

Abstract
Listeria monocytogenes is a major cause of mortality resulting from food poisoning in the United States. In mice, the fifth complement component (C5) has been genetically linked to host resistance to listeriosis. Despite this genetic association, it remains poorly understood how C5 and its activation products, C5a and C5b, confer host protection to this Gram-positive intracellular bacterium. Here we show in a systemic infection model that the major receptor for C5a, C5aR1, is required for a normal robust host immune response against L. monocytogenes. In comparison to WT mice, C5aR1−/− mice had reduced survival and increased bacterial burden in their livers and spleens. Infected C5aR1−/− mice exhibited a dramatic reduction in all major subsets of splenocytes, which was associated with elevated caspase-3 activity and increased TUNEL staining. As Type 1 IFN has been reported to impede the host response to L. monocytogenes through the promotion of splenocyte death, we examined the effect of C5aR1 on type 1 IFN expression in vivo. Indeed, serum levels of IFN-α and IFN-β were significantly elevated in L. monocytogenes-infected C5aR1−/− mice. Similarly, the expression of TRAIL, a type 1 IFN target gene and a pro-apoptotic factor was elevated in NK cells isolated from infected C5aR1−/− mice. Treatment of C5aR1−/− mice with a type 1 IFN receptor blocking antibody resulted in near complete rescue of L. monocytogenes-induced mortality. Thus, these findings reveal a critical role for C5aR1 in host defense against L. monocytogenes through the suppression of type 1 IFN expression.
Introduction
Listeria monocytogenes is a major cause of foodborne illness in the United States. A Gram positive, facultative intracellular pathogen, L. monocytogenes is commonly found in soil, sewage and ground water and thus contaminates unpasteurized dairy products, raw meat and produce. Its threat to the food supply is enhanced by its ability to survive and proliferate at low temperatures and under osmotic stress (1). The elderly and immune compromised are particularly susceptible to the development of systemic infections that can result in sepsis, meningitis and/or death. In pregnant woman L. monocytogenes may cross the placenta and infect the fetus, causing miscarriage, stillbirth or neonatal meningitis. Although cases of listeriosis are relatively uncommon, the mortality rate (20–30%) is much higher than that of more common foodborne illnesses like salmonellosis. As a consequence, L. monocytogenes is the third leading cause of death from food poisoning in the United States (2).
Ultimate clearance of L. monocytogenes is dependent on CD4+ and CD8+ lymphocytes (3, 4). However, a robust innate immune reaction must precede the lymphocyte response to provide early containment and initiate adaptive immunity. For example, studies in knockout mice have revealed that TNF-α, IFN-γ and IL-6 are required early in the course of infection for mobilization and activation of neutrophils, monocytes and macrophages (5–8). Although the type 2 interferon, IFN-γ, is critical in providing early host protection to L. monocytogenes, type 1 interferons are reported to impair the host response and create a permissive microenvironment to support L. monocytogenes growth. For example, mice deficient in the type 1 IFN receptor IFNAR1 are highly protected against L. monocytogenes, containing substantially less bacteria in their spleens 72 hours post infection (9–11). Mechanistically, this difference has been linked to the ability of type 1 IFN to sensitize lymphocytes to apoptosis (10, 12). Substantial amounts of TUNEL+ cells are seen in the spleens of infected WT mice, whereas IFNAR1−/− mice have relatively few. Apoptotic cells reportedly induce the expression of the anti-inflammatory cytokine IL-10 in macrophages, which in turn represses innate immunity (12, 13). Macrophages have also been observed to undergo cell death in response to L. monocytogenes in a type 1 IFN-dependent fashion (14). A major downstream target of type 1 IFN is TNF-related apoptosis-inducing ligand (TRAIL). A member of the TNF superfamily, TRAIL is a well-recognized IFN response gene (15). It induces cell death by binding to the death receptors DR4 and DR5. TRAIL expression is induced during L. monocytogenes infection in a type 1 IFN-dependent fashion primarily on the surface of NK cells (9, 16, 17). TRAIL−/− mice resemble IFNAR−/− mice in their enhanced containment of L. monocytogenes and reduced splenocyte depletion (16, 17).
An ancient and powerful arm of innate immunity is the complement system. L. monocytogenes triggers the alternative pathway of complement activation, resulting in its opsonization by C3b and release of the complement anaphylatoxins C3a and C5a (18–20). Several studies have shown an important role for C3 and its cleavage polypeptides in the host response to L. monocytogenes (19–24). In contrast, little is known about the contribution of C5 and its major activation fragments C5a and C5b. The A/J mouse is one of the most susceptible strains to infection with L. monocytogenes (25). This susceptibility is largely due to the absence of C5 protein caused by a 2-bp gene deletion in the 5′-exon of the structural gene encoding murine C5 (Hc locus) (26). The C5b fragment that initiates the formation of the C5b-9 complex is unlikely to be a factor in this susceptibility as Gram positive bacteria are protected against membrane attack complex (MAC)-mediated lysis by their thick layer of peptidoglycan (27). C5a is a 74 amino acid peptide that exerts its biological effects through a G-protein coupled receptor, C5aR1 (28). Classically described as an anaphylatoxin because of its ability to cause vasodilatation, histamine release and smooth muscle contraction, C5a is widely considered to be a pro-inflammatory molecule. This stems from its anaphylactic and chemotactic properties as well as its ability to enhance the expression of inflammatory cytokines like TNF-α, IL-6 and IL-1β (29–31). Accordingly, it seemed plausible that C5a might provide protection against L. monocytogenes by promoting the expression of cytokines needed for the early cellular immune response. To test this hypothesis we utilized a model of systemic L. monocytogenes infection in WT and C5aR1−/− mice. Surprisingly, we found that while C5aR1−/− mice are highly susceptible to L. monocytogenes, C5aR1 was not required for the early production of protective cytokines, including IFN-γ and TNF-α. Instead, C5a/C5aR1 protects the host from L. monocytogenes systemic infection through a previously unknown function of C5aR1--the suppression of type 1 IFN expression.
Materials and Methods
Mice
The C5aR1−/− mice used for these studies have been previously described (32). They were backcrossed for over ten generations onto the C57BL/6 background. Age-matched C57BL/6 mice from our colony served as WT controls. All mice were housed in HEPA-filtered Techniplast cages in a pathogen-free barrier facility. Male mice between 11 to 14 weeks of age were used in these studies. All mouse protocols followed institutional guidelines for animal care and welfare.
Bacterial infection
Listeria monocytogenes ATCC strain 13932 (MicroBioLogics, Inc.), a clinical isolate, was used for all studies. Bacteria were cultured in Bacto brain heart infusion (BHI) broth at 37°C to mid-logarthmic phase, pelleted by centrifugation, washed with PBS, and resuspended in PBS. Mice were infected i.v. with 1 × 105 bacteria in 100 μl PBS. Control mice received 100 μl PBS. The number of bacteria present in the inoculum was verified by culturing serial dilutions of the inoculum on Bacto BHI agar plates.
Survival study
Mice were infected i.v. with 5 × 104 L. monocytogenes and were observed every 6 hours. Mice that showed signs of severe morbidity were euthanized. For rescue experiments mice were injected i.p. with 1 mg of either the IFNAR blocking antibody, MAR1-5A3 (BioXCell), or an isotype control antibody, MOPC-21 (BioXCell), 4 hours before infection. Survival curves were generated using GraphPad Prism software, and statistical significance was assessed using the Logrank test.
Bacterial burden in the liver and spleen
Following exsanguination from the inferior vena cava, the liver and spleen were dissected from mice either 24 h or 72 h, rinsed in PBS and then placed in 2 ml HBSS. Organs were homogenized using a PRO200 homogenizer (ProScientific) on medium speed and were then placed on ice. Serial dilutions were plated on BHI agar plates to determine bacterial numbers per organ. Data are expressed as mean CFU per organ ± SEM.
Spleen histology
The whole spleen was dissected at 72 h, rinsed in PBS and fixed in 10% neutral buffered formalin for at least 24 h at 4°C. Organs were dehydrated, embedded in paraffin, cut into 5-μm sections and stained with hematoxylin & eosin. Brightfield images were taken using Spot Advanced software and a Zeiss Axioskop microscope (Carl Zeiss) equipped with a SPOT-RT digital camera (Diagnostic Instruments). For spleen histology a 20X objective was used for a final magnification of 200x.
Measurement of caspase-3 activity
Caspase-3 activity was measured in spleen homogenates using the CaspACE Assay System (Promega). Briefly, dissected spleens were cut in half. One half was used to enumerate the number of cells in the spleen, while the other half was homogenized as described above. After clearing the homogenate by centrifugation, caspase-3 activity was measured as per manufacturer’s instructions. The measured activity was normalized by the number of cells per spleen (per 107 cells) and is reported as mean absorbance (A405) ± SEM.
TUNEL staining
TUNEL staining was quantified from single cell suspensions of splenocytes using the HT TiterTACS Colorimetric Assay Kit (R&D Systems). Briefly, spleens were removed from mice at 72 h and were dissociated into single cell suspensions using a GentleMACS Dissociator (Miltenyi Biotec). Suspensions were filtered successively through 70 and 40 μM filters. Erythrocytes were then lysed with ACK lysis buffer (Lonza). Total live cell numbers were determined by cell counts with a hemocytometer using trypan blue exclusion. An equal number of cells were then used for the TUNEL assay according to the manufacturer’s instructions, with the exception that the assay was performed in 1.7 ml tubes instead of a 96 well plate. The samples were transferred to a 96 well plate at the detection step. The data is reported as mean absorbance (A450) ± SEM.
Flow Cytometry
Spleens were dissociated into single cell suspensions using a GentleMACS Dissociator. Suspensions were filtered successively through 100 and 40 μM filters. Erythrocytes were then lysed with ACK lysis buffer. Total live cell numbers were determined by cell counts with a hemocytometer using trypan blue exclusion. Fc receptors were blocked by incubation with an anti-CD16/32 antibody (BD Pharmingen). Cells were subsequently stained with antibodies for CD4 (GK1.5), CD8 (53-6.7), CD19 (6D5), NK1.1 (PK136), CD11b (M1/70), CD11c (N418), Ly6G (1A8), Ly6C (HK1.4), and/or TRAIL (N2B2) (Biolegend). During the final wash step DAPI (Invitrogen) was added as a viability dye. A minimum of 50,000 events were collected on a FACSAria (BD Biosciences) flow cytometer. For TRAIL expression studies 1,000,000 events were collected. Data analysis was done using the Kaluza program (Beckman Coulter). Dead cells were excluded from the analysis by gating on DAPI negative cells. Data are expressed as mean cell number per organ ± SEM.
Cytokine measurements
Most cytokines and chemokines were measured in sera or clarified liver homogenates taken at 24 & 72 h by the Milliplex Mouse Cytokine/Chemokine 22-plex kit (Millipore #MPXMCYTO70KPMX22) on the Luminex 200 system. Serum IFN-α and IFN-β levels at 24 h were measured using the VeriKine Mouse IFN Alpha ELISA kit and VeriKine Mouse IFN Beta ELISA kit (R&D Systems), respectively, as per manufacturer’s instructions.
Statistical analysis
Statistical analysis was done with GraphPad Prism 5. All values are expressed as mean values with the SEM as error bars. For experiments involving two groups, data were analyzed via unpaired two-tailed t test. In experiments involving multiple groups one-way ANOVA with the Tukey post-test was used to determine significance. Survival curves were analyzed by the log-rank (Mantel-Cox) test. P values less than 0.05 were considered significant.
Results
C5aR1 deficiency results in increased susceptibility to L. monocytogenes
We began our assessment of the role of C5aR1 in host defense against L. monocytogenes with a survival experiment. WT and C5aR1−/− mice were injected i.v. with 5×104 CFU of L. monocytogenes and then followed for two weeks. At this dose, no mortality was observed in WT mice (9 of 9 survived). In contrast, approximately 60% of C5aR1−/− mice succumbed to the infection within the first week (3 of 7 survived, p = 0.0103) (Fig. 1). To determine if C5aR1 contributes to the control of L. monocytogenes we infected WT and C5aR1−/− mice and then harvested livers and spleens at 24 and 72 h. At 24 h a modest two-fold elevation of L. monocytogenes was observed in the spleens of C5aR1−/− mice compared with WT mice (p = 0.0276) (Fig. 2A). No difference in bacterial burden was observed in the liver between the two genotypes at this time (Fig. 2A). By 72 h a marked difference in CFUs was observed in both organs (Fig. 2B). C5aR1−/− mice had approximately 6-fold more bacteria in their spleens (p < 0.0001) and 26-fold more in their livers (p = 0.0010) than WT mice (Fig. 2B). This elevation of bacterial burden roughly coincides with the onset of mortality in C5aR1−/− mice. Therefore, C5aR1 is important for the containment and survival of L. monocytogenes infection.
Figure 1. Impaired survival in C5aR1−/− mice during L. monocytogenes infection.

For survival studies WT and C5aR1−/− mice were infected i.v. with 5 × 104 CFU of L. monocytogenes and followed for two weeks. The data are pooled from two independent experiments. n = 9 for WT and n = 7 for C5aR1−/− mice, p = 0.0110 by the Log-rank test.
Figure 2. Defective bacterial clearance in C5aR1−/− mice during L. monocytogenes infection.

WT and C5aR1−/− mice were infected i.v. with 1 × 105 CFU of L. monocytogenes, and at 24 h (A) and 72 h (B) spleens and livers were dissected, homogenized and CFU per organ was determined. The data are pooled from two independent experiments and are presented as mean CFU per organ ± SEM. n = 9–12 mice per group per time point, n.s. = not significant, * p = 0.0276, ** p = 0.0010, *** p < 0.0001 by t test.
Greater spleen pathology in C5aR1−/− mice
To better understand the susceptibility of C5aR1−/− mice to L. monocytogenes, we next examined the histology of the spleen in infected WT and C5aR1−/− mice. L. monocytogenes initially enters the spleen through macrophages and CD8α+ dendritic cells of the red pulp (23, 33, 34). From there L. monocytogenes migrates into the white pulp via dendritic cells, establishes infective foci, and causes lymphocyte and myeloid cell depletion in a type 1 IFN-dependent fashion (9, 10). By H&E staining no obvious differences could be seen between the spleens of PBS-treated WT and C5aR1−/− mice (Fig. 3A). However, on infection the appearance of the spleens were markedly different between WT and C5aR1−/− mice at 72 h. In comparison with L. monocytogenes-infected WT spleens, infected C5aR1−/− spleens were strikingly hypocellular (Fig. 3A). This was most apparent in the splenic follicles of C5aR1−/− mice which lacked the typical densely packed appearance still observable in WT mice. To expand upon these histological observations, we next assessed the total number of viable cells in the spleens of uninfected and infected mice. No difference in splenocyte numbers was observed between uninfected WT and C5aR1−/− mice (Fig. 3B). Similarly, at 24 h splenocyte numbers were not significantly different from uninfected mice; moreover, at this time point no difference existed between WT and C5aR1−/− infected mice (Fig. 3B). In contrast, by 72 h there was a 43% reduction in splenocyte numbers in infected WT mice compared to uninfected WT controls (p=0.0097). In accord with the histolological data, at 72 h the infected C5aR1−/− mice had approximately 80% fewer splenocytes than infected WT mice (p=0.0007) (Fig. 3B).
Figure 3. Elevated spleen pathology and cell death in C5aR1−/− mice during L. monocytogenes infection.

(A) WT and C5aR1−/− mice were injected i.v. with PBS or 1 × 105 CFU of L. monocytogenes, and their spleens were removed 72 h and prepared for histology. Sections were stained with H&E and are representative of 4 mice per group (200X magnification). (B) WT and C5aR1−/− mice were infected i.v. with 1 × 105 CFU of L. monocytogenes, and their spleens were removed 24 h and 72 h for determination of total viable cell counts. Spleens from uninfected animals were used as controls. Data are representative of two independent experiments and are presented as mean cells per spleen +/− SEM. n = 3 per genotype for controls, n= 6 per each experimental time point and per each genotype. **p < 0.0097, ***p = 0.0007 by ANOVA with the Tukey post-test. (C) Spleen homogenates were prepared from PBS treated and L. monocytogenes-infected WT and C5aR1−/− mice at 72 h, and caspase-3 activity was measured. Data are presented as mean caspase-3 activity per 107 cells ± SEM. n = 4 per condition and genotype, *** p < 0.0001 by ANOVA with the Tukey post-test. (D) Splenocytes were harvested at 72 h, and apoptosis was quantitated using the TUNEL method. Data are presented as mean TUNEL+ staining (A450) ± SEM. n = 4 per genotype, *** p = 0.0002 by t test.
The depletion of splenocytes during listeriosis results primarily from caspase-3 dependent apoptotic cell death (9, 10, 17, 35). Caspase-3 is the key executioner caspase that initiates apoptosis and is often used to quantify apoptosis. Therefore, to test whether C5aR1 protects splenocytes from L. monocytogenes-induced apoptosis, we examined caspase-3 activity in spleen homogenates from PBS treated and infected WT and C5aR1−/− mice. As expected, PBS-treated animals had little caspase-3 activity (Fig. 3C). Following infection with L. monocytogenes both WT and C5aR1−/− exhibited significant increases in caspase-3 activity. Consistent with the data shown in Figs. 3A and 3B, infected C5aR1−/− mice had significantly more caspase-3 activity on a per cell basis than infected WT mice (Fig. 3C) (p < 0.0001). In addition, splenocytes taken from infected C5aR1−/− mice were also significantly more TUNEL+ than those from infected WT mice at 72 h (Fig. 3D) (p = 0.0002). Taken together, these results indicate that C5aR1 protects against the destruction of splenocytes during listeriosis by limiting L. monocytogenes-induced apoptosis.
C5aR1 broadly protects splenocytes in L. monocytogenes-infected mice
The spleen consists of many types of immune cells that play different roles in the course of an infection. C5aR1 is thought to be expressed by many of them, albeit with considerable variation in expression levels. Recent work indicated a role for C5aR1 in promoting T cell survival both in vitro and in vivo (36, 37). Therefore, it seemed important to determine if the increased cell loss observed in C5aR1−/− mice was specific for particular subsets of splenocytes. To test this we dissociated spleens, counted the number of viable cells, and then used cell surface staining along with a viability dye to identify the major subsets of live lymphocytes and myeloid cells. Similar numbers of B cells (CD19+), CD4+ & CD8+ T cells, NK cells (NK1.1+), neutrophils (CD11b+, Ly6G+/Ly6C+), monocytes (CD11b+, Ly6G-/Ly6C+) and dendritic cells (CD11c+) were observed in uninfected WT and C5aR1−/− mice (Fig. S1) and in infected mice at 24 h (Fig. 4A). However, by 72 h every cell type examined was significantly reduced in C5aR1−/− mice relative to their WT counterparts (Fig. 4B). C5aR1 thus broadly protects against splenocyte depletion in listeriosis.
Figure 4. Splenocyte depletion in C5aR1−/− mice during L. monocytogenes infection.
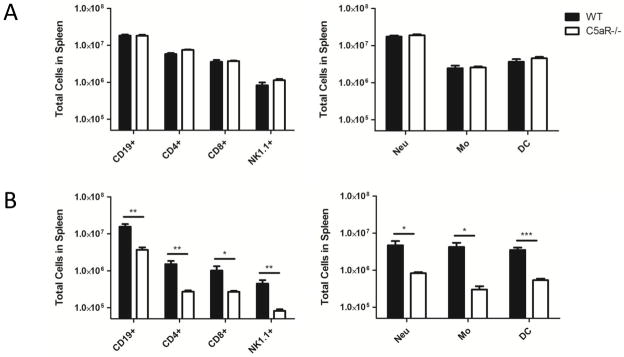
WT and C5aR1−/− mice were infected i.v. with 1 × 105 CFU of L. monocytogenes, and their spleens were removed 24 h and 72 h for determination of total viable cell counts. Splenocytes from infected mice were stained with various markers and the viability dye DAPI to determine the number of live splenocyte cell types at 24 h (A) and 72 h (B). Cells were defined as follows: Neu = neutrophils, CD11b+ Ly6G+ Ly6C+, Mo = monocytes, CD11b+ Ly6G− Ly6C+, DC = dendritic cells, CD11c +, B cells = CD19+, T cells = CD4+ or CD8+, and NK cells = NK1.1+. The data are pooled from two independent experiments. Data are presented as mean cells per spleen ± SEM. n = 3 per genotype for controls, 6 per time point and genotype. * p ≤ 0.0444, ** p ≤ 0.0074, *** p = 0.0003 by t test.
C5aR1 is not required for the expression of protective inflammatory cytokines and chemokines
Early resistance to L. monocytogenes infection has been attributed to the production of IFN-γ and TNF-α as well as other interleukins, cytokines and chemokines important for the recruitment and activation of monocytes/macrophages and neutrophils (5–8). As C5a potentiates inflammatory cytokine and chemokine expression in many models, we anticipated that the production of IFN-γ and TNF-α and other cytokines/chemokines protective during listeriosis might be defective in C5aR1−/− mice. We therefore examined serum cytokine and chemokine levels in WT and C5aR1−/− mice at 24 and 72 h through the Luminex platform. Contrary to our expectations, the expression of protective interleukins, cytokines and chemokines were either similar or elevated in C5aR1−/− mice compared with WT mice as early as 24 h (Fig. 5). Cytokine and chemokine levels in the liver showed a similar pattern as those in the serum (Fig. S2). Currently, it is not clear why the absence of C5aR1 during L. monocytogenes infection resulted in increased production of pro-inflammatory interleukins, cytokines, and chemokines. Possibly, the increased L. monocytogenes infection that occurred in the C5aR1−/− mice caused elevated inflammation that was mediated by other compensatory pro-inflammatory receptors. In any event, these data clearly indicate that there is no reduction of critical interleukins, cytokines, or chemokines that would cause the increased L. monocytogenes infection observed in the C5aR1−/− mice.
Figure 5. Protective cytokines and chemokines are either elevated or unchanged in C5aR1−/− mice during L. monocytogenes infection.

WT and C5aR1−/− mice were injected i.v. with 1 × 105 CFU of L. monocytogenes or PBS, and serum was isolated from the mice 24 h and 72 h. Cytokine and chemokine levels were measured using the Luminex platform. Sera from PBS injected animals had little to no detectable cytokines and chemokines, and no differences were observed between the genotypes (data not shown). Data from two independent experiments were combined and are presented as mean pg/ml ± SEM. n = 10–12 mice per genotype per time point. * p ≤ 0.0388, ** p ≤ 0.0086, *** p < 0.0001 by t test.
C5a represses type 1 IFN expression in vivo
Bacterial counts and spleen pathologies of L. monocytogenes-infected C5aR1−/− mice were in many ways completely opposite to those observed in L. monocytogenes-infected IFNAR−/− mice (9–12). For example, IFNAR−/− mice have significantly enhanced bacterial clearance (less CFUs) at 72 h. In contrast, at 72 h C5aR1−/− mice exhibited a dramatic increase in bacterial counts compared to WT mice. In addition, IFNAR−/− mice are protected against splenocyte death, whereas C5aR1−/− mice have greatly enhanced splenocyte depletion. These opposing parallels led us to suspect that C5a/C5aR1 might inhibit the type 1 IFN pathway in listeriosis. We therefore examined type 1 IFN expression in L. monocytogenes-infected WT and C5aR1−/− mice. There are two major types of type 1 IFN, IFN-α and IFN-β. In L. monocytogenes models IFN-α expression is partially dependent on IFN-β, and type 1 IFN expression peaks at 24 h (38–40). At this time point both IFN-α and IFN-β were significantly elevated in the serum of C5aR1−/− mice (p ≤ 0.0004) (Fig. 6A). As in prior reports, serum IFN-β levels were quite low (40, 42). However, a consistent difference was seen between WT and C5aR1−/− mice.
Figure 6. Type 1 IFN and its target TRAIL are elevated in C5aR1−/− mice during L. monocytogenes infection.

WT and C5aR1−/− mice were injected i.v. with 1 × 105 CFU of L. monocytogenes or PBS, and serum was isolated from the mice 24 h. (A) IFN-α and IFN-β were measured by ELISA. Sera from PBS injected animals had no detectable type 1 IFN (data not shown). Data from two independent experiments were combined and are presented as mean pg/ml ± SEM. n = 9–11 per genotype, *** p ≤ 0.0004 by t test. (B and C) WT and C5aR1−/− mice were treated with PBS or infected with L. monocytogenes and their spleens removed at 72 h. Splenocytes were stained with the viability dye DAPI, TRAIL-PE, and NK1.1-APC to determine the percentage of live TRAIL+ NK cells. Representative plots of TRAIL expression in DAPI-, NK1.1+ cells are shown (B). The percentage of live TRAIL+ NK cells in mice from two independent experiments are depicted (C). n = 6 per group, ** p = 0.0055, *** p = 0.0001 by ANOVA with the Tukey post-test.
TRAIL is a type 1 IFN response gene and an important mediator of splenocyte depletion during L. monocytogenes infection (15–17). A member of the TNF superfamily recognized for its ability to trigger apoptosis in immune cells, TRAIL expression is restricted to the surface of NK cells at 72 h (16). To determine whether the elevation of type 1 IFN observed in C5aR1−/− mice was biologically significant, we examined TRAIL expression on NK1.1+ NK cells by flow cytometry. In PBS-treated animals little to no TRAIL+ NK cells were observed (Fig. 6B,C). By 72 h a small fraction of NK cells was TRAIL+ in WT animals. The percentage of TRAIL+ NK cells was significantly higher in C5aR1−/− mice (p = 0.0001) (Fig. 6B,C). These data therefore show that C5aR1 inhibits type 1 IFN expression and its downstream target TRAIL during listeriosis.
Blockade of the type 1 interferon receptor rescues C5aR1−/− mice
IFN-α and IFN-β bind and signal through a common receptor comprised of the subunits IFNAR1 and IFNAR2 (41). If dysregulation of type 1 IFN expression is indeed responsible for the mortality observed in C5aR1−/− mice, then inhibition of the type 1 IFN axis should rescue them. To test this hypothesis we administered either an isotype antibody or an IFNAR1 blocking antibody (MAR1-5A3) i.p. 4 hours before i.v. infection (41). Similar to our earlier survival study, at a dose of 5×104 CFU, WT mice showed no mortality (12 of 12 survived) while C5aR1−/− mice were highly susceptible to L. monocytogenes infection (Fig. 7). C5aR1−/− mice given MAR1-5A3 were almost completely rescued (13 of 14 survived), whereas an isotype antibody failed to rescue them (3 of 14 survived) (p < 0.0001). Taken together, our data suggest that C5a protects mice during listeriosis through the repression of type 1 IFN expression.
Figure 7. Blocking IFNAR1 rescues C5aR1−/− mice from L. monocytogenes-induced mortality.

4 hours before infection C5aR1−/− mice were administered 1 mg of either the IFNAR1 blocking antibody MAR1-5A3 or an isotype antibody control MOPC-21 i.p. in PBS. WT and C5aR1−/− mice were then infected i.v. with 5 × 104 CFU of L. monocytogenes and followed for two weeks. The data are pooled from two independent experiments. n = 12–14 mice per condition, p < 0.0001 by the Log-rank test.
Discussion
Here we have demonstrated that the complement anaphylatoxin receptor C5aR1 protects mice against L. monocytogenes through the inhibition of type 1 IFN expression. The absence of C5aR1 in mice resulted in increased mortality and bacterial burden in the liver and spleen. Splenocyte depletion, a major feature of listeriosis that impedes the host response to L. monocytogenes, was sharply elevated in L. monocytogenes-infected C5aR1−/− mice. This splenocyte depletion was associated with increased caspase-3 activity in the spleens of C5aR1−/− mice, suggesting that it resulted from elevated L. monocytogenes-induced apoptosis. As L. monocytogenes causes lymphocyte apoptosis in a type 1 IFN dependent fashion, we examined serum IFN-α & β levels at 24 h and found that both were significantly elevated in C5aR1−/− mice in comparison with WT mice. This elevation was associated with increased NK expression of TRAIL, a type 1 IFN-response gene and a major driver of L. monocytogenes-induced splenocyte depletion. Finally, we showed that blockade of IFNAR rescued C5aR1−/− mice from L. monocytogenes-induced mortality, thereby demonstrating that the elevation of type 1 IFN seen in C5aR1−/− mice is responsible for their increased susceptibility to L. monocytogenes.
During the past few years, there has been growing evidence that C5a is critical in protecting cells from damage during tissue regeneration by providing pro-survival/anti-apoptotic signals via C5aR1 (42). For example, when C5aR1-deficient mice are subjected to liver injury, they exhibit severe hepatic apoptosis, preventing normal liver regeneration (43). In this model, C5a protects regenerating hepatocytes from apoptotic death indirectly by acting as an upstream mediator that increases STAT-3-dependent IL-6 and TNF-α gene expression via activation of C5aR1 bearing Kupffer cells (44). TNF-α and IL-6 are crucial regulators of the priming phase of liver regeneration, and IL-6 in particular is a major pro-survival factor for regenerating hepatocytes via the P13K/AKT/mTOR pathway (45). C5a has also received considerable attention recently as an important mediator of T-cell survival during the immune response. In vitro, constitutive signaling through C5aR1 was reported as necessary for optimal T cell survival (36). Similarly, T cell activation and expansion in vivo have been reported to require C5aR1 signaling, in part because C5aR1 signaling inhibits activation-induced T cell apoptosis (36, 37). Other in vivo studies that support a role for C5aR1 signaling in T-cell survival include a mouse model of influenza where the absence or antagonism of C5aR1 caused a reduction in the numbers of CD8+ T cells specific for influenza type A virus (46) and a mouse model of GVHD where the absence or antagonism of C5aR1 impaired T cell expansion (47). These in vitro and in vivo investigations collectively make a strong compelling case for the importance of C5aR1 in providing pro-survival signals to activated T-cells. Our investigations also support the importance of C5a/C5aR1 in providing anti-apoptotic signals to activated T-cells; but in contrast to the findings of Strainic et al (36), we did not observe any reduction in the number of T-cells in the spleens of naïve uninfected C5aR1−/− mice (Fig. 3B, 4A and Fig. S1). The C5aR1−/− mice used in our studies and those of Strainic et al originated from distinct colonies of founder C5aR1 knock-out mice (32, 48); therefore, possible genetic variability affecting survival of naïve T-cells (distinct or in concert with C5aR1 deficiency) may account for the different results obtained in these two investigations.
Similar to regenerating hepatocytes, the pro-survival/anti-apoptotic effect of C5a on T-cells is thought to occur indirectly through C5aR1 mediated production of cytokines that affect the P13K/AKT/mTOR pathway (36, 37). In addition to indirect protection, it has been reported that C5aR1 may also provide pro-survival signals directly to T-cells expressing C5aR1 (36, 37). However, the possibility of direct protection by C5a has been challenged by recent studies indicating that T-cells (naïve or activated) do not express C5aR1 (49). Here we have discovered a novel means by which C5aR1 can provide pro-survival activity through another indirect mechanism. Instead of bolstering the expression of protective cytokines such as TNF-α and IL-6 as in liver injury, C5aR1 protects against L. monocytogenes-induced splenocyte loss through the inhibition of type 1 IFN expression.
While generally thought of as a pro-inflammatory molecule, C5a can also adopt a regulatory role in certain contexts. The first demonstration of this came over a decade ago in two papers showing that C5a inhibits the expression of IL-12 in human macrophages in response to LPS and Staphylococcus aureus (50, 51). Since then this C5a inhibitory activity has been extended to include most members of the IL-12 cytokine family and additional stimuli such as CD40 activation and the intracellular bacteria Porphyromonas gingivalis (52, 53). Beyond the IL-12 family, C5a also represses the production of IL-17A in LPS-activated macrophages in vitro and in a mouse model of endotoxemia in vivo (54). Furthermore, the inhibitory effects of C5a are not limited to cytokines, as C5aR1 also suppresses the expression of the chemokines CCL17 and CCL22 in DCs during allergic asthma models (55). Pathogens even exploit the regulatory activity of C5a for their own gain. The gingival pathogen Porphyromonas gingivalis actively cleaves C5 to trigger crosstalk between C5aR1 and TLR2 that in turn inhibits the release of nitric oxide (56). While the effects of C5a/C5aR1 on a variety of cytokines have been examined, to date no one has examined how they regulate the type 1 IFNs. This report therefore adds type 1 IFNs to the scope of C5a/C5aR1’s regulatory functions for the first time.
It is increasingly appreciated that type 1 IFN can impair the host response to bacteria (57, 58). Studies with Salmonella typhimurium, Chlamydia muridarium, Brucella abortus, and L. monocytogenes have illustrated that type 1 IFN broadly promotes macrophage and lymphocyte death during intracellular bacterial infections (9, 10, 59–61). Beyond this, type 1 IFN can also repress anti-bacterial activity in other ways. For example, type 1 IFN appears to dampen the responsiveness of macrophages to IFN-γ during L. monocytogenes infection (62). Although type 1 IFN is believed to induce IL-10 expression during listeriosis through lymphocyte apoptosis, in other models type 1 IFN directly triggers IL-10 expression in macrophages and lymphocytes (57). Compared to WT infected mice, IL-10 serum levels in infected C5aR1−/− mice were increased by approximately 200-fold at 72 h post infection (Fig. 5), providing further evidence that increased levels of type 1 IFNs result in increased IL-10 production. Furthermore, type 1 IFN suppresses the expression of IL-17, a key anti-bacterial cytokine, in both innate γδ T cells and Th17 cells (63–65). C5aR1 may therefore have developed a regulatory role for type 1 IFN expression in order to limit detrimental effects during intracellular bacterial infections.
In summary, this study reveals in a mouse model the previously unknown, yet important function of C5aR1 in providing host defense against L. monocytogenes systemic infection through the impairment of L. monocytogenes-induced apoptosis of both myeloid and lymphoid cells required for ultimate clearance of this intracellular bacterium. Moreover, C5aR1 impairs cellular apoptosis during listeriosis not by increasing the production of pro-survival/anti-apoptotic cytokines such as TNF-α and IL-6, which are important in C5aR1-mediated liver regeneration and T-cell activation, but rather by suppressing the expression of Type 1 interferons and their downstream target TRAIL.
Supplementary Material
Acknowledgments
This work was supported by a National Institutes of Health Public Service Grant RO1 AI025011 (to RAW). Support was also provided by the Hans J. Muller-Eberhard and Irma Gigli Distinguished Chair in Immunology.
We thank Dr. Amy Hazen of the Brown Foundation Institute for Molecular Medicine Flow Cytometry Service Laboratory for her advice and assistance with flow cytometry. This work was performed by D.G.C. in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Sciences, The University of Texas Health Science Center at Houston Graduate School of Biomedical Sciences MD/PhD Program. We thank the members of Dr. Calame’s PhD advisory committee, Drs. Terry Walters, Brian Davis, Barrett Harvey, and Amber Luong.
Abbreviations
- C5aR1
C5a receptor
- BHI
brain heart infusion
References
- 1.Cossart P. Illuminating the landscape of host-pathogen interactions with the bacterium Listeria monocytogenes. Proc Natl Acad Sci USA. 2011;108:19484–19491. doi: 10.1073/pnas.1112371108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.CDC. Vital Signs: Listeria Illnesses, Deaths, and Outbreaks – United States, 2009–2011. MMWR Morb Mortal Wkly Rep. 2013;62:448–452. [PMC free article] [PubMed] [Google Scholar]
- 3.Unanue ER. Studies in listeriosis show the strong symbiosis between the innate cellular system and the T-cell response. Immunological Reviews. 1997;158:11–25. doi: 10.1111/j.1600-065x.1997.tb00988.x. [DOI] [PubMed] [Google Scholar]
- 4.Parmer EG. Immune responses to Listeria monocytogenes. Nature Reviews Immunology. 2004;4:812–823. doi: 10.1038/nri1461. [DOI] [PubMed] [Google Scholar]
- 5.Rothe J, Werner L, Lotscher H, Lang Y, Koebel P, Kontgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H. Mice lacking the tumor necrosis factor receptor 1 are resistant to IMF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature. 1993;364:798–802. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- 6.Pfeffer K, Matsuyama T, Kundig TM, Wakeham A, Kishihara K, Shahinian A, Wiegmann K, Ohashi PS, Kronke M, Mak TW. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to Listeria monocytogenes infection. Cell. 1993;73:457–467. doi: 10.1016/0092-8674(93)90134-c. [DOI] [PubMed] [Google Scholar]
- 7.Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, Vilcek J, Zinkernagel RM, Aguet M. Immune response in mice that lack the interferon-gamma receptor. Science. 1993;259:1742–1745. doi: 10.1126/science.8456301. [DOI] [PubMed] [Google Scholar]
- 8.Dalrymple SA, Lucian LA, Slattery R, McNeil T, Aud DM, Fuchino S, Lee F, Murray R. Interleukin-6-deficient mice are highly susceptible to Listeria monocytogenes infection: correlation with inefficient neutrophilia. Infect Immun. 1995;63:2262–2268. doi: 10.1128/iai.63.6.2262-2268.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, Perry AK, Nguyen BO, Lane TF, Taniguchi T, Miller JF, Cheng G. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med. 2004;200:437–445. doi: 10.1084/jem.20040712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carrero JA, Calderon B, Unanue ER. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J Exp Med. 2004;200:535–540. doi: 10.1084/jem.20040769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Auerbach V, Brockstedt DG, Meyer-Morse N, O’Riordan M, Portnoy DA. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med. 2004;200:527–433. doi: 10.1084/jem.20040976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carrero JA, Calderon B, Unanue ER. Lymphocytes are detrimental during the early innate immune response against Listeria monocytogenes. J Exp Med. 2006;203:933–940. doi: 10.1084/jem.20060045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dai WJ, Kohler G, Brombacher F. Both innate and acquired immunity to Listeria monocytogenes infection are increased in IL-10-deficient mice. J Immunol. 1997;158:2259–2267. [PubMed] [Google Scholar]
- 14.Stockinger S, Materna T, Stoiber D, Bayr L, Steinborn R, Kolbe T, Unger H, Chakraborty T, Levy DE, Muller M, Decker T. Production of type I IFN sensitizes macrophages to cell death induced by Listeria monocytogenes. J Immunol. 2002;169:6522–6529. doi: 10.4049/jimmunol.169.11.6522. [DOI] [PubMed] [Google Scholar]
- 15.Sato K, Hida S, Takayanagi H, Yokochi T, Kayagaki N, Takeda K, Yagita H, Okumura K, Tanaka N, Taniguchi T, Ogasawara K. Antiviral response by natural killer cells through TRAIL gene induction by IFN-α/β. Eur J Immunol. 2001;31:3138–3146. doi: 10.1002/1521-4141(200111)31:11<3138::aid-immu3138>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 16.Zheng S, Wang P, Tsabary G, Chen YH. Critical roles of TRAIL in hepatic cell death and hepatic inflammation. J Clin Invest. 2004;113:58–64. doi: 10.1172/JCI200419255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng S, Jiang J, Shen H, Chen YH. Reduced apoptosis and ameliorated listeriosis in TRAIL-null mice. J Immunol. 2004;173:5652–5658. doi: 10.4049/jimmunol.173.9.5652. [DOI] [PubMed] [Google Scholar]
- 18.Baker LA, Campbell PA, Hollister JR. Chemotaxigenesis and complement fixation by Listeria monocytogenes cell wall fractions. J Immunol. 1977;119:1723–1726. [PubMed] [Google Scholar]
- 19.Croize J, Arvieux J, Berche P, Colomb MG. Activation of the human complement alternative pathway by Listeria monocytogenes: evidence for direct binding and proteolysis of the C3 component on bacteria. Infect Immun. 1993;61:5134–5139. doi: 10.1128/iai.61.12.5134-5139.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drevets DA, Campbell PA. Roles of complement and complement receptor type 3 in phagocytosis of Listeria monocytogenes by inflammatory peritoneal macrophages. Infect Immun. 1991;59:2645–2652. doi: 10.1128/iai.59.8.2645-2652.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Helmy KY, Katschke KJ, Gorgani NN, Klijavin NM, Elliott JM, Diehl L, Scales SJ, Ghilardi N, van Lookeren Campagne M. CRIg: A macrophage complement receptor required for phagocytosis of circulating pathogens. Cell. 2006;124:915–927. doi: 10.1016/j.cell.2005.12.039. [DOI] [PubMed] [Google Scholar]
- 22.Nakayama Y, Kim SI, Kim EH, Lambris JD, Sandor M, Suresh M. C3 promotes expansion of CD8+ and CD4+ T cells in a Listeria monocytogenes infection. J Immunol. 2009;183:2921–2931. doi: 10.4049/jimmunol.0801191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verschoor A, Neuenhahn M, Navarini AA, Graef P, Plaumann A, Seidlmeier A, Nieswandt B, Massberg S, Zinkernagel RM, Hengartner H, Busch DH. A platelet-mediated system for shuttling blood-borne bacteria to CD8alpha+ dendritic cells depends on glycoprotein GPIb and complement C3. Nat Immunol. 2011;12:1194–1201. doi: 10.1038/ni.2140. [DOI] [PubMed] [Google Scholar]
- 24.Mueller-Ortiz SL, Morales JE, Wetsel RA. The receptor for the complement C3a anaphylatoxin (C3aR) provides host protection against Listeria monocytogenes-induced apoptosis. J Immunol. 2014;193:1278–1289. doi: 10.4049/jimmunol.1302787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gervais F, Stevenson M, Skamene E. Genetic control of resistance to Listeria monocytogenes: regulation of leukocyte inflammatory responses by the Hc locus. J Immunol. 1984;132:2078–2083. [PubMed] [Google Scholar]
- 26.Wetsel RA, Fleischer DT, Haviland DL. Deficiency of the murine fifth complement component (C5). A 2-base pair gene deletion in a 5′-exon. J Biol Chem. 1990;265:2435–2440. [PubMed] [Google Scholar]
- 27.Berends ET, Dekkers JF, Nijland R, Kuipers A, Soppe JA, van Strijp JA, Rooijakkers SH. Distinct localization of the complement C5b-9 complex on Gram-positive bacteria. Cell Microbiol. 2013;15:1955–1968. doi: 10.1111/cmi.12170. [DOI] [PubMed] [Google Scholar]
- 28.Wetsel RA, Kildsgaard J, Haviland DL. Complement anaphylatoxins (C3a, C4a, C5a) and their receptors (C3aR, C5aR/CD88) as therapeutic targets in inflammation. In: Lambris JD, Holers VM, editors. Contemporary Immunology: Therapeutic Interventions in the Complement System. Humana Press; Totowa, NJ: 2000. pp. 113–154. [Google Scholar]
- 29.Okusawa S, Yancey KB, van der Meer JW, Endres S, Lonnemann G, Hefter K, Frank MM, Burke JF, Dinarello CA, Gelfand JA. C5a stimulates secretion of tumor necrosis factor from human mononuclear cells in vitro. Comparison with secretion of interleukin 1 beta and interleukin 1 alpha. J Exp Med. 1988;168:443–448. doi: 10.1084/jem.168.1.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schindler R, Gelfand JA, Dinarello CA. Recombinant C5a stimulates transcription rather than translation of interleukin-1 (IL-1) and tumor necrosis factor: translational signal provided by lipopolysaccharide or IL-1 itself. Blood. 76:1631–1638. [PubMed] [Google Scholar]
- 31.Zhang X, Kimura Y, Fang C, Zhou L, Sfyroera G, Lambris JD, Wetsel RA, Miwa T, Song W. Regulation of Toll-like receptor-mediated inflammatory response by complement in vivo. Blood. 2007;110:228–236. doi: 10.1182/blood-2006-12-063636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hollmann TJ, Mueller-Ortiz SL, Braun MC, Wetsel RA. Disruption of the C5a receptor gene increases resistance to acute Gram-negative bacteremia and endotoxic shock: Opposing roles of C3a and C5a. Mol Immunol. 2008;45:1907–1915. doi: 10.1016/j.molimm.2007.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aoshi T, Carrero JA, Konjufca V, Koide Y, Unanue ER, Miller MJ. The cellular niche of Listeria monocytogenes infection changes rapidly in the spleen. Eur J Immunol. 2009;39:417–425. doi: 10.1002/eji.200838718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edelson BT, Bradstreet TR, Hildner K, Carrero JA, Frederick KE, Wumesh KC, Belizaire R, Aoshi T, Schreiber RD, Miller MJ, Murphy TL, Unanue ER, Murphy KM. CD8α+ dendritic cells are an obligate cellular entry point for productive infection by Listeria monocytogenes. Immunity. 35:236–248. doi: 10.1016/j.immuni.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Merrick JC, Edelson BT, Bhardwaj V, Swanson PE, Unanue ER. Lymphocyte apoptosis during early phase of Listeria infection in mice. Am J Pathol. 1997;151:785–792. [PMC free article] [PubMed] [Google Scholar]
- 36.Strainic MG, Liu J, Huang D, An F, Lalli PN, Mugim N, Shapiro VS, Dubyak GR, Heeger PS, Medof ME. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naïve CD4+ T cells. Immunity. 2008;28:425–435. doi: 10.1016/j.immuni.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, Heeger PS. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood. 2008;112:1759–1766. doi: 10.1182/blood-2008-04-151068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stockinger S, Kastner R, Kernbauer E, Pilz A, Westermayer S, Reutterer B, Soulat D, Stengl G, Vogl C, Frenz T, Waibler Z, Taniguchi T, Rulicke T, Kalinke U, Muller M, Decker T. Characterization of the interferon-producing cell in mice infected with Listeria monocytogenes. PLoS Pathog. 2009;5:e1000355. doi: 10.1371/journal.ppat.1000355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Solodova E, Jablonska J, Weiss S, Lienenklaus S. Production of IFN-β during Listeria monocytogenes infection is restricted to monocyte/macrophage lineage. PLoS One. 2011;6:e18543. doi: 10.1371/journal.pone.0018543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dresing P, Borkens S, Kocur M, Kropp S, Scheu S. A fluorescence reporter model defines “Tip-DCs” as the cellular source of interferon β in murine listerosis. PLoS One. 2010;5:e15567. doi: 10.1371/journal.pone.0015567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sheehan KCF, Lai KS, Dunn GP, Bruce AT, Diamond MS, Heutel JD, Dungo-Arthur C, Carrero JA, White JM, Hertzog PJ, Schreiber RD. Blocking monoclonal antibodies specific for mouse IFN-α/β receptor subunit 1 (IFNAR-1) from mice immunized by in vivo hydrodynamic transfection. J Interferon Cytokine Res. 2006;26:804–819. doi: 10.1089/jir.2006.26.804. [DOI] [PubMed] [Google Scholar]
- 42.Mastellos DC, Deangelis RA, Lambris JD. Complement-triggered pathways orchestrate regenerative responses throughout phylogenesis. Semin Immunol. 2013;25:29–38. doi: 10.1016/j.smim.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strey CW, Markiewski M, Mastellow D, Tudoran R, Spruce LA, Greenbaum LE, Lambris JD. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J Exp Med. 2003;198:913–923. doi: 10.1084/jem.20030374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Markiewski MM, DeAngelis RA, Strey CW, Foukas PG, Gerard C, Gerard N, Wetsel RA, Lambris JD. The regulation of liver cell survival by complement. J Immunol. 2009;182:5412–5418. doi: 10.4049/jimmunol.0804179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hong F, Nguyen VA, Shen X, Kunos G, Gao B. Rapid activation of protein kinase B/Akt has a key role in anti-apoptotic signaling during liver regeneration. Biochem Biophys Res Commun. 2000;279:974–979. doi: 10.1006/bbrc.2000.4044. [DOI] [PubMed] [Google Scholar]
- 46.Kim AH, Dimitriou ID, Holland MC, Mastellos D, Mueller YM, Altman JD, Lambris JD, Katsikis PD. Complement C5a receptor is essential for the optimal generation of antiviral CD8+ T cell responses. J Immunol. 2004;173:2524–2529. doi: 10.4049/jimmunol.173.4.2524. [DOI] [PubMed] [Google Scholar]
- 47.Kwan WH, Hashimoto D, Paz-Artal E, Ostrow K, Greter M, Raedler H, Medof ME, Merad M, Heeger PS. Antigen-presenting cell-derived complement modulates graft-versus-host disease. J Clin Invest. 2012;122:2234–2238. doi: 10.1172/JCI61019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hopken UE, Lu B, Gerard NP, Gerard C. The C5a chemoattractant receptor mediates mucosal defense to infection. Nature. 1996;383:86–89. doi: 10.1038/383086a0. [DOI] [PubMed] [Google Scholar]
- 49.Dunkelberger J, Zhou L, Miwa T, Song W-C. C5aR expression in a novel GFP reporter gene knockin mouse: implications for the mechanism of action of C5aR signaling in T cell immunity. J Immunol. 2012;188:4032–4042. doi: 10.4049/jimmunol.1103141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Braun MC, Lahey E, Kelsall BL. Selective suppression of IL-12 production by chemoattractants. J Immunol. 2000;164:3009–3017. doi: 10.4049/jimmunol.164.6.3009. [DOI] [PubMed] [Google Scholar]
- 51.Wittmann M, Zwirner J, Larsson V, Kirchhoff K, Begemann G, Kopp A, Gotze O, Werfel T. C5a suppresses the production of IL-12 by IFN-γ-primed and lipopolysaccharide-challenged human monocytes. J Immunol. 1999;162:6763–6769. [PubMed] [Google Scholar]
- 52.Hawlisch H, Belkaid Y, Baelder R, Hildeman D, Gerard C, Kohl J. C5a negatively regulates toll-like receptor 4-induced immune responses. Immunity. 2005;22:415–426. doi: 10.1016/j.immuni.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 53.Liang S, Krauss JL, Domon H, McIntosh ML, Hosur KB, Qu H, Li F, Tzekou A, Lambris JD, Hajishengallis G. The C5a receptor impairs IL-12-dependent clearance of Porphyromonas gingivalis and is required for induction of periodontal bone loss. J Immunol. 2010;186:869–877. doi: 10.4049/jimmunol.1003252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bosmann M, Sarma JV, Atefi G, Zetoune FS, Ward PA. Evidence for anti-inflammatory effects of C5a on the innate IL-17A/IL-23 axis. FASEB J. 2012;26:1640–1651. doi: 10.1096/fj.11-199216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kohl J, Baelder R, Lewkowich IP, Pandey MK, Hawlisch H, Wang L, Best J, Herman NS, Sproles AA, Zwirner J, Whitsett JA, Gerard C, Sfyroera G, Lambris JD, Wils-Karp M. A regulatory role for the C5a anaphylatoxin in type 2 immunity in asthma. J Clin Invest. 2006;116:783–796. doi: 10.1172/JCI26582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang M, Krauss JL, Domon H, Housr KB, Liang S, Magotti P, Triantafilou M, Triantafilou K, Lambris JD, Hajishengallis G. Microbial hijacking of complement-toll-like receptor crosstalk. Sci Signal. 2010;16:ra11. doi: 10.1126/scisignal.2000697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trinchieri G. Type I interferon: friend or foe? J Exp Med. 2010;207:2053–2063. doi: 10.1084/jem.20101664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carrero JA. Confounding roles for type I interferons during bacterial and viral pathogenesis. Int Immunol. 2013;25:663–669. doi: 10.1093/intimm/dxt050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol. 2012;13:954–962. doi: 10.1038/ni.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qiu H, Fan Y, Joyee AG, Wang S, Han X, Bai H, Jiao L, Rooijen NV, Yang X. Type I IFNs enhance susceptibility to Chlamydia muridarum lung infection by enhancing apoptosis of local macrophages. J Immunol. 2008;181:2092–2102. doi: 10.4049/jimmunol.181.3.2092. [DOI] [PubMed] [Google Scholar]
- 61.de Almeida LA, Carvalho NB, Oliveira FS, Lacerda TLS, Vasconcelos AC, Nogueira L, Bafica A, Silva AM, Oliveira SC. MyD88 and STING signaling pathways are required for IRF3-mediated induction in response to Brucella abortus infection. PLoS One. 2011;6:e23135. doi: 10.1371/journal.pone.0023135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rayamajhi M, Humann J, Penheiter K, Andreasen K, Lenz LL. Induction of IFN-αβ enables Listeria monocytogenes to suppress macrophage activation by IFN-γ. J Exp Med. 2010;207:327–337. doi: 10.1084/jem.20091746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Henry T, Kirimanjeswara GS, Ruby T, Jones JW, Peng K, Perret M, Ho L, Sauer J, Iwakura Y, Metzger DW, Monack DM. Type I IFN signaling constrains IL-17A/F secretion by γδ T Cells during bacterial infections. J Immunol. 2010;184:3755–3767. doi: 10.4049/jimmunol.0902065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meeks KD, Sieve AN, Kolls JK, Ghilardi N, Berg RE. IL-23 is required for protection against systemic infection with Listeria monocytogenes. J Immunol. 2009;183:8026–8034. doi: 10.4049/jimmunol.0901588. [DOI] [PubMed] [Google Scholar]
- 65.Guo B, Chang EY, Cheng G. The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J Clin Invest. 2008;118:1680–1690. doi: 10.1172/JCI33342. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.