

Abstract
A novel, sensitive locus-specific touchdown-multiplex polymerase chain reaction (TMPCR), which is based on two-stage amplification pertaining to multiplex PCR and conditional touchdown strategy, was used in detecting and differentiating Vibrio cholerae serogroups. A panel of molecular marker-based TMPCR method generates reproducible profiles of V. cholerae-specific (588 bp) amplicons derived from ompW gene encoding the outer membrane protein and serogroup-specific amplicons, 364 bp for the O1 and 256 bp for the O139, authentically copied from rfb genes responsible for the lipopolysaccharide biosynthesis. The TMPCR amplification efficiency yields either equally or unequally detectable duplex DNA bands of the O1 (588 and 364 bp) and O139 (588 and 256 bp) or a DNA fragment of non-O1/non-O139 (588 bp) while providing no false positive identifications using the genomic DNA templates of the other vibrios and Enterobacteriaceae. The reciprocal analysis of two-template combinations demonstrated that, using V. cholerae O1, O139, or equally mixed O1 and O139, the TMPCR had a detection limit of as low as 100 pg of the O1, O139, or non-O1/non-O139 in reactions containing unequally or equally mixed gDNAs. In addition, the O serogroup-specific TMPCR method had 100% agreement with the serotyping method when examined for the serotyped V. cholerae reference strains and those recovered from clinical samples. The potential benefit of using this TMPCR tool would augment the serotyping method used in epidemiological surveillance and monitoring of V. cholerae serogroups, O1, O139, and non-O1/non-O139 present in clinical and environmental samples.
1. Introduction
Vibrio cholerae is a water-borne agent causing cholera and is autochthonous to aquatic habitats in coastal and estuarine ecosystems to which symbiosis with zooplanktons for its survival and multiplication is related [1–5]. Vibrio spp., including V. cholerae serogroups [6] and Vibrio parahaemolyticus [7, 8], normally grow in the natural environments and can enter into viable-but-nonculturable (VBNC) state. The significant roles of the toxigenic O1 and O139 serogroups of V. cholerae, as well as the non-O1/non-O139 serogroups, have been studied in several ways. For instance, the VBNC V. cholerae O1 in microcosms or from aquatic environments are converted to culturable state through animal passage [9]. The VBNC O139 and non-O1/non-O139 can also resuscitate when cocultured with several animal cell lines [10, 11]. More recently, the classical biotype of V. cholerae O1 retains viability but loses culturability when cocultured with the El Tor biotype [12]. This might suggest that the emergence of the El Tor biotype of V. cholerae O1 relates to displacement of the existing classical biotype as the predominant cause of epidemic cholera. Similar to the toxigenic O1 and O139 serogroups that possess virulence-association genes [13–18], the other non-O1 and non-O139 serogroups recovered from aquatic environments or clinical specimens have also been epidemiologically linked with the pathogenic and epidemic potential [18–20]. Thus, public health surveillance and monitoring of cholera cases require the systems used in the national surveillance for notifiable diseases, public health laboratory, and environmental surveillance [21–24]. Molecular detection techniques such as molecular marker-based polymerase chain reaction (PCR) methods developed for probing these eccentric V. cholerae microorganisms have been so far proven useful in diagnosis and surveillance for the outbreaks or epidemic investigations worldwide.
In the Gulf of Bengal, South and Southeast Asia, it has been suggested that such outbreaks of cholera pertaining to cross-contamination and spread of V. cholerae after the passage(s) from susceptible persons to aquatic environments are likely to be epidemiologically linked with the interconnections of sanitation with poorly chlorinated waters, contamination in seafood/food commodities and seawaters, and direct fecal-oral contact among food handlers or seafood preprocessing plant workers [2, 14–16, 21, 22, 24]. Standard culture methods used in routine diagnosis and surveillance for V. cholerae by public health reference laboratories depend on handling the samples of which V. cholerae culture is recovered. Regarding this, V. cholerae cultures under study investigation were recovered from V. cholerae-contracted or suspected patients with acute or severe diarrhea whose stools or rectal swabs had been collectively obtained by clinical laboratory settings during the outbreaks over two past decades between 1994 and 2010 in Central Thailand.
We proposed that the genomic DNA archived from the preserved stock cultures could serve as the template when examined for the O1, O139, and non-O1/non-O139 serogroups by PCR methods as this can also permit a reciprocal detection of the O1 and O139 serogroups from clinical specimens associated with the past outbreaks. In this study, we have successfully developed a novel sensitive, specific, semiquantitative touchdown-multiplex PCR (TMPCR) for detection and differentiation of V. cholerae O1, O139, and non-O1/non-O139 serogroups. TMPCR employs the useful molecular markers originally derived from the DNA locus involved in de novo O-antigen biosynthesis of V. cholerae [25–28] and the outer membrane protein (ompW) encoding gene specific for V. cholerae [29, 30]. To achieve the goal of the study, we analyzed the performance efficiency (specificity and sensitivity) of this O serogroup-specific TMPCR in detecting and differentiating the O1, O139, and non-O1/non-O139 genomes, which were empirically determined using reference strains of V. cholerae and unrelated Vibrio strains. We then explored its usefulness in differentiating the O serogroups present in those V. cholerae stock cultures originally isolated from clinical specimens. Additionally, the advancement of TMPCR that tested V. cholerae present in environmental samples was also discussed.
2. Materials and Methods
2.1. Bacterial Cultures and Laboratory Classification
Sixty-nine V. cholerae strains that served as target gDNA templates included reference and wild-type strains of the O1 (including El Tor/Classical Ogawa and El Tor/Classical Inaba biotypes), O139, and non-O1/non-O139. The 31 other bacterial strains used as nonspecific gDNA templates included 8 Vibrio spp., 16 Gram-negative bacteria, and 7 Gram-positive bacteria. For the batch propagation of V. cholerae, the 0.5 mL overnight culture of the O1 569B or O139 MO45 strains was inoculated into a 100 mL Luria-Bertani (LB) broth supplemented with 1% sodium chloride (NaCl) solution and then incubated at 37°C for 3 h with vigorous shaking. The cell culture with an optical density of 0.8 was used for drop plate method and, subsequently, enumerated by using plate count (PC) agar (Difco, Michigan, USA, or Eiken, Japan). The PC agar was also supplemented with 1% NaCl. The non-O1/non-O139 strains including O22 and O155 were cultured as before.
Subcultures of V. cholerae and other Vibrio spp. were grown on selective thiosulfate citrate bile salt sucrose (TCBS) agar (Merck, Darmstadt, Germany) and, subsequently, they were biochemically and serologically characterized according to the methods described elsewhere [31, 32]. Briefly, biochemical tests included triple sugar iron (TSI), motility indole-lysine (MIL), oxidase, urea, MR, Voges-Proskauer (VP), citrate, lysine, ornithine, arginine, lactose, sucrose, mannose, arabinose, mannitol, glucose/gas, inositol, aesculin, and salt tolerance of 0%, 3%, 6%, 8%, and 10% NaCl solution. All reagents were purchased from Difco Laboratory (Difco, Michigan, USA). The serogroups O1, O139, and non-O1/non-O139 were tested on the basis of agglutination reaction using commercially available O serogroup-specific antisera (antiserum VcO1/O139 Polyvalent SAP, S & A Reagents Lab, Bangkok, Thailand). The non-O1/non-O139 was characterized into two nonagglutinable (NAG) groups (NAG I and II) according to Heiberg's reaction. In addition, PC agar was used for viable count and enumeration of all the bacterial strains tested in this study.
2.2. V. cholerae Cultures from Clinical Specimens and Serotyping
A total of 148 stock cultures of V. cholerae that corresponded to 108 stools and 40 rectal swabs were collectively obtained from V. cholerae-contracted or suspected patients with acute or severe diarrhea during the outbreaks in Central Thailand between 1994 and 2010. By using anonymous system, the cultures initially isolated by clinical laboratory settings were performed on isolation source and time of collection and the corresponding TCBS stock cultures were prepared. All were subsequently subcultured and kept in semisolid agar medium containing 1% NaCl at the Department of Microbiology, Faculty of Public Health, Mahidol University, Thailand. For serotyping the O1, O139, and non-O1/non-O139, the agglutination of the 148 subcultured V. cholerae isolates in individuals was blindly performed as before. In addition, only the V. cholerae O1 serogroup whose subcultures agglutinated with polyvalent O1 antiserum was characterized using monovalent Ogawa or Inaba antiserum. For biotyping, the phenotypic testing of the strains was performed using hemolysis of sheep blood, agglutination of chicken erythrocytes, Voges-Proskauer, and sensitivity to polymyxin B. Meanwhile, the genomic DNAs of the same V. cholerae isolates were prepared according to the rapid boiling method described below or using QIAamp DNA minikit (Qiagen, Hilden, Germany). The stock solution of purified gDNA extracts was initially quantified and then stored according to the method described below.
2.3. Genomic DNA Extraction
A modified miniprep gDNA extraction method was used for both target and nontarget gDNAs as follows. As mentioned above, the bacterial culture (1.5 mL) that was grown overnight at 37°C with vigorous shaking was spun down at 12,000 rpm for 1 min. The cell pellet was suspended in 582 μL of TE buffer (10 mM Tris-HCl, 1 mM EDTA), pH 8.0. The suspension was lysed with an addition of 15 μL 20% sodium dodecyl sulfate solution and 3 μL 20 mg/mL proteinase K, mixed thoroughly, and then left at 37°C for 1 h. A 10 μL of 10 mg/mL RNase A was added, mixed thoroughly, and incubated at 37°C for 30 min. The 100 μL 5 M sodium chloride (NaCl) solution and 80 μL 10% hexadecyl trimethyl ammonium bromide (CTAB) in 0.7 M NaCl solution were added, mixed thoroughly, and incubated at 65°C for 10 min. Then, a 0.4x volume of chloroform/isoamyl alcohol (24 : 1) was added, mixed gently for 5 min, and centrifuged at 12,000 rpm for 5 min. The aqueous phase containing gDNA was twice treated with an equal volume of phenol-chloroform and followed by centrifugation as before. An equal volume of isopropanol was added to recovered DNA solution, mixed thoroughly, and DNA pellet was centrifuged as before. After dehydration of an extreme volume of 70% ice-cold ethanol and further centrifugation, it was dissolved in TE buffer pH 7.6.
In addition, using rapid boiling method, the same overnight culture was spun down at 12,000 rpm for 1 min. The cell pellet was suspended well in 500 μL of TE buffer pH 8.0 and incubated at 56°C for 30 min. Immediately after being boiled at 100°C for 10 min, the suspension was stood on ice for 10 min and then centrifuged at 12,000 rpm for 3 min to pellet cell debris; the clear gDNA supernate was obtained. Finally, the purified gDNA extracts as well as crude extracts (boiled lysate) were qualitatively and quantitatively analyzed by spectrophotometer at a wavelength ratio of 260/280 nm or by agarose gel electrophoresis. The aliquots of varying concentration of 30 to 50 μg/mL (a 260/280 OD greater than 1.7) used throughout the study were kept at −20°C until use.
2.4. Primer Design
The homologous primer sets specific for V. cholerae serogroups (Table 1) were originally derived from target DNA sequences of V. cholerae O1, a 22 kb rfb DNA locus [25], and of V. cholerae O139, a 46 kb rfb DNA locus [28]. These rfb gene homologs are involved in lipopolysaccharide (LPS) biosynthesis for the O1 and O139 antigens. The rfbV gene responsible for the rhs element involved in LPS biosynthesis was designed for V. cholerae O1-specific primers to generate 364 bp amplicon. The wbfR gene encoding asparagine synthetase was designated as V. cholerae O139-specific primers (256 bp amplicon). The ompW gene encoding the outer membrane protein was used for V. cholerae species-specific or universal primers that amplify 588 bp amplicons [29, 30]. Analysis of their unanimous homology at DNA and protein levels was performed on the online available BLAST Programs (http://www.ncbi.nlm.nih.gov/blast/), BLASTN, BLASTx, and BLASTP, for sequence similarity algorithms. The multiple sequence alignments were analyzed for consensus sequences using the ClustalW Program (http://www.ebi.ac.uk/clustalW/). The aliquots of these synthesized multiplex primer sets at a stock concentration of 100 μM in TE buffer pH 8.0 used throughout the study were stored at −80°C until use.
Table 1.
Primer sequencesa used in touchdown-multiplex PCR.
| Primer set | Sequence (5′ to 3′) | Tm (°C) | Direction | Amplicon size (bp) | Reference |
|---|---|---|---|---|---|
| Universal primersb | |||||
| MVCO1 | CAC CAA GAA GGT GAC TTT ATT GTG | 68 | Forward | 588 | Nandi et al., 2000 [30] |
| MVCO2 | GAA CTT ATA ACC ACC CGC G | 58 | Reverse | ||
| O1-specific primersc | |||||
| MVCO3 | GAC TGT CAG CTG GCG GAA | 58 | Forward | 364 | This study |
| MVCO4 | GTT GGC GTA TTA CGG TAC | 54 | Reverse | ||
| O139-specific primersd | |||||
| MVCO5 | GCG TTT ATC GCC GGT CGA C | 62 | Forward | 256 | This study |
| MVCO6 | GTA ACT TGG TAC AAT CTC G | 54 | Reverse |
aAll were originally derived from the nucleotide sequences (positions) available from the GenBank accession numbers bAE003853 (819612–820199), bCP000626 (404919–405506), cAE003852 (264415–264778), cCP000627 (2789549–2789912), and dU47057 (902–1157); from the EMBL accession numbers bX51948 (221–808), cX59554 (18246–18609), cY07788 (199–562), and dY07786 (11878–12133); or from the DDBJ accession number dAB012956 (32652–32907).
2.5. Touchdown-Multiplex PCR Amplification
Initially, two-template combinations of the V. cholerae O1 and O139 were tested under the simultaneous locus-specific amplification conditions [33, 34] using a 96-well MyCycler Thermal Cycler (BIORAD, USA) or PCR gradient thermal cycler (ThermoHybraid Px2, Ashford, UK), in which two combined factors such as concentrations of primer sets and gDNA templates were empirically determined in a 25 μL PCR mixture consisting of 1x PCR buffer (50 mM KCl, 1.5 mM MgCl2, and 10 mM Tris-HCl pH 9.0), 200 μM each dNTP (dATP, dTTP, dCTP, and dGTP), 1 U of AmpliTaq (Perkin Elmer, USA), or Taq DNA polymerase (Promega, USA). The amplification conditions were performed on predenaturation at 95°C for 5 min and followed by 30 cycles of denaturation at 94°C for 1 min and extension at 72°C for 1 min, except that primer annealing temperature decrements (70 → 50°C) were used each for 1 min. The last extension was done at 72°C for 10 min. First, using a constant concentration of 1 μM each primer set, reactions employed mixed O1 569B and O139 MO45 gDNAs (ng) at equal ratios of 10 : 10, 1 : 1, and 0.1 : 0.1 or at unequal ratios of 1 : 10 and 0.1 : 10 and vice versa. Second, using mixed gDNAs (10 ng each), the primer sets (universal : O1-specific : O139-specific) at concentration ratios (μM) of 1 : 1 : 1, 1 : 0.5 : 0.5, 1.0 : 0.2 : 0.2, 0.5 : 1.0 : 1.0, 0.5 : 0.5 : 1.0, 0.2 : 0.2 : 1.0, and 0.1 : 0.1 : 1.0 were analyzed. Under these circumstances, the primer binding specificity in amplifying target loci depended upon annealing temperature algorithms to which primer-template combinations were restricted. These algorithms permitted the development of thermocycling TM-PCR protocol as described below.
Finally, a 25 μL optimized TMPCR mixture was similar except that V. cholerae O1 and O139 gDNA templates, 10 ng each, and three multiplex primer sets (0.4, 0.4, and 1.0 μM each, resp.) were used instead. The optimized amplification condition was performed on one cycle of predenaturation at 94°C for 5 min, followed by the 5 cycles with successive annealing temperature decrements that one °C changed from 57°C to 53°C in every cycle versus time increments (30 sec in the first three cycles and 35 sec in the last two cycles). The reaction was denatured at 94°C for 1 min, followed by annealing at this temperature algorithm and polymerization at 72°C for 1 min. Subsequently, the 20 cycles of denaturation at 94°C for 1 min, annealing at 52°C for 40 sec, and elongation at 72°C for 1 min were employed. The last primer extension step was done at 72°C for 10 min. In similar fashion, the purified V. cholerae O22 or O155 gDNA was used instead of the O139. Either Escherichia coli gDNA template as negative control (NC) or nuclease-free water, instead of gDNA template, as internal control (IC) was used in all the experiments.
2.6. Semiquantitative TMPCR
The reciprocal detection of V. cholerae O1 versus O139 was aimed at analyzing the amplification efficiency of the O serogroup-specific TMPCR under carefully controlled experiments that utilized gDNA template ratios whether unequally or equally. That is, the detectable level of O1 gDNA could be amplified in reactions containing high amount of other related O139 gDNAs, and vice versa.
The unequally reciprocal detection of V. cholerae O1 versus O139 (Table 2) was done as follows. In experiment A, the reactions contained the amount of the 100 ng O1 gDNA versus the amount of 10-fold serially diluted O139 gDNA (100 ng to 1 pg), or the amount ratios of the O1 : O139 varied from onefold to 105-fold, and vice versa. In experiment B, the reactions contained the amount ratios of the O1 : O139 gDNAs as 100 ng to 1 pg, 10 ng to 10 pg, and 1 ng to 100 pg or the amount of the O1 as 105-, 103-, and 10-fold greater than the O139, and vice versa. Also, the amount of the 100 pg O139 gDNA was tested against the varying amounts of the O1 gDNA: 100 ng, 50 ng, 25 ng, 10 ng, 1 ng, and 100 pg.
Table 2.
The unequally reciprocal detection of V. cholerae O1 : O139 by the O serogroup-specific TMPCR.
| O1 | O139 | |||||
|---|---|---|---|---|---|---|
| 0.001 | 0.01 | 0.1 | 1 | 10 | 100 | |
| 100 | 105 | 104 | 103 | 102 | 101 | 1 |
| 10 | 103 | 101 | ||||
| 1 | 101 | 102 | ||||
| 0.1 | 101 | 103 | ||||
| 0.01 | 103 | 104 | ||||
| 0.001 | 105 | |||||
The contents (ng) of purified O1 : O139 gDNAs were used in this assay.
The equally reciprocal detection of V. cholerae O1 versus O139 (Table 3) was done as follows. In experiment C, the equal amounts of the O1 : O139 gDNAs were tested at a serial 10-fold dilution, 100 ng to 1 pg. For the internal controls (experiments D and E), the same amounts of the 10-fold serially diluted gDNA templates, O1 or O139, were used. Also, the purified V. cholerae O22 or O155 gDNA template was used instead of O139. All experiments were performed in triplicate.
Table 3.
The equally reciprocal detection of V. cholerae O1 : O139 by the O serogroup-specific TMPCR.
| O1 | O139 | |||||
|---|---|---|---|---|---|---|
| 100 | 10 | 1 | 0.1 | 0.01 | 0.001 | |
| 100 | 100 | |||||
| 10 | 100 | |||||
| 1 | 100 | |||||
| 0.1 | 100 | |||||
| 0.01 | 100 | |||||
| 0.001 | 100 | |||||
The contents (ng) of purified O1 : O139 gDNAs were used in this assay.
2.7. 16S rDNA Amplification
For the quality control of the TMPCR amplification, the 16s rDNA amplification that served as internal control reaction was performed in triplicate with all the reactions containing target or nontarget DNA template throughout the study. In addition, the primers that are specific for microbial DNA (16S rDNA locus) included forward primer 5′-CGG TGA AAT GCG TAG AGA T-3′ and reverse primer 5′-TTA CTA GCG ATT CCG AGT TC-3′ [35]. A 25 μL optimized PCR mixture contained 1X PCR buffer and 200 μM each dNTP as before except that forward and reverse primers, Taq DNA polymerase, and gDNA template were used at concentration of 0.5 μM each, 0.5 U, and 1 μL of undiluted template, respectively. The optimized amplification was performed on predenaturing at 94°C for 5 min and followed by 30 cycles of denaturing at 94°C for 45 sec, annealing at 55°C for 45 sec, and extension at 72°C for 45 sec. Finally, the last extension was at 72°C for 5 min. In this regard, the amplicons with expected size of 663 bp were obtained from all reactions containing microbial gDNA templates.
2.8. Analysis of PCR Products
One-fifth of the amplicons mixed with 6x loading dye solution were electrophoresed through the 1.5% to 2.0% agarose gels at constant voltage of 10 V/cm in 1x TBE buffer (89 mM Tris-HCl, 89 mM Borate, 2 mM EDTA) (Ameresco, Ohio, USA). The ethidium bromide- (EtBr-) stained DNA gel was used to analyze the amplicon sizes in base pairs as compared to DNA standard size marker (500 ng of each 100 bp ladder) (Phamacia Biotech, USA). The intensity of EtBr-stained DNA bands was visualized and photographed under an ultraviolet (UV) wavelength using DNA gel documentation (Fotodyne, Hartland, USA).
In addition, analysis of amplification specificity, sensitivity, and fidelity of the O serogroup-specific TMPCR was done as follows. In reactions using target gDNAs, intense EtBr-stained duplex bands were detected, either equally or unequally, for O1 (588 bp and 364 bp) and for O139 (588 bp and 258 bp). An intense 588 bp band was detected for the non-O1 (both NAG I and NAG II). In reactions using nontarget gDNAs or nuclease-free water instead of DNA, neither expected amplicon sizes nor nonspecific DNA bands were observed.
3. Results
Primer-template specificity binding was empirically determined in reactions of which optimized PCR parameters were analyzed upon the amplification efficiency of three multiplex primer sets that specifically bind to V. cholerae gDNAs of the O1, O139, or O22/O155. We initially optimized the separate reactions containing the gDNA content of V. cholerae O1 569B or O139 MO45 alone, or equally mixed. Most reliable amplification conditions were reproducibly performed on the reactions using a 10 ng each of V. cholerae O1 569B or O139 MO45 per reaction assay, or equally mixed. The resulting algorithm upon annealing temperature above 55°C was likely to decrease amplification yields of the specific amplicons with expected sizes (data not shown). All reactions containing O22 or O155 gDNA putatively yielded a 588 bp DNA band (data not shown). Optimum annealing temperatures ranged from 53°C to 57°C. It was clear that PCR factors underlying the annealing temperatures and primer set concentrations were more likely to influence the amplification efficiency whether the O1, O139, or equally mixed gDNAs were used. As a result, optimized multiplex PCR amplification depended on annealing temperature at 55°C and primer set concentrations of 0.4 (universal), 0.4 (O1-specific), and 1.0 (O139-specific) μM (Figure 1).
Figure 1.
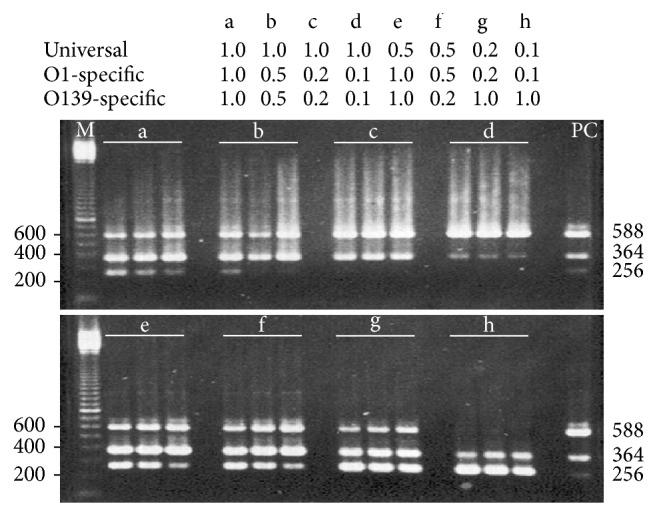
Agarose gel electrophoresis analysis of the PCR algorithm upon annealing temperatures and primer set concentrations that had effects on yields of three specific amplicons. In a 25 μL PCR assay using 10 ng each of purified V. cholerae O1 and O139 gDNAs, amplification was performed in triplicate with varying concentration of three primer sets (μM) (a to h) and annealing temperature increments (°C) at 54.2, 55.5, and 56.9. The 100 bp molecular marker (lane M) and serogroup-specific PCR amplicons as positive control (lane PC) were used for base pair size comparisons.
We further employed the stringent protocol developed for TMPCR because the multiplex PCR provided a ladder-like problem of amplification background of most reactions containing nontarget gDNAs. Similarly, the multiplex PCR yielded spurious products even in some reactions containing V. cholerae O1, O139, or non-O1/non-O139 gDNAs prepared by CTAB/phenol-chloroform extraction method or by rapid boiling method (data not shown). As a result of optimized O serogroup-specific TMPCR with loose touchdown strategy, thermal cycling condition employed annealing temperature decrements at an interval of 57°C to 53°C versus time increments to minimize nonlagging products during initial geometric amplification cycles. As compared to the 16S rDNA amplification, this proven TMPCR had 100% specificity or gave putatively positive amplifications in reactions containing V. cholerae genomes but not other nontarget DNAs (Table 4). Of the 21 V. cholerae O1 serving as target DNAs tested by the TMPCR, it was clear that consistently positive amplifications with EtBr-intense duplex DNA bands (588 and 364 bp) were observed in reactions containing reference strains (8), Eltor Inaba (5), Eltor Ogawa (4), Classical Inaba (2), and Eltor Ogawa (2). Similarly, other 16 V. cholerae O139 strains yielded the amplified duplex DNA fragments (588 and 256 bp), while both NAG I and NAG II gave only a single 588 bp DNA band as expected. Only the representative target and nontarget gDNAs amplified by O serogroup-specific TMPCR, as compared to that amplified by 16S rDNA PCR, are shown in Figure 2. In addition, there were no differences of specificity and sensitivity of TMPCR when using V. cholerae O1, O139, and non-O1/non-O139 gDNA templates whether purified or boiled.
Table 4.
Amplification specificity of TMPCR and 16S rDNA-specific PCR.
| Organism | Strains tested (n = 100) |
Specificity (%) | |
|---|---|---|---|
| TMPCRa | 16S rDNA PCRb | ||
| V. cholerae O1 | 21 | 100 | 100 |
| V. cholerae O139 | 16 | 100 | 100 |
| V. cholerae NAG Ic | 16 | 100 | 100 |
| V. cholerae NAG IId | 16 | 100 | 100 |
| Other vibrios | 8 | 0 | 100 |
| Gram-negative bacteria | 16 | 0 | 100 |
| Gram-positive bacteria | 7 | 0 | 100 |
aThe specificity (%) = strains showing a positive amplification of expected amplicon sizes by TMPCR as having no cross-hybridization with negative controls/a total of strains tested, multiplied by 100.
bAs for 16S rDNA-specific PCR, no cross-hybridization occurred in reactions containing nuclease-free water or no DNA.
cNAG I non-O1/non-O139 growing in semisolid agar in the presence of sucrose and mannose but not arabinose.
dNAG II non-O1/non-O139 growing in semisolid agar only supplemented with sucrose.
Figure 2.
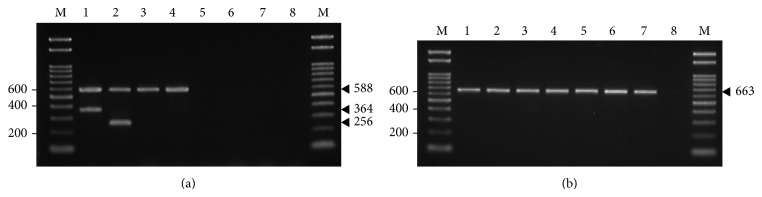
Agarose gel electrophoresis of the amplification specificity of optimized TMPCR (a) and 16S rDNA PCR (b), using representative target and nontarget gDNAs. In lanes 1 to 8, amplification reactions contained V. cholerae O1, V. cholerae O139, V. cholerae NAG I, V. cholerae NAG II, Vibrio parahaemolyticus, Vibrio vulnificus, Escherichia coli, and nuclease-free water, respectively. The expected sizes of specific amplicons (base pairs) were compared to the 100 bp standard ladder marker (lane M).
To analyze the amplification sensitivity of the TMPCR, we determined a reciprocal amplification pertaining to the algorithms that used the amount of ratios of mixed V. cholerae gDNAs (O1 : O139), in comparison to those reactions containing each gDNA template serially diluted. The resulting TMPCR experiments in which CTAB/phenol-chloroform extracted V. cholerae gDNA whether O1, O139, or O22/O155 was amplified are shown in Figure 3. As for Figure 3(a), the TMPCR could detect O serogroup-specific DNA fragments authentically derived from the O1 or the O139 in reciprocal reactions. The detection limit of as low as 1 ng of the O1 was obtained in the presence of the higher amount of O139 or 100-fold greater than the O1. Meanwhile, the detection limit of as low as 10 ng of the O139 was obtained in the presence of the higher amount of the O1 or 10-fold greater than the O139. Figure 3(b) shows that its detection limit was at 100 pg of the O1 or the O139 in inverse reactions containing unequal amount ratios of the 100 pg O1 to 1 ng O139, and vice versa. Again, the TMPCR was likely to show detection limit of as low as 100 pg each of gDNAs in the reaction containing equal amount ratio of the O1 to O139. It was clear to note that the TMPCR had the detection limit of as low as 100 pg gDNA of the O1 or O139 when examined for the equal amount ratio of the serially diluted O1 and O139 templates (Figure 3(c)). Similarly, it also had the same detection limit of as low as 100 pg gDNA of the O1 or O139 when separately copied (Figures 3(d) and 3(e)). The amount of as low as 100 pg O22 or O155 gDNA was also detected (data not shown).
Figure 3.

Agarose gel electrophoresis analysis of the amplification efficiency of semiquantitative TMPCR. A reciprocal amplification algorithm of detectable levels of the O1 to O139 gDNAs (experiments (a) to (e)) was described in the text, while E. coli gDNA template serving as negative control (NC) was included. (a) Lanes 1 to 6, the 100 ng O1 gDNA versus a 10-fold serially diluted O139 gDNA, 1 pg to 100 ng, and vice versa (lanes 7 to 12). (b) Lanes 1 to 3, unequal amount ratios of the O1 : O139, 100 ng to 1 pg, 10 ng to 10 pg, and 1 ng to 100 pg, and vice versa (lanes 4 to 6). Lanes 7 to 12, the 100 pg O139 gDNA versus the O1 gDNA contents of varying 100 ng, 50 ng, 25 ng, 10 ng, and 1 ng to 100 pg. (c) Lanes 1 to 6, the equal amount ratios of 10-fold serially diluted O1 to O139 gDNAs, 100 ng to 1 pg. (d) Lanes 1 to 6, 10-fold serially diluted O1 gDNA contents, 100 ng to 1 pg. (e) Lanes 1 to 6, 10-fold serially diluted O139 gDNA contents, 100 ng to 1 pg. A 100 bp DNA ladder marker (lane M) was used in comparison of the amplicons with expected size (bp).
Lastly, we tested the amplification fidelity of O serogroup-specific TMPCR by using target gDNA samples archived from the stock cultures of the 148 V. cholerae clinical isolates. Overall, the TMPCR exhibited 100% concordance with serotyping method as reference whether V. cholerae cultures were obtained from stools or rectal swabs (Table 5). All the 108 cultures from the stools tested gave positive TMPCR results: 65 (60.2%) of the O1 serotype concordant with the O1 serogroup-specific TMPCR (588 and 364 bp), 15 (13.9%) of the O139 serotype concordant with the O139 serogroup-specific TMPCR (588 and 256 bp), and 28 (25.9%) of the non-O1/non-O139 serotype concordant with the O serogroup-specific TMPCR (588 bp). Meanwhile, all the 40 cultures from the rectal swabs were also consistently positive with the tests: 39 (97.5%) accordant with the O1 serotype and the O1 serogroup-specific TMPCR and only one (2.5%) accordant with the non-O1/non-O139 serotype and 588 bp specific TMPCR. Only the representative gDNAs archived from the clinically isolated V. cholerae cultures are shown in Figure 4. All samples tested by O serogroup-specific TMPCR were consistently positive with the 16S rDNA PCR.
Table 5.
Agreement of O serogroup-specific TMPCR for detecting and differentiating gDNAs of the O1, O139, and non-O1/non-O139 serogroups archived from clinical samples (n = 148), using serotyping method as reference.
| Source | Serogroupa | TMPCR | ||
|---|---|---|---|---|
| ompW | rfbV | wbf | ||
| Stool n = 108 |
O1 (n = 65) | 65 | 65 | 0 |
| O139 (n = 15) | 15 | 0 | 15 | |
| Non-O1/non-O139 (n = 28) | 28 | 0 | 0 | |
| Total | 108 | 65 | 15 | |
|
| ||||
| Rectal swab n = 40 |
O1 (n = 39) | 39 | 39 | 0 |
| O139 (n = 0) | 0 | 0 | 0 | |
| Non-O1/non-O139 (n = 1) | 1 | 0 | 0 | |
| Total | 40 | 39 | 0 | |
aAll laboratory settings yielded 100% concordant V. cholerae cultures whether rectal swabs or stools examined using standard serotyping method as described in the text.
Figure 4.
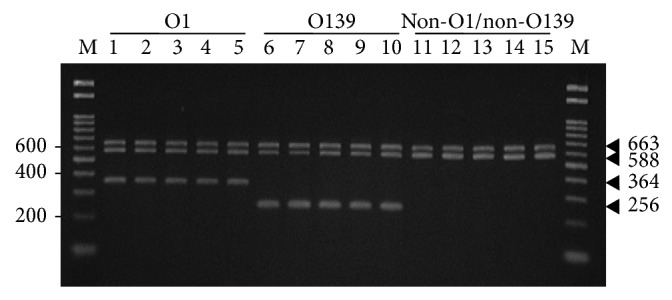
Agarose gel electrophoresis of the O serogroup-specific TMPCR and 16S rDNA PCR profiles using representative V. cholerae gDNAs archived from the clinical specimens. PCR products obtained by both TMPCR (5 μL) and 16S rDNA PCR (5 μL) that were separately performed are confined to the same lane: lanes 1–3, the O1 from stools; lanes 4-5, the O1 from rectal swabs; lanes 6–10, the O139 from stools; and lanes 11–15, the non-O1/non-O139 from stools. In each TMPCR employing the O1, O139, or non-O1/non-O139 gDNA, the reliable reaction represents duplex DNA fragments (588 bp and 364 bp from the O1 or 588 bp and 256 bp from the O139) and a single 588 bp DNA band from the non-O1/non-O139, as compared to 16S rDNA amplicons (663 bp) and 100 bp DNA ladder marker (lane M).
Additionally, TMPCR provided reliable testing results when examined for V. cholerae present in environmental samples such as water samples and seafood, as compared to the culture method/serotyping (Table 6). Particularly when examined for V. cholerae present in aquatic environments, TMPCR provided positive result for V. cholerae non-O1/non-O139 in a water sample, whereas culture method did not.
Table 6.
Advancement of TMPCR for V. cholera detection using environmental samples.
| Sourcea | Sample number | Culture method and serotyping | TMPCR | ||||
|---|---|---|---|---|---|---|---|
| O1 | O139 | Non-O1/Non-O139 | O1 | O139 | Non-O1/Non-O139 | ||
| Water samples | 50 | 0 | 0 | 0 | 0 | 0 | 1 |
| Seafood | 18 | 5 | 0 | 8 | 5 | 0 | 8 |
aAll were collectively obtained during the outbreaks. The individual samples enriched in broth were used in V. cholerae detection and differentiation using the culture method/serotyping and TMPCR.
4. Discussion
The conventional culture technique is fundamental to recover the V. cholerae culture from clinical samples of V. cholerae-contracted or cholera-like patients. However, the approach relies upon the ability of the V. cholerae propagating in vitro culture and expressing normally phenotypic characteristics under physiological conditions whether morphologically, biochemically, or serologically. This classical microbiological approach is not just to grow viable microbial cells onto a plate count agar or in an enrichment culture medium which constitutes the entire nutrients essential for growth of the target microbe(s), but also to incubate the propagated cultures under conditions favoring their optimal growth. Such VBNC V. cholerae as well as some VBNC vibrios can exist in nature in aquatic habitats such that they are not as yet cultured in the laboratory and hence have not been characterized using routine microbiological methods due to the limits of recovery of resuscitated cultures from environmental samples. However, the V. cholerae once recovered from those isolation sources needs to be logically analyzed to determine their property whether phenotypically or genotypically.
In the present study, we provided the proof that all the serotypes of V. cholerae both reference strains and clinical isolates were consistently positive with the O serogroup-specific TMPCR method, showing the concordance of serotypes and O serogroup-specific genotypes. In other words, the TMPCR method that employed the primer sets specific for the O1, O139, and non-O1/non-O139 could simultaneously yield amplicons authentically derived from the target DNA sequences only in reactions containing the low amounts of target gDNAs but not the unrelated gDNAs. This implied that the TMPCR had high performance efficiency (specificity and sensitivity) in the detection and identification of the O1, O139, and non-O1/non-O139 serogroups of V. cholerae reference strains and clinical isolates. The O serogroup-specific primer sets did not cross-hybridize against nontarget DNA sequences neither in reactions containing target gDNA templates nor in reactions containing unrelated gDNA templates. As for the quantitatively direct determination assessment tool, the TMPCR had the detection limit of as low as 100 pg of V. cholerae gDNAs of the O1, O139, or non-O1/non-O139. When mimic conditions of the mixed gDNAs of the O1 and O139 were analyzed, the TMPCR method exhibited high sensitivity. The reaction assays that employed the equally or unequally mixed O1 and O139 gDNAs could compare well with the findings of the reactions that used the single source gDNAs. This might suggest that the O serogroup-specific TMPCR assay could augment the efficiency of the serotyping method when examined for the qualitative and quantitative analyses of the serotyped V. cholerae cultures recovered from any isolation sources whether clinically or environmentally. As compared to the culture method, only the TMPCR gave positive result when target DNA of V. cholerae non-O1/non-O139 could be detected in a water sample in this study. That is, the TMPCR can provide the proof that V. cholerae non-O1/non-O139 present in a water sample is the VBNC state. This promising technique can be used as similar to the molecular markers-based PCR methods that permit the detection and identification of V. cholerae O1, O139, and non-O1/non-O139 isolates from clinical patients or aquatic environments [13–24].
Additionally, the TMPCR requires the procedures for the gDNA preparation, target gene amplification, and postanalysis of PCR product. However, compared to that of the serotyping method, the technique becomes easier in terms of process and time required to run a lot of the sampled V. cholerae cultures. Similar to that of other touchdown or multiplex PCR methods developed for probing a variety of microorganisms [33, 36, 37], the performance efficiency of the TMPCR tested in this study was achieved by a two-stage amplification incorporating multiplex PCR with conditional touchdown strategies, which were suitable for simultaneous amplification of target DNA sequences with high fidelity and workable rate. This improved multiplex PCR comprises a simultaneous PCR and a specific PCR, and either one or both of the amplification steps are performed with a touchdown strategy, of which loose touchdown strategy is applied with a temperature lower than the optimized annealing temperature, and stringent touchdown strategy is applied with a temperature higher than the optimized annealing temperature. The TMPCR approach to circumvent the problems of multiplex PCR assays has been successfully used for simultaneous amplification of target DNA sequences originally derived from the O1, O139, and non-O1/non-O139 serogroups with the corresponding touchdown-PCR parameters.
Acknowledgments
The authors were grateful to the Faculty of Tropical Medicine, Mahidol University, and the Armed Forces Research Institute of Medical Sciences, Bangkok, Thailand, for providing the O1 569B and O139 MO45 strains. Wild strains of the O1 (including El Tor/Classical Ogawa and El Tor/Classical Inaba biotypes) and O139 were kindly provided by Bamrasnaradura Infectious Diseases Hospital, Nonthaburi. The non-O1 were from the Faculty of Public Health, Mahidol University. Other reference strains of the O22 and O155 were kindly provided by Professor Toshio Shimada, the National Institute of Infectious Diseases, Tokyo, Japan.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publishing of this paper.
References
- 1.Huq A., Small E. B., West P. A., Huq M. I., Rahman R., Colwell R. R. Ecological relationships between Vibrio cholerae and planktonic crustacean copepods. Applied and Environmental Microbiology. 1983;45(1):275–283. doi: 10.1128/aem.45.1.275-283.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Faruque S. M., Albert M. J., Mekalanos J. J. Epidemiology, genetics, and ecology of toxigenic Vibrio cholerae . Microbiology and Molecular Biology Reviews. 1998;62(4):1301–1314. doi: 10.1128/mmbr.62.4.1301-1314.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gil A. I., Louis V. R., Rivera I. N. G., et al. Occurrence and distribution of Vibrio cholerae in the coastal environment of Peru. Environmental Microbiology. 2004;6(7):699–706. doi: 10.1111/j.1462-2920.2004.00601.x. [DOI] [PubMed] [Google Scholar]
- 4.Constantin de Magny G., Colwell R. R. Cholera and climate: a demonstrated relationship. Transactions of the American Clinical and Climatological Association. 2009;120:119–128. [PMC free article] [PubMed] [Google Scholar]
- 5.Lutz C., Erken M., Noorian P., Sun S., McDougald D. Environmental reservoirs and mechanisms of persistence of Vibrio cholerae . Frontiers in Microbiology. 2013;4, article 375 doi: 10.3389/fmicb.2013.00375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chaiyanan S., Huq A., Maugel T., Colwell R. R. Viability of the nonculturable Vibrio cholerae O1 and O139. Systematic and Applied Microbiology. 2001;24(3):331–341. doi: 10.1078/0723-2020-00032. [DOI] [PubMed] [Google Scholar]
- 7.Baffone W., Citterio B., Vittoria E., et al. Retention of virulence in viable but non-culturable halophilic Vibrio spp. International Journal of Food Microbiology. 2003;89(1):31–39. doi: 10.1016/s0168-1605(03)00102-8. [DOI] [PubMed] [Google Scholar]
- 8.Wong H. C., Wang P. Induction of viable but nonculturable state in Vibrio parahaemolyticus and its susceptibility to environmental stresses. Journal of Applied Microbiology. 2004;96(2):359–366. doi: 10.1046/j.1365-2672.2004.02166.x. [DOI] [PubMed] [Google Scholar]
- 9.Alam M., Sultana M., Balakrish Nair G., et al. Viable but nonculturable Vibrio cholerae O1 in biofilms in the aquatic environment and their role in cholera transmission. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(45):17801–17806. doi: 10.1073/pnas.0705599104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Senoh M., Ghosh-Banerjee J., Ramamurthy T., et al. Conversion of viable but nonculturable Vibrio cholerae to the culturable state by co-culture with eukaryotic cells. Microbiology and Immunology. 2010;54(9):502–507. doi: 10.1111/j.1348-0421.2010.00245.x. [DOI] [PubMed] [Google Scholar]
- 11.Senoh M., Ghosh-Banerjee J., Ramamurthy T., et al. Conversion of viable but nonculturable enteric bacteria to culturable by co-culture with eukaryotic cells. Microbiology and Immunology. 2012;56(5):342–345. doi: 10.1111/j.1348-0421.2012.00440.x. [DOI] [PubMed] [Google Scholar]
- 12.Pradhan S., Mallick S. K., Chowdhury R. Vibrio cholerae classical biotype is converted to the viable non-culturable state when cultured with the El Tor biotype. PLoS ONE. 2013;8(1) doi: 10.1371/journal.pone.0053504.e53504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoshino K., Yamasaki S., Mukhopadhyay A. K., et al. Development and evaluation of a multiplex PCR assay for rapid detection of toxigenic Vibrio cholerae O1 and O139. FEMS Immunology and Medical Microbiology. 1998;20(3):201–207. doi: 10.1016/s0928-8244(98)00014-5. [DOI] [PubMed] [Google Scholar]
- 14.Chakraborty S., Khanam J., Takeda Y., Nair G. B. Application of PCR for detection of toxigenic Vibrio cholerae O1 in water samples during an outbreak of cholera. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1999;93(5):527–528. doi: 10.1016/s0035-9203(99)90366-8. [DOI] [PubMed] [Google Scholar]
- 15.Pal B. B., Khuntia H. K., Samal S. K., Das S. S., Chhotray G. P. Emergence of Vibrio cholerae O1 biotype El Tor serotype Inaba causing outbreaks of cholera in Orissa, India. Japanese Journal of Infectious Diseases. 2006;59(4):266–269. [PubMed] [Google Scholar]
- 16.Alam M., Sultana M., Nair G. B., et al. Toxigenic Vibrio cholerae in the aquatic environment of Mathbaria, Bangladesh. Applied and Environmental Microbiology. 2006;72(4):2849–2855. doi: 10.1128/aem.72.4.2849-2855.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rivera I. N. G., Lipp E. K., Gil A., Choopun N., Huq A., Colwell R. R. Method of DNA extraction and application of multiplex polymerase chain reaction to detect toxigenic Vibrio cholerae O1 and O139 from aquatic ecosystems. Environmental Microbiology. 2003;5(7):599–606. doi: 10.1046/j.1462-2920.2003.00443.x. [DOI] [PubMed] [Google Scholar]
- 18.Rivera I. N. G., Chun J., Huq A., Sack R. B., Colwell R. R. Genotypes associated with virulence in environmental Isolates of Vibrio cholerae . Applied and Environmental Microbiology. 2001;67(6):2421–2429. doi: 10.1128/aem.67.6.2421-2429.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bakhshi B., Mohammadi-Barzelighi H., Adabi M., Lari A. R., Pourshafie M. R. A molecular survey on virulence associated genotypes of non-O1 non-O139 Vibrio cholerae in aquatic environment of Tehran, Iran. Water Research. 2009;43(5):1441–1447. doi: 10.1016/j.watres.2008.12.025. [DOI] [PubMed] [Google Scholar]
- 20.Bakhshi B., Mohammadi-Barzelighi H., Sharifnia A., Dashtbani-Roozbehani A., Rahbar M., Pourshafie M. R. Presence of CTX gene cluster in environmental non-O1/O139 Vibrio cholerae and its potential clinical significance. Indian Journal of Medical Microbiology. 2012;30(3):285–289. doi: 10.4103/0255-0857.99487. [DOI] [PubMed] [Google Scholar]
- 21.Sharma C., Thungapathra M., Ghosh A., et al. Molecular analysis of non-O1, non-O139 Vibrio cholerae associated with an unusual upsurge in the incidence of cholera-like disease in Calcutta, India. Journal of Clinical Microbiology. 1998;36(3):756–763. doi: 10.1128/jcm.36.3.756-763.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ang G. Y., Yu C. Y., Balqis K., et al. Molecular evidence of cholera outbreak caused by a toxigenic Vibrio cholerae O1 El Tor variant strain in Kelantan, Malaysia. Journal of Clinical Microbiology. 2010;48(11):3963–3969. doi: 10.1128/jcm.01086-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alam M., Nusrin S., Islam A., et al. Cholera between 1991 and 1997 in Mexico was associated with infection by classical, El Tor, and El Tor variants of Vibrio cholerae . Journal of Clinical Microbiology. 2010;48(10):3666–3674. doi: 10.1128/jcm.00866-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mishra A., Taneja N., Sharma M. Environmental and epidemiological surveillance of Vibrio cholerae in a cholera-endemic region in India with freshwater environs. Journal of Applied Microbiology. 2012;112(1):225–237. doi: 10.1111/j.1365-2672.2011.05191.x. [DOI] [PubMed] [Google Scholar]
- 25.Fallarino A., Mavrangelos C., Stroeher U. H., Manning P. A. Identification of additional genes required for O-antigen biosynthesis in Vibrio cholerae O1. Journal of Bacteriology. 1997;179(7):2147–2153. doi: 10.1128/jb.179.7.2147-2153.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stroeher U. H., Parasivam G., Kate Dredge B., Manning P. A. Novel Vibrio cholerae O139 genes involved in lipopolysaccharide biosynthesis. Journal of Bacteriology. 1997;179(8):2740–2747. doi: 10.1128/jb.179.8.2740-2747.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stroeher U. H., Jedani K. E., Manning P. A. Genetic organization of the regions associated with surface polysaccharide synthesis in Vibrio cholerae O1, O139 and Vibrio anguillarum O1 and O2: a review. Gene. 1998;223(1-2):269–282. doi: 10.1016/s0378-1119(98)00407-7. [DOI] [PubMed] [Google Scholar]
- 28.Yamasaki S., Shimizu T., Hoshino K., et al. The genes responsible for O-antigen synthesis of Vibrio cholerae O139 are closely related to those of Vibrio cholerae O22. Gene. 1999;237(2):321–332. doi: 10.1016/s0378-1119(99)00344-3. [DOI] [PubMed] [Google Scholar]
- 29.Jalajakumari M. B., Manning P. A. Nucleotide sequence of the gene, ompW, encoding a 22 kDa immunogenic outer membrane protein of Vibrio cholerae . Nucleic Acids Research. 1990;18(8):p. 2180. doi: 10.1093/nar/18.8.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nandi B., Nandy R. K., Mukhopadhyay S., Nair G. B., Shimada T., Ghose A. C. Rapid method for species-specific identification of Vibrio cholerae using primers targeted to the gene of outer membrane protein Omp W. Journal of Clinical Microbiology. 2000;38(11):4145–4151. doi: 10.1128/jcm.38.11.4145-4151.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heiberg B. The biochemical reaction of vibrios. Journal of Hygiene. 1936;36:114–117. doi: 10.1017/s0022172400043473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kelly M. T., Hickman-Brenner F. W., Farmer J. J. Vibrio. In: Balow A., Hauler W. J. Jr., Herrman K. L., Isenberg H. D., Shadorny H. J., editors. Manual of Clinical Microbiology. Washington, DC, USA: ASM; 1991. pp. 384–395. [Google Scholar]
- 33.Quaglia N. C., Normanno G., Dambrosio A., et al. Multiplex-touchdown PCR assay for the detection and genotyping of Helicobacter pylori from artificially contaminated sheep milk. Journal of Food Protection. 2005;68(10):2136–2139. doi: 10.4315/0362-028x-68.10.2136. [DOI] [PubMed] [Google Scholar]
- 34.Dieffenbach C. W., Dveksler G. S. PCR Primer: A Laboratory Manual. Cold Spring Harbor, NY, USA: Cold Spring Harbor Laboratory; 1995. [Google Scholar]
- 35.Tarr C. L., Patel J. S., Puhr N. D., Sowers E. G., Bopp C. A., Strockbine N. A. Identification of Vibrio isolates by a multiplex PCR assay and rpoB sequence determination. Journal of Clinical Microbiology. 2007;45(1):134–140. doi: 10.1128/jcm.01544-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nhung P. H., Ohkusu K., Miyasaka J., Sun X. S., Ezaki T. Rapid and specific identification of 5 human pathogenic Vibrio species by multiplex polymerase chain reaction targeted to dnaJ gene. Diagnostic Microbiology and Infectious Disease. 2007;59(3):271–275. doi: 10.1016/j.diagmicrobio.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 37.Pechgit P., Intarapuk A., Pinyoowong D., Bhumiratana A. Touchdown-touchup nested PCR for low-copy gene detection of benzimidazole-susceptible Wuchereria bancrofti with a Wolbachia endosymbiont imported by migrant carriers. Experimental Parasitology. 2011;127(2):559–568. doi: 10.1016/j.exppara.2010.10.022. [DOI] [PubMed] [Google Scholar]