

Abstract
High-fat diet (HFD) and inflammation are the key contributors to insulin resistance and type 2 diabetes (T2D). Previous study shows fatty acid-induced accumulation of damaged, reactive oxygen species (ROS)-generating mitochondria, and this in turn activates the NLRP3 inflammasome interference with insulin signaling. Our previous research shows NLRP3 inflammasome activation signal originates from defects in autophagy. Yet how the fatty acid related to mitophagy alteration leads to the activation of NLRP3-ASC inflammasome has not been considered. Here we demonstrated that palmitate (PA) induced mitophagy deficiency, leading to damaged mitochondrion as characterized by mito-ROS production and loss of membrane potential. Antioxidant APDC or Ca2+ signaling inhibitor Nifedipine blocked PA-induced NLRP3 inflammasome activation. Further, we provided evidences that PA reduced the expression of Ras homolog enriched in brain (Rheb) and disrupted Rheb recruitment to the mitochondrial outer membrane. In addition, sustained PA caused disassociation of kinesin family member 5B (KIF5B) from binding with mitochondria via Ca2+-dependent effects. Disruption of Rheb and KIF5B interaction with mitochondria blocked mitochondrial degradation along with IL-1β dependent insulin resistance, which was majorly attenuated by Rheb/KIF5B overexpression. In a consequence, defective mitophagy led to the accumulation of damaged-ROS-generating mitochondria, down pathway of NLRP3-ASC-Caspase 1 activation, and subsequently, insulin resistance. These findings provide insights into the association of inflammation, mitophagy and T2D.
Keywords: Mitophagy, Proinflammatory response, Rheb, KIF5B
Abbreviations: HFD, high-fat diet; BMM, bone marrow-derived macrophage; MCP-1, monocyte chemotactic protein-1; PA, palmitic acid; T2D, type 2 diabetes; ROS, reactive oxygen species; Lamp, lysosome-associated membrane protein; LC3I/II, light chain 3I/II; DLP1, dynamin-like protein; NLR, NOD-like receptors; CHX, cycloheximide; LPS, lipopolysaccharide; rheb, Ras homolog enriched in brain; KIF5B, kinesin family member 5B
Highlights
-
•
PA induced disruption of KIF5B-mediated mitochondrial motility and loss of Rheb-dependent mitophagy.
-
•
Defective mitophagy led to NLRP3-ASC-Caspase 1 activation, and subsequently, insulin resistance.
-
•
Antioxidant APDC or Ca2+ signaling inhibitor nifidipine blocked PA-induced NLRP3 inflammasome activation.
Graphical abstract
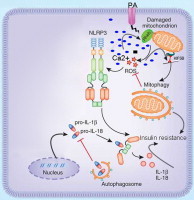
Introduction
The prevalence of T2D has increased markedly both in developing and developed countries. It is a low-grade inflammatory disease with insulin resistance caused by glucolipotoxicity in islets of diabetic patients and animal models. The discovery of NOD-like receptors (NLRs) as essential components of the immune system triggered significant interest in the study of their contributions to be a key regulator of glucose and insulin homeostasis [1–3]. This association between metabolic stress and inflammation suggests a causal link, which is now supported by experimental data from in vitro and in vivo. in vitro, palmitic acid (PA) induces proinflammatory responses in many cell types such as macrophage, rat insulinoma line, and islets [4–6]. PA involves in the production of reactive oxygen species (ROS), which in turn induces stress kinases. ROS may also lead to the formation of NLRP3 inflammasomes, which activates the IL-1 system. in vivo, several studies using genetically modified mice that lack inflammasome components NLRP3, ASC, and Caspase-1 displaying improved insulin sensitivity provides initial evidence that activation of the NLRP3 inflammasome is a key mechanism that induces metabolic inflammation and insulin resistance [2,7–9]. Taken together, previous research data suggests that HFG triggers aberrant activation of the NLRP3-ASC-Caspase 1 immune system in metabolic disorders, and the mitochondrion appears to be the common pathway for these events.
Mitochondria in beta cells plays an essential role by metabolizing nutrients and generating signals required for both triggering and amplifying pathways of insulin secretion respond to changes in extra-cellular nutrients. Sustained HFD-induced stress leads to damaged mitochondria within cells [10]. The elimination of cytosolic components inside cells such as damaged organelles can occur through autophagy. Mitophagy is a specific form of autophagy in which damaged mitochondria are specifically targeted for autophagic degradation by the lysosomes to dampen the proinflammatory activation [11,12]. The premise that mitophagy dysfunction plays a main role in HFD-induced NLRP3 dependent proinflammatory responses and later on T2D initiates a growing interest on this topic in order to validate the hypothesis [13]. Our previous research showed that PA induced NLRP3 dependent proinflammatory responses dependent of autophagy activation status [5]. However, how the mitophagy machinery responds to metabolic stress imposed by PA linking to the NLRP3-ASC-Caspase 1 proinflammatory response is far from clear.
In this report, we demonstrated that PA-induced disruption of kinesin family member 5B (KIF5B)-mediated mitochondrial motility and loss of the Ras homolog enriched in brain (Rheb)-dependent mitophagy resulted in defective mitophagy, leading to the accumulation of damaged-mitochondria-producing-ROS, and down pathway of NLRP3 dependent proinflammatory responses, and subsequently, caused IL-1β dependent insulin resistance. Our results therefore provided a possible mechanism for mitophagy deficiency under PA stress in T2D.
Materials and methods
Cell culture and stimulation
Rat Insulinoma INS-1E cells were grown in RPMI 1640 medium as described previously [48]. Bone marrow-derived macrophages (BMMs) were generated in the presence of L-929 conditional medium and granulocyte-macrophage colony-stimulating factor. After pretreatment with ultrapure lipopolysaccharide (LPS) (200 ng/ml) for 3 h, BMMs were stimulated with PA-BSA (500 μM) for 24 h as indicated. Where indicated, chemical inhibitors were added 30 min before cell stimulation (2.5 h after LPS priming). Supernatant and cell lysate were collected for ELISA and immunoblot analysis.
PA-BSA solution preparation
Sodium PA was dissolved in 95% ethanol at 60 °C to yield a stock concentration of 10 mM and kept at −20 °C. Nitrogen gas was used to completely remove ethanol from PA stock solution. PA was then conjugated with fatty acid-free BSA at a 3:1 molar ratio.
Cell transfection
Islets or BMMs were transfected as appropriate described in the text with Lipofectamine 2000 reagent according to the manufacturer’s suggested protocol. In experiments studying caspase inhibition in PA-induced inflammasome activation, BMMs were transfected with siNLRP3. The transfection efficiency for islets and BMMs was 80% by jetPEI kits (Polyplus). In the experiments studying the mitophagy deficiency in IL-1β dependent insulin secretion, islets were transfected with siRheb or siKIF5B or siAtg7 or PCMV-GFP-Rheb or PCMV-GFP-KIF5B as appropriately described in the text.
Confocal microscopy
Fixed and permeabilized BMMs or INS-1E or islets were used for confocal microscopy. Cells were grown and transfected as described above on one thickness 2-well Lab-Tek chambered cover glass. After 24 h, confocal microscopy of live cells was performed by incubation with 20 ng/ml of a mitochondrion-specific dye and/or 1–3 M staurosporine as indicated in individual experiments. Images were collected on an LSM 410 microscope with a 401.2 NA Apochromat objective. 568 nm lines of a krypton/argon laser were used for fluorescence excitation of Mitotracker red CMXRos. For immunofluorescent labeling, the cells were washed with PBS followed by fixation with 2% paraformaldehyde and permeabilization with 0.04% saponin. The fixed cells were blocked with normal goat serum, probed with mouse anti-Rheb or KIF5B monoclonal antibodies and stained with Texas red-conjugated goat anti-mouse IgG antibodies. The cover glasses finally were washed, mounted, and examined using the confocal laser microscope.
ELISA
Cytokines in culture supernatants were measured with ELISA kits.
Western blot analysis
Cell lysates were separated by electrophoresis prior to transfer to PVDF membranes. The membranes were then probed with interest antibody and immunoreactive bands were detected by chemiluminescence. Images were captured and data were analyzed using a BioRad ChemiDoc™ XRS + imaging system. Data were normalized relative to actin. The following primary antibodies were used for Western blots and immunostaining: rabbit anti-LC3 (Sigma, L8918); Rat monoclonal to LAMP2 (Abcam, ab13524); anti-β actin antibody (Abcam, ab3280); rabbit polyclonal anti-Atg7 (Cell Signaling Technology, 2631); Anti-IL-1β antibody (Abcam, ab2105); Anti-IL6 antibody (Abcam, ab6672); Anti-MCP1 antibody (Abcam, ab9669); Anti-TNF alpha antibody (Abcam, ab9635); guinea pig polyclonal anti-p62 (ProGen, GP62-N); anti-Rheb(Abcam, ab2587); anti-Tom20 (SantaCruz, sc-17764); anti-calregulin (SantaCruz, sc-6468-R); anti-Gapdh (Abcam, ab9483); anti-Golgi 58 (Abcam, ab27043); Alexa Fluor-conjugated antibodies (Molecular probes, A21057, A21076, A21096) were used as secondary antibodies. Data for LC3 Western blot analysis was obtained in the presence of an autophagic flux inhibitor bafilomycin (10 nM) to exclude the possibility that defective autophagy/inhibited flux increased LC3-II.
Mitochondrial isolation
Mitochondria were isolated using mitochondrial isolation kit according to the manufacturer’s instructions (Pierce). Briefly, cells were homogenized in a dounce homogenizer and then centrifuged at 750gfor 10 min at 4 °C. The supernatant was further centrifuged at 12,000g for 15 min at 4 °C. The pellet was then washed and kept as the mitochondrial fraction. The supernatant was further centrifuged at 100,000gfor 1 h at 4 °C and designated as the cytosolic fraction.
Mitochondrial morphology
Cells were grown on poly-d-lysine coated glass coverslips. Mitochondria were labeled with Mitotracker Red (50 nM, Invitrogen) at 37 °C for 30 min and fixed with 4% paraformaldehyde according to the manual’s instructions. Coverslips were subsequently mounted with Prolong Gold Antifade Reagent with DAPI (Invitrogen). Fluorescence images were captured at room temperature using a camera mounted to an inverted epifluorescence microscope. Overall contrast and brightness of acquired images were adjusted using Adobe Photoshop CS6.
Detection of reactive species
Treated islets were washed in PBS and placed in PBS containing 10 µM 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (DHR), di(acetoxymethyl ester) for 30 min at 37 °C. Following a PBS wash and re-incubation in PBS for 1 h at 37 °C, 10 µM Hydrogen Peroxide (H2O2) for 2 h was used as a positive control. Fluorescence was measured on a SpectraMax M5 plate reader; an emission wavelength 480 nm; excitation wavelength 530 nm.
Islet insulin secretion and insulin content measurements
Before experiments, islets were cultured for 1 h in RPMI-1640 medium. Isolated islets were distributed into 12-well plates (6 islets/well in triplicates) and incubated for 2 h in 1 ml RPMI complete medium containing 3 mM glucose. After pretreatment with ultrapure LPS for 3 h, they were then washed and preincubated for 40 min at 37 °C in 1 ml Krebs-Ringer bicarbonate buffer containing 10 mM HEPES (KRBH; pH 7.4), 0.5% defatted BSA, and 3 mM glucose plus 0.4 mM PA in the presence of Rheb or KIF5B or siControl or siRheb or siKIF5B. The islets were then preincubated for 45 min at 37 °C in 0.5 ml Krebs-Ringer bicarbonate buffer containing 16.7 mM glucose. Supernatant was collected and cells were lysed by 0.15% HCl. Secreted insulin and cellular insulin content was monitored by RIA using the rat insulin RIA kit (Millipore). Insulin secretion index (16.87 mmol/l GSIS over 3 mmol/l GSIS) was calculated. Total intracellular insulin content was extracted by the acid/ethanol method. Briefly, cells were incubated in 1% hydrochloric acid alcohol (ethanol/H2O/HCl, 14:57:3) overnight at 4 °C. The insulin in the supernatant was detected by RIA (Linco, Research, St. Charles, MO), and normalized to total protein content as described previously [49].
Flow cytometry
MitoSOX, MitoTracker Green and Red, and DCFDA staining were done according to manufacturer’s instructions (Invitrogen). Data were acquired with a FACSCalibur flow cytometer (BD Biosciences).
Statistics
The data are presented as the mean ± SD of three independent experiments unless otherwise noted. Differences between means were analyzed using either one-way or two-way ANOVA followed by Newman–Keuls post-hoc testing for pair-wise comparison using SPSS. The null hypothesis was rejected when the p-value < 0.05.
Results
Sustained PA + LPS blocks mitochondrial degradation along with proinflammatory responses
PA is one of the most abundant saturated fatty acids in plasma and is substantially elevated following HFD. Our previous research demonstrated that 0.5 mM PA induced autophagy without causing necrosis cell death in INS-1 cells [5], and data from other groups showed that exposing cells to lipopolysaccharide (LPS) induced the transcription and translation of pro-IL-1β and pro-IL-18 [14], and autophagy [15] in macrophages. Exposing cells to LPS and then to PA conjugated to fatty acid-free medium induced NLRP3-ASC-Caspase 1 proinflammatory responses [4]. Based on this, we treated LPS-primed macrophages with 0.5 mM PA for examining the relationship between mitophagy and proinflammatory responses. As showed in Fig. 1, PA induced IL-1β and IL-18 secretion (Fig. 1A and B) (***, p < 0.001). In contrast to IL-1β production, the secretion of IL-6 or TNF-α, which depended on Toll-like receptor signaling only, was not significantly increased by PA (p = 0.172; Fig. 1C and D), indicating specificity of the PA effect on cytokines production. To investigate whether the PA effect on the proinflammatory responses was driven by mitophagy dysfunction, we first measured mitochondrial proteins degradation in LPS-primed macrophages at 0, 24 h treatment by 0.5 mM PA. We examined the rate of protein degradation by treating macrophage cells with the protein synthesis inhibitor, cycloheximide (CHX). As a result, cells with LPS demonstrated a higher rate of mitochondrial protein degradation compared to LPS + 0.5 mM PA. Effect of CHX treatment on protein levels of other cellular compartments such as Golgi (Golgi-58), lysosome (Lamp2), endoplasmic reticulum (calregulin), or cytosol (Gapdh, Actin) were independent of PA effects (*, p < 0.05; Fig. 1E) suggesting that solely mitochondrial protein degradation rate was impacted under PA stress.
Fig. 1.
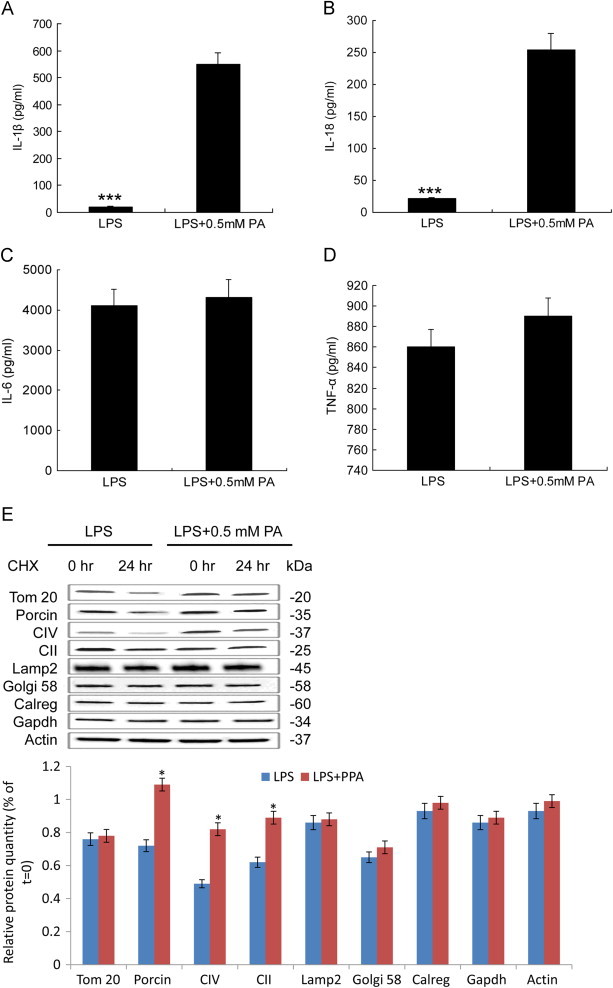
PA activates pro-inflammatory cytokines expression. (A–C) ELISA for IL-1β (A), IL-18 (B), and IL-6 (C), and TNF-α (D) in supernatants of LPS primed BMMs stimulated with PA. (E) Protein degradation rates were measured in macrophage cells grown under LPS and LPS + 0.5 mM PA conditions at 0 and 24 h of treatment with 10 mg/ml cycloheximide (CHX). Level of the mitochondrial proteins was analyzed using antibodies against SDHA subunit of complex II (CII), ATP synthase (CV), porin, and Tom20. Golgi apparatus, cytosol, lysosomes, and endoplasmic reticulum were analyzed using Golgi58, Gapdh, Lamp2, and calregulin (calreg) antibodies. Quantification of protein level after 24 h CHX treatment on macrophage cells was showed in down panel. Black bars are LPS, and gray bars are in LPS + 0.5 mM PA condition. Values are percentage of respective protein amount at t = 0 (n = 3). Dashed line represents value at t = 0 and normalized to 100% for each protein. *p < 0.05, ***p < 0.001. Data are presented as mean ± SD.
Inhibition of mitochondrial protein degradation was due to mitophagy deficiency dependent of Ca2+
We proposed that this blockage of mitochondrial proteins degradation was due to defective mitophagy. To test this, we examined mitophagy using a combination of immunoblot, microscopy, and pharmacological inhibition analyses. First, we analyzed the proteolytic conversion of the microtubule-associated protein light chain 3 (LC3-I) into the LC3-II isoform, which was conjugated to phosphatidylethanolamine and targeted to autophagic membranes upon the induction of autophagy. We found that the activation of autophagy was inhibited by PA in macrophage characterized by reduction of LC3II/LC3I, Lamp2 and elevation of P62 after 24 h of incubation LPS + PA in contrast to the control (Fig. 2A). These results were further confirmed by quantification of LC3II/Actin demonstrated in the right panel (**, p < 0.01; Fig. 2A) and immunofluorescence staining of LC3II, Lamp2 and P62 in LPS + PA group compared with LPS group (Fig. 2B). To test whether the mitophagic deficiency was responsible for the accumulation of mitochondrial proteins upon LPS + PA treatment, we analyzed the effect of autophagic inhibition on the mitochondrial degradation. Inhibition of autophagic/lysosomal degradation using protease inhibitors PepA/E64D or by silencing the autophagic protein 7 (Atg7) hampered mitochondrial degradation (Fig. 2C and D). We assumed that inhibition of mitophagy led to the accumulation of ROS-producing damaged mitochondria dependent of Ca2+, and as a consequence, to the activation of the inflammasome. To test this, we treated BMMs with antioxidant APDC and Ca2+ signaling inhibitor Nifedipine. As expected, inhibition of mitophagy by PA resulted in increased concentrations of mitochondrial ROS (*, p < 0.05, Fig. 2E, up) and PA -induced ROS generation was paralleled by augmented secretion of IL-1β upon mitophagy inhibition (Fig. 1A), which was blocked by the antioxidant APDC or Ca2+ signaling inhibitor Nifedipine (**, p < 0.01, Fig. 2E, down).
Fig. 2.
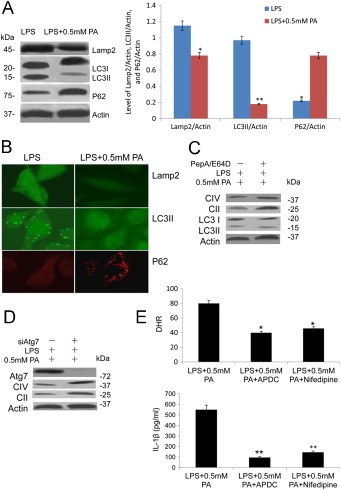
PA blocked proper mitophagy activation in macrophage cells. (A) PA stimulated autophagy activation associated proteins LC3II/LC3I, P62, Lamp2, and CTSB expression were analyzed by immunoblotting, β-actin as a control; analysis of Lamp2, LC3II, and P62 levels from macrophage cells were on the right panel (*, p < 0.05; **, p < 0.01). (B) Immunofluorescence staining of PA stimulated autophagy activation associated proteins Lamp2, LC3II, and P62 in macrophage cells under LPS and LPS + PA treatment. (C and D) Effect of autophagy inhibition on mitochondrial degradation assayed by treatment protease inhibitors, pepstatin/E64D (C) or by ATG7 silencing (D). (E) PA induced increased concentrations of mito-ROS detected by dichlorodihydrofluorescein (DHR) and was blocked by ROS antioxidant APDC or Ca2+ signaling inhibitor nifedipine (*, p < 0.05, up). PA-induced augmented secretion of IL-1β was blocked by the antioxidant APDC or nifedipine. Data are presented as mean ± SD (**, p < 0.01, down).
PA-induced mitophagy deficiency responsible for damaged mitochondrion-producing-mtROS mediated NLRP3 dependent proinflammatory responses
Caspase-1 is one of cysteine proteases that initiate or execute cellular programs, leading to inflammation or cell death. Its catalytic activity is tightly regulated by signal-dependent autoactivation within the NLRP3 inflammasome that mediates caspase-1-dependent processing of IL-1β. To examine the role of NLRP3 inflammasome components in the activation of caspase-1 caused by mitophagy inhibition, NLRP3 in macrophage cells was depleted by siRNA. Compared to non-target siRNA control, protein levels of NLRP3 knockdown cells were totally inhibited (Fig. 3A). NLRP3 and nontarget control knockdown cells were then challenged with additional treatments, and caspase-1 activation was revealed by the appearance of caspase-1 and IL-1β in Western blots. When compared to non-target control cell group, depletion of NLRP3 caused distinct reduction for IL-1β in (0.5 mM PA + LPS+siNLRP3) group (Fig. 3B). Concomitantly, distinct reduction of caspase-1 levels was also observed (Fig. 3C). Taken together, the results showed that NLRP3 was required for mitophagy-deficiency-induced caspase-1 activation and IL-1β processing in macrophage cells. To investigate PA-induced mitophagy deficiency induced damaged mitochondrion-producing ROS, we measured mito-ROS production by three types of mitochondria-specific labels that distinguish respiring (Mitotracker deep red), total (Mitotracker green) and ROS-generating mitochondria (MitoSOX). We found that PA stress resulted in robust ROS production and loss of mitochondrial membrane potential (Fig. 3D and E), and these efforts were blocked by ROS inhibitor APDC or Ca2+ signaling inhibitor Nifedipine. Because these processes were required for NLRP3 inflammasome activation, these findings suggested that the critical role of Ca2+ signaling, at least during PA stimulation, was to mediate mitochondrial damage.
Fig. 3.
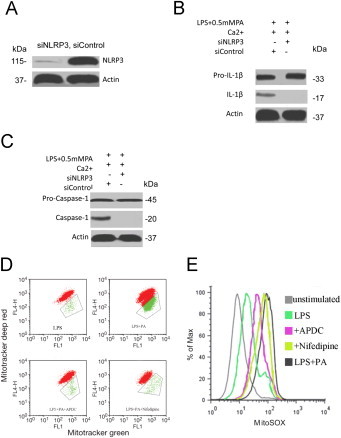
NLRP3 knockdown blocked mitophagy deficiency induced proinflammatory responses. (A) Macrophage cells were stably transfected with siRNAs that targeted NLRP3, and protein levels of NLRP3 knockdown cells were analyzed by Western blot analysis. NLRP3, or non-target control (siRNA Ctrl) knockdown cells were treated with 0.5 mM PA + LPS. (B) Secreted IL-1β and (C) activated caspase-1 were examined by Western blot analysis. (D) and (E) Macrophage cells were stimulated with LPS, LPS + 0.5 mM PA, LPS + PA + APDC, and LPS + PA+Nifedipine for 6 h and then stained with Mitotracker green and Mitotracker deep red (D) or MitoSOX (E) for 30 min and analyzed by flow cytometry.
Rheb decreased expression and translocation from the mitochondria contributed to PA-induced mitophagy deficiency
Previous research showed that small GTPase Rheb was recruited to the mitochondrial outer membrane to regulate mitochondrial energetic status-induced mitophagy [16]. To test whether Rheb participated in the PA-induced mitophagy deficiency, we analyzed the effect of PA on Rheb expression. As shown in Fig. 4A, the decreased expression of Rheb over 48 h in cells grown with PA was concomitant with the decreased activation of LC3. This was further confirmed by immunostaining analysis in groups treated with LPS and LPS + PA (Fig. 4B). Then we investigated PA effect on Rheb distribution within INS-1E cells, the co-localization of Rheb with Mitotracker was observed using confocal microscopy. Under non-PA condition, the much larger size of localization of Rheb was found to localize to mitochondrial structure and distribute throughout cytoplasm of INS-1E cell (Fig. 4C). The promoting translocation of Rheb from the mitochondria in the presence of PA was further confirmed by Western blot; Hsp60 was a loading control. The ratio of Rheb-cytosol/Hsp60 or Rheb-mitochondria/Hsp60 was shown in Fig. 4D. The cytosolic concentration of Rheb (Rheb-cytosol) was markedly higher in the group treated PA while Rheb-mitochondria concentration was significantly decreased in PA compared with a group treated by LPS (**, p < 0.01; Fig. 4D). Given that Rheb is known to be essential for Rheb-dependent mitophagy contributes to the maintenance of optimal mitochondrial energy production, loss of this Rheb-dependent mitophagy damages mitochondria under sustained PA stress and may initiate and integrate inflammatory responses.
Fig. 4.
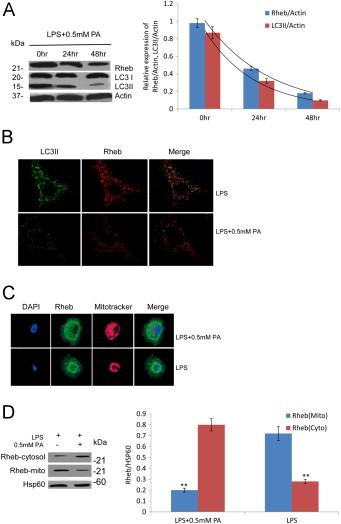
Rheb participated in the PA-induced mitophagy deficiency. (A) The decrease expression of rheb over 48 h in cells grown in PA was concomitant with the decrease activation of LC3 by immunostaining analysis in groups treated with LPS and LPS + PA respectively. (B) The co-localization of rheb with LC3II was observed using confocal microscopy in groups treated with LPS and LPS + PA respectively. (C) The co-localization of rheb with Mitotracker was observed using confocal microscopy. Rheb were disassociated from mitochondria and distributed throughout cytoplasm of INS-1E cell in presence of PA. (D) The promoting translocation of rheb from the mitochondria in the presence of PA was further confirmed by Western blot; Hsp60 was a loading control. The ratio of rheb-cytosol/Hsp60 or rheb-mitochondria/Hsp60 was showed on the right panel. Data are expressed as the mean ± S.D. (**, p < 0.01; Fig. 4D) (n = 6).
PA-induced disassociation of KIF5B from outer membrane of mitochondria contributed to mitophagy deficiency
Previous research along with our unpublished data shows that the protein Miro1 links mitochondria to KIF5B motor proteins, allowing mitochondria to move along microtubules [17]. This linkage is inhibited by micromolar levels of Ca2+ binding to Miro1. Exposure to the PA has been linked to ROS-induced mitochondrial Ca2+ overload and damage [18]. To test whether the PA-induced rise of Ca2+ played efforts on KIF5B, the distribution of the KIF5B within macrophage cells was analyzed. The co-localization of KIF5B with the mitochondria was observed in the absence/presence of the PA. Using confocal microscopy to examine the localization of KIF5B and Mitotracker in BMMs, we found that before exposure to PA, KIF5B and mitochondria showed diffused patterns in the cytosol that closely colocalized. After exposure to PA, they accumulated in the nucleus with partly overlap in their distribution (Fig. 5A). The promoting translocation of KIF5B from the mitochondria was further confirmed by Western blot (Fig. 5B); Hsp60 was a loading control. The ratio of KIF5B-cytosol/Hsp60 or KIF5B-mitochondria/Hsp60 was shown on the lower panel. The cytosolic concentration of KIF5B (KIF5B-cytosol) was markedly higher in group treated with PA than group in the presence of LPS alone. KIF5B-mitochondria concentration was significantly decreased in state of both PA and LPS compared with the group treated with either PA or LPS (**, p < 0.01; Fig. 5B). Given that KIF5B is known to be essential for mitochondrial transport in mammalian cells, loss of this KIF5B-dependent transport pathway has been showed to be involved in mitochondrial fragmentation and following depletion of mitochondria in neurons [19].
Fig 5.
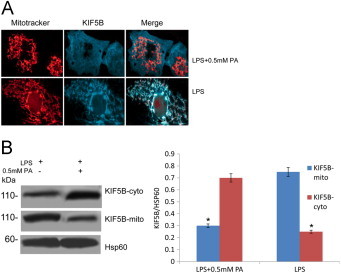
Ca2+-mediated disassociation of KIF5B from outer membrane of mitochondria under PA stress. (A) The distribution of the KIF5B within macrophage cells primed with LPS was analyzed using confocal microscopy in the absence/presence of the PA. (B) The promoting translocation of KIF5B from the mitochondria was further confirmed by Western blot; Hsp60 was a loading control. The ratio of KIF5B-cytosol/Hsp60 or KIF5B-mitochondria/Hsp60 were showed on the right panel. Data are expressed as the mean ± S.D. (*, p < 0.05; **, p < 0.01) (n = 6).
Overexpression of KIF5B or Rheb rescued glucose-stimulated insulin secretion impairment in rat islets
To determine the effect of KIF5B or Rheb on the insulin secretion function in rat islets, the insulin secretion of cells under different treatments was measured by a RIA kit and then insulin secretion indexes were calculated. In the presence of 16.7 mM glucose, the insulin release in rats islet exposed to LPS + PA group was markedly reduced compared with control group to LPS (*, p < 0.05, **, p < 0.01; Fig. 6A and B), and this effort was rescued by KIF5B or Rheb overexpression. There was a similar insulin release between PA + LPS+siRheb group and PA + LPS+siKIF5B, but these groups showed a significantly lower insulin secretion compared with control group (**, p < 0.01, Fig. 6A and B). LPS + PA GSIS of high glucose (16.7 mM glucose) was markedly decreased (*, p < 0.05, Fig. 6B) compared with control. In PA + LPS+siRheb and PA + LPS+siKIF5B treated groups, the GSIS in rat islet cells was severely decreased compared with control (**, p < 0.01, Fig. 6B) while there was no difference between LPS + PA + Rheb/KIF5B and control group. These results suggested that KIF5B or Rheb inhibition was deleterious for impairing GSIS, and this effort could be attenuated by Rheb or KIF5B overexpression in rat islet cells. To further determine the toxic effects of KIF5 or Rheb, we next investigated. After exposure for 24 h, the groups treated with PA + LPS and PA + LPS+siRheb or siKIF5B robustly reduced the total insulin content in rat islet cells by 35.6%, 39.6% and 39.5% compared with control group (0 mM PA) (Fig. 6C) (**, p < 0.01) respectively. Rheb or KIF5B overexpression rescued this effort, suggesting that rat islet cells were damaged by the inhibition expression of KIF5B or Rheb. To confirm the PA-induced IL-1β dependent insulin resistance, the co-localization of Rheb/KIF5B and insulin was observed under immunostaining. LPS group showed larger beta-cell size, highly expressed, and diffusely distributed Rheb/KIF5B and insulin throughout the cytoplasm. When cells were under PA stress, the decreased expression of both Rheb/KIF5B and insulin was distributed near nuclear (Fig. 6D). Thus, Rheb and KIF5B appear to be important for the mitophagy machinery in response to metabolic stress linking to the IL-1β dependent insulin resistance. Hereby, schematic mechanism to explain the data: sustained PA induced Ca2+-dependent effect disrupted Rheb and KIF5B interaction with mitochondria. Defective mitophagy led to the accumulation of damaged-ROS-generating mitochondria, down the pathway of NLRP3 dependent proinflammatory responses, and subsequently, insulin resistance (Fig. 6E).
Fig. 6.
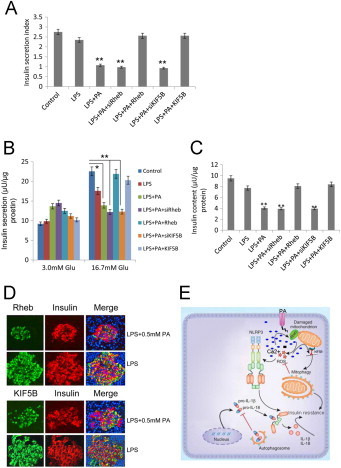
The effect of expression level of KIF5B or rheb on the insulin secretion function in rat islets. After equilibration at 3.3 mM glucose, islets cells were stimulated with high glucose (16.7 mM) for 1 h (A). The glucose-stimulated insulin secretion in islets cells was measured by RIA, and then the insulin secretion index was calculated (insulin release at high glucose/insulin release at basal glucose) in control, LPS, LPS + PA, LPS + PA+siRheb, LPS + PA + rheb, LPS + PA + siKIF5B, LPS + PA + KIF5B groups (**, p < 0.01) (n = 6). (B) The insulin secretion in islets cells in response to 3.0 and 16.7 mM glucose stimulation was examined at above-mentioned groups (*, p < 0.05; **, p < 0.01) (n = 6). (C) Total insulin content in islets cells was extracted by the acid/ethanol method and detected using an RIA kit. Data are expressed as the mean ± S.D. (error bars) (**, p < 0.01) (n = 6). (D) Rheb/KIF5B and insulin partially co-localize following treated by LPS + PA in islets. Confocal microscopy of islets primed with LPS for 2 h and stimulated with/without PA, and immunostained for rheb/KIF5B and insulin. Single and merged images from representative cells are shown. Scale bars shown are 10 μM. (E) A proposal mechanism of PA induced Ca2+-mediated mitophagy deficiency, leading to NLRP3 dependent proinflammatory responses and subsequently, insulin resistance. PA induced rise of Ca2+ concentration targeted KIF5B and rheb, the disruption of KIF5B-mediated mitochondrial motility and loss of the rheb-dependent mitophagy resulted in defective mitophagy, leading to the accumulation of damaged-mitochondria-producing-ROS, and down pathway of NLRP3 dependent IL-1β and IL-18 production, and subsequently, caused IL-1β dependent insulin resistance.
Discussion
Over the last decades, substantial progress has been made in defining the physiological function of mitochondria in controlling insulin action and the potential defects that lead to insulin resistance and later on T2D. Mitophagy is a highly conserved cellular pathway designed to degrade damaged mitochondria during times of metabolic stress. It is also an important part of immune cell function and shapes subsequent immune responses under HFD-induced NLRP3 dependent proinflammatory activation [20,21]. The purpose of present study is to define the role of mitophagy in the PA-induced NLRP3 proinflammatory responses and insulin resistance. Our findings showed that mitophagy deficiency driven by malfunction of Rheb and KIF5B under PA stress contributed to damaged-mitochondria ROS-producing NLRP3 dependent inflammasome activation and later on insulin resistance in LPS primed macrophage cell model. Similar conclusions have also been drawn from other studies in both cell lines [10] and animals [1] that mitophagy dysfunction contributes to the inflammasome activation and later on etiology of T2D.
ROS has been shown to activate the NLRP3 inflammasome upon treatment with fatty acids, leading to the release of active IL-1β and production of IL-1-dependent cytokines and chemokines [3,8,22–34]. Consistent with these previous studies, our findings showed that PA treatment induced damaged-mitochondrial ROS generation in LPS primed macrophage cells. These efforts led to an increased expression of cytokines IL-18, IL-1β without affecting TNF-α, IL-6 expression, which was prevented by antioxidant APDC or Ca2+ signaling inhibitor Nifedipine. In addition, we provided several lines of evidence showing that mitophagy was damaged under metabolic stress. First, we demonstrated that increased mitochondrial metabolic stress accompanied decreased mitochondrial degradation. Second, we showed that this degradation occurred through the formation of autophagosomes that tethered and eliminated damaged mitochondria. Finally, mitochondrial damage characterized by robust mito-ROS production and loss of membrane potential linked to NLRP3 inflammasome activation, and manipulations that blocked any of these processes reduced inflammasome activation. In sum, our data suggested that this identified PA-ROS-NLRP3 pathway was strongly associated with mitophagy function. In agreement with previous report that HFG-induced proinflammatory responses, which then ultimately resulted in insulin resistance [4], our findings confirmed that PA led to inflammasome activation interfering with insulin signaling pathway, and inhibition of this pathway with siRheb, siKIF5B aggravated insulin impairment by PA.
Mitophagy has been mainly regarded as an acute degradation process that is triggered by local and severe mitochondrial damages under pathological conditions [35,36] or as part of a developmental process for the removal of excess mitochondria under physiological conditions [36,37]. The protein Rheb enriched in brain is a Ras-like small GTPase. Rheb binds GTP after stimulation by nutrients such as amino acids and glucose, and is regulated by insulin [38]. Rheb has been found recruited to the mitochondrial outer membrane upon metabolic stress and regulates mitochondrial energetic status-induced mitophagy [16]. In congruent with these data, our results indicated that PA-induced mitophagy deficiency was driven by Rheb decreased expression and disassociation from mitochondria. In Rheb KO mouse models, Rheb knockdown led to a strong accumulation of mitochondria in liver without a rise of mitochondrial activity, suggesting a pathological situation. Mouse model overexpressing Rheb in beta-cells showed increased beta-cell size and improved glucose tolerance with higher insulin secretion and prevention of hyperglycemia [39], consistent with our study. Given that Rheb plays an important role in Rheb-dependent mitophagy contributions to the maintenance of optimal mitochondrial energy production, our data combined with others demonstrate that altered mitophagy regulation damages mitochondria dynamic in beta-cells in response to sustained PA, and in a consequence, may integrate proinflammatory responses.
Kinesin superfamily protein KIF5B, is the molecular motor conveying cargos along microtubules, known to be involved in mitochondrial movement. The resultant rise in cytosolic Ca2+ concentration in response to PA primarily triggering insulin exocytosis [40,41] was also found to regulate mitochondrial movements [17,42,43]. This regulation of mitochondrial dynamics is Ca2+-dependent effects on the KIF5B in pancreatic beta cells [44,45]. Miro1 has been showed to participate in this process by linking mitochondria to KIF5B motor protein, which was regulated by changes of Ca2+ concentrations. Elevated Ca2+ concentration causes the Miro1 to lose its association with microtubules [17,46]. This was consistent with our results that KIF5B was disassociated from mitochondria and distributed throughout the cytoplasm macrophage cell under PA-induced increase of Ca2+ condition. Given that KIF5B is known to be essential for mitochondrial transport in mammalian cells, loss of this KIF5B-dependent transport pathway enhances mitochondrial fragmentation and following depletion of damaged mitochondria under conditions of PA stress before mitophagy damaged.
Mitophagy in conjunction with mitochondrial biogenesis, regulates the changes in steady-state mitochondrial number that is required to meet metabolic demand under PA stress. Damaged mitochondria are selectively removed by mitophagy to maintain quality control. Defective autophagic machinery operated through Rheb and KIF5B-mediated pathway thereby resulted in proinflammatory responses and subsequently, damaged insulin secretion. Recent findings indicate that Ca2+ is a key molecular regulator of the NLRP3 inflammasome that has critical roles in the pathogenesis of auto-inflammatory disease [47]. Yet how the Ca2+ as an important regulator of mitophagy under PA stress needs to be fully elucidated in the future. Thorough understanding of mitophagy alterations under PA stress is significant towards development of better therapies to combat inflammatory malignancies in T2D.
Author contributions
C.X. and S.L. researched data. S.Y. wrote the manuscript and researched data. L.L. and L.Z. contributed to discussion and reviewed/edited manuscript. Y.H. researched data and contributed to discussion. S.Y. and L.L. reviewed the manuscript.
Conflict of interest
All authors declared that there was no conflict of interest between them.
Funding
This study was supported by the “National Natural Science Foundation of China (J1103507), Jiangsu NSF (BK2011784)” and “The Priority Academic Program Development of Jiangsu Higher Education Institutions”. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgements
We would like to thank Dr. Long Chen for kind assistances in preparing this manuscript.
References
- 1.Vandanmagsar B., Youm Y.H., Ravussin A., Galgani J.E., Stadler K., Mynatt R.L., Ravussin E., Stephens J.M., Dixit V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nature Medicine. 2011;17:179–188. doi: 10.1038/nm.2279. 21217695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stienstra R., van Diepen J.A., Tack C.J., Zaki M.H., van de Veerdonk F.L., Perera D., Neale G.A., Hooiveld G.J., Hijmans A., Vroegrijk I., van den Berg S., Romijn J., Rensen P.C., Joosten L.A., Netea M.G., Kanneganti T.D. Inflammasome is a central player in the induction of obesity and insulin resistance. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:15324–15329. doi: 10.1073/pnas.1100255108. 21876127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee H.M., Kim J.J., Kim H.J., Shong M., Ku B.J., Jo E.K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62:194–204. doi: 10.2337/db12-0420. 23086037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wen H., Gris D., Lei Y., Jha S., Zhang L., Huang M.T., Brickey W.J., Ting J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nature Immunology. 2011;12:408–415. doi: 10.1038/ni.2022. 21478880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruan Y., Mo M., Joss-Moore L., Li Y.Y., Yang Q.D., Shi L., Zhang H., Li R., Xu W.H. Increased waist circumference and prevalence of type 2 diabetes and hypertension in Chinese adults: two population-based cross-sectional surveys in Shanghai, China. British Medical Journal Open. 2013;3:e003408. doi: 10.1136/bmjopen-2013-003408. 24165029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boni-Schnetzler M., Boller S., Debray S., Bouzakri K., Meier D.T., Prazak R., Kerr-Conte J., Pattou F., Ehses J.A., Schuit F.C., Donath M.Y. Free fatty acids induce a proinflammatory response in islets via the abundantly expressed interleukin-1 receptor I. Endocrinology. 2009;150:5218–5229. doi: 10.1210/en.2009-0543. 19819943 [DOI] [PubMed] [Google Scholar]
- 7.Jourdan T., Godlewski G., Cinar R., Bertola A., Szanda G., Liu J., Tam J., Han T., Mukhopadhyay B., Skarulis M.C., Ju C., Aouadi M., Czech M.P., Kunos G. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nature Medicine. 2013;19:1132–1140. doi: 10.1038/nm.3265. 23955712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dixit V.D. Nlrp3 inflammasome activation in type 2 diabetes: is it clinically relevant? Diabetes. 2013;62:22–24. doi: 10.2337/db12-1115. 23258906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masters S.L., Dunne A., Subramanian S.L., Hull R.L., Tannahill G.M., Sharp F.A., Becker C., Franchi L., Yoshihara E., Chen Z., Mullooly N., Mielke L.A., Harris J., Coll R.C., Mills K.H., Mok K.H., Newsholme P., Nunez G., Yodoi J., Kahn S.E., Lavelle E.C., O’Neill L.A. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nature Immunology. 2010;11:897–904. doi: 10.1038/ni.1935. 20835230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou R., Yazdi A.S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. 21124315 [DOI] [PubMed] [Google Scholar]
- 11.East D.A., Campanella M. Ca2+ in quality control: an unresolved riddle critical to autophagy and mitophagy. Autophagy. 2013;9:1710–1719. doi: 10.4161/auto.25367. 24121708 [DOI] [PubMed] [Google Scholar]
- 12.Campanella M., Klionsky D.J. Keeping the engine clean:a mitophagy task for cellular physiology. Autophagy. 2013;9:1647. doi: 10.4161/auto.26915. 24162014 [DOI] [PubMed] [Google Scholar]
- 13.Deretic V., Jiang S., Dupont N. Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends in Cell Biology. 2012;22:397–406. doi: 10.1016/j.tcb.2012.04.008. 22677446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fang H., Pengal R.A., Cao X., Ganesan L.P., Wewers M.D., Marsh C.B., Tridandapani S. Lipopolysaccharide-induced macrophage inflammatory response is regulated by SHIP. Journal of Immunology (Baltimore, Md.: 1950) 2004;173:360–366. doi: 10.4049/jimmunol.173.1.360. 15210794 [DOI] [PubMed] [Google Scholar]
- 15.Sanjuan M.A., Dillon C.P., Tait S.W.G., Moshiach S., Dorsey F., Connell S., Komatsu M., Tanaka K., Cleveland J.L., Withoff S., Green D.R. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–1257. doi: 10.1038/nature06421. 18097414 [DOI] [PubMed] [Google Scholar]
- 16.Melser S., Chatelain E.H., Lavie J., Mahfouf W., Jose C., Obre E., Goorden S., Priault M., Elgersma Y., Rezvani H.R., Rossignol R., Benard G. Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metabolism. 2013;17:719–730. doi: 10.1016/j.cmet.2013.03.014. 23602449 [DOI] [PubMed] [Google Scholar]
- 17.Macaskill A.F., Rinholm J.E., Twelvetrees A.E., Arancibia-Carcamo I.L., Muir J., Fransson A., Aspenstrom P., Attwell D., Kittler J.T. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009;61:541–555. doi: 10.1016/j.neuron.2009.01.030. 19249275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giorgi C., Agnoletto C., Bononi A., Bonora M., De Marchi E., Marchi S., Missiroli S., Patergnani S., Poletti F., Rimessi A., Suski J.M., Wieckowski M.R., Pinton P. Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion. 2012;12:77–85. doi: 10.1016/j.mito.2011.07.004. 21798374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cagalinec M., Safiulina D., Liiv M., Liiv J., Choubey V., Wareski P., Veksler V., Kaasik A. Principles of the mitochondrial fusion and fission cycle in neurons. Journal of Cell Science. 2013;126:2187–2197. doi: 10.1242/jcs.118844. 23525002 [DOI] [PubMed] [Google Scholar]
- 20.Deretic V., Saitoh T., Akira S. Autophagy in infection, inflammation and immunity. Nature Reviews. Immunology. 2013;13:722–737. doi: 10.1038/nri3532. 24064518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levine B., Mizushima N., Virgin H.W. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. 21248839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Subramanian N., Natarajan K., Clatworthy M.R., Wang Z., Germain R.N. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153:348–361. doi: 10.1016/j.cell.2013.02.054. 23582325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan Y., Jiang W., Spinetti T., Tardivel A., Castillo R., Bourquin C., Guarda G., Tian Z., Tschopp J., Zhou R. Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity. 2013;38:1154–1163. doi: 10.1016/j.immuni.2013.05.015. 23809162 [DOI] [PubMed] [Google Scholar]
- 24.Kagan J.C., Horng T. NLRP3 inflammasome activation: CD36 serves double duty. Nature Immunology. 2013;14:772–774. doi: 10.1038/ni.2668. 23867926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Misawa T., Takahama M., Kozaki T., Lee H., Zou J., Saitoh T., Akira S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nature Immunology. 2013;14:454–460. doi: 10.1038/ni.2550. 23502856 [DOI] [PubMed] [Google Scholar]
- 26.Mishra B.B., Rathinam V.A., Martens G.W., Martinot A.J., Kornfeld H., Fitzgerald K.A., Sassetti C.M. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1beta. Nature Immunology. 2013;14:52–60. doi: 10.1038/ni.2474. 23160153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rayamajhi M., Miao E.A. Just say NO to NLRP3. Nature Immunology. 2013;14:12–14. doi: 10.1038/ni.2493. 23238751 [DOI] [PubMed] [Google Scholar]
- 28.Sheedy F.J., Grebe A., Rayner K.J., Kalantari P., Ramkhelawon B., Carpenter S.B., Becker C.E., Ediriweera H.N., Mullick A.E., Golenbock D.T., Stuart L.M., Latz E., Fitzgerald K.A., Moore K.J. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nature Immunology. 2013;14:812–820. doi: 10.1038/ni.2639. 23812099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sokolovska A., Becker C.E., Ip W.K., Rathinam V.A., Brudner M., Paquette N., Tanne A., Vanaja S.K., Moore K.J., Fitzgerald K.A., Lacy-Hulbert A., Stuart L.M. Activation of caspase-1 by the NLRP3 inflammasome regulates the NADPH oxidase NOX2 to control phagosome function. Nature Immunology. 2013;14:543–553. doi: 10.1038/ni.2595. 23644505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heneka M.T., Kummer M.P., Stutz A., Delekate A., Schwartz S., Vieira-Saecker A., Griep A., Axt D., Remus A., Tzeng T.C., Gelpi E., Halle A., Korte M., Latz E., Golenbock D.T. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–678. doi: 10.1038/nature11729. 23254930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leavy O. Inflammasome: turning on and off NLRP3. Nature Reviews. Immunology. 2013;13:1. doi: 10.1038/nri3366. 23197112 [DOI] [PubMed] [Google Scholar]
- 32.Bruchard M., Mignot G., Derangere V., Chalmin F., Chevriaux A., Vegran F., Boireau W., Simon B., Ryffel B., Connat J.L., Kanellopoulos J., Martin F., Rebe C., Apetoh L., Ghiringhelli F. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nature Medicine. 2013;19:57–64. doi: 10.1038/nm.2999. 23202296 [DOI] [PubMed] [Google Scholar]
- 33.Negash A.A., Ramos H.J., Crochet N., Lau D.T., Doehle B., Papic N., Delker D.A., Jo J., Bertoletti A., Hagedorn C.H., Gale M., Jr. IL-1beta production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathogens. 2013;9 doi: 10.1371/journal.ppat.1003330. 23633957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Green D.R., Galluzzi L., Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science (New York, N.Y.) 2011;333:1109–1112. doi: 10.1126/science.1201940. 21868666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lupfer C., Thomas P.G., Anand P.K., Vogel P., Milasta S., Martinez J., Huang G., Green M., Kundu M., Chi H., Xavier R.J., Green D.R., Lamkanfi M., Dinarello C.A., Doherty P.C., Kanneganti T.D. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nature Immunology. 2013;14:480–488. doi: 10.1038/ni.2563. 23525089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang J. Autophagy and mitophagy in cellular damage control. Redox Biology. 2013;1:19–23. doi: 10.1016/j.redox.2012.11.008. 23946931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhatia-Kissova I., Camougrand N. Mitophagy:a process that adapts to the cell physiology. International Journal of Biochemistry & Cell Biology. 2013;45:30–33. doi: 10.1016/j.biocel.2012.07.006. 22801005 [DOI] [PubMed] [Google Scholar]
- 38.Avruch J., Hara K., Lin Y., Liu M., Long X., Ortiz-Vega S., Yonezawa K. Insulin and amino-acid regulation of mTOR signaling and kinase activity through the rheb GTPase. Oncogene. 2006;25:6361–6372. doi: 10.1038/sj.onc.1209882. 17041622 [DOI] [PubMed] [Google Scholar]
- 39.Hamada S., Hara K., Hamada T., Yasuda H., Moriyama H., Nakayama R., Nagata M., Yokono K. Upregulation of the mammalian target of rapamycin complex 1 pathway by Ras homolog enriched in brain in pancreatic beta-cells leads to increased beta-cell mass and prevention of hyperglycemia. Diabetes. 2009;58:1321–1332. doi: 10.2337/db08-0519. 19258434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dezaki K., Hosoda H., Kakei M., Hashiguchi S., Watanabe M., Kangawa K., Yada T. Endogenous ghrelin in pancreatic islets restricts insulin release by attenuating Ca2+ signaling in beta-cells: implication in the glycemic control in rodents. Diabetes. 2004;53:3142–3151. doi: 10.2337/diabetes.53.12.3142. 15561944 [DOI] [PubMed] [Google Scholar]
- 41.Hellman B., Hallgren R., Abrahamsson H., Bergsten P., Berne C., Gylfe E., Rorsman P., Wide L. The dual action of glucose on the cytosolic Ca2+ activity in pancreatic beta-cells. Demonstration of an inhibitory effect of glucose on insulin release in the mouse and man. Biomedica Biochimica Acta. 1985;44:63–70. 3888211 [PubMed] [Google Scholar]
- 42.Gergalova G., Lykhmus O., Kalashnyk O., Koval L., Chernyshov V., Kryukova E., Tsetlin V., Komisarenko S., Skok M. Mitochondria express α7 nicotinic acetylcholine receptors to regulate Ca2+ accumulation and cytochrome c release: study on isolated mitochondria. PloS One. 2012;7:e31361. doi: 10.1371/journal.pone.0031361. 22359587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doczi J., Turiak L., Vajda S., Mandi M., Torocsik B., Gerencser A.A., Kiss G., Konrad C., Adam-Vizi V., Chinopoulos C. Complex contribution of cyclophilin D to Ca2+-induced permeability transition in brain mitochondria, with relation to the bioenergetic state. Journal of Biological Chemistry. 2011;286:6345–6353. doi: 10.1074/jbc.M110.196600. 21173147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X., Schwarz T.L. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136:163–174. doi: 10.1016/j.cell.2008.11.046. 19135897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cui J., Wang Z., Cheng Q., Lin R., Zhang X.M., Leung P.S., Copeland N.G., Jenkins N.A., Yao K.M., Huang J.D. Targeted inactivation of kinesin-1 in pancreatic beta-cells in vivo leads to insulin secretory deficiency. Diabetes. 2011;60:320–330. doi: 10.2337/db09-1078. 20870970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Spronsen M., Mikhaylova M., Lipka J., Schlager M.A., van den Heuvel D.J., Kuijpers M., Wulf P.S., Keijzer N., Demmers J., Kapitein L.C., Jaarsma D., Gerritsen H.C., Akhmanova A., Hoogenraad C.C. TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron. 2013;77:485–502. doi: 10.1016/j.neuron.2012.11.027. 23395375 [DOI] [PubMed] [Google Scholar]
- 47.Lee G.S., Subramanian N., Kim A.I., Aksentijevich I., Goldbach-Mansky R., Sacks D.B., Germain R.N., Kastner D.L., Chae J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. 2012;492:123–127. doi: 10.1038/nature11588. 23143333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Merglen A., Theander S., Rubi B., Chaffard G., Wollheim C.B., Maechler P. Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology. 2004;145:667–678. doi: 10.1210/en.2003-1099. 14592952 [DOI] [PubMed] [Google Scholar]
- 49.Delghingaro-Augusto V., Nolan C.J., Gupta D., Jetton T.L., Latour M.G., Peshavaria M., Madiraju S.R., Joly E., Peyot M.L., Prentki M., Leahy J. Islet beta cell failure in the 60% pancreatectomised obese hyperlipidaemic Zucker fatty rat: severe dysfunction with altered glycerolipid metabolism without steatosis or a falling beta cell mass. Diabetologia. 2009;52:1122–1132. doi: 10.1007/s00125-009-1317-8. 19294363 [DOI] [PubMed] [Google Scholar]