data sets for a panel of 15 transcription factors important for ESC self-renewal and differentiation (22). From this comparison, we observed a highly significant overlap between Bptf-dependent NFRs and binding sites for the ubiquitous multivalent factor Ctcf (Fig. 3A). Using previously published independent ChIP-Seq data sets from ESCs, MEFs, and DPs, we confirmed a significant overlap between Bptf-dependent NFRs and Ctcf but also revealed overlap with subunits of the cohesin complex (Smc1a, Smc3, Rad21, SA1, and SA2) (Fig. 3B) (23–25). We did not observe as significant an overlap with the cohesin-associated and enhancer-binding factors Nipbl, Med1, and Med21, suggesting that the Bptf-dependent NFRs correlate with cohesin only when in context with Ctcf (Fig. 3B) (24). To confirm that the correlations are not due to changes in protein levels, we performed Western blotting and observed similar expression of Ctcf, Smc1a, Smc3, Rad21, and SA2 proteins between control and Bptf KO cells (Fig. 3C).
FIG 3.
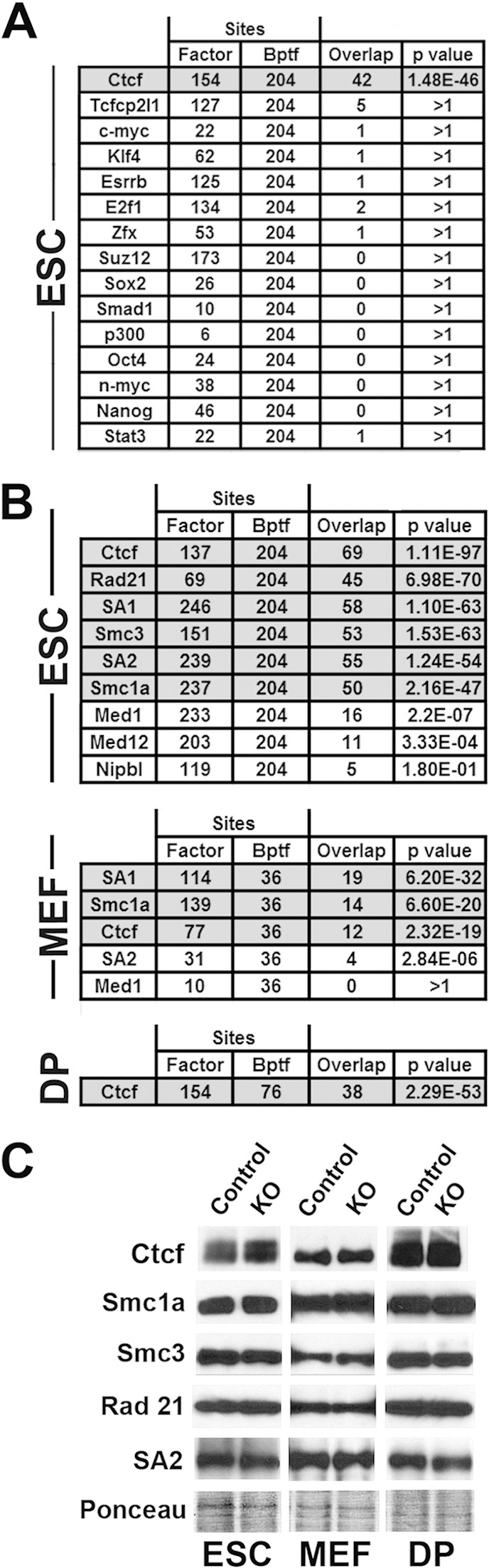
Bptf-dependent NFRs correlate with Ctcf and cohesin occupancy. (A) Summary of overlap of peaks of transcription factor occupancy identified by previously published ChIP-Seq data (22) and Bptf-dependent NFRs across 3.3 Mb of the genome sampled by our tiling arrays. The numbers of transcription factor peaks, the numbers of Bptf-dependent NFRs, and overlaps between the two are shown. Corrected Fisher's exact test P values for significance of overlap are listed in the “P value” column. (B) Summary of overlap of peaks of enrichment for Ctcf, subunits of cohesin, and other associated factors as determined by previously published ChIP-Seq data (23–25) and Bptf-dependent NFRs across 3.3 Mb of the genome sampled by our tiling arrays. Columns are as described for panel A. (C) Western blot analysis of Ctcf and cohesin subunit protein expression in Bptf KO and control ESCs, MEFs, and DP thymocytes using Ponceau S as a loading control.
Ctcf localization in chromatin usually correlates with an NFR (15). To study the effects of Bptf KO on NFRs located at Ctcf binding sites, we utilized aggregation plots of nucleosome occupancy measurements, as shown in Fig. 2C. For these plots we first preselected Ctcf-occupied sites located on our tiling arrays from each cell type. Nucleosome occupancy profiles were centered on the x axis at the Ctcf consensus sequence and then grouped by cell type and their requirement for Bptf to maintain the NFR prior to aggregation. This analysis revealed a more pronounced NFR at Ctcf binding sites with a Bptf-dependent NFR (Fig. 4A, top row) than at Ctcf binding sites with a Bptf-independent NFR (Fig. 4A, bottom row). As with the bulk of Bptf-dependent NFRs, Bptf KO usually increased nucleosome occupancy at Ctcf binding site NFRs (see the numbers in the aggregation plots in Fig. 4A). Together, these results demonstrate that Bptf is required for maintaining pronounced NFRs found at a subset of Ctcf binding sites in the mammalian genome.
FIG 4.

Bptf maintains pronounced NFRs at Ctcf binding sites with some cell type specificity. (A) Aggregate plot of nucleosome occupancy measurements across occupied Ctcf binding sites located on our tiling arrays with Bptf-dependent NFRs (top row) and Ctcf binding sites with Bptf-independent NFRs (bottom row) from ESCs, MEFs, and DP thymocytes. Nucleosome occupancy is displayed on the y axis, as described for Fig. 2C. Nucleosome occupancy maps are centered (0 bp) along the x axis at the consensus Ctcf DNA binding sequence. The numbers of Ctcf sites with an increase or decrease in nucleosome occupancy at the NFR with Bptf KO are given in the plots. (B) Venn diagram analysis showing overlap of Bptf-dependent NFRs between cell types. Compared by Venn diagram analysis are all Bptf-dependent NFRs, NFRs which are not occupied by Ctcf in either cell type (−Ctcf), and NFRs which are occupied by Ctcf in either of the cell types (+Ctcf). The significance of the overlap calculated by Fisher's exact test is shown below each of the Venn diagrams.
Previous analyses showed that Bptf regulates the NFR at a subset of Ctcf binding sites in each cell type (for ESCs, ∼27 to 50% of Ctcf sites depending on data set; for MEFs, ∼16% of Ctcf sites; for DP thymocytes, ∼25% of Ctcf sites) (Fig. 3A and B). From these analyses it is unclear if Bptf regulates the NFR of a Ctcf binding site occupied in ESCs, MEFs, or DP thymocytes or if Bptf regulates the same Ctcf binding sites occupied across several of these cell types. To clarify this, we determined the overlap of Ctcf sites which require Bptf to maintain the NFR between each of the cell types. As controls, we performed a similar comparison for the entire Bptf-dependent NFR data sets and those which are not occupied by Ctcf (Fig. 4B). From this, we observe that for two of the comparisons (ESCs versus MEFs and MEFs versus DP thymocytes), Ctcf binding sites with Bptf-dependent NFRs were less frequently found in common between the cell types than all Bptf-dependent NFRs. Conversely, we observed a similar overlap for Ctcf binding sites with Bptf-dependent NFRs and all Bptf-dependent NFRs in the ESC-versus-DP thymocyte comparison. To various degrees, we observed some overlap of Ctcf sites with Bptf-dependent NFRs between the cell types, but when Bptf maintained the NFR of a Ctcf binding site, it usually did so in only one of the two cell types.
NURF directly interacts with Ctcf and the cohesin subunit SA2.
To look for physical interactions between Bptf and Ctcf, we performed coimmunoprecipitation (co-IP) experiments from total ESC extracts. From these experiments, we observed selective co-IP of Bptf with antibodies to Ctcf but not with antibodies to Oct1, Sp1, and Nfatc1 (Fig. 5A). Similar experiments using antibodies to Ctcf and the cohesin subunits Smc1a, Smc3, Rad21, and SA2 confirmed co-IP of Bptf with antibodies to Ctcf but also revealed interactions with SA2 (Fig. 5B). Reciprocal pulldown experiments using an antibody to Bptf revealed co-IP of both Ctcf and SA2, providing further evidence for their interaction in vivo (Fig. 5C). Previous reports document physical interactions between Ctcf and SA2, suggesting that one of the observed interactions to Bptf could be indirect (26). To assay for direct interactions between Ctcf, SA2, and NURF, we performed in vitro GST pulldown assays. From these experiments we observe pulldown of a recombinant human NURF complex with human GST-CTCF and GST-SA2 (Fig. 5D). A domain analysis discovered interactions between NURF, the zinc finger DNA binding domain of CTCF, and the N-terminal end of SA2 (Fig. 5E and F). Together, these results suggest that the NURF chromatin-remodeling complex directly interacts with Ctcf and SA2.
FIG 5.
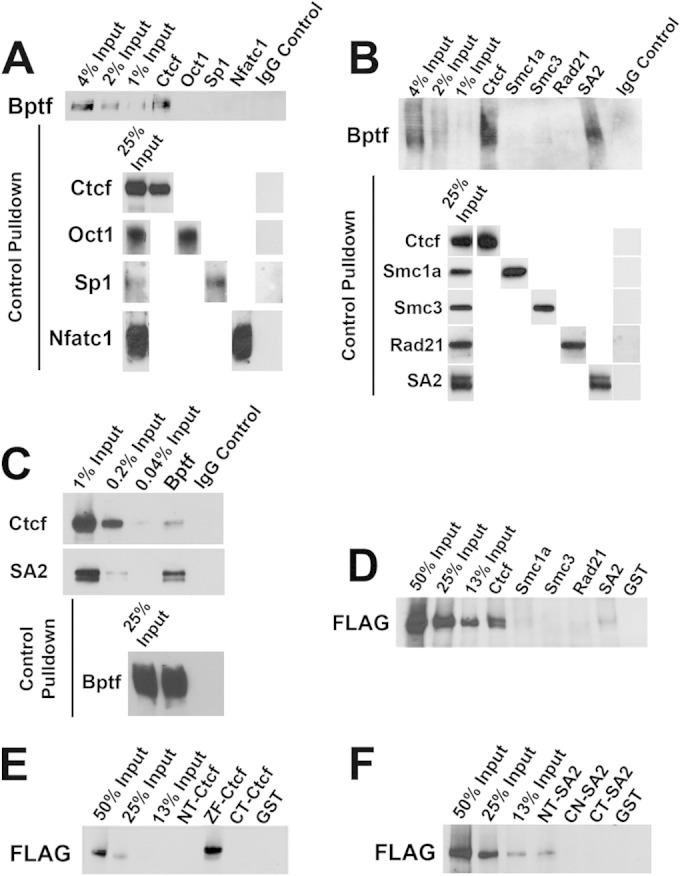
NURF directly interacts with Ctcf and the cohesin subunit SA2. (A) Results of a representative in vivo co-IP experiment from total ESC extracts using antibodies to Ctcf, Oct1, Sp1, and Nfatc1 and a control IgG. Bptf co-IP with Ctcf pulldown was detected by Western blotting with an antibody to Bptf. Ctcf, Oct1, Sp1, and Nfatc1 IP was confirmed by Western blotting with the antibodies used for pulldown. (B) Results of a representative in vivo co-IP experiment from total ESC extracts using antibodies to Ctcf, Smc1a, Smc3, Rad21, and SA2 and a control IgG. Bptf co-IP with Ctcf and SA2 pulldown was detected by Western blotting with an antibody to Bptf. Ctcf, Smc1a, Smc3, Rad21, and SA2 IP was confirmed by Western blotting with the antibodies used for pulldown. (C) Results of a representative in vivo co-IP experiment from total ESC extracts using an antibody to Bptf and a control IgG. Co-IP of Ctcf and SA2 were detected by Western blotting with antibodies to Ctcf and SA2, respectively. Bptf IP was confirmed by Western blotting using same antibody for pulldown. (D) Results of a representative in vitro pulldown experiment of purified recombinant human NURF complex (containing a FLAG-Bptf subunit) by resin-bound human GST-CTCF, GST fusions to the human cohesin subunits SMC1a, SMC3, RAD21, and SA2, or a GST control. Pulldown of FLAG-BPTF was detected by Western blotting with an antibody to FLAG. (E) Representative in vitro pulldown of recombinant human NURF complex (containing a FLAG-BPTF subunit) by resin-bound GST N-terminal (NT-CTCF), zinc finger (ZF-CTCF), and C-terminal (CT-CTCF) Ctcf fusions. Pulldown of FLAG-BPTF was detected by Western blotting with an antibody to FLAG. (F) Results of a representative pulldown of recombinant human NURF complex (containing a FLAG-BPTF subunit) by resin-bound GST fusions with the N-terminal (NT-SA2), center (CN-SA2), and C-terminal (CT-SA2) regions of SA2. FLAG-BPTF was detected by Western blotting with an antibody to FLAG.
Ctcf, H3, and H2AZ occupancy changes at Ctcf binding sites with Bptf-dependent NFRs.
In addition to an NFR, Ctcf binding sites are correlated with the histone variants H2A.Z/H3.3 and the cohesin complex (15, 27). To determine if Bptf influences the localization of these factors, we measured their occupancy using ChIP at several Ctcf binding sites with Bptf-dependent NFRs (D1 to D6; “D” signifies “Bptf dependent”). As a control, we used Ctcf binding sites with Bptf-independent NFRs (I1 to I5; “I” signifies “Bptf independent”). From these experiments, we observed changes in Ctcf occupancy at some Ctcf binding sites (3 of 6 sites) with Bptf-dependent NFRs in Bptf KO ESCs (Fig. 6A). In contrast to Ctcf, cohesin occupancy does not significantly change at Ctcf binding sites with a Bptf-dependent NFR (Fig. 6A). How Bptf affects Ctcf occupancy is unknown but could be influenced by several factors which vary in importance at individual sites (see Discussion). More consistently correlated with Bptf-dependent NFRs are an increase in histone H3 occupancy and a decrease in H2A.Z occupancy (Fig. 6B). The increase in H3 occupancy at D1 to D6 with Bptf KO validates our findings from the tiling arrays, which showed an increase in nucleosome occupancy at each of these NFRs. The inverse relationship between histone H3 and H2A.Z occupancy could be related, because an NFR is important for H2A.Z deposition into chromatin (28, 29). To investigate if the observed changes in Ctcf, H3, and H2A.Z occupancy could be due to direct effects of NURF, we performed Bptf ChIP at the same sites. Bptf ChIP showed enrichment at Ctcf sites with a Bptf-dependent NFR (D1 to D6) over Ctcf sites with a Bptf-independent NFR (I1 to I5) (Fig. 6C). To test the model that Ctcf is required for Bptf localization, we performed Bptf ChIP at D1 to D6 in Ctcf KD ESCs (Fig. 6D). These experiments show that Bptf occupancy at five of these sites is significantly reduced, with Ctcf KD supporting a model where Bptf localization to chromatin is dependent on Ctcf (Fig. 6E). Together, these results confirm that Bptf KO results in a significant change in nucleosome occupancy at a subset of Ctcf binding sites which correlate with decreased H2A.Z incorporation (D1 to D6) and in some cases changes in Ctcf occupancy (D3, D4, and D5). These results also demonstrate that Bptf occupancy at Ctcf occupied sites usually (for 5 of 6 sites tested) requires Ctcf.
FIG 6.

Ctcf, H3, and H2AZ occupancy changes at Ctcf binding sites with Bptf-dependent NFRs. (A) Measurement of Ctcf and Smc1a occupancy by ChIP in ESCs at Ctcf binding sites with a Bptf-dependent NFR (D1 to D6) and Ctcf binding sites which do not require Bptf to maintain the NFR (I1 to I5) (t test; *, P < 0.05; data are representative of 3 biological replicates). (B) Measurement of histone H3 and H2A.Z occupancy by ChIP in ESCs at Ctcf binding sites with a Bptf-dependent NFR (D1 to D6) and Ctcf binding sites which do not require Bptf to maintain the NFR (I1 to I5). Occupancy for histone H3 is expressed as a percentage of input. H2A.Z occupancy is expressed as a ratio of H2A.Z to H3 occupancy, measured as a percentage of input (t test; *, P < 0.05; data are representative of 3 biological replicates). (C) ChIP analysis of Bptf occupancy at Ctcf binding sites with a Bptf-dependent NFR (D1 to D6). Bptf occupancy is expressed as a ratio of input from control to that from Bptf KO ESCs (t test; *, P < 0.05; data are representative of 3 biological replicates). (D) Western blot analysis of Ctcf and Bptf from total cell extracts from control or Bptf KO ESCs expressing a control or Ctcf KD shRNA introduced by retrovirus. Ponceau S staining was used as a loading control. (E) Bptf occupancy was measured by ChIP at Ctcf binding sites with a Bptf-dependent NFR (D1 to D6) in control and Bptf KO ESCs expressing either control or Ctcf shRNA KD sequences (t test; *, P < 0.05; data are representative of 3 biological replicates).
Ctcf binding sites with Bptf-dependent NFRs require Bptf for regulatory activity.
To discover possible regulatory activities for Ctcf binding sites, we used integrating reporter vectors with Ctcf binding sites cloned either upstream, downstream (between the promoter and enhancer), or in place of the promoter of a neomycin resistance reporter gene (Neo) to measure barrier, enhancer blocking, or promoter activity, respectively (Fig. 7A) (30). Silencer and enhancer activities which occur when a DNA fragment has similar effects on Neo expression when positioned at the upstream or downstream cloning sites can also be detected with these assays. With these assays, the reporter vector is randomly integrated into the genome of a cell population, and the number of colonies which survive G418 selection is proportional to Neo reporter activity. For these experiments, we used Bptf KO ESCs (Fig. 1A) and Bptf shRNA knockdown (KD) NIH 3T3 fibroblasts with controls (Fig. 7B). We chose NIH 3T3 fibroblasts because they are a cell type comparable to MEFs, with similar Bptf protein levels (Fig. 7C), which can form colonies using the integrating reporter assay. Using these reporters, we measured Bptf-dependent regulatory activity at 4 Ctcf binding sites with a Bptf-dependent NFR (D1 to D4) in both ESCs and MEFs and at 3 randomly chosen Ctcf binding sites with a Bptf-independent NFR (I1 to I3) as controls (Fig. 7D). From these experiments, we observed that the activity of each fragment with a Bptf-dependent NFR (D1 to D4) but not that of control fragments (I1 to I3) was significantly changed with Bptf KO/KD. Activities included a Bptf-dependent promoter activity (D1), barrier activity (or silencer and enhancer activity) (D2 and D3), and enhancer blocking activity (D2, D3, and D4) (Fig. 7E and F). Bptf-dependent activities for 3 of 4 fragments varied with cell type, including promoter activity for D1 in ESCs but not NIH 3T3 cells (Fig. 7E), barrier (or silencer and enhancer activity) or enhancer blocking activity for D2 in ESCs or NIH 3T3 cells, respectively, and more pronounced barrier activity (or silencer/enhancer activity) for D3 in ESCs over NIH 3T3 cells (Fig. 7F). In total these results demonstrate that the effect of Bptf KO/KD on the regulatory activity of Ctcf binding sites is largely restricted to sites with a Bptf-dependent NFR and that the effects of Bptf KO/KD on their activity vary with cell type.
FIG 7.

Bptf depletion alters the regulatory activity of Ctcf binding sites with Bptf-dependent NFRs. (A) Diagram showing integrating reporter vectors used to assay DNA fragments for promoter (pNI-P-MCS), barrier, or enhancer blocking activity (pNI-MCS). The base reporter vector contains the chicken globin insulator (Ins), human γ-globin promoter (Pro), mouse β-globin enhancer (Enh), and neomycin resistance gene (Neo). Hatched blocks show where DNA fragments are cloned for functional assays: promoter, barrier, or enhancer blocking. (B) Efficiency of Bptf knockdown (KD) was confirmed by Western blotting of Bptf from NIH 3T3 cell total cell extracts after Bptf shRNA KD using Ponceau S as a loading control. (C) Bptf expression was determined by Western blotting of Bptf from control MEFs and NIH 3T3 total cell extracts using Ponceau S as a loading control. (D) Summary of DNA fragments assayed using integrating reporter vectors. DNA fragment location relative to the coding sequence (location), its distance to the closest gene, cell types where Ctcf occupies the DNA fragment, and cell types where the Ctcf site has a Bptf-dependent NFR are shown. (E) Results of promoter activity assay for a Ctcf binding site with a Bptf-dependent NFR located in a promoter (D1) and a control promoter containing a Ctcf binding site with a Bptf-independent NFR (I1). Relative activity of the reporter plasmid is expressed as a ratio of Neo-resistant colonies observed for Bptf KD/KO cells to those observed for control cells (t test; *, P < 0.05; data are representative of 3 biological replicates). (F) Results from insulator and enhancer blocking assays for Ctcf binding sites with a Bptf-dependent NFR (D2 to D4) and control Ctcf binding sites containing a Bptf-independent NFR (I2 and I3). Relative activity is expressed as a ratio of Neo-resistant colonies from cells transfected with the reporter containing the Ctcf binding site to those from cells transfected with the pNI-MCS control plasmid (t test; *, P < 0.05; data are representative of 3 biological replicates). (G) Repeat of integrating reporter assays using reporters assayed for panels E and F after deletion of the Ctcf binding site in DNA fragments which show significant changes in regulatory activity with Bptf KO/KD (t test; *, P < 0.05; data are representative of 3 biological replicates).
To determine if the activity observed for fragments D1 to D4 with Bptf KO/KD require Ctcf, we deleted the Ctcf binding site from each DNA fragment and repeated the integrating reporter assays. These experiments show that deleting the Ctcf binding site abolishes activity observed with Bptf KO/KD. These activities include Bptf-dependent promoter activity for D1, enhanced barrier activity for D2 and D3, and Bptf-dependent enhancer blocking activity for D2, D3 and D4 (Fig. 7G). The sum of these results supports a functional link between Ctcf and Bptf during gene regulation by demonstrating that the Ctcf binding sites are required for the observed activity of these fragments with Bptf KO/KD.
Bptf regulates Klf4 binding near a Ctcf binding site upstream of H2-K1.
In a step toward uncovering how Bptf influences the regulatory activity of Ctcf binding sites, we focused on site D2 from our integrating reporter assays (Fig. 7D, F, and G). Ctcf binding site D2 resides ∼3.2 kb upstream of H2-K1 (Fig. 8A and B). H2-K1 is an MHC class I receptor which is expressed in many tissues of the early embryo, is required for fetal growth (31–33), and when deregulated could contribute to the phenotypes observed in Bptf KO mouse embryos (17, 34). Our microarray data sets show that H2-K1 expression is Bptf dependent in ESCs but not MEFs and DP thymocytes, which was confirmed by quantitative reverse transcription-PCR (qRT-PCR) (Fig. 8C; also, see Table S1 in the supplemental material) (17, 18). Previously published ChIP-Seq data sets demonstrate that Ctcf occupies D2 in ESCs and to a lesser extent MEFs, as well as a site downstream of H2-K1 in both cell types (Fig. 8A) (19). We observed a significant Bptf-dependent NFR at D2 and the promoter in ESCs and MEFs but not at the downstream Ctcf binding site (Fig. 8A). We next used integrating reporter assays to determine if the Ctcf binding sites require Bptf for regulatory activity. Our previous reporter assays on D2 demonstrate an enhanced barrier activity in ESCs and a reduced enhancer-blocking activity in NIH 3T3 cells with Bptf KO (Fig. 7F and G). Additional integrating reporter assays showed that the full-length H2-K1 promoter (bp −1077 to +57) and the downstream Ctcf binding site are Bptf independent in both ESCs and NIH 3T3 fibroblasts (Fig. 8D). In summary, these preliminary studies discovered an enhanced gene-regulatory activity for D2 with Bptf KO/KD (Fig. 7F) but not the H2-K1 promoter or a downstream Ctcf binding site (Fig. 8D).
FIG 8.

A Ctcf binding site upstream of H2-K1 has a Bptf-dependent NFR and requires Bptf for regulatory activity. (A) Diagram showing the position (MM9 build coordinates) of significant changes in nucleosome occupancy in ESCs and MEFs, normalized nucleosome occupancy measurements, and Ctcf occupancy measurements from previously published ChIP-Seq data sets (19) and the position of D2 relative to the H2-K1 gene. (B) Higher magnification of the significant changes in nucleosome occupancy and normalized nucleosome occupancy measurements at the upstream Ctcf binding site D2. (C) Results from qRT-PCR experiments measuring H2-K1 transcripts in ESCs normalized to GAPDH (t test; *, P < 0.05; n = 4 biological replicates). (D) Results from the assay of the full-length H2-K1 promoter or the downstream Ctcf binding site using integrating vector reporter assays in ESCs and NIH 3T3 fibroblasts as described for Fig. 7.
To further explore for connections between Bptf, the upstream Ctcf binding site, and H2-K1 expression, we created a D2 deletion series in the previously utilized integrating reporter vector (Fig. 7A). Assay of this deletion series in ESCs identified a minimal 44-bp fragment necessary for its activity in ESCs with Bptf KO (Fig. 9A and B). The MatInspector program identified a GKLF transcription factor binding site in the minimal 44-bp fragment (35). GKLF, also known as Klf4, is a transcription factor with important roles in ESC pluripotency and cellular reprogramming (36). Deletion of the GKLF consensus sequence from D2 abolishes the enhanced reporter activity with Bptf KO in ESCs, demonstrating that it is necessary for this activity (Fig. 9A and B).
FIG 9.

Bptf regulates Klf4 binding to DNA sequences adjacent to Ctcf with possible consequences for H2-K1 expression. (A) Cartoon of integrating reporter constructs used to identify DNA sequences in D2 (see Fig. 8A and B) with Bptf-dependent regulatory activity using the pNI-MCS integrating reporter vector. The minimal 44-bp fragment required for enhanced Neo reporter expression with Bptf KO is defined with vertical bars. Site-directed mutagenesis was used to delete the GKLF DNA binding site in D2 and is designated by an X. (B) Assay of deletion constructs described for panel A identifies a minimal 44-bp DNA element which is required for enhanced reporter activity with Bptf KO. Site-directed mutagenesis of a GKLF consensus site (delGKLF) in the full-length D2 abolishes enhanced reporter activity with Bptf KO (t test; *, P < 0.05; data are representative of 3 biological replicates). (C) Cartoon of integrating reporter constructs used to assay D2 fragment for Bptf-dependent activity in context with the full-length H2-K1 promoter. Site-directed mutagenesis was used to mutate the GKLF (delGKLF) and Ctcf (delCTCF) DNA consensus sequences in the D2 DNA fragment, as designated by an X. (D) Results from assaying integrating the reporter constructs described for panel C in ESCs and NIH 3T3 fibroblasts. Repression of the H2-K1 promoter by D2 was observed in ESCs with Bptf KO and requires the GKLF and Ctcf binding sites (t test; *, P < 0.05; data are representative of 3 biological replicates). (E) Bptf ChIP at the endogenous D2 site in both control and Bptf KO ESCs and MEFs (t test; *, P < 0.05; data are representative of 3 biological replicates). (F) Klf4 ChIP at the endogenous D2 site in both control and Bptf KO ESCs and MEFs (t test; *, P < 0.05; data are representative of 3 biological replicates). (G) Klf4 ChIP at the endogenous D2 site in both control and Bptf KO ESCs expressing either control of Ctcf KD shRNA expression constructs (t test; *, P < 0.05; data are representative of 3 biological replicates). (H) Western analysis of Klf4 expression in ESCs, MEFs, and DP thymocytes total cell extracts using Ponceau S as a loading control. (I) Western analysis of Klf4 expression in control and Bptf KO from total ESC extracts using Ponceau S as a loading control. (J) Results of a representative in vivo co-IP experiment from total ESC extracts using antibodies to Ctcf and Klf4 or normal IgG. Western analysis for Bptf was performed using an antibody to Bptf. Ctcf and Klf4 IP was controlled by Western blotting using the same antibodies for pulldown. (K) Model for NURF function at Ctcf binding sites. Generalized from our nucleosome occupancy measurements, NURF regulates the NFR at a fraction of Ctcf binding sites. At these sites, NURF could regulate the ability of cell-type-restricted transcription factors (TF) like Klf4 to bind adjacent DNA sequences, with consequences for cell-type-restricted gene expression.
Our initial integrating reporter assays utilized a construct containing regulatory elements from the globin genes, including the human γ-globin promoter (Pro) and the mouse β-globin enhancer (Enh) (Fig. 7A). As a result, these constructs assay D2, a DNA fragment found upstream of the H2-K1 promoter, in the context of nonphysiological globin-regulatory elements, possibly influencing the results. To study the regulatory activity of D2 in a more physiological context, we created an integrating reporter vector controlled by the full-length H2-K1 promoter and lacking the mouse β-globin enhancer (Enh) (this modified reporter also lacks the chicken globin insulator [Ins], which has been replaced by D2; see the barrier assay configuration in Fig. 7A). Assay of D2 with this construct shows that it represses Neo gene expression with Bptf KO in ESCs and that the repression requires both the GKLF and Ctcf binding sites (Fig. 9C and D). The ability of D2 to repress the H2-K1 promoter with Bptf KO is consistent with the observed repression of endogenous H2-K1 expression in Bptf KO ESCs. These results suggest functional connections between Klf4, Ctcf, and Bptf during H2-K1 expression.
We next looked for a possible functional relationship between Bptf, Ctcf, and Klf4 at the endogenous D2 sequence. Bptf ChIP shows that it occupies the endogenous D2 sequence in ESCs but not MEFs (Fig. 9E) and that Bptf occupancy at D2 in ESCs requires Ctcf (see the results for D2 in Fig. 6E). We next used ChIP to measure Klf4 occupancy at the endogenous D2 in these same cell types. With these experiments, we show that Klf4 occupancy is elevated in Bptf KO ESCs but not MEFs (Fig. 9F) and that the elevated Klf4 occupancy requires Ctcf (Fig. 9G). Klf4 enrichment at D2 in Bptf KO ESCs is likely due to its restricted expression in ESCs and not due to elevated protein levels with Bptf KO (Fig. 9H and I). Bptf occupancy at D2 is likely independent of Klf4, because in vivo IP experiments using antibodies to Klf4 do not co-IP Bptf (Fig. 9J). Together, these results suggest a model where Ctcf is required for NURF localization to the endogenous D2 sequence. Once recruited, NURF prevents the cell-type-specific transcription factor Klf4 from binding adjacent to Ctcf and repressing H2-K1 expression abnormally (Fig. 9K).
DISCUSSION
To better understand how chromatin-remodeling complexes regulate gene expression, we measured transcript levels genome-wide in ESCs, MEFs, and DP thymocytes lacking Bptf, the largest subunit of the NURF complex (3). Our studies document that Bptf, and likely by extension NURF, regulates the expression of a largely nonoverlapping set of genes between cell types, many of which change in expression >5-fold between cell types. These results are in contrast to those obtained with the SWI/SNF family of complexes, which have more global roles in regulating gene expression through interactions with RNA polymerase (6, 37, 38).
Known functions for NURF during gene regulation include coregulator activities for cell-type-specific transcription factors which could regulate cell-type-specific gene expression (5). Specificity could also result from the incorporation of cell-type-specific subunits into the NURF complex. The SWI/SNF family of chromatin-remodeling complexes incorporate cell-type-specific paralogous subunits to confer a degree of cell type specificity during gene expression (39). NURF purifications from Drosophila embryos and human cells revealed highly homologous complexes, suggesting little variation in subunit composition (3, 40), but caution must be used when generalizations are made from a limited number of cell types. It is also likely that differences in NURF abundance between cell types contribute to its effects on gene expression. For example, differences in Bptf expression vary in adult mouse tissues and during thymocyte differentiation (17, 18).
In an attempt to understand how NURF chromatin-remodeling activities could contribute to regulated expression, we interrogated for changes in nucleosome occupancy which occur with Bptf KO. Similar approaches performed on ISWI family members in flies and Saccharomyces cerevisiae have documented subtle changes at the TSS or TTS (41–43) or more dramatic and dispersed effects across the genome (44). From our experiments, we observed that most changes in nucleosome occupancy occurred at an NFR located >1 kb away from a TSS or TTS. These results are similar to what was observed by Moshkin et al. (44), suggesting that NURF functions predominantly at distal regulatory elements. If our observations are scalable, an expansion of our nucleosome occupancy maps from 3.3 Mbp to the entire genome should reveal ∼1,000-fold more Bptf-dependent changes in nucleosome occupancy for each cell type (∼204,000 for ESCs, ∼76,000 for DP thymocytes, and ∼36,000 for MEFs). If realized, these results would establish NURF as a major regulator of chromatin structure in mammals drawing parallels to those observed for Drosophila ISWI (44). Which of these changes are directly due to NURF function is unknown, and this is an ongoing focus of our laboratory.
Using previously published ChIP-Seq data sets, we discovered that many Bptf-dependent NFRs colocalized with Ctcf and the subunits of cohesin. Functional interactions between NURF and Ctcf/cohesin seem plausible, because both Ctcf and cohesin are known to physically interact with chromatin-remodeling complexes (6, 7, 45) and colocalize to regions of chromatin with evidence of remodeling activity—a pronounced NFR flanked by an organized array of nucleosomes (15). Functional interactions between NURF, Ctcf, and cohesin are supported by both in vivo and in vitro physical interactions and colocalization of Bptf and Ctcf by ChIP. Taken together, these results suggest that Ctcf/cohesin could localize NURF to a subset of its binding sites to remodel surrounding nucleosomes. Why Bptf regulates the NFR at a subset of Ctcf binding sites is unknown. Beyond interactions with Ctcf and SA2, specificity could involve interactions with adjacent transcription factors, or the posttranslational modification of several factors, including histones, NURF subunits, Ctcf, or cohesin. Expanding these preliminary studies to the entire genome could provide the statistical power necessary to discover these factors.
Physical interactions between NURF and the Ctcf zinc fingers place it in close proximity to H2A.Z/H3.3 variant nucleosomes at Ctcf sites. The observed reduction of H2A.Z occupancy at Ctcf sites with Bptf KO, in context with an increase in H3 occupancy, suggests that NURF could indirectly regulate the incorporation of H2A.Z into nucleosomes, by maintaining the NFR. Reduced H2A.Z occupancy at Ctcf sites with Bptf KO is similar to what is observed at the TSS NFR with RSC KO in yeast. In yeast, the RSC chromatin-remodeling complex is proposed to create an NFR at a TSS through its remodeling activity, which in turn promotes the incorporation of H2A.Z into flanking nucleosomes by the SWR1 family of remodeling complexes (28, 29). A similar pathway may operate through the NURF complex at Ctcf binding sites in mammals.
While Bptf regulates nucleosome occupancy at the NFR at a subset of Ctcf binding sites, its importance for Ctcf binding is variable, as measured by ChIP. The ability of Ctcf to bind to DNA is dependent on the position and occupancy of nucleosomes, the presence of other DNA-binding proteins, and DNA methylation (16, 46). Bptf KO could positively or negatively affect one or more of these factors to influence Ctcf binding. Determining which of these factors could be Bptf dependent and influence Ctcf binding is likely to require a detailed investigation of individual Ctcf binding sites and was beyond the scope of this work.
The observation of physical and functional links to Ctcf significantly broadens potential functions for NURF to diverse regulatory elements bound by Ctcf, including enhancers, silencers, promoters, and barrier and enhancer-blocking elements (10). Using an integrating reporter controlled by the H2-K1 promoter, we discovered a silencer activity in Bptf KO ESCs for a Klf4 binding site-adjacent Ctcf binding site. Follow-up experiments support a model where NURF maintains H2-K1 expression in ESCs by preventing Klf4 from binding DNA sequences adjacent to an upstream Ctcf binding site. This model is consistent with known functions for NURF as a regulator of transcription factor binding to chromatin (5). More broadly, this model predicts that NURF could regulate gene expression by influencing cell-type-specific transcription factor binding to DNA sequences adjacent to Ctcf across the genome (Fig. 9K). In support of this model, recent work on the estrogen receptor proposed that its binding to sequences adjacent to Ctcf is important for cell-type-specific gene expression (47). It is therefore plausible that interfering with the binding of cell-type-specific transcription factors to sequences adjacent to Ctcf could have consequences for genes that are differentially expressed between cell types and could explain some of our observations.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Landry, Lloyd, Lichten, and Wu labs for helpful comments, and Kevin Hogan for editorial assistance. We thank Carl Wu for support during the initial stages of this project.
This work was supported by grants from the V Foundation for Cancer Research, Jeffress Foundation, VCU School of Medicine, and VCU Massey Cancer Center.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00553-14.
REFERENCES
- 1.Clapier CR, Cairns BR. 2009. The biology of chromatin remodeling complexes. Annu Rev Biochem 78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- 2.Ho L, Crabtree GR. 2010. Chromatin remodelling during development. Nature 463:474–484. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barak O, Lazzaro MA, Lane WS, Speicher DW, Picketts DJ, Shiekhattar R. 2003. Isolation of human NURF: a regulator of Engrailed gene expression. EMBO J 22:6089–6100. doi: 10.1093/emboj/cdg582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao H, Sandaltzopoulos R, Wang HM, Hamiche A, Ranallo R, Lee KM, Fu D, Wu C. 2001. Dual functions of largest NURF subunit NURF301 in nucleosome sliding and transcription factor interactions. Mol Cell 8:531–543. doi: 10.1016/S1097-2765(01)00345-8. [DOI] [PubMed] [Google Scholar]
- 5.Alkhatib SG, Landry JW. 2011. The nucleosome remodeling factor. FEBS Lett 585:3197–3207. doi: 10.1016/j.febslet.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Euskirchen GM, Auerbach RK, Davidov E, Gianoulis TA, Zhong G, Rozowsky J, Bhardwaj N, Gerstein MB, Snyder M. 2011. Diverse roles and interactions of the SWI/SNF chromatin remodeling complex revealed using global approaches. PLoS Genet 7:e1002008. doi: 10.1371/journal.pgen.1002008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ishihara K, Oshimura M, Nakao M. 2006. CTCF-dependent chromatin insulator is linked to epigenetic remodeling. Mol Cell 23:733–742. doi: 10.1016/j.molcel.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 8.Lai AY, Wade PA. 2011. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat Rev Cancer 11:588–596. doi: 10.1038/nrc3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trotter KW, Archer TK. 2008. The BRG1 transcriptional coregulator. Nucl Recept Signal 6:e004. doi: 10.1621/nrs.06004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Phillips JE, Corces VG. 2009. CTCF: master weaver of the genome. Cell 137:1194–1211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, Tsutsumi S, Nagae G, Ishihara K, Mishiro T, Yahata K, Imamoto F, Aburatani H, Nakao M, Imamoto N, Maeshima K, Shirahige K, Peters JM. 2008. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 451:796–801. doi: 10.1038/nature06634. [DOI] [PubMed] [Google Scholar]
- 12.Faure AJ, Schmidt D, Watt S, Schwalie PC, Wilson MD, Xu H, Ramsay RG, Odom DT, Flicek P. 2012. Cohesin regulates tissue-specific expression by stabilizing highly occupied cis-regulatory modules. Genome Res 22:2163–2175. doi: 10.1101/gr.136507.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmidt D, Schwalie PC, Ross-Innes CS, Hurtado A, Brown GD, Carroll J S, Flicek P, Odom DT. 2010. A CTCF-independent role for cohesin in tissue-specific transcription. Genome Res 20:578–588. doi: 10.1101/gr.100479.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zuin J, Dixon JR, van der Reijden MI, Ye Z, Kolovos P, Brouwer RW, van de Corput MP, van de Werken HJ, Knoch TA, van Ijcken WF, Grosveld FG, Ren B, Wendt KS. 2014. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc Natl Acad Sci U S A 111:996–1001. doi: 10.1073/pnas.1317788111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu Y, Sinha M, Peterson CL, Weng Z. 2008. The insulator binding protein CTCF positions 20 nucleosomes around its binding sites across the human genome. PLoS Genet 4:e1000138. doi: 10.1371/journal.pgen.1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weth O, Renkawitz R. 2011. CTCF function is modulated by neighboring DNA binding factors. Biochem Cell Biol 89:459–468. doi: 10.1139/o11-033. [DOI] [PubMed] [Google Scholar]
- 17.Landry J, Sharov AA, Piao Y, Sharova LV, Xiao H, Southon E, Matta J, Tessarollo L, Zhang YE, Ko MS, Kuehn MR, Yamaguchi TP, Wu C. 2008. Essential role of chromatin remodeling protein Bptf in early mouse embryos and embryonic stem cells. PLoS Genet 4:e1000241. doi: 10.1371/journal.pgen.1000241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Landry JW, Banerjee S, Taylor B, Aplan PD, Singer A, Wu C. 2011. Chromatin remodeling complex NURF regulates thymocyte maturation. Genes Dev 25:275–286. doi: 10.1101/gad.2007311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, Ren B. 2012. A map of the cis-regulatory sequences in the mouse genome. Nature 488:116–120. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weiner A, Hughes A, Yassour M, Rando OJ, Friedman N. 2010. High-resolution nucleosome mapping reveals transcription-dependent promoter packaging. Genome Res 20:90–100. doi: 10.1101/gr.098509.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang C, Pugh BF. 2009. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet 10:161–172. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, Loh YH, Yeo HC, Yeo ZX, Narang V, Govindarajan KR, Leong B, Shahab A, Ruan Y, Bourque G, Sung WK, Clarke ND, Wei CL, Ng HH. 2008. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 23.Ebert A, McManus S, Tagoh H, Medvedovic J, Salvagiotto G, Novatchkova M, Tamir I, Sommer A, Jaritz M, Busslinger M. 2011. The distal V(H) gene cluster of the Igh locus contains distinct regulatory elements with Pax5 transcription factor-dependent activity in pro-B cells. Immunity 34:175–187. doi: 10.1016/j.immuni.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 24.Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, Taatjes DJ, Dekker J, Young RA. 2010. Mediator and cohesin connect gene expression and chromatin architecture. Nature 467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nitzsche A, Paszkowski-Rogacz M, Matarese F, Janssen-Megens EM, Hubner NC, Schulz H, de Vries I, Ding L, Huebner N, Mann M, Stunnenberg HG, Buchholz F. 2011. RAD21 cooperates with pluripotency transcription factors in the maintenance of embryonic stem cell identity. PLoS One 6:e19470. doi: 10.1371/journal.pone.0019470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiao T, Wallace J, Felsenfeld G. 2011. Specific sites in the C terminus of CTCF interact with the SA2 subunit of the cohesin complex and are required for cohesin-dependent insulation activity. Mol Cell Biol 31:2174–2183. doi: 10.1128/MCB.05093-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parelho V, Hadjur S, Spivakov M, Leleu M, Sauer S, Gregson HC, Jarmuz A, Canzonetta C, Webster Z, Nesterova T, Cobb BS, Yokomori K, Dillon N, Aragon L, Fisher AG, Merkenschlager M. 2008. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 132:422–433. doi: 10.1016/j.cell.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 28.Hartley PD, Madhani HD. 2009. Mechanisms that specify promoter nucleosome location and identity. Cell 137:445–458. doi: 10.1016/j.cell.2009.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ranjan A, Mizuguchi G, FitzGerald PC, Wei D, Wang F, Huang Y, Luk E, Woodcock CL, Wu C. 2013. Nucleosome-free region dominates histone acetylation in targeting SWR1 to promoters for H2A.Z replacement. Cell 154:1232–1245. doi: 10.1016/j.cell.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bell AC, West AG, Felsenfeld G. 1999. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell 98:387–396. doi: 10.1016/S0092-8674(00)81967-4. [DOI] [PubMed] [Google Scholar]
- 31.Arcellana-Panlilio MY, Schultz GA. 1994. Temporal and spatial expression of major histocompatibility complex class I H-2K in the early mouse embryo. Biol Reprod 51:169–183. doi: 10.1095/biolreprod51.2.169. [DOI] [PubMed] [Google Scholar]
- 32.Hedley ML, Drake BL, Head J R, Tucker PW, Forman J. 1989. Differential expression of the class I MHC genes in the embryo and placenta during midgestational development in the mouse. J Immunol 142:4046–4053. [PubMed] [Google Scholar]
- 33.Madeja Z, Yadi H, Apps R, Boulenouar S, Roper SJ, Gardner L, Moffett A, Colucci F, Hemberger M. 2011. Paternal MHC expression on mouse trophoblast affects uterine vascularization and fetal growth. Proc Natl Acad Sci U S A 108:4012–4017. doi: 10.1073/pnas.1005342108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goller T, Vauti F, Ramasamy S, Arnold HH. 2008. Transcriptional regulator BPTF/FAC1 is essential for trophoblast differentiation during early mouse development. Mol Cell Biol 28:6819–6827. doi: 10.1128/MCB.01058-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. 2005. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 21:2933–2942. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- 36.Yamanaka S. 2007. Strategies and new developments in the generation of patient-specific pluripotent stem cells. Cell Stem Cell 1:39–49. doi: 10.1016/j.stem.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 37.Armstrong JA, Papoulas O, Daubresse G, Sperling AS, Lis JT, Scott MP, Tamkun JW. 2002. The Drosophila BRM complex facilitates global transcription by RNA polymerase II. EMBO J 21:5245–5254. doi: 10.1093/emboj/cdf517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson CJ, Chao DM, Imbalzano AN, Schnitzler GR, Kingston RE, Young RA. 1996. RNA polymerase II holoenzyme contains SWI/SNF regulators involved in chromatin remodeling. Cell 84:235–244. doi: 10.1016/S0092-8674(00)80978-2. [DOI] [PubMed] [Google Scholar]
- 39.Wu J I, Lessard J, Crabtree GR. 2009. Understanding the words of chromatin regulation. Cell 136:200–206. doi: 10.1016/j.cell.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsukiyama T, Wu C. 1995. Purification and properties of an ATP-dependent nucleosome remodeling factor. Cell 83:1011–1020. doi: 10.1016/0092-8674(95)90216-3. [DOI] [PubMed] [Google Scholar]
- 41.Sala A, Toto M, Pinello L, Gabriele A, Di Benedetto V, Ingrassia AM, Lo Bosco G, Di Gesu V, Giancarlo R, Corona DF. 2011. Genome-wide characterization of chromatin binding and nucleosome spacing activity of the nucleosome remodelling ATPase ISWI. EMBO J 30:1766–1777. doi: 10.1038/emboj.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Whitehouse I, Rando OJ, Delrow J, Tsukiyama T. 2007. Chromatin remodelling at promoters suppresses antisense transcription. Nature 450:1031–1035. doi: 10.1038/nature06391. [DOI] [PubMed] [Google Scholar]
- 43.Yen K, Vinayachandran V, Batta K, Koerber RT, Pugh BF. 2012. Genome-wide nucleosome specificity and directionality of chromatin remodelers. Cell 149:1461–1473. doi: 10.1016/j.cell.2012.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moshkin YM, Chalkley GE, Kan TW, Reddy BA, Ozgur Z, van Ijcken WF, Dekkers DH, Demmers J A, Travers AA, Verrijzer CP. 2012. Remodelers organize cellular chromatin by counteracting intrinsic histone-DNA sequence preferences in a class-specific manner. Mol Cell Biol 32:675–688. doi: 10.1128/MCB.06365-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hakimi MA, Bochar DA, Schmiesing J A, Dong Y, Barak OG, Speicher DW, Yokomori K, Shiekhattar R. 2002. A chromatin remodelling complex that loads cohesin onto human chromosomes. Nature 418:994–998. doi: 10.1038/nature01024. [DOI] [PubMed] [Google Scholar]
- 46.Wang H, Maurano MT, Qu H, Varley KE, Gertz J, Pauli F, Lee K, Canfield T, Weaver M, Sandstrom R, Thurman RE, Kaul R, Myers RM, Stamatoyannopoulos JA. 2012. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res 22:1680–1688. doi: 10.1101/gr.136101.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ross-Innes CS, Brown GD, Carroll JS. 2011. A co-ordinated interaction between CTCF and ER in breast cancer cells. BMC Genomics 12:593. doi: 10.1186/1471-2164-12-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.