

Abstract
Factor XIII (FXIII) is necessary for cross linking of fibrin strands and generation of stable fibrin clot. FXIII Val34Leu is a common genetic single nucleotide polymorphism that has been associated with accelerated fibrin stabilization and reduced rate of fibrinolysis. The contribution of Val34Leu to long term risk of recurrent myocardial infarction (MI) in patients with coronary stenting has not been conclusively established. The objective of the study was to examine the effects of Val34Leu on fibrin generation, platelet aggregation, and long term clinical outcomes in patients with coronary artery disease treated with dual antiplatelet therapy. Patients with angiographically documented coronary artery disease who were treated with aspirin and clopidogrel were enrolled (n = 211). Light transmittance aggregometry and plasma fibrin clot formation using thrombelastography (TEG) were determined. Genotyping of Val34Leu was performed using Taqman assay. Clinical events during follow up were recorded. Homozygous carriers of 34Leu variant had significantly shorter fibrin clot formation time as compared to wild type individuals (TEG K: 1.27 ± 0.3 vs. 1.68 ± 1.1 min, p = 0.011). The Val34Leu variant was associated with gene dose dependent increased risk of MI (log rank, p = 0.002) or occurrence of composite of MI and CV death (log rank, p = 0.005) with highest event rates observed in homozygous carriers of 34Leu. In summary, FXIII Val34Leu polymorphism was associated with increased rate of fibrin stabilization in homozygous carriers of the variant and may increase risk of recurrent MI and death in patients with angiographically established coronary artery disease treated with dual antiplatelet therapy.
Keywords: Acute myocardial infarction, Genetic polymorphism, Coagulation, Platelet function tests, Thrombelastography
Introduction
Despite advances in medical therapy, recurrent myocardial infarction (MI) remains a significant contributor to long term morbidity and mortality in patients with coronary artery disease. In patients treated with clopidogrel after coronary stenting, high residual platelet reactivity induced by adenosine diphosphate (ADP) has been associated with increased risk of stent thrombosis and recurrent MI [1–3]. During acute coronary syndromes (ACS), activation of coagulation and thrombin generation is a central step preceding coronary thrombosis and treatment with heparin or low molecular weight heparin is standard of care in the initial in-hospital management of ACS [4]. Risk associated with continued prothrombotic changes related to vascular inflammation may be an important mediator of long term recurrent risk of ischemic events in patients with coronary stenting [5–10]. While large scale efforts have been directed at identifying genetic variants causally linked to the development of coronary artery disease, fewer efforts have been directed specifically at identifying single nucleotide polymorphisms (SNPs) associated with recurrent ischemic risk in patients with established coronary artery disease [11].
Coagulation factor XIII (FXIII) plays an important role in the final stages of the coagulation cascade and the regulation of fibrinolysis, and it is essential in the formation of stable and fibrinolysis resistant fibrin clot [12]. FXIII is a glycoprotein tetramer (A2B2) consisting of two catalytic A subunits encoded by the F13A1 gene mapping to 6p25.3–p24.3, and two carrier/inhibitory B subunits encoded by the F13B gene mapping to 1q31–q32.1. When activated, FXIII covalently cross-links fibrin forming a stiff fibrin network. The F13A1 gene is characterized by significant allelic heterogeneity, and several polymorphisms have been identified with varied functional importance [12]. The F13A1 c.103G>T (p.Val35Leu) rs5985 SNP, hereinafter referred to as its legacy name of Val34Leu, is critically located near the thrombin activation site (R37-G38) of FXIII-A and has been demonstrated to affect the rate of FXIII activation peptide cleavage by thrombin, resulting in faster FXIII activation and more rapid fibrin clot formation. This variant is present in approximately 40 % of white subjects [13–15]. A large proportion of circulating FXIII-A is stored in platelets and released upon platelet activation. The activated FXIII (FXIIIa) has been shown to support platelet activation and enhance thrombus formation by matrix proteins through interactions with the platelet glycoprotein αIIbβ3 receptor and integrin αVβ3 [16]. However, the effect of Val34Leu polymorphism on clopidogrel platelet inhibition in subjects with coronary artery disease has not been examined [12].
We hypothesized that the Val34Leu polymorphism is a associated with faster fibrin stabilization and increased risk of recurrent MI during follow up in patients with established coronary artery disease treated with dual antiplatelet therapy.
Materials and methods
Patients
The study protocols were approved by Indiana University School of Medicine’s Institutional Review Board for Research. Informed consent was obtained from all subjects. Subjects with established angiographic diagnosis of coronary artery disease who were treated with dual antiplatelet therapy after coronary stenting or ACS with clopidogrel and aspirin 81–325 mg daily were enrolled. To be included in this study, subjects either had to have been taking clopidogrel 75 mg daily for at least 5 days prior to enrollment or had received a clopidogrel 600 mg loading dose at least 6 h prior to blood draws. Exclusion criteria included a history of drug or alcohol abuse, bleeding disorder, myelodysplastic or myeloproliferative disorders, chronic liver disease, current warfarin use, pregnancy, thrombocytopenia (platelet count less than 100,000/mm3), and glycoprotein IIb/IIIa antagonist use.
Blood samples
Peripheral venous blood samples were drawn at least 6 h after administration of a clopidogrel loading dose or prior to the next clopidogrel dose in patients on clopidogrel maintenance dosing for platelet assays. All blood samples were directly transferred into vacutainer tubes containing 3.2 % Na-citrate. Citrate plasma was obtained from blood samples obtained prior to percutaneous coronary interventions and prior to administration of anticoagulants by centrifugation at 2,000×g for 15 min and was stored at −80 °C until analysis.
Platelet aggregation studies
Ex vivo platelet function was assessed by light transmittance aggregometry (LTA) at 37 °C with an Optical Lumi-Aggregometer (Model 700 with AggroLink 8 software, Chrono-Log Corporation, Havertown, PA, USA). Platelet rich plasma (PRP) and platelet poor plasma were obtained by differential centrifugation of citrate blood as previously described [2, 17]. Platelet aggregation in PRP was induced with ADP at 5, 10 and 20 μM, with thrombin receptor activating peptide (TRAP) at 15 and 25 μM, and arachidonic acid (AA) 1 mM.
Thrombelastography (TEG)
TEG was performed using citrate plasma samples obtained prior to percutaneous coronary intervention (PCI) according to the manufacturer’s instructions (TEG® 5000 Thrombelastograph® Hemostasis Analyzer System, Haemonetics, MA, USA). Citrate plasma was mixed with kaolin, inverted five times, and then loaded in a heparinase coated cup containing 20 μl of CaCl2. TEG was stopped after maximal fibrin clot strength was recorded. Time to fibrin formation (R), clot formation time (K), angle (α), and maximal clot strength [G (dyn/cm2)] was recorded.
Genotyping
Genomic DNA was extracted from whole-blood with the use of Qiagen’s QIAamp® DNA Blood Midi Kit (Germantown, MD, USA). Factor XIII Val34Leu (rs5985) SNP was analyzed using Bio-Rad Laboratories real-time PCR system. Sequence specific primers were used to amplify the alleles of interest, along with two allele-specific TaqMan probes, purchased by Applied Biosystems, (Foster City, CA, USA). Allelic discrimination was used to determine individual genotypes (Optical system 3.1 software, Bio-Rad Laboratories, CA, USA).
ELISA
Citrated plasma was analyzed by ELISA assay according to manufacturer’s instructions after appropriate dilutions for fibrinogen (Alpco Diagnostics, USA) and FXIIIa concentrations (Abnova, USA).
Clinical endpoints
MI was defined according to the universal definition for MI, as a rise of biomarkers of myocardial injury elevated above the 99th percentile of the upper reference limit (URL) in subjects presenting with symptoms of ischemia [18]. PCI related MI was defined as a rise of creatinine kinase MB fraction above 3 × 99th percentile of URL when associated with prolonged symptoms suggestive of myocardial ischemia, new ischemic ECG changes, angiographically documented coronary occlusion, or imaging evidence of loss of viable myocardium. Stent thrombosis was defined as probable or definite according to Academic Research Consortium criteria [1]. Bleeding was defined as moderate or severe bleeding according to GUSTO criteria [19]. Electronic medical records were examined for the occurrence of clinical events as defined.
Statistics
SPSS statistics software package (Version 21, IBM, USA) was used for analysis. Continuous variables are expressed as mean ± SD. Categorical data are presented as percent frequency and groups were compared using the Chi square test. Student’s t test was used to compare normally distributed continuous data between two groups and for genotype group comparisons one-way analysis of variance was used.
Kaplan–Meier survival analysis was performed for combined cardiovascular (CV) death/MI and MI alone, according to Val34Leu genotypes with log-rank method. Outcomes were censored at time of last clinical follow up. Cox regression analysis was performed with adjustment for age, gender, race, diabetes mellitus, PCI, MI, coronary artery bypass grafting (CABG), hyperlipidemia, peripheral vascular disease (PVD), smoking, body mass index (BMI), and clopidogrel pretreatment in a forward stepwise model to calculate hazard ratios associated with Val34Leu genotypes and clinical outcomes (Table 3.)
Table 3.
Clinical event rates for death, cardiovascular (CV) death, myocardial infarction, stent thrombosis, and bleeding according to Val34Leu genotypes
| Clinical events | Total | Val/Val | Val/Leu | Leu/Leu | Val/Leu vs. Val/Val HR (95 % CI); p value |
Leu/Leu vs. Val/Val HR (95 % CI); p value |
Leu carriers vs. Val/Val HR (95 % CI); p value |
|---|---|---|---|---|---|---|---|
| Death | 12/211 (5.7 %) | 4/114 (3.5 %) | 7/86 (8.1 %) | 1/11 (9.1 %) | 2.6 (0.75–9.2); 0.13 | 1.1 (0.11–11.3); 0.92 | 2.0 (0.6–7); 0.25 |
| Cardiovascular Death | 7/211 (3.3 %) | 3/114 (2.6 %) | 3/86 (3.5 %) | 1/11 (9.1 %) | 1.4 (0.3–7.2); 0.66 | 2.0 (0.2–20); 0.56 | 1.5 (0.3–6.9); 0.57 |
| Myocardial Infarction | 24/211 (11.4 %) | 7/114 (6.1 %) | 12/86 (14 %) | 5/11 (45.5 %) | 2.6 (1.0–6.6); 0.046 | 9.2 (2.9–29); <0.001 | 3.3 (1.4–7.9); 0.008 |
| CV Death or Myocardial Infarction | 28/211 (13.3 %) | 9/114 (7.9 %) | 14/86 (16.2 %) | 5/11 (45.5 %) | 2.4 (1.0–5.5); 0.042 | 7.3 (2.4–22.1); <0.001 | 2.9 (1.3–6.4); 0.009 |
| Death or Myocardial Infarction | 31/211 (14.7 %) | 10/114 (8.8 %) | 16/86 (18.6 %) | 5/11 (45.5 %) | 2.4 (1.1–5.4); 0.027 | 6.2 (2.1–18); 0.001 | 2.9 (1.3–6.1); 0.006 |
| Stent Thrombosis | 5/211 (2.4 %) | 3/114 (2.6 %) | 1/86 (1.2 %) | 1/11 (9.1 %) | 0.47 (0.05–4.4); 0.5 | 3.1 (0.3–30); 0.31 | 0.81 (0.14–4.9); 0.82 |
| Bleeding | 11/211 (5.2 %) | 5/114 (4.4 %) | 6/86 (6.9 %) | 0/11 (0 %) | 2.4 (0.65–8.6); 0.19 | 0 (0); 0.98 | 2.1 (0.6–7.8) 0.26 |
Cox regression hazard ratios (HR) are presented for all carriers of 34Leu variant and individual genotypes after adjustment for clinical variables and demographics
Results
Baseline characteristics of the study subjects are described according to FXIII Val34Leu polymorphisms in (Table 1). A total of 211 patients were enrolled. The patients mean age was 57.3 ± 10 years. The majority was male, white and obese (Table 1), although a significant proportion of women and African Americans were also enrolled. Among 211 patients in this study, 114 (54 %) were carriers of the Val/Val genotype, 86 (40.8 %) were heterozygous allele carriers (Val/Leu) and 11(5.2 %) were homozygous Leu/Leu genotype carriers. Genotype frequency distribution was consistent with Hardy–Weinberg equilibrium (p = 0.31). The majority of patients had undergone coronary stenting (82 %), while the remainder was treated with dual anti-platelet therapy after an acute coronary syndrome (18 %).
Table 1.
Demographics and clinical variables of subjects included in the study according to Val34Leu genotype
| Clinical variables | Total | Val/Val | Val/Leu | Leu/Leu | p value |
|---|---|---|---|---|---|
| Number of patients | 211 (100 %) | 114 (54 %) | 86 (40.8 %) | 11 (5.2 %) | |
| Age (mean ± SD) (years) | 57.3 ± 10 | 57 ± 9.7 | 58.4 ± 10 | 52 ± 10.7 | 0.13 |
| Gender (%) | |||||
| Male | 121 (57 %) | 63 (55 %) | 50 (56 %) | 8 (73 %) | 0.53 |
| Female | 90 (43 %) | 51 (45 %) | 36 (44 %) | 3 (27 %) | |
| Race (%) | |||||
| White | 151 (72 %) | 77 (68 %) | 67 (78 %) | 7 (64 %) | 0.7 |
| Black | 56 (27 %) | 34 (30 %) | 18 (21 %) | 4 (36 %) | |
| Others | 4 (2 %) | 3 (3 %) | 1 (1 %) | 0 (0 %) | |
| Smoking (%) | |||||
| Non-smoker | 115 (55 %) | 61 (54 %) | 51 (59 %) | 3 (27 %) | 0.13 |
| Current smoker | 96 (45 %) | 53 (46 %) | 35 (41 %) | 8 (73 %) | |
| Weight (kg) | 95.2 ± 25 | 91.8 ± 21 | 98.1 ± 25 | 112.3 ± 47 | 0.16 |
| Height (cm) | 170.4 ± 11 | 169.3 ± 9 | 171.5 ± 10 | 173.9 ± 14 | 0.45 |
| BMI (kg/m2) | 32.8 ± 9 | 32.3 ± 6.7 | 32.9 ± 7.3 | 36.8 ± 7.5 | 0.3 |
| Previous myocardial infarction (%) | 143 (68 %) | 82 (72 %) | 54 (63 %) | 7 (64 %) | 0.3 |
| Previous CABG (%) | 46 (22 %) | 24 (21 %) | 22 (26 %) | 0 (0 %) | 0.15 |
| Coronary stent placement (%) | 167 (82 %) | 94 (82 %) | 70 (81 %) | 9 (81 %) | 0.98 |
| Left ventricular ejection fraction (EF) (%) | 49.7 ± 17 | 48.2 ± 20 | 51.7 ± 15 | 47.5 ± 15 | 0.59 |
| DM (%) | 91 (43 %) | 49 (43 %) | 39 (45 %) | 3 (27 %) | 0.52 |
| Hypertension (%) | 196 (93 %) | 104 (91 %) | 81 (94 %) | 11 (100 %) | 0.46 |
| Hyperlipidemia (%) | 186 (94 %) | 108 (95 %) | 79 (92 %) | 11 (100 %) | 0.48 |
| PVD (%) | 29 (14 %) | 19 (17 %) | 10 (12 %) | 0 (0 %) | 0.24 |
| ACE-I/ARB (%) | 165 (78 %) | 90 (79 %) | 65 (76 %) | 10 (91 %) | 0.23 |
| Betablocker (%) | 196 (93 %) | 106 (93 %) | 80 (93 %) | 10 (91 %) | 0.69 |
| Calcium channel blocker (%) | 50 (24 %) | 30 (26 %) | 18 (21 %) | 2 (18 %) | 0.61 |
| Proton pump inhibitor (%) | 66 (31 %) | 34 (30 %) | 28 (33 %) | 4 (36 %) | 0.86 |
| Statin (%) | 176 (83 %) | 98 (86 %) | 70 (81 %) | 8 (73 %) | 0.36 |
| Clopidogrel loading dose (%) | 94 (50 %) | 56 (49 %) | 45 (52 %) | 4 (36 %) | 0.6 |
ACEI angiotensin converting enzyme inhibitor, ARB angiotensin receptor blocker. Values represent mean ± SD
Overall clinical variables were well balanced among genotype groups with no significant differences in clinical variables. Importantly, major established CV risk factors such as diabetes mellitus (DM), and smoking were not significantly different among groups.
Platelet aggregation induced by ADP demonstrated wide inter-individual variability during therapy with clopidogrel, as previously documented [2, 3]. Among the three genotypes, there was no significant difference in ADP induced on treatment platelet aggregation during clopidogrel therapy before and after adjustment for DM, obesity, smoking and proton pump inhibitor therapy (Table 2). Similarly, PAR-1 mediated aggregation stimulated by thrombin receptor agonist peptide (TRAP) was not significantly different among Val34Leu genotypes (Table 2). With the exception of two individuals of Val34Val wild type, all subjects were found to be aspirin responders as defined by maximal platelet aggregation (MPA) <20 % by AA.
Table 2.
MPA induced by ADP and TRAP measured by LTA according to Val34Leu genotypes
| MPA ADP 5 μM (%) |
MPA ADP 10 μM (%) |
MPA ADP 20 μM (%) |
MPA TRAP 15 μM (%) |
MPA TRAP 25 μM (%) |
|
|---|---|---|---|---|---|
| Val/Val (n = 114) | 33 ± 15 | 41.6 ± 16 | 47 ± 16 | 77.4 ± 9 | 79.3 ± 10 |
| Val/Leu (n = 86) | 32.3 ± 16 | 39.2 ± 18 | 45.1 ± 18 | 75.6 ± 12 | 79.2 ± 11 |
| Leu/Leu (n = 11) | 34.2 ± 12 | 37.4 ± 15 | 49.1 ± 11 | 78 ± 5 | 81 ± 4 |
| p value | 0.9 | 0.53 | 0.63 | 0.67 | 0.81 |
p value for linear regression analysis after multivariate adjustment for DM, smoking, proton pump inhibitor use, and weight
The three genotype groups did not differ significantly in TEG-R, TEG-G, and TEG-α measurements (Fig. 1). However, TEG-K (clot formation time) significantly decreased with increasing number of 34Leu alleles and a statistically significant difference between the wild type (Val/Val) and the homozygous genotype (Leu/Leu) could be detected (1.68 ± 1.1 vs. 1.27 ± 0.3 min, p = 0.011; mean difference 0.41 (95 % CI 0.1–0.71) (Fig. 1). TEG-G was not significantly different between homozygous carriers of the 34Leu allele and wild type individuals (3,263 ± 996 vs. 2,893 ± 1,088 dynes/cm2, p = 0.29) (Fig. 1). Fibrinogen concentration (7.4 ± 4 vs. 9.2 ± 6 mg/ml; p = 0.13) and FXIIIa levels (22.2 ± 9 vs. 24.1 ± 11 ng/ml; p = 0.5) were not significantly different between carriers of Val34Leu and wild type subjects.
Fig. 1.

Thrombelastography measurements grouped according to Val34Leu genotypes. a TEG-K: fibrin clot formation time. b TEG-angle. c TEG-G: maximal fibrin clot strength. d TEG-R: time to clot formation
The mean duration of follow up was 3.0 ± 1.9 years. Kaplan–Meier survival curves for MI and combined CV death and MI endpoints are shown according to Val34Leu genotypes (Fig. 2). Event free survival was highest in individuals of 34Val wild type, with incrementally higher event rates in heterozygous and homozygous carriers of the 34Leu variant. Cox regression analysis was performed after adjustment for clinical variables and demographics (supplemental online material). Clinical events are listed according to genotypes (Table 3). Homozygous 34Leu was associated with the highest hazard ratio (HR) for recurrent MI (HR = 6.7; 95 % CI 2.1–21; p = 0.001) as compared to wild type individuals, with a relatively smaller risk in all carriers of the 34Leu allele (HR = 2.8; 95 % CI 1.2–6.8; p = 0.02) (Table 3). Twelve patients (5.7 %) died during follow-up, with half of deaths due to non-CV causes. Deaths due to CV causes were evenly distributed and the risk of all cause death was not significantly different across genotypes (Table 3). Homozygous 34Leu was associated with a HR of 6.2 (95 % CI 2.1–18; p = 0.001) for recurrent MI or death as compared to wild type individuals, whereas for all carriers of the 34Leu allele the HR was 2.9 (95 % CI 1.3–6.1; p = 0.006) (Table 3). Stent thrombosis and bleeding events were not significantly different among genotypes (Table 3). The primary combined endpoint of CV death and MI occurred less frequently in patients who had been treated with PCI as compared to medical treatment after ACS (HR = 0.5, 95 % CI 0.2–1.2; p = 0.1).
Fig. 2.
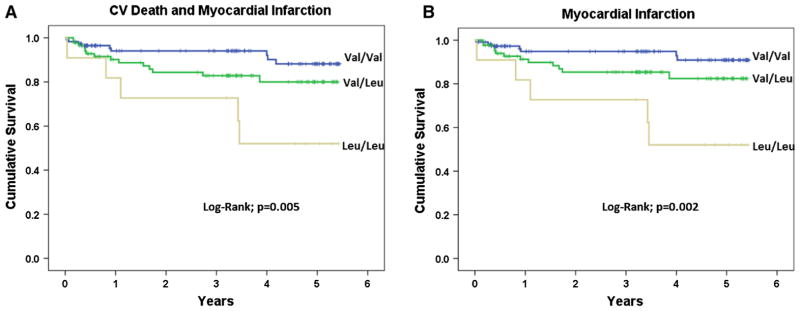
Kaplan-Meier event free survival curves for cardiovascular death or myocardial infarction (a) and myocardial infarction alone (b), grouped according to Val34Leu genotypes
Discussion
In regard to the association of Val34Leu polymorphism with coronary artery disease, there have been conflicting reports, and the results of our study are in contrast to the conclusions of some previous studies. In essence, some case–control studies examining the prevalence of 34Leu allele in survivors of acute MI found a reduced number of patients with 34Leu alleles as compared with matched healthy controls [14, 20–24]. Although more recent reports demonstrated neutral results in larger patient series, 34Leu allele has been suggested to be protective for the development of coronary artery disease and early MI. In contrast, carriers of 34Leu have been shown to exhibit resistance to fibrinolysis administered during ST elevation MI with reduced lytic success, worse reperfusion, and worse clinical outcome [25, 26]. Since many of the case–control studies that examined the prevalence of Val34Leu allele recruited MI survivors during the thrombolytic era prior to widespread adoption of primary PCI in acute MI, it is possible that the protective effect of 34Leu allele may at least in part be due to survival bias [23]. In addition, despite the confirmation of a modest protective effect of 34Leu on incidence of MI in a meta-analysis, publication bias could not be excluded by the authors of the meta-analysis [23]. Sylvain et al. [14] prospectively followed 228 young survivors of acute MI and could not demonstrate a significant difference in survival between carriers of Val34Leu variant and wild type individuals using a dominant model of inheritance. Event rates were relatively low despite long term follow-up, with a non-significant trend towards increased risk in carriers of 34Leu allele as compared to wild type subjects (HR 1.09; 95 % CI 0.46–2.6; p = 0.85) [14].
While questions remain regarding contributions of haemostatic genes to the de-novo incidence of MI in case–control studies, the implantation of coronary stents in coronary arteries changes the risk equation in regards to the relative importance of small differences in prothrombotic phenotype. This is evidenced by the example of increased protection derived from P2Y12 inhibition by clopidogrel in subjects treated with coronary stenting as compared to patients treated medically after an acute coronary syndrome [27, 28]. In theory, a similar analogy could explain differences of gene-risk association observed with Val34Leu in patients with coronary artery disease treated medically as compared to PCI.
Overall, the findings of our study suggest a dose dependent effect of 34Leu allele on ischemic risk in subjects with coronary artery disease with highest risk observed in homozygous carriers of the allele. Therefore, a dose genotype model appears preferable over a dominant model in examining thrombotic risk after coronary stenting in future studies. Ischemic events included a significant number of late and very late stent thrombosis as well as spontaneous MIs.
Since FXIII has been implicated in platelet adhesion to fibrin polymer, we intended to examine the association of Val34Leu with clopidogrel platelet inhibition, a potential confounder of ischemic risk after coronary stenting. Importantly, we could not find an association of Val34Leu with ADP induced platelet reactivity, a measure consistently associated with higher rates of stent thrombosis after coronary stenting. Limitations of our study include the relatively small number of subjects homozygous for the minor allele variant, and possible unidentified confounding factors in carriers of 34Leu variants.
In conclusion, among patients with coronary artery disease treated with dual antiplatelet therapy, carriers of Val34Leu variant demonstrate increased risk of recurrent MI and the combined endpoint of CV death and MI as compared to patients of wild type. Homozygous individuals for 34Leu exhibit faster rate of fibrin clot formation measured by TEG and increased risk for MI and the combined endpoint of CV death and MI as compared to heterozygous subjects or subjects of wild type. Further validation in large prospective studies examining ischemic events rates after coronary stenting is warranted.
Supplementary Material
Acknowledgments
This study was supported in part, by the Indiana Clinical and Translational Sciences Institute funded, in part by Grant Number (RR025761) from the National Institutes of Health, National Center for Research Resources, Clinical and Translational Sciences Award, as well as the Indiana University Health Values Grant, the Indiana University Health—Indiana University School of Medicine Strategic Research Initiative, and internal funding from the Department of Medicine, Indiana University School of Medicine, Indianapolis.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s11239-014-1059-4) contains supplementary material, which is available to authorized users.
Disclosure YJ is currently an employee of Eli & Lilly, Co. The other authors have no conflicts of interest to declare.
Contributor Information
Rolf P. Kreutz, Email: rkreutz@iu.edu, Krannert Institute of Cardiology, Indiana University School of Medicine, 1800 N. Capitol Ave, ME-400, Indianapolis, IN 46202, USA. Division of Clinical Pharmacology, Indiana University School of Medicine, Indianapolis, IN, USA
Abbas Bitar, Krannert Institute of Cardiology, Indiana University School of Medicine, 1800 N. Capitol Ave, ME-400, Indianapolis, IN 46202, USA.
Janelle Owens, Division of Clinical Pharmacology, Indiana University School of Medicine, Indianapolis, IN, USA.
Zeruesenay Desta, Division of Clinical Pharmacology, Indiana University School of Medicine, Indianapolis, IN, USA.
Jeffrey A. Breall, Krannert Institute of Cardiology, Indiana University School of Medicine, 1800 N. Capitol Ave, ME-400, Indianapolis, IN 46202, USA
Elisabeth von der Lohe, Krannert Institute of Cardiology, Indiana University School of Medicine, 1800 N. Capitol Ave, ME-400, Indianapolis, IN 46202, USA.
Anjan Sinha, Krannert Institute of Cardiology, Indiana University School of Medicine, 1800 N. Capitol Ave, ME-400, Indianapolis, IN 46202, USA.
Matteo Vatta, Krannert Institute of Cardiology, Indiana University School of Medicine, 1800 N. Capitol Ave, ME-400, Indianapolis, IN 46202, USA. Department of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, IN, USA.
Perry Nystrom, Division of Clinical Pharmacology, Indiana University School of Medicine, Indianapolis, IN, USA.
Yan Jin, Division of Clinical Pharmacology, Indiana University School of Medicine, Indianapolis, IN, USA.
David A. Flockhart, Division of Clinical Pharmacology, Indiana University School of Medicine, Indianapolis, IN, USA
References
- 1.Holmes DR, Jr, Kereiakes DJ, Garg S, Serruys PW, Dehmer GJ, Ellis SG, Williams DO, Kimura T, Moliterno DJ. Stent thrombosis. J Am Coll Cardiol. 2010;56:1357–1365. doi: 10.1016/j.jacc.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 2.Gurbel PA, Bliden KP, Guyer K, Cho PW, Zaman KA, Kreutz RP, Bassi AK, Tantry US. Platelet reactivity in patients and recurrent events post-stenting: results of the prepare post-stenting study. J Am Coll Cardiol. 2005;46:1820–1826. doi: 10.1016/j.jacc.2005.07.041. [DOI] [PubMed] [Google Scholar]
- 3.Price MJ, Berger PB, Teirstein PS, Tanguay JF, Angiolillo DJ, Spriggs D, Puri S, Robbins M, Garratt KN, Bertrand OF, Stillabower ME, Aragon JR, Kandzari DE, Stinis CT, Lee MS, Manoukian SV, Cannon CP, Schork NJ, Topol EJ GRAVITAS Investigators. Standard- vs. high-dose clopidogrel based on platelet function testing after percutaneous coronary intervention: the GRAVITAS randomized trial. JAMA. 2011;305:1097–1105. doi: 10.1001/jama.2011.290. [DOI] [PubMed] [Google Scholar]
- 4.Jneid H, Anderson JL, Wright RS, Adams CD, Bridges CR, Casey DE, Jr, Ettinger SM, Fesmire FM, Ganiats TG, Lincoff AM, Peterson ED, Philippides GJ, Theroux P, Wenger NK, Zidar JP, Anderson JL. 2012 ACCF/AHA focused update of the guideline for the management of patients with unstable angina/Non-ST-elevation myocardial infarction (updating the 2007 guideline and replacing the 2011 focused update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2012;26:875–910. doi: 10.1161/CIR.0b013e318256f1e0. [DOI] [PubMed] [Google Scholar]
- 5.Libby P, Ridker PM, Hansson GK. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol. 2009;54:2129–2138. doi: 10.1016/j.jacc.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Emerging Risk Factors Collaboration. Kaptoge S, Di Angelantonio E, Lowe G, Pepys MB, Thompson SG, Collins R, Danesh J. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet. 2010;375:132–140. doi: 10.1016/S0140-6736(09)61717-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park DW, Yun SC, Lee JY, Kim WJ, Kang SJ, Lee SW, Kim YH, Lee CW, Kim JJ, Park SW, Park SJ. C-reactive protein and the risk of stent thrombosis and cardiovascular events after drug-eluting stent implantation. Circulation. 2009;120:1987–1995. doi: 10.1161/CIRCULATIONAHA.109.876763. [DOI] [PubMed] [Google Scholar]
- 8.Tantry US, Bliden KP, Suarez TA, Kreutz RP, Dichiara J, Gurbel PA. Hypercoagulability, platelet function, inflammation and coronary artery disease acuity: results of the thrombotic risk progression (TRIP) study. Platelets. 2010;21:360–367. doi: 10.3109/09537100903548903. [DOI] [PubMed] [Google Scholar]
- 9.Gurbel PA, Bliden KP, Kreutz RP, Dichiara J, Antonino MJ, Tantry US. The link between heightened thrombogenicity and inflammation: pre-procedure characterization of the patient at high risk for recurrent events after stenting. Platelets. 2009;20:97–104. doi: 10.1080/09537100802687666. [DOI] [PubMed] [Google Scholar]
- 10.Kreutz RP, Owens J, Breall JA, Lu D, von der Lohe E, Bolad I, Sinha A, Flockhart DA. C-reactive protein and fibrin clot strength measured by thrombelastography after coronary stenting. Blood Coagul Fibrinol. 2013;24:321–326. doi: 10.1097/MBC.0b013e32835cc193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deloukas P, Kanoni S, Willenborg C, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2012;45:25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bagoly Z, Koncz Z, Hársfalvi J, Muszbek L. Factor XIII, clot structure, thrombosis. Thromb Res. 2012;129:382–387. doi: 10.1016/j.thromres.2011.11.040. [DOI] [PubMed] [Google Scholar]
- 13.Schroeder V, Chatterjee T, Kohler HP. Influence of blood coagulation factor XIII and FXIII Val34Leu on plasma clot formation measured by thrombelastography. Thromb Res. 2001;104:467–474. doi: 10.1016/s0049-3848(01)00395-4. [DOI] [PubMed] [Google Scholar]
- 14.Silvain J, Pena A, Vignalou JB, Hulot JS, Galier S, Cayla G, Bellemain-Appaix A, Barthélémy O, Beygui F, Bal-dit-Sollier C, Drouet L, Weisel JW, Montalescot G, Collet JP. FXIII-A Leu34 genetic variant in premature coronary artery disease: a genotype–phenotype case control study. Thromb Haemost. 2011;106:511–520. doi: 10.1160/TH11-01-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ariëns RA, Philippou H, Nagaswami C, Weisel JW, Lane DA, Grant PJ. The factor XIII V34L polymorphism accelerates thrombin activation of factor XIII and affects cross-linked fibrin structure. Blood. 2000;96:988–995. [PubMed] [Google Scholar]
- 16.Magwenzi SG, Ajjan RA, Standeven KF, Parapia LA, Naseem KM. Factor XIII supports platelet activation and enhances thrombus formation by matrix proteins under flow conditions. J Thromb Haemost. 2011;9:820–833. doi: 10.1111/j.1538-7836.2011.04234.x. [DOI] [PubMed] [Google Scholar]
- 17.Kreutz RP, Tantry US, Bliden KP, Gurbel PA. Inflammatory changes during the ‘common cold’ are associated with platelet activation and increased reactivity of platelets to agonists. Blood Coagul Fibrinol. 2007;18:713–718. doi: 10.1097/MBC.0b013e328201c77e. [DOI] [PubMed] [Google Scholar]
- 18.Thygesen K, Alpert JS, White HD. Joint ESC, ACCF, AHA, WHF Task Force for the redefinition of myocardial infarction, universal definition of myocardial infarction. Circulation. 2007;116:2634–2653. doi: 10.1161/CIRCULATIONAHA.107.187397. [DOI] [PubMed] [Google Scholar]
- 19.Rao SV, O’Grady K, Pieper KS, Granger CB, Newby LK, Mahaffey KW, Moliterno DJ, Lincoff AM, Armstrong PW, Van de Werf F, Califf RM, Harrington RA. A comparison of the clinical impact of bleeding measured by two different classifications among patients with acute coronary syndromes. J Am Coll Cardiol. 2006;47:809–816. doi: 10.1016/j.jacc.2005.09.060. [DOI] [PubMed] [Google Scholar]
- 20.Aleksic N, Ahn C, Wang YW, Juneja H, Folsom AR, Boerwinkle E, Wu KK. Factor XIIIA Val34Leu polymorphism does not predict risk of coronary heart disease: the Atherosclerosis Risk in Communities (ARIC) Study. Arterioscler Thromb Vasc Biol. 2002;22:348–352. doi: 10.1161/hq0202.102874. [DOI] [PubMed] [Google Scholar]
- 21.Atherosclerosis Thrombosis, and Vascular Biology Italian Study Group . No evidence of association between prothrombotic gene polymorphisms and the development of acute myocardial infarction at a young age. Circulation. 2003;107:1117–1122. doi: 10.1161/01.cir.0000051465.94572.d0. [DOI] [PubMed] [Google Scholar]
- 22.Bereczky Z, Balogh E, Katona E, Pocsai Z, Czuriga I, Széles G, Kárpáti L, Adány R, Edes I, Muszbek L. Modulation of the risk of coronary sclerosis/myocardial infarction by the interaction between factor XIII subunit A Val34Leu polymorphism and fibrinogen concentration in the high risk Hungarian population. Thromb Res. 2007;120:567–573. doi: 10.1016/j.thromres.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 23.Vokó Z, Bereczky Z, Katona E, Adány R, Muszbek L. Factor XIII Val34Leu variant protects against coronary artery disease: a meta-analysis. Thromb Haemost. 2007;97:458–463. [PubMed] [Google Scholar]
- 24.Smith NL, Bis JC, Biagiotti S, Rice K, Lumley T, Kooperberg C, Wiggins KL, Heckbert SR, Psaty BM. Variation in 24 hemostatic genes and associations with non-fatal myocardial infarction and ischemic stroke. J Thromb Haemost. 2008;6:45–53. doi: 10.1111/j.1538-7836.2007.02795.x. [DOI] [PubMed] [Google Scholar]
- 25.Marín F, González-Conejero R, Lee KW, Corral J, Roldán V, López F, Sogorb F, Caturla J, Lip GY, Vicente V. A pharmacogenetic effect of factor XIII valine 34 leucine polymorphism on fibrinolytic therapy for acute myocardial infarction. J Am Coll Cardiol. 2005;45:25–29. doi: 10.1016/j.jacc.2004.09.051. [DOI] [PubMed] [Google Scholar]
- 26.Hernández-Romero D, Marín F, Lee KW, Roldán V, Caturla J, Corral J, Valdés M, Lip GY, Vicente V, González-Conejero R. Synergism between factor XII-4C>T and factor XIII Val34Leu polymorphisms in fibrinolytic therapy in acute myocardial infarction. Thromb Haemost. 2010;104:650–652. doi: 10.1160/TH10-01-0022. [DOI] [PubMed] [Google Scholar]
- 27.Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK. Clopidogrel in unstable angina to prevent recurrent events trial investigators, effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med. 2001;345:494–502. doi: 10.1056/NEJMoa010746. [DOI] [PubMed] [Google Scholar]
- 28.Iakovou I, Schmidt T, Bonizzoni E, Ge L, Sangiorgi GM, Stankovic G, Airoldi F, Chieffo A, Montorfano M, Carlino M, Michev I, Corvaja N, Briguori C, Gerckens U, Grube E, Colombo A. Incidence, predictors, and outcome of thrombosis after successful implantation of drug-eluting stents. JAMA. 2005;293:2126. doi: 10.1001/jama.293.17.2126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.