

Abstract
We describe a novel method of drug discovery using MLSD and drug repositioning, with cancer target STAT3 being used as a test case. Multiple drug scaffolds were simultaneously docked into hot spots of STAT3 by MLSD, followed by tethering to generate virtual template compounds. Similarity search of virtual hits on drug database identified Celecoxib as a novel inhibitor of STAT3. Furthermore, we designed two novel lead inhibitors based on one of the lead templates and Celecoxib.
Introduction
New drug development still presents grand challenges. Conventional high throughput screening (HTS) drug discovery approach identifies many hits, but few of them can be developed into drugs. Currently, there are less than 1500 FDA-approved drugs. Poor efficacy and safety of hits are the major attritions of drug development. It is suggested that poor drug-space, low structural diversity and poor drug ADMET properties of compounds in HTS libraries may contribute to both false positives and negatives. Over the past decade, fragment-based drug design (FBDD) has emerged as a successful alternative to drug discovery using biophysical methods like NMR and X-ray crystallography. For computational FBDD, conventional single fragment docking has problems of non-specific binding and poor ranking power due to weak binding of small fragments. Recently, we have developed multiple ligand simultaneous docking (MLSD) to simulate the interplay of multiple molecules binding to the protein binding site(s).1 In a test case, MLSD identified the correct binding modes of multiple fragments of drug lead 4-[4-[(4'-Chloro[1,1'-biphenyl]-2-yl)methyl]-1-piperazinyl]-N-[[4-[[(1R)-3-(dimethylamino)-1-[(phenylthio)methyl]propyl]amino]-3-nitrophenyl]sulfonyl]benzamide (ABT-737)1 in the respective sub-pockets of the binding groove of cancer target Bcl-xL, whereas single-fragment docking failed to do so due to energetic and dynamic coupling among the fragments.2 The results suggest potential applications of MLSD to improve fragment-based docking screening. On the other hand, to reuse existing drugs for new targets, a drug repositioning concept has been proposed recently.3 Previous analysis revealed that more than 30% of drugs share building blocks.4 We hypothesize that FBDD using privileged drug scaffolds would help to generate lead compounds with improved ADMET properties.
To meet the challenge of drug discovery, we present here a novel approach for drug lead discovery using MLSD, drug scaffolds and drug repositioning. Cancer target signal transducer and activator of transcription 3 (STAT3), an oncogene being constitutively activated in numerous cancers, was used as a test case in our study.5–7 Currently there is no report of an approved drug to target STAT3, although a number of small molecule inhibitors of STAT3 have been discovered via HTS and virtual docking.8–15 Figure 1 shows our drug discovery methodology. It proceeds as follows: 1. A small library of drug scaffolds is identified for the binding hot spots of STAT3 SH2 domain; 2. MLSD screening of the privileged drug scaffolds is then performed to identify optimal fragment combination(s); 3. Linking of the fragment hits generates possible hit compounds as templates; 4. Similarity search of template compounds in drug databases identifies existing drugs as possible inhibitors of the protein target of interest.
Figure 1.
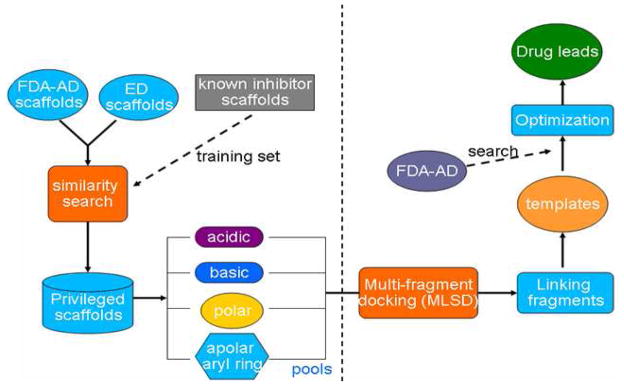
Scheme of drug discovery using MLSD and drug repositioning
Results and Discussion
Identifying privileged drug scaffolds for STAT3
It has been reported that the STAT3 pathway is activated upon the phosphorylation of tyrosine 705, followed by dimerization, nuclear translocation and DNA binding. The druggable binding cleft of the STAT3 SH2 domain (PDB code 1BG1) consists of 3 sub-pockets: pTyr705 (pY705) binding site, Leu706 binding site (L706) and a side pocket (Ile597, Leu607, Thr622 and Ile634). The main pTyr705 binding site is polar and basic, while the Leu706 and side pocket are hydrophobic. We built a small library of feature fragments from a collection of small molecule inhibitors of STAT3 SH2 in previous reports.4–11 To avoid fragments with undesired drug ADMET properties, drug scaffolds structurally or chemically similar to the obtained feature fragments were identified by similarity search on a drug scaffold database. Figure 2 lists a small library of drug scaffolds identified, which were grouped into 2 pools: polar and nonpolar. The polar scaffolds in Pool 1 favor binding to the polar and basic pY705 site, and the relatively nonpolar scaffolds in Pool 2 are for the L706 site or side pocket.
Figure 2.

Privileged drug scaffolds for STAT3 SH2. Pool 1 is for pY705 site, and pool 2 is for L706 site or side pocket.
Simultaneous docking of 3 fragments to binding hot spots of STAT3 SH2
So far, there has been no report of a fragment-based design approach to identify inhibitors of STAT3. Docking modeling showed that previously reported inhibitors bound to 2 of the 3 sub-pockets of the STAT3 SH2 domain. To improve binding affinity, we applied MLSD to dock multiple drug scaffolds in a concerted way to the 3 binding hot spots of STAT3, like fitting the right piece into the right place in jigsaw puzzle (Figure 3). Briefly, three drug fragments, one from pool 1 and the other two from pool 2, were used as inputs for the MLSD docking screening. The combination of drug scaffolds in the two pools generated a diverse set. Figure 3 shows that hits H1 (f1, f2 and f3) and H2 (f1, f4 and f5) docked to the hot spots of STAT3 SH2, with a predicted binding energy of -12.5 kcal/mol and -12.1 kcal/mol, respectively. In both hits, the polar fragment f1 (phenylsulfonamide) occupied the main pY705 binding pocket formed by surrounding residues Arg609, Lys591, Glu612, and Ser613. In hit H1, fragment f2 (1-phenylethanol) occupied the L706 sub-pocket, and f3 (2-phenylpropane) docked to the side pocket. In hit H2, f4 and f5 bound to the L706 and sub-pocket, respectively.
Figure 3.

Scheme of MLSD screening of drug scaffolds for binding hot spots of STAT3 SH2. A. In hit1 (H1), fragments f1 (sulfonamide), f2 (1-phenylethanol), and f3 (2-phenylpropane) occupied the 3 hot spots (pY705, L706 and side pocket) of STAT3 with a binding energy of -12.5 kcal/mol. B. In hit2 (H2), fragments f1 (sulfonamide), f4 (1,4-Dimethylbenzene), and f5 (1-Bromo-4-methylbenzene) docked into 3 hot spots with a binding energy of -12.1 kcal/mol. Scaffolds are shown in stick-ball and colored with atom type.
Interestingly, predicted binding energies of hit H1 and H2 are very close. Further binding mode cluster analysis revealed that with similar binding energies, the binding modes of f2, f3, f4, f5 in the L706 and side pocket could be different and dynamic (Figure S2 of Supplemental Materials). The binding affinity of aromatic fragments f2 and f3 was similar to that of f4 and f5. Docking simulation results suggest that the polar pY705 site is essential and sulfonamide f1 moiety is a key fragment for binding. The docked fragments and their binding modes could be used as a blueprint to design possible inhibitors for STAT3. Our results show for the first time 3 fragments docked into the 3 hot spots of STAT3 SH2.
Linking fragments for hits and virtual template compounds design
To generate possible lead candidates, we linked the three fragments in hits H1 and H2 using different chemical tethers such as amide, amine, ether and olefin. The structures of linked compounds and their docking energies are listed in Table S1 of Supplemental Materials. The virtual templates had docking binding energies in the range of -8.2 to -12.0 kcal/mol. All 15 virtual compounds had relatively good binding to all 3 hot spots of STAT SH2 in docking modeling. Figure 4A shows the docking models of template compounds T1 and T2. The top hit compound T1 (green) demonstrated binding modes and binding energy (ΔG of −12.0 kcal/mol) very close to that of the docked f1, f2 and f3 in hit H1 (ΔG of −12.5 kcal/mol). Compound T2 (red color) also bound relatively well to the 3 hot spots of STAT3, with binding modes similar to that of fragments f1, f4 and f5 in H2. The linked molecule T2 gave a lower binding affinity as compared to the 3 docked fragments (f1, f4 and f5) possibly due to the short linker between f1 and f5 in T2, which could lead to the tilt of f1 and shallow binding of f5. Also the strain and single bond entropic penalty of linker upon binding as compared to the free fragments could result in a weaker binding energy. We found it is very difficult for a linked compound to exactly match binding modes and orientations of the docked fragments. One possible reason is that the docking simulation gives a cluster of binding modes with very similar binding energies. Fragment binding in the L706 and side pocket is dynamic to some degree as shown in Figure S2 of Supplemental Materials.
Figure 4.

A. Linking the docked fragments to obtain virtual template compounds. Tethering f1, f2 and f3 in hit H1 gave T1 (green stick-ball), and Linking f1, f4 and f5 of H2 generated T2 (red stick-ball). Docked fragments of f1, f2 and f3 are represented by the thin green line, and f1, f4 and f5 are represented by the thin red line. Re-docking of T1 and T2 to STAT3 resulted in binding energies of -12.0 kcal/mol and -8.6 kcal/mol, respectively. B. Drug Celecoxib was identified as a novel inhibitor of STAT3 SH2 by similarity searching for T1, T2 and other virtual compounds in DrugBank. Docking of Celecoxib (atomic coloring stick-ball) to STAT3 SH2 showed that phenylsulfonamide and phenylmethyl bound to the pY705 site and side pocket, respectively.
Drug repositioning
The docked fragments define the blueprint and main pharmacophore of possible binders. This would enable us to search for template compounds on a drug database to match compounds with a similar binding pharmacophore. We performed a similarity search of all 15 template compounds on an FDA approved drug set in DrugBank. 17 13 out of the 15 (87%) virtual compounds identified Celecoxib as a top hit (Tanimoto similarity coefficient >= 0.6). Both compound T1 and T2 matched Celecoxib as a top hit in DrugBank. Docking Celecoxib to STAT3 SH2 resulted in a major binding cluster (80% probability), where the polar phenylsulfonamide occupied the pY705 site and non-polar phenylmethyl occupied the side pocket (Figure 4B). In addition, docking modeling of Celecoxib to STAT3 indicated the binding modes and binding energy were comparable to most of the known inhibitors, which bound to the main pY705 site and only one of the L706 and side-pocket hot spots. 4–11 Docking Celecoxib gave a lower binding affinity (-6.9 kcal/mol) than that of T1 (-12.0 kcal/mol) and T2 (−8.6 kcal/mol). Comparing binding modes of Celecoxib with that of T1 and T2 indicates that the lower binding affinity is likely due to the missing 3rd fragment in Celecoxib for the L706 binding sub-pocket.
Virtual hit compounds T2 and T3 as more potent inhibitors of STAT3
To investigate whether binding to all 3 sub-pockets of STAT3 could improve inhibition on STAT3, template compounds were selected for synthesis based on ease of synthetic procedure and predicted binding modes and energies. The synthesis of top hit compound T1 is challenging and the effort to synthesize it is ongoing. T2 was synthesized. T3 is an optimization of celecoxib to strength side pocket binding and to add L706 binding. The docking modeling of T2 and T3 shows that both T2 and T3 bind relatively well to all 3 sub-pockets of STAT3 and can effectively compete with native Phospho-Tyrosine peptide binding (Figure 5). As compared to Celecoxib, T2 and T3 had better binding modes and an improved binding energy of -8.6 kcal/mol and -9.2 kcal/mol, respectively. The stronger binding is likely due to an extra fragment in T2 and T3 for binding to the L706 sub-pocket. The modeling would suggest that binding the L706 sub-pocket helps increase inhibition of STAT3. Cancer cell (HCT-116) assays also demonstrated that T2 and T3 are more potent inhibitors of STAT3 than Celecoxib (Fig 6A and Table 1). The modeling results are consistent with the inhibition activities of Celecoxib, T2 and T3.
Figure 5.
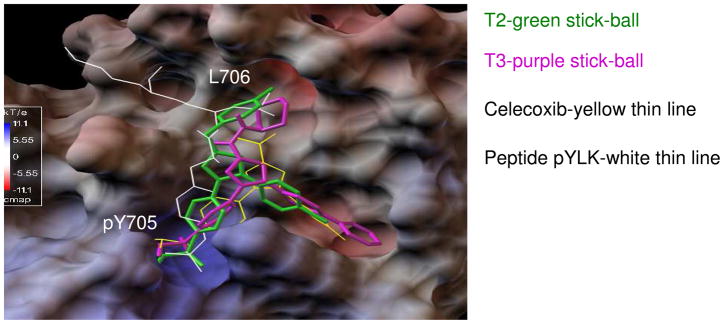
Docking modeling of Celecoxib (yellow thin line), T2 (green stick-ball) and T3 (purple stick-ball) to STAT3. Native pTyr peptide (pYLK) binding is shown as a thin white line. Both compounds T2 and T3 have an extra hydrophobic group binding to the L706 site, which results in better binding energies of -8.6 kcal/mol and -9.2 kcal/mol, respectively, as compared to Celecoxib (-6.9 kcal/mol). The predicted binding modes and energies show good correlations with inhibition activities of Celecoxib, T2 and T3 on STAT3 in HCT-116 cell based assays.
Figure 6.

Selective inhibition of STAT3 by Celecoxib, T2 and T3. A. Celecoxib, T2 and T3 inhibited STAT3 phosphorylation (Y705). Compounds T2 and T3 were more potent than Celecoxib at inhibiting P-STAT3. B. Celecoxib showed inhibition of STAT3 phosphorylation induced by IL-6 but not STAT1 phosphorylation by Interferon-γ.
Table 1.
Docked binding energies and IC50 of Celecoxib, T2 and T3 in HCT-116 cell viability assays
| compound | Docking ΔG (kcal/mol) | IC50 (μM) | Binding modes | MW | CLogP | PSA(Å2) |
|---|---|---|---|---|---|---|
| Celecoxib | −6.9 | 43.3 | pY705 and side pocket | 381.4 | 3.6 | 78.0 |
| T2 | −8.6 | 9.7 | pY705, L706 and side pocket | 482.4 | 5.5 | 78.0 |
| T3 | −9.2 | 10.1 | pY705, L706 and side pocket | 501.6 | 3.2 | 110.3 |
Cancer cell line assays: Celecoxib and compounds T2 and T3 down-regulate STAT3 phosphorylation in a dose-dependent manner
The drug Celecoxib is commonly known as a cyclooxygenase-2 (COX-2) inhibitor. To exclude interference of COX-2, human colon cancer cell line (HCT-116) that expresses constitutively activated STAT3, but not COX-2, was used in our study.18–19 To examine the inhibition of STAT3 phosphorylation, a Western blot was performed to detect the amount of phosphorylated STAT3 (P-STAT3) after HCT-116 cells were treated with Celecoxib, T2 and T3 (10μM to 50μM). As shown in Figure 6, the amount of P-STAT3 decreased in HCT-116 cells with increasing doses of compounds Celecoxib, T2 and T3. STAT3 is phosphorylated through the gp130 cytoplasmic pTyr loop binding to the STAT3 SH2 domain, thus blocking of STAT3 SH2 by inhibitors stops STAT3 phosphorylation. The expression of total STAT3 remained constant, which indicates that the decrease of P-STAT3 was not due to a constitutional decrease of total STAT3 protein. The results also show that both T2 and T3 have more potent inhibitory effects on P-STAT3 than Celecoxib (Fig 6A). This is consistent with the docking prediction.
Celecoxib inhibits Interleukin-6 (IL-6) induced STAT3 phosphorylation but not STAT1
We investigated whether Celecoxib could inhibit IL-6 induced STAT3 phosphorylation in PANC-1 cancer cells. PANC-1 cells were cultured in serum free medium for 24 hours and were pretreated with 25μM or 50μM of Celecoxib for 2 hours. Then the cells were treated with 50 ng/ml of IL-6 or interferon- for 30 minutes. We observed that Celecoxib inhibited IL-6 induced STAT3 phosphorylation but had little effect on STAT1 phosphorylation induced by IFN-γ (Figure 6B).
Cell Viability Assay
Cell viability assay was used to measure inhibitory effects of Celecoxib, T2 and T3 on human colon cancer cells (HCT-116). The results are listed in Table 1. IC50 values are as follows: Celecoxib (43.3 μM); T2 (9.7μM); T3 (10.1μM). T2 and T3 showed more potent inhibition of P-STAT3 than Celecoxib. The results are consistent with the modeling prediction.
Conclusion
In summary, our results show that combinations of three drug fragments found through MLSD bind to the three sub-pockets of STAT3 SH2. Linking of the docked fragments gave virtual hit compounds with potentially improved potency and ADMET properties. Similarity searching of virtual compounds in DrugBank identified Celecoxib as a novel inhibitor of STAT3. Also two novel hit compounds were designed to bind all 3 sub-pockets, which demonstrated more potent inhibition of STAT3 with IC50 values in the low μM range in HCT-116 cancer cell line assays. The proposed computational method, which uses privileged drug fragments, MLSD and drug repositioning, can potentially be applied for drug discovery and fragment-based drug design for other targets.
Experimental section
Privileged drug scaffolds preparation for STAT3
436 drug scaffolds from FDA approved drugs (AD) as reported by Wang et. al. were used for a drug scaffold database.14 The top 50 AD scaffolds in the database covered 52.6% of all FDA approved drugs. To prepare the drug scaffold library for STAT3, a collection of known inhibitors of STAT3 in previous reports were used to generate a set of feature fragments for the binding hot spots of STAT3 SH2. These known inhibitors were fragmented by a retro-synthetic approach. Drug scaffolds for STAT3 were then identified by similarity search of the obtained feature fragments on drug scaffold database. The privileged drug scaffold library for STAT3 was used for multi-fragment docking screening.
Multi-fragment docking screening and drug repositioning
The crystal structure of STAT3 SH2 domain (PDB code 1BG1) was used as the receptor for docking. MLSD program was employed for multi-fragment docking.12, 16 Privileged drug scaffolds were used as fragments for MLSD screening. In multi-fragment docking, 2 or 3 fragments from the 2 pools of the drug scaffolds were used to probe binding sub-pockets of STAT3 SH2. Systematic multi-fragment docking screening with the combinations of drug scaffolds was ranked by the predicted binding energy. The docked fragments with a predicted binding energy < −8.2 kcal/mol (dissociation constant Kd in sub μM range) were considered for further visual inspection of binding modes and selected as hits. Previously reported procedure and parameter settings were used for multi-fragment docking.12 Lamarckian Genetic Algorithm (LGA) and Particle Swarm Optimization (PSO) were used as a searching method depending on the dimensionality of search space. PSO, a searching method inspired by bird flocking, was used for MLSD with relatively high dimensions of conformational space. Virtual template compounds were obtained by linking docked fragments using various types of tethers. The candidates were optimized for drug properties: octanol-water partition coefficient (logP) and polar surface area (PSA). Drug properties were calculated using Molinspiration molecular property service (http://www.molinspiration.com). The linked compounds were re-docked to STAT3 SH2 and ranked by binding energies and binding modes to generate hits. To apply the drug repositioning concept, similarity searches for template compounds in DrugBank (http://www.drugbank.ca/) were performed to identify potential drug analogs of hit compounds.17 SMILES encoding and Tanimoto similarity coefficient cut-off of 0.5 were used for similarity search. The identified hit compounds were verified by computational re-docking before selection for synthesis or purchase to perform cell line assays.
Cell lines and cell culture
Human colon cancer cell line (HCT-116) and human pancreatic cancer cell line (PANC-1) were purchased from the American Type Culture Collection (ATCC). Cancer cell lines were cultured in DMEM medium supplemented with 10% FBS and 100U/ml penicillin/streptomycin B, in a humidified 37°C incubator with 5% CO2.
Western blot analysis
HCT-116 cells were treated with Celecoxib, T2 and T3 (10μM to 50μM) or DMSO control at 60–80% confluence in the presence of 10% Fetal Bovine Serum (FBS) for 24 hours and lysed in cold RIPA lysis buffer containing a cocktail of protease inhibitors to prepare whole-cell extracts.11 Lysates were then centrifuged at 14,000 rpm for 10 minutes to remove insoluble materials. 30–100 protein samples were separated by SDS-PAGE, transferred onto a PVDF membrane. After being blocked with 5% nonfat milk, the proteins were immunoblotted overnight at 4 °C with 1:1000 dilution of primary antibodies (Cell Signaling Technology) against phospho-STAT3 (pTyr705), STAT3 and GAPDH, respectively, and 1:10,000 dilution of HRP conjugated secondary antibody for 1 hour at room temperature. The target proteins were visualized by chemiluminescence (Cell Signaling Technology).
Cell Viability Assay
HCT-116 cells were seeded in 96-well plates at a density of 3,000 cells per well. Escalating concentrations (5–160 μM) of Celecoxib, and (1–75 μM) of T2 and T3 were added in triplicate to the plates in the presence of 10% FBS. The cells were incubated at 37°C for a period of 72 hours. 3-(4,5-Dimethylthiazolyl)-2,5-diphenyltetrazolium bromide (MTT) viability assay was done according to manufacturer’s protocol (Roche Diagnostics, Mannheim, Germany). The absorbance was read at 595 nm. Half-maximal inhibitory concentrations (IC50) were determined using Sigma Plot 9.0 Software (Systat Software Inc., San Jose, CA).
Synthesis of template compounds T2 and T3
All chemical reagents and solvents were purchased from commercial sources and used without further purification. Thin-layer chromatography (TLC) was performed on silica gel plates. Column chromatography was performed sing silica gel (Hailang, Qingdao), 200-300 mesh. 1H NMR spectra were recorded with a Bruker AV-400 spectrometer with chemical shifts expressed in parts per million (in DMSO-d6 or CDCl3, Me4Si as internal standard). The HRMS were collected at the Mass Instrumentation Facility of Analysis and Research Center of ECUST. The purities of all newly synthesized compounds were analyzed by HPLC, with the purity all being higher than 95%. Analytical HPLC was performed on a Hewlett-Packard 1100 system chromatograph equipped with photodiode array detector using a Zorbax RX-C18 5 μm, 250 mm×4.6 mm column (reverse phase) to detect the purity of the products. The mobile phase was a gradient of 70–100% MeCN (solvent 1) and 10.8 mM NH4OAc in water (pH 6.0) (solvent 2) at a flow rate of 1.0 mL/min. 0–11 min, 50%–70% solvent 1; 11–15 min, 70% solvent 1; 15.0–20.0 min, 70–100% solvent 1). Scheme S3 shows the synthesis of T2, and scheme S4 shows the synthesis of T3.
4-[5-(4-bromophenyl)-3-(4-methylphenylmethyl)pyrazol-1-yl]benzenesulfonamide (T2)
To a solution of KOBu-t (265 mg, 2.1 mmol) in THF, under ice-cooling and argon, was added 1- (4-bromophenyl)ethanone (398 mg, 2 mmol) in 5 mL THF dropwise. After 30 min, methyl 2-p-tolylacetate (328 mg, 2 mmol) in 3 mL THF was added and the reaction mixture was stirred at room temperature for 48 h. After work-up, crude compound 1 (1-(4-bromophenyl)-3-[(4-methylphenyl) methyl]propane-1,3-dione) was obtained as red oil.
A mixture of compound 1 (165 mg, 0.5 mmol) and p-Sulfonamide-phenylhydrazine hybrochloride (0.6 mmol) in ethanol was refluxed for overnight. Work-up and recrystallization of reaction mixture gave compound T2 as white solid; 1H NMR (400MHz, DMSO-d6) δ ( ppm ): 7.89 (d, J = 8.0 Hz, 2H), 7.48-7.43 (m, 4H), 7.26 (d, J = 7.6 Hz, 2H), 7.16 (d, J = 7.6 Hz, 2H), 7.07 (d, J = 8.0 Hz, 2H), 6.25 (s, 1H), 4.98 (s, 2H), 4.01 (s, 2H), 2.36 (s, 3H); HRMS (m/e), found 482.0535 (M+H+), calc. 482.0538.
3-(Piperidylaminocarbonyl)-5-biphenyl)-1-(4-aminosulfonylphenyl)-1H-pyrazole (T3)
To a solution of 5-biphenyl-1-(4-aminosulfonylphenyl)pyrazole-3-Carboxylic acid (4) (84 mg, 0.2 mmol) (Prepared by Scheme S4 of Supplemental Materials) in 10 mL DMF was added N-aminopiperidine (30 mg, 0.3 mmol), HATU (114 mg, 0.3 mmol) and DIPEA. The reaction stirred at room temperature for 9h. The reaction mixture was then concentrated and purified by flash column (EA/PE, 1/1) to give T3 (40 mg, 40%) as red solid. 1H NMR (400MHz, DMSO-d6) δ ( ppm ): 9.24 (s, 1H), 7.89 (d, J = 8.8 Hz, 2H), 7.71(t, J = 8.0 Hz, 4H), 7.60 (d, J = 8.4 Hz, 2H), 7.49–7.45 (m, 4H), 7.40–7.37 (m, 3H), 7.08 (s, 1H), 2.82 (t, J = 5.2 Hz, 4H), 1.60 (t, J = 4.4 Hz, 4H), 1.37 (d, J = 8.4 Hz, 2H); MS (ESI), 502.1 (M+H+) and 500.1 (M-H+).
Supplementary Material
Acknowledgments
The work was partially supported by a NIH R21 grant (1R21 CA 133652-01A1) to J. Lin and C. Li, and the Ohio Supercomputer Center Glenn cluster computing resources.
Abbreviations
- FBDD
fragment-based drug design
- HTS
high throughput screening
- MLSD
multiple ligand simultaneous docking
- DAPI
4',6-diamidino-2-phenylindole
- IL-6
interleukin 6
- DMSO
dimethyl sulfoxide
- logP
octanol-water partition coefficient
- PSA
polar surface area
- STAT3
signal transducer and activator of transcription 3 (STAT3)
- ADMET
Absorption, Distribution, Metabolism, Excretion, and Toxicity
Footnotes
Supporting Information Available: Virtual docking results of template compounds, binding mode cluster of docked fragments and scheme of synthesis of T2 and T3. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Li H, Li C. Multiple ligand simultaneous docking (MLSD): Orchestrated dancing of ligands in binding sites of protein. J Comput Chem. 2010;31:2014 –2022. doi: 10.1002/jcc.21486. [DOI] [PubMed] [Google Scholar]
- 2.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O'Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 3.Chong CR, Sullivan DJ. New uses for old drugs. Nature. 2007;448:645–646. doi: 10.1038/448645a. [DOI] [PubMed] [Google Scholar]
- 4.Wang J, Hou TJ. Drug and drug candidate building block analysis. Chem Inf Model. 2010;50:55–67. doi: 10.1021/ci900398f. [DOI] [PubMed] [Google Scholar]
- 5.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr Stat3 as an Oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 6.Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, Niu G, Kay H, Mulé J, Kerr WG, Jove R, Pardoll D, Yu H. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11:1314–1321. doi: 10.1038/nm1325. [DOI] [PubMed] [Google Scholar]
- 7.Costantino L, Barlocco D. STAT 3 as a target for cancer drug discovery. Curr Med Chem. 2008;15:834–843. doi: 10.2174/092986708783955464. [DOI] [PubMed] [Google Scholar]
- 8.Xu X, Kasembeli MM, Jiang X, Tweardy BJ, Tweardy DJ. Chemical Probes that Competitively and Selectively Inhibit Stat3 Activation. PLoS ONE. 2009;4(3):e4783. doi: 10.1371/journal.pone.0004783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu J, Cole DC, Chang CP, Ayyad R, Asselin M, Hao W, Gibbons J, Jelinsky SA, Saraf KA, Park K. Inhibition of the signal transducer and activator of transcription-3 (STAT3) signaling pathway by 4-oxo-1-phenyl-1,4-dihydroquinoline-3-carboxylic acid esters. J Med Chem. 2008;51:4115–4121. doi: 10.1021/jm701271y. [DOI] [PubMed] [Google Scholar]
- 10.Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: A Small-Molecule Inhibitor of STAT3 Activation and Dimerization. Chemistry & Biology. 2006;13:1235–1242. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 11.Song H, Wang R, Wang S, Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc Natl Acad Sci US A. 2005;102:4700–4705. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, Yip ML, Jove R, McLaughlin MM, Lawrence NJ, Sebti SM, Turkson J. Selective chemical probe inhibitor of stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci USA. 2007;104:7391–7396. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siddiquee KA, Gunning PT, Glenn M, Katt WP, Zhang S, Schrock C, Sebti SM, Jove R, Hamilton AD, Turkson J. An oxazole-based small-molecule Stat3 inhibitor modulates Stat3 stability and processing and induces antitumor cell effects. ACS Chem Biol. 2007;2:787–798. doi: 10.1021/cb7001973. [DOI] [PubMed] [Google Scholar]
- 14.Bhasin D, Cisek K, Pandharkar T, Regan N, Li C, Pandit B, Lin J, Li PK. Design, synthesis, and studies of small molecule STAT3 inhibitors. Bioorg Med Chem Lett. 2008;18:391–395. doi: 10.1016/j.bmcl.2007.10.031. [DOI] [PubMed] [Google Scholar]
- 15.Lin L, Hutzen B, Li PK, Ball S, Zuo M, DeAngelis S, Foust E, Sobo M, Friedman L, Bhasin D, Cen L, Li C, Lin JA. Novel Small Molecule, LLL12, Inhibits STAT3. Phosphorylation and Activities and Exhibits Potent Growth-Suppressive Activity in Human Cancer Cells. Neoplasia. 2010;12:39–50. doi: 10.1593/neo.91196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639. [Google Scholar]
- 17.Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, Tzur D, Gautam B, Hassanali M. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008;36(Database issue):D901–6. doi: 10.1093/nar/gkm958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agarwal B, Swaroop P, Protiva P, Raj SV, Shirin H, Holt PR. Cox-2 is needed but not sufficient for apoptosis induced by Cox-2 selective inhibitors in colon cancer cells. Apoptosis. 2003;8:649–654. doi: 10.1023/A:1026199929747. [DOI] [PubMed] [Google Scholar]
- 19.Molina MA, Sitja-Arnau M, Lemoine MG, Frazier ML, Sinicrope FA. Increased Cyclooxygenase-2 Expression in Human Colon Carcinomas and Cell Lines: Growth Inhibition by Nonsteroidal Anti-Inflammatory Drugs. Cancer Research. 1999;59:4356–4362. [PubMed] [Google Scholar]
- 20.Wei D, Le X, Zheng L, Wang L, Frey JA, Gao AC, Peng Z, Huang S, Henry Q, Xiong HQ, Abbruzzese JL, Xie K. Stat3 activation regulates the expression of vascular endothelial growth factor and human colon cancer angiogenesis and metastasis. Oncogene. 2003;22:319–329. doi: 10.1038/sj.onc.1206122. [DOI] [PubMed] [Google Scholar]
- 21.Lin L, Hutzen B, Zuo M, Ball S, Deangelis S, Foust E, Pandit B, Ihnat MA, Shenoy SS, Kulp S, Li PK, Li C, Fuchs J, Lin J. Novel STAT3 phosphorylation inhibitors exhibit potent growth-suppressive activity in colon and breast cancer cells. Cancer Res. 2010;70:2445–2454. doi: 10.1158/0008-5472.CAN-09-2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.