

Abstract
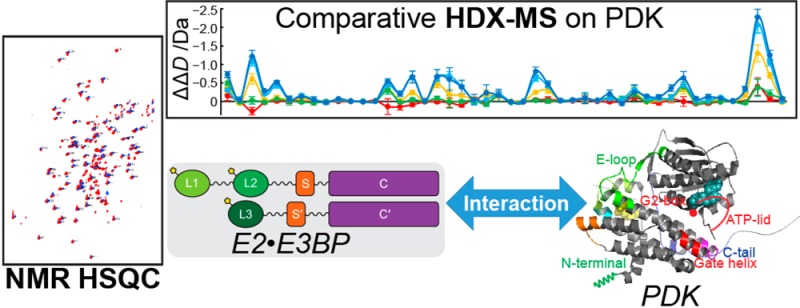
The human pyruvate dehydrogenase complex (PDC) comprises three principal catalytic components for its mission: E1, E2, and E3. The core of the complex is a strong subcomplex between E2 and an E3-binding protein (E3BP). The PDC is subject to regulation at E1 by serine phosphorylation by four kinases (PDK1–4), an inactivation reversed by the action of two phosphatases (PDP1 and -2). We report H/D exchange mass spectrometric (HDX-MS) and nuclear magnetic resonance (NMR) studies in the first attempt to define the interaction loci between PDK1 and PDK2 with the intact E2·E3BP core and their C-terminally truncated proteins. While the three lipoyl domains (L1 and L2 on E2 and L3 on E3BP) lend themselves to NMR studies and determination of interaction maps with PDK1 and PDK2 at the individual residue level, HDX-MS allowed studies of interaction loci on both partners in the complexes, PDKs, and other regions of the E2·E3BP core, as well, at the peptide level. HDX-MS suggested that the intact E2·E3BP core enhances the binding specificity of L2 for PDK2 over PDK1, while NMR studies detected lipoyl domain residues unique to interaction with PDK1 and PDK2. The E2·E3BP core induced more changes on PDKs than any C-terminally truncated protein, with clear evidence of greater plasticity of PDK1 than of PDK2. The effect of L1L2S paralleled HDX-MS results obtained with the intact E2·E3BP core; hence, L1L2S is an excellent candidate with which to define interaction loci with these two PDKs. Surprisingly, L3S′ induced moderate interaction with both PDKs according to both methods.
The human pyruvate dehydrogenase complex (PDC) catalyzes the oxidative decarboxylation of pyruvate with formation of acetyl-CoA, CO2, and NADH (H+) and occupies a key position at the intersection of glycolysis and the citric acid cycle.1−3 In mammals, PDC plays a gatekeeper’s role in the metabolism of pyruvate to maintain glucose homeostasis during the fed and fasting states. The highly evolved 9.5 MDa PDC comprises multiple copies of a minimum of six proteins: 20–30 copies of thiamin diphosphate (ThDP)-dependent pyruvate dehydrogenase (E1, α2β2 heterotetramer), 48 copies of dihydrolipoamide transacetylase (E2), 12 copies of dihydrolipoamide dehydrogenase (E3), 12 copies of unique E3-binding protein (E3BP), and two regulatory enzymes, pyruvate dehydrogenase kinase (PDK, four human isoforms)3−8 and pyruvate dehydrogenase phosphatase (PDP, two human isoforms).9−11 The 48 copies of E2 and 12 copies of E3BP form the core of the PDC according to a “substitution” model, to which the peripheral components E1 and E3, PDKs, and PDPs are bound noncovalently.5,12 The entire complex exhibits icosahedral symmetry. E2 has a multidomain structure, comprising (from the N-terminal end) two lipoyl domains, the outer (L1) and the inner lipoyl domain (L2) approximately 9 kDa each, a peripheral subunit-binding domain (PSBD or S, 4 kDa), and the acetyltransferase or core domain (C) (28 kDa), separated by 25–30-amino acid flexible linkers (Scheme 1). Human E3BP is composed of three linker-connected domains similar to E2 (L3 and S′ in Scheme 1), but with a transacetylation-incompetent core domain (C′). The structures of some individual components of PDC have been determined by X-ray.13−18 The structures of the E2 catalytic domain,19 reconstituted full-length E2·E3BP core, and a full-length E2·E3BP-E3 core were determined by cryoelectron microscopy.20 The flux of pyruvate through PDC is tightly regulated in different tissues under different metabolic conditions by the reversible phosphorylation of E1, involving dedicated PDKs and PDPs.3,9,21,22 Three sites in E1 are phosphorylated in vivo at different rates and with different specificities by four PDKs.23−25 Site 1 is preferentially phosphorylated, and sites 2 and 3 are sequentially phosphorylated.25,26 Starvation and diabetes induce PDK2 and PDK4 activity in different tissues, inducing phosphorylation and inactivation of PDC.27−30 PDC is also implicated to play a role in neurodegenerative diseases, obesity, and other diseases.31−33 More recently, a large body of evidence has been published indicating that PDC is a target for regulating glucose oxidation in cancer cells leading to the Warburg effect (aerobic glycolysis),34−42 where the pyruvate is converted to lactate, partially because of upregulation of gene expression of PDK1,43−45 PDK2,46 and PDK3.47,48 Recent studies also revealed that targeting of PDK could serve as a novel therapeutic approach in oncology. Dichloroacetate (an analogue of pyruvate), a well-known inhibitor for PDKs, shifts cancer metabolism from aerobic glycolysis to oxidative phosphorylation, leading to inhibition of tumor growth.49−51 However, dichloroacetate is a nonspecific inhibitor that binds to the allosteric site in the N-terminal domain of PDKs.52,53 Among known inhibitors of PDKs, the glucose-lowering compound AZD7545 (AstraZeneca), a mimic of dihydrolipoamide, exhibited efficient inhibition of PDK2 and PDK1 activities, but not of PDK4.54−56 Also, the secondary amides SDZ048–619 and their substituted analogues displayed efficient inhibition of PDKs, but these compounds did not lower glucose levels in diabetic animal models.57
Scheme 1. Structure of the Domains of the Pyruvate Dehydrogenase E2·E3BP Core and Their C-Terminally Truncated Proteins.
The structures of human PDK1 and PDK3 with AZD7545, dichloroacetate, and radicicol (antitumor compound) revealed that AZD7545 binds to the lipoyl-binding pocket in the N-terminal region of PDK1 and prevents PDK1 from binding to the E2·E3BP core,52 while radicicol binds to the nucleotide-binding site of PDK3 and inhibits its activity by competing with ATP for the nucleotide-binding site.52 A structure-based approach demonstrated that PDK inhibitors could be created by targeting the lipoyl-binding pocket of PDK2 and PDK3 (but were not effective)53,54 and the ATP-binding pocket of PDK2 and PDK4.58
The goals of our study are to examine PDK1 and PDK2 in parallel using two different approaches, H/D exchange mass spectroscopy (HDX-MS) and NMR: (i) to uncover loci of interaction between PDK1 and PDK2 (as important representatives of the four) with the E2·E3BP core and their C-terminally truncated proteins and (ii) to reveal differential binding loci between the particular PDK and E2·E3BP core as a first step in the design of kinase-specific inhibitors. While crystal structures of PDK1,52 the PDK2–L2 complex,58−60 the PDK3–L2 complex,61,62 and PDK463 have been reported, the premise of earlier structural work was that interactions between PDC and the PDKs are dominated by interactions with L2. The HDX-MS and NMR studies presented in this paper on loci of interactions clearly revealed that the interactions are multipoint, demonstrating that studies with intact components are valuable for understanding these important issues in the absence of a high-resolution structure on intact PDC.
The most important conclusion from these studies is that there are differences in the binding loci of the two kinases with the E2·E3BP core. Mapping of binding-induced changes between the E2·E3BP core and PDK1 and PDK2 creates a rich source of starting points to better define critical regions that could serve as targets for future drug design.
Experimental Procedures
Bacterial Strains, Plasmids, Overexpression, and Purification
The following recombinant C-terminally truncated proteins of the E2·E3BP core were studied (Scheme 1): (i) L1, containing the outer lipoyl domain and linker (residues 1–98), (ii) the L2S didomain containing the inner lipoyl domain L2, second hinge region, peripheral subunit-binding domain (PSBD or S), and third hinge region (residues 128–330), (iii) the L1L2S tridomain, which comprises L1, L2, both hinge regions, and the subunit-binding domain (S, residues 1–330),15,21,22 and (iv) the L3S′ didomain of E3BP (residues 1–230). The E2·E3BP core was expressed and purified as reported previously.5,24 Recombinant rat PDK1 and rat PDK2 were used for the studies. The sequences of mammalian PDK isozymes are highly conserved for each of the four isoforms (>94% identical between human and rat).67 The PDKs were overexpressed and purified to 90–95% purity according to a literature protocol64 with some modifications.21
Expression and Purification of 15N- and 13C-Labeled C-Terminally Truncated E2·E3BP Proteins
For the production of the doubly labeled proteins, overnight cultures were grown in LB supplemented with kanamycin (50 μg mL–1) at 37 °C. Cells were then diluted (1:100) in M9 minimal medium supplemented with 1 g/L (w/v) 15NH4Cl and 4 g/L (w/v) [U-13C]glucose as the sole sources of nitrogen and carbon, respectively, kanamycin (50 μg mL –1), and 0.3 mM d,l-lipoic acid to achieve lipoylation of L1–L3. Cells were grown at 37 °C to an OD600 of 0.6–0.8, and protein expression was induced by 1 mM IPTG for 20 h at 25 °C. The proteins were purified using sequentially Ni affinity, anion exchange, and size exclusion chromatography.
HDX-MS Studies
HDX-MS analysis was conducted as described using a 7T Bruker Daltonics FT-MS instrument.68,69 The E2·E3BP core, L1, L2S, L1L2S, and L3S′, PDK1, and PDK2 were analyzed individually and in their binary complexes. For sample preparation, all proteins were exchanged into 10 mM KH2PO4 (pH 7.5) containing 50 mM KCl. Prior to deuterium labeling, individual protein samples were prepared in aqueous buffer, including five individual proteins derived from the E2·E3BP core (80 μM), PDK1, and PDK2 (80 μM), in the absence and presence of 400 μM ATP. Samples of the complexes were prepared from the E2·E3BP core and its C-terminally truncated proteins with PDK1 or PDK2 (80 μM each, in the absence and presence of 400 μM ATP). Subunit concentrations are given. Hence, 40 μM PDK (homodimer) and 1.33 μM E248·E3BP12 were used for each protein sample. The samples were allowed to equilibrate for 1 h at 25 °C. The deuterium labeling reaction was initiated by diluting 2 μL of the sample into 38 μL of labeling buffer [10 mM KH2PO4 (pD 7.5) containing 50 mM KCl and 99.8% D2O], followed by incubation at 25 °C, yielding a final concentration of 94.8% D2O. ATP (400 μM) was added to the labeling buffer as needed. After incubation for 3 min, a 30 μL aliquot from each labeling reaction mixture was rapidly quenched into 30 μL of ice-cold quench buffer [0.2 M KH2PO4 and 4 M guanidine hydrochloride (pH 2.0)], achieving a final pH of 2.5 to minimize the rate of H/D exchange. The samples were immediately frozen in liquid nitrogen and stored at −80 °C for no more than 2 weeks before being analyzed. Nondeuterated samples were generated following the same procedure except that protein samples were diluted into aqueous buffer instead of labeling buffer. The frozen deuterated sample was rapidly thawed and loaded with an ice-cold syringe into a 20 μL sample loop inside the refrigeration system. The sample analysis was similar to that reported previously68,69 using Bruker Daltonics DataAnalysis 4.0 for spectrum analysis and data treatment. Peptides were identified from nondeuterated samples by a customized program DXgest.70 H/D exchange data for each individual peptide at various time points were processed using HX-Express.71 No back exchange correction was needed for the purpose of comparative analysis. The number of exchangeable backbone amides (maxD) of a peptide was calculated as the total number of residues excluding proline residues and two fast exchangeable N-terminal residues.72,73 The percentage of deuterium incorporation (without back exchange correction) of each peptide was calculated from the equation ΔD% = ΔD/(maxD × 1.0063 × 0.948) × 100%, where 1.0063 is the atomic mass difference between deuterium and hydrogen and 0.948 represents the fractional D2O content of the labeling reaction mixture. Butterfly and difference plots were produced with Microsoft Excel. Bar graphs were created with Origin (OriginLab, Northampton, MA).
NMR Spectroscopy
NMR spectra were recorded using a 0.5–0.8 mM solution of uniformly 13C- and 15N-labeled L1, L2S, L1L2S, and L3S′, in 20 mM KH2PO4 (pH 6.5–7.0) with 150 mM NaCl. All NMR samples contained 10% D2O for a lock signal. All NMR data for backbone resonance assignments were recorded at 15–37 °C on a Varian INOVA 600 MHz spectrometer equipped with a 5 mm HCN triple-resonance room-temperature probe. Sequence-specific resonance assignments for all labeled proteins were determined using triple-resonance NMR experiments74 from the Varian standard bio pack pulse sequence library. Spectra were processed with NMR Pipe 2.11275 and transferred to CARA 3.10676 for further analysis. 1H chemical shifts were referenced to the water line at 4.7 ppm, and nitrogen chemical shifts were referenced indirectly to liquid NH3.77 For measurements of interactions by the chemical shift mapping methodology, 0.3 mM C-terminally truncated labeled E2·E3BP protein was mixed with 0.1 mM unlabeled PDK1 or PDK2 (based on a monomer mass of ∼45 kDa) in 20 mM KH2PO4 (pH 7.0) containing 150 mM NaCl, 5% glycerol, and 10% D2O. The 15N–1H HSQC TROSY spectra were recorded in the absence and presence of unlabeled PDK1 and PDK2. Chemical shift deviations of peak positions were extracted from the 15N–1H HSQC TROSY spectra.
Isothermal Titration Calorimetry (ITC)
The heats of interaction of L2S, L1L2S, and L3S′ with PDK2 were determined using a VP microcalorimeter (MicroCal, Northampton, MA). All proteins were dialyzed exhaustively against a buffer containing 20 mM KH2PO4 (pH 7.4), 150 mM NaCl, 2.0% ethylene glycol, and 2.5 mM dithiothreitol (DTT). The concentration of each protein was determined by the Bradford assay. For each titration, 0.4 mM L2S, L1L2S, or L3S′ in the syringe was injected in 10 μL increments (28 injections at 3 min intervals) into the reaction cell containing 1.4 mL of 0.02 mM PDK2 at 30 °C. The binding isotherm was calculated by integration of the thermogram using a single-site binding model (Origin version 7.0) to derive the three nonlinear parameters for the interactions: the association constant (Ka), the molar ratio (n), and the enthalpy change (ΔH).
Results and Discussion
Identification of Loci of Interaction between the E2·E3BP core and PDK1 and PDK2 by HDX-MS
General Information about Deuterium Incorporation
To the best of our knowledge, this is the first instance that the interaction of the entire E2·E3BP core with PDK1 and PDK2 was analyzed at peptide resolution by comparative HDX-MS. A total of 153 peptides were selected (Tables S1–S4 of the Supporting Information), including 31 peptides from E2, 31 from E3BP, 46 from PDK1, and 45 from PDK2, for HDX-MS analysis with sequence coverages of 97, 95, 99, and 98%, respectively, generated by MS Tools.78 The following information was revealed upon digestion of the E2·E3BP core. (1) From among the lipoyl domains, two peptides, 42VETDKATVGF51 and 169IETDKATIGF178, from E2 (L1 and L2, respectively, in Table S1 of the Supporting Information) and one peptide, 44KAVVTL49, from E3BP (L3 in Table S2 of the Supporting Information), all three containing the sequence for ligation of lipoic acid to lipoyllysine, were identified. L1–L3 displayed moderate deuterium uptake at 25–47%. In control experiments, full lipoylation of all three lipoyl domains was confirmed by FT-MS. (2) The subunit-binding domains (S in E2 and S′ in E3BP) and the hinges between domains [H1–H5 (Figure 1A)] experienced the highest percentage of deuterium uptake (67–81% in 3 min of exchange at 25 °C, without back exchange correction), reflecting the highly flexible and unstructured behavior of these regions. (3) From the two core domains (C from E2 and C′ from E3BP), the deuterium uptake patterns were largely divided, with certain regions showing ΔD% values of >70%, while some were less than 5% or nearly zero. The “hindered regions” in the E2 core domain are peptides Met351–Asp365, Phe393–Leu400, Val416–Asp424, Ile436–Phe441, Phe485–Leu502, and Phe549–Thr560 (Figure 1) that contain highly conserved residues in prokaryotic and eukaryotic E2’s as deduced from sequence alignments (Figure S1 of the Supporting Information). The “hindered regions” in E3BP are peptides Phe278–Ala284, Phe370–Glu374, and Val416–Leu429 from E3BP (Table S2 of the Supporting Information). The “exposed regions” in the E2 core domain (ΔD% > 40%) are peptides Thr335–Leu350, Leu373–Asp392, Val425–Leu435, and Val455–Phe484 and in the E3BP core peptides Thr223–Asn229, Lys261–Asp277, Val312–Glu369, and Lys397–Thr415. Peptide Leu373–Asp392 from the E2 core exhibited a decreased level of deuterium uptake of 0.5 Da induced by the presence of ATP, which may be due to the binding of ATP in the CoA-binding pocket.
Figure 1.

Comparative HDX-MS analysis of the interaction of the E2·E3BP core with PDK1 and PDK2. (A) Relative deuterium incorporation percentage (ΔD%, y axis, deuterons exchanged/maximal exchangeable amides × 100%) of peptic peptides from the E2·E3BP core (x axis, listed peptic peptides from the N- to C-terminus, from E2 followed by peptic peptides from E3BP) in its free form (black) and in complexes with PDKs (with or without ATP, in various colors). The domain organization of the human E2·E3BP core is shown in Scheme 1. The termini of the domains were drawn according to the peptic peptides on the x axis. (B) Difference plot showing the changes in deuterium incorporation (ΔΔD, y axis, deuterons exchanged in the presence of PDK minus deuterons exchanged in the absence of PDK) of peptic peptides from the E2·E3BP core upon its complexation with PDK1 and PDK2. Each data point represents the mean ± the standard deviation from three independent experiments. Arrows show statistically significant changes.
Changes in Deuterium Uptake Observed in the E2·E3BP Core upon Addition of PDK1 and PDK2
Changes in Deuterium Uptake Observed in L2
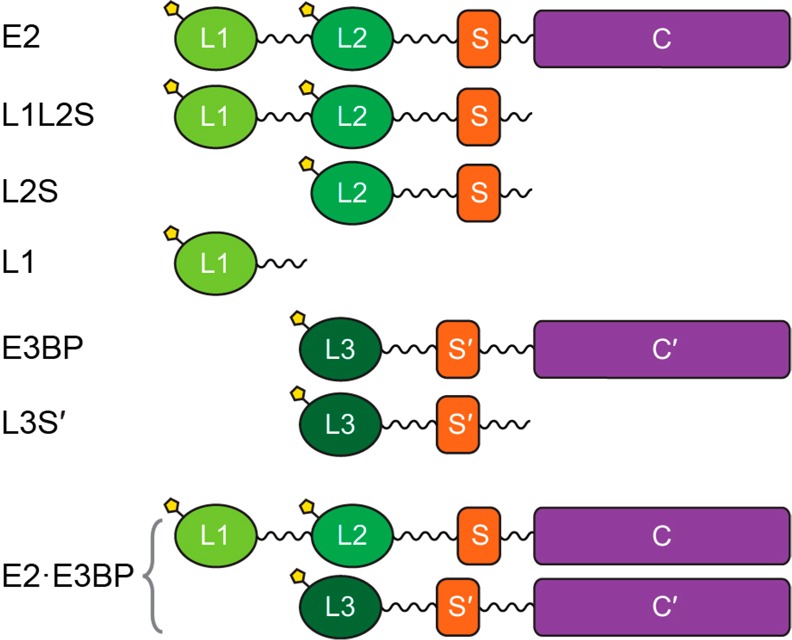
It is believed that L2 of the E2·E3BP core is a docking site for association of PDK2 with PDC that activates kinase and provides regulation.52,53,61 Upon incubation of the E2·E3BP core with PDK2, the most significant HDX-MS changes were observed in the N-terminal region of L2 for peptide Gln134–Leu165, which experienced a ΔΔD of ∼1.0 Da. Upon comparison of the deuterium incorporation (ΔD) by this peptide of 8.62 ± 0.16 Da in the absence of PDKs, with 8.23 ± 0.0 Da in the presence of PDK1 (ATP present) and 7.65 ± 0.02 Da in the presence of PDK2 (ATP also present), it has become evident that PDK2 has an interaction with the N-terminal region of L2 stronger than that of PDK1 (Figure 2), indicating a stronger binding between PDK2 and L2 (please note that, throughout, stronger retardation of deuterium exchange is indicative of stronger interaction). In contrast, the same Gln134–Leu165 peptide in L2S and L1L2S displayed very similar deuterium uptake changes upon interaction with PDK1 and PDK2; again, deuterium uptake was decreased in the presence of ATP (Figure 2). The comparison of deuterium uptake described above suggests that the intact E2·E3BP core enhances the specificity of L2 for PDK2 over PDK1, while addition of ATP has a more pronounced effect for binding to both PDK1 and PDK2. The C-terminal region of L2, i.e., peptides Glu179–Leu186 and Ala187–Cys205, experienced ΔΔD values of ∼0.3 and ∼0.5 Da, respectively (Figure 1B). These values were not affected by the C-terminal truncation of E2, by PDK1 or PDK2, or by ATP activation, indicating global changes induced upon their binding.
Figure 2.
Comparison of the changes in deuterium uptake by peptide Gln134–Leu165 in C-terminally truncated E2·E3BP core-derived proteins upon complexation with PDK1 and PDK2.
Changes Observed in the E2 Core Domain
A small reduction in ΔD (∼0.3) was also apparent in the E2 core domain, particularly for peptide Ala503–Met525, located on an internal hairpin loop of the E2 core domain according to cryo-electron microscopy.19
Changes Observed in E3BP
Two peptides, one from the N-terminal region of L3 and one from the linker region connecting L3 and the E3-binding domain in E3BP, displayed deuterium uptake changes upon interaction with PDK1 and PDK2. For N-terminal peptide Gly1–Leu37, a decrease in deuterium uptake of ∼0.27 Da on binding to PDK1 and of ∼0.47 Da on binding of PDK2 was detected. For peptide Trp85–Phe129 from the linker region, these values were 0.30 Da for PDK1 and 0.25 Da for PDK2. The presence of ATP did not affect the results. While previous studies were focused on structural insight into interactions of E3 and the E3-binding domain with L2,15,16 the HDX-MS studies presented here not only detected peptides originated from L3 but also revealed changes in deuterium uptake by L3 peptides upon interaction with PDK1 and PDK2, a change more pronounced with PDK2 than with PDK1.
The HDX-MS results described above are in good agreement with the X-ray structure of L2 bound to PDK2.60 According to the X-ray structure, residues of L2 that form extensive contacts with PDK2 (one PDK2 dimer binds two L2’s) are Ala139–Pro142, Glu153, Glu162–Leu165, Glu170–Thr175, Glu179, and Arg196, in good agreement with an essential peptide (Gln134–Leu165) identified by HDX-MS upon interaction of the E2·E3BP core with PDK2. Among L2 residues identified by X-ray, Leu140, Lys173, Ile176, and, to a lesser extent, Glu179, Asp164, Asp172, and Ala174 were all suggested to be essential for recognition of PDK2 according to site-directed mutagenesis studies.55 In the X-ray structure of the PDK2–L2 complex, residues Leu137–Thr145 are positioned on the L2 recognition loop located between strands β1 and β2,60 and Glu162 forms an ionic bond to Lys376 on PDK2. Although Lys376 is conserved in all PDKs, there is no ionic bond apparent in the crystal structure of the PDK3–L2 complexes.61,62
Changes Observed on PDK1 and PDK2 in the Presence of the E2·E3BP Core and Their C-Terminally Truncated Proteins
Among four PDK isozymes, PDK2 is the most abundant64,65 and the most studied isoform. Crystal structures for mammalian PDK1 and PDK2 are known, including rat PDK2,59 the rat PDK2–L2 complex,60 human PDK2 in free form and in complexes with ATP, ADP, inhibitors in the N-terminal (R) domain,66 and inhibitors in the ATP-binding pocket.58 Published crystal structures of PDK1 include the apo and holo forms bearing lipoate or pyruvate analogues.52
Because residues from both the N-terminal (R) and C-terminal (K) domains of PDKs are involved in interaction with L2 according to the X-ray structure, we present a detailed analysis of changes in deuterium uptake in both domains of PDKs upon interaction with the E2·E3BP core and their C-terminally derived proteins. The descriptive names of the structural elements of PDKs used in this paper are from their published structures.
Changes in the N-Terminal (regulatory) Domain of PDK1 and PDK2 upon Addition of the E2·E3BP Core and Its C-Terminally Truncated Proteins
The following N-terminal regions of both PDKs are affected by interaction with the E2·E3BP core.
(i) In rat PDK1, the deuterium uptake in peptide Asp29–Phe48, and especially in peptide Arg51–Met59 (the hinge between helices α1 and α2), was significantly affected (ΔΔD values of ∼0.65 and ∼1.34 Da, respectively) (Figure 3). Because the ΔΔD from peptide Val42–Phe48 was negligible, most of the observed ΔΔD changes were due to peptide Asp29–Gly41. For peptide Arg51–Met59 the decreasing order of effectiveness of interaction was in the following order: E2·E3BP (∼1.34 Da) > L1L2S (∼1.23 Da) > L2S (∼0.62 Da) > L3S′ (∼0.17 Da) ∼ L1 (∼0.16 Da) (Figures 3 and 4). In human PDK1, the N-terminal peptide Ser29–Arg40 (corresponding residues in rat PDK1 are Asp29–Ser40) was not seen in the crystal structures, and this region was thus considered to be disordered.52 However, according to HDX-MS, peptide Asp29–Gly41 displayed a moderate ΔD% of 37% after exchange for 3 min, indicating that this N-terminal region may be partially folded, but its function in PDK remains unknown. It is noteworthy that these residues are unique to PDK1 among the four PDK isozymes.
Figure 3.

Comparative HDX-MS analysis of the interaction between PDK1 and the E2·E3BP core. (A) Butterfly plot representing relative deuterium incorporation percentage (ΔD%, y axis, deuterons exchanged/maximal exchangeable amides × 100%) of peptic peptides from rat PDK1 (x axis, listed peptic peptides from the N- to C-terminus) in its free form (black) and in complex with human C-terminally truncated E2·E3BP core-derived proteins (in various colors), and in the absence (top) and presence of ATP (bottom). Overlapping peptides were capped. (B) Butterfly plot showing the changes in deuterium incorporation (ΔΔD, y axis, deuterons exchanged in the presence of each C-terminally truncated E2·E3BP core-derived protein minus deuterons exchanged in its absence) of peptic peptides from rat PDK1 upon its complexation with C-terminally truncated E2·E3BP core-derived proteins (in various colors) in the absence (top) and presence of ATP (bottom). (C) Difference plot showing the changes in deuterium incorporation (ΔΔD, y axis, deuterons exchanged in the presence of ATP minus deuterons exchanged in the absence of ATP) of peptic peptides from rat PDK1 [in its free form (black) and complexed with C-terminally truncated E2·E3BP core-derived proteins (in various colors)] in the presence of ATP. Each data point represents the mean ± the standard deviation from three independent experiments. Arrows show statistically significant changes.
Figure 4.

Changes in deuterium uptake for peptic peptides in PDK1 and PDK2 upon complexation with various C-terminally truncated E2·E3BP core-derived proteins. The exchange-active residues represent the results from subtraction of ΔΔD of overlapped peptides in Figure 3 and Figure S2 of the Supporting Information excluding two fast exchangeable N-terminal residues.
As for PDK2, peptides Lys9–Leu13 and His22–Met33 from the N-terminus were detected by HDX-MS (Figure S2 of the Supporting Information). However, neither peptide displayed a statistically significant ΔΔD upon complexation with the E2·E3BP core, even in the presence of ATP. This is in accord with X-ray structural data. Peptide His22–Met33 containing half of helix α1 and the hinge region between helices α1 and α2 displayed only a weak change according to HDX-MS (ΔΔD ∼ 0.18) (Figure S2 of the Supporting Information), unlike the hinge in PDK1, where a ΔΔD of ∼1.34 Da was determined for the corresponding peptide Arg51–Met59.
Helix α2
In PDK2, peptide Phe36–Glu46, containing part of helix α2, showed a ΔΔD of ≤0.6 Da (Figure S2 of the Supporting Information). Peptide Lys47–Phe50 was also affected (ΔΔD ∼ 0.3 Da); the decreasing order of effectiveness of interaction was as follows: E2·E3BP > L1L2S ∼ L2S > L3S′. L1 had no effect (Figure S2 of the Supporting Information). In PDK1, peptide Phe62–Ser67 also showed a ΔΔD of 0.5 Da, which was affected by complexation with L1L2S and the E2·E3BP core only (Figure 3).
Hinge between Helices α5 and α6 and the N-Terminus of Helix α6
Peptide Phe124–Glu137 in PDK1 (ΔΔD ∼ 0.55 Da) and the corresponding Asp100–Gln111 peptide in PDK2 (ΔΔD ∼ 0.3) were modestly affected by complexation with the E2·E3BP core (Figure 4).
Gate Helix near the ATP Lid
Part of the gate helix, peptide Ala157–Glu162 (ΔΔD = 0.66 Da) in PDK1, and peptide Ala131–Glu136 (ΔΔD = 0.62 Da) in PDK2 were modestly affected by binding to the E2·E3BP core (Figure 4). These peptides from PDK1 and PDK2 exhibit distinct behavior in the presence of ATP: peptide Ala157–Glu162 in PDK1 showed an increased ΔD up to 0.67 Da (Figure 3C), while peptide Ala131–Glu136 in PDK2 displayed a reduced ΔD up to 0.86 Da (Figure S2C of the Supporting Information). The relative effects in decreasing order in PDK1 were as follows: E2·E3BP ∼ L1L2S > L2S > L3S′ = L1 = 0. In PDK2, without ATP present, they were as follows: E2·E3BP > L2S > L1L2S ∼ L3S′ > L1 = 0. With ATP present, they were as follows: E2·E3BP > L1L2S > L2S > L1 > L3S′ (Figure 4).
Changes in the E-Loop of PDK1 and PDK2
According to X-ray structural data, the E-loop in PDK2 (Gly178–Pro185) undergoes a disorder-to-order transition upon complexation with L2.60 In the PDK2 structure, this loop connects helices α7 and α8 and forms the back wall of the E1-binding cavity. It was suggested that ordering of the E-loop in PDK2 upon L2 binding is responsible for the greater affinity for E1p.59,60 On complexation with the E2·E3BP core, the E-loop of PDK1 peptide Asn196–Leu201 (ΔΔD of ∼0.72 Da in the presence and ∼0.80 Da in the absence of ATP) and peptide Gly204–Asp222 (ΔΔD of ∼0.53 Da in the presence and ∼0.47 Da in the absence of ATP) revealed reduced ΔD values upon complexation. On complexation with L1L2S, similar changes were observed, but not on complexation with L1, L2S, and L3S′ (Figure 4). The corresponding E-loop peptide Asn170–Asp199 in PDK2 behaved similarly (ΔΔD of ∼0.65 Da in the presence and ∼0.63 Da in the absence of ATP). With this PDK2 peptide, all C-terminally truncated E2·E3BP core-derived proteins, with the exception of L1, behaved like the E2·E3BP core.
Changes in the C-Terminal Nucleotide-Binding Domain of PDK1 and PDK2
Changes in the C-Tail of PDK1 and PDK2
The C-terminal regions in PDK1 (peptides Ile385–Ser394 and Ile395–Asp417) and corresponding C-terminal regions in PDK2 (peptide Leu363–Asp390) were affected by binding to the E2·E3BP core. Complexation of PDK1 with the E2·E3BP core causes a reduction of ΔD of up to 0.3 Da in peptide Ile385–Ser394 and a major reduction in ΔΔD of ∼2.0 Da in peptide Ile395–Asp417, with the relative effect decreasing in the following order: E2·E3BP (1.94 Da) > L1L2S (1.74 Da) > L2S (1.13 Da) > L3S′ (0.41 Da) = L1 (0.42 Da) (Figure 4). Data also indicate that the major reduction in deuterium uptake originates from L1L2S in the E2·E3BP core. Upon complexation of the E2·E3BP core with the corresponding peptide Leu363–Asp390 in PDK2, a moderate change in ΔΔD of only ∼0.9 Da was detected, compared with a ΔΔD of 2.28 Da for PDK1 for the corresponding peptide. The HDX-MS results suggest significant ordering of the carboxyl-terminal region of the K domain of PDK1 upon complexation with the E2·E3BP core. According to the X-ray structure of PDK3, the C-terminal part of the K domain was suggested to be involved in forming the L2-binding site.52,61
Changes in the Conserved G1-Box Motif Involved in Binding the Adenine Moiety of Nucleotide
In PDK1, a 0.3–0.6 Da decrease in ΔD was found in peptide 313KMSDRGGGVPLRKIDRLFN331 containing the conserved G1-box motif (DXGXG) upon ATP binding (residues from the G1-box are underlined) (Figure 3). A 0.2 Da decrease in ΔD was found in the corresponding 287KMSDRGGGVPLRKIERL303 peptide in PDK2 (Figure S2 of the Supporting Information).
Changes in the G2-Box Characteristic Motif Involved in Stabilization of Phosphate Groups of the Nucleotide
The central Gly residues of the G2-box motif (GXGXG, corresponding to residues GFGYG in PDK) are Gly354 in PDK1 and Gly327 in PDK2 and displayed reduced ΔD values of ≤0.5 Da in the presence of C-terminally truncated E2·E3BP proteins. As shown in Figure 4, Gly354 in PDK1 was affected in a statistically significant manner by most of the C-terminally truncated E2·E3BP proteins (L2S, L1L2S, L3S′, and E2·E3BP) in the absence of ATP, while in PDK2, Gly327 was affected by all C-terminally truncated E2·E3BP proteins (L1, L2S, L1L2S, L3S′, and intact E2·E3BP) regardless of the presence or absence of ATP. A single L1 lipoyl domain was sufficient to induce a statistically significant ΔΔD on Gly327 of PDK2, but not on Gly354 of PDK1.
Changes in the ATP Lid
No changes in ΔD were found in peptide Thr336–Leu362 of PDK1 upon ATP binding, while the corresponding Thr310–Leu335 peptide of PDK2 revealed a small decrease in ΔD in the presence of ATP. According to the crystal structures (Figure 5), the ATP lid is ordered in PDK1 but disordered in other PDKs.52,60−63,66 This fact may have contributed to the observed difference in HDX-MS.
Figure 5.

Peptic peptides of PDK1 and PDK2 whose deuterium uptake responds to complexation with E2·E3BP/ATP are highlighted on the crystal structures of the rat PDK2–L2 complex (Protein Data Bank entry 3crl)60 and human PDK1 (Protein Data Bank entry 2q8g).52 The figures were produced by PyMOL.
Additional Changes Observed
In PDK2, peptides 213DKY215 (0.4 Da reduction with both the E2·E3BP core and its C-terminally truncated proteins), Ala218–Glu223 (0.3 Da reduction with any E2 protein), and Asn255–Ser267 (0.3–0.4 Da reduction with L3S′ and L1L2S) showed decreased ΔD values upon ATP binding. However, the corresponding peptides Val223–Leu240, Tyr241–Leu250, and Phe279–Leu310 in PDK1 did not show any changes in ΔD (Figure 3).
All peptic peptides of PDK1 and PDK2 identified by HDX-MS as responding to changes in deuterium uptake upon complexation with the E2·E3BP core are highlighted on the X-ray structures of human PDK1 and rat PDK2 in Figure 5.
Identification of Loci of Interaction between C-Terminally Truncated E2·E3BP Proteins and PDK1 and PDK2 by NMR
Backbone Resonance Assignment of C-Terminally Truncated E2·E3BP Proteins
Complete backbone resonance assignment was only possible for the single outer lipoyl domain (L1). Partial assignments of the backbone resonances were made for the larger L2S, L1L2S, and L3S′ proteins, all of which contain a subunit-binding domain. The PSBD is the smallest globular protein domain known without disulfide bridges, metal ions, or prosthetic groups. Its surface is characterized by the presence of a large number of positively charged residues, and they exhibit varying degrees of ionic strength-dependent characteristics.79,80 We have only limited assignment details available for the S and S′ domains. All four proteins, L1, L2S, L1L2S, and L3S′, displayed reasonable signal dispersion in the proton–nitrogen correlation map at 600 MHz. Peak positions were adjusted on the basis of HNCO strips. HN(CO)CACB and HNCACB spectra were used to link sequential amide groups via matching pairs of Cα/Cβ peaks, and sequence-specific backbone (1HN, 15N, 1Hα, 13Cα, 1Hβ, 13Cβ, and 13C′) resonance assignments were obtained. Even though there is high degree of sequence similarity between the lipoyl domains, the residues from the L1 and L2 domains do not overlap, and their resonances could be assigned using doubly 15N- and 13C-labeled L1L2S proteins. A brief summary of assigned peaks is presented in Table S5 of the Supporting Information, while the sequence-specific resonance assignments for L1, L2S, L1L2S, and L3S′ are presented in Tables S6–S9 of the Supporting Information.
Interaction of C-Terminally Truncated E2·E3BP Proteins with PDK1 and PDK2 by NMR
With the available sequence-specific resonance assignments to the four C-terminally truncated E2·E3BP core-derived proteins, L1, L2S, L1L2S, and L3S′, we undertook studies of interaction of these proteins with PDK1 and PDK2 to provide more insight into the role of the individual domains and the combination of domains in their interactions. This study would help to identify some differences in the ways these proteins recognize each other and how they interact. On the basis of our studies using chemical shift mapping, it was indicated that both PDK1 and PDK2 interact with several residues in all four truncated E2·E3BP core-derived proteins. However, differences in sensitivity were observed between the different proteins. There were some residues that contribute to the specific recognition. A summary of the interaction of the four C-terminally truncated E2·E3BP core-derived proteins with PDK1 and PDK2 is presented in Table 1. Whether these interactions are strong enough to help bind the PDKs to the phosphorylation site of E1 is not answered by these studies.
Table 1. Summary of the NMR Data upon Interaction of C-Terminally Truncated E2·E3BP Core-Derived Proteins with PDK1 and PDK2.
| protein | domain or region | peaks that disappeared upon complexation with PDK1 | peaks that disappeared upon complexation with PDK2 |
|---|---|---|---|
| L1 | L1 + linker | S14, T16, Q18, D45, K46, A47, N92 | T16, Q18, D45, A47, N92 |
| L2S | L2 + linker | M133, T143, M146, T148, Q150, L166, T171, D172, A174, F178, V180, A187, I189, D197, G201, L204, I206 | T143, T148, L166, T171, D172, F178, V180, L204 |
| S + linker | Y220, V232 | Y220, V232 | |
| L1L2S | L1 + linker | T16, E41, T44, A60, I62, L63, A99 | E41, T44, A99 |
| L2 + linker | V135, L137, L140, T143, M146, G147, Q150, W152, S161, G163, I169, E170, T171, I176, F178, E179, G184, Y185, A187, I189, E193, G201, T202, V208 | V135, L136, L137, L140, T143, G147, Q150, W152, G157, S161, G163, I169, E170, T171, I176, G177, F178, E179, G184, A187, I189, E193, G201, I207, V208 | |
| S + linker | I214, S215, F217, Y220, A279, I303 | I214, S215, Y220, A279, I303 | |
| L3S′ | L3 + linker | I4, T14, E16, G18, V21, L24, G28, G34, I40, T42, D43, V47, L49, I56, A58, I60, L71, G72, I75, G76, G82, W85 | I4, T14, E16, L24, T42, D43, V47, A58, I60, L71, G72, I75, G76, E80, G82, W85 |
| S + linker | G149, I157, G173 | G149, I157, G173 |
Interaction of the Outer Lipoyl Domain L1 with PDK1 and PDK2
The 15N–1H HSQC TROSY spectra (Figure 6A,D) of L1 recorded in the presence of PDK1 and PDK2 cause the disappearance of a few L1 residues because of overbroadening (Table 1). The two spectra gave essentially identical results. The residues at the dominant interaction site are in the surface loop region close in space to the lipoyllysine β-turn and in the lipoyllysine β-turn itself.81 The chemical shift changes are not very specific, many of the peaks arising from the entire lipoyl domain that underwent a change in chemical shift (>0.01 ppm). However, the chemical shift changes observed with the L1–PDK1 complex were larger than those observed with PDK2 (Figure 8). Residue Ser14, which disappeared in the L1–PDK1 interaction, showed a significant change of 0.07 ppm with PDK2, implying its interaction with PDK2, as well. Also, residues Ile39 and Asp96 showed chemical shift changes of >0.05 ppm with both PDKs. Residues Thr48 and Leu54 show unique chemical shift changes of >0.05 ppm with PDK1.
Figure 6.

Overlaid two-dimensional 15N–1H HSQC TROSY spectra of the C-terminally truncated E2-derived proteins L1, L2S, and L1L2S in the absence (red peaks) and presence of PDK1 (blue peaks) and PDK2 (green peaks). The C-terminally truncated E2-derived protein (0.3 mM) was mixed with unlabeled PDK1 (0.1 mM based on monomer) or PDK2 in 20 mM KH2PO4 (pH 7.0) containing 150 mM NaCl, 5% glycerol, and 10% D2O, and spectra were recorded at 25 °C. Spectra A–C represent interaction of L1, L2S, and L1L2S with PDK1, respectively, and spectra D–F represent interaction of L1, L2S, and L1L2S with PDK2, respectively. Assigned peaks are indicated with the one-letter amino acid code and residue number.
Figure 8.

Chemical shift differences of the 1H and 15N nuclei upon complexation of L1, L1L2S, and L3S′ with PDK1 and PDK2. Chemical shift deviations of peak positions were extracted from the 15N–1H HSQC TROSY spectra. The chemical shift differences were calculated using the formula Δδ = {Δ(1H)2 + [αN Δ(15N)]2}1/2 with a scaling factor (αN) of 0.2.
Interaction of the L2S Didomain with PDK1 and PDK2
Chemical shift mapping studies of uncomplexed L2S and L2S bound to PDK1 or PDK2 signaled overbroadening of several L2 domain peaks that can no longer be detected in the 15N–1H HSQC TROSY spectrum in the complex (Figure 6B,E). On interaction with PDK1, several resonances corresponding to amino acid residues Met133, Thr143, Met146, Thr148, Gln150, Leu166, Thr171, Asp172, Ala174, Phe178, Val180, Ala187, Ile189, Asp197, Gly201, Leu204, Ile206, Tyr220, Val232 disappeared. Among these residues Met133, Met146, Gln150, Ala174, Ile189, Asp197, Gly201, Ile206 were overbroadened only upon complexation with PDK1, but not with PDK2. The remaining residues were affected equally on interaction with PDK1 and PDK2 (Table 1). In case of the outer lipoyl domain L1, the interacting residues are mostly limited to surface loop and lipoyl β-turn region, while those in the inner L2 domain were widely distributed over the entire lipoyl domain, especially over the loop regions. Most of these residues are hydrophobic, while some are acidic.
Interaction of L1L2S tridomain with PDK1 and PDK2
The largest E2 protein studied was L1L2S, and its interactions with the two kinases led to the chemical shift perturbation maps presented in Figure 6C and 6F. Both PDKs interact with L1L2S at a number of residues, but most importantly, there are clear differences: interaction of two residues (Glu35 and Val198) with PDK1, and two residues (Glu153 and Glu209) with PDK2 are unique. These provide clear “hotspots” to test further and perhaps to design inhibitors against (Figure 8). Apart from these chemical shift changes, the interaction also caused disappearance of a few residues in the L1 region and several residues in the L2 region in the [15N, 1H] - HSQC TROSY spectra (Figure 6C,F and Table 1). Also, this L1L2S tridomain reveals a larger number of interacting residues in the L2 region with the kinases than the same region in the L2S didomain does (Table 1).
Interaction of L3S′ with PDK1 and PDK2
Upon interaction of the two PDKs with the L3S′ didomain, the apparent chemical shift differences are not as clear, although interaction with PDK1 is strongly implied, suggesting some hot spots, as well (Figure 8). The overlaid 15N–1H HSQC TROSY spectra of L3S′ in the absence and presence of PDKs are presented in Figure 7. In these interactions, resonances corresponding to several residues of L3 over both halves of the antiparallel β-sheet disappeared upon complexation with PDKs. However, with L3S′, more resonances disappeared (overbroadened and corresponding to residues located on the β-sheet encompassing the lipoylated Lys residue) upon interaction with PDK1 than with PDK2, further implying its stronger interaction with PDK1. Resonances corresponding to L3S′ residues that are too broad to be detected upon complexation with PDKs are listed in Table 1.
Figure 7.
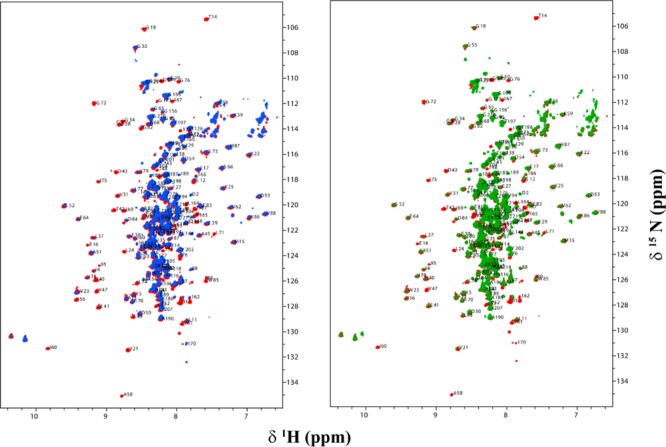
Overlaid two-dimensional 15N–1H HSQC TROSY spectra of the C-terminally truncated E3BP-derived protein L3S′ in the absence (red peaks) and presence of PDK1 (blue peaks) and PDK2 (green peaks). L3S′ (0.3 mM) was mixed with unlabeled PDK1 or PDK2 (0.10 mM based on monomer) in 20 mM KH2PO4 (pH 7.0) containing 150 mM NaCl, 5% glycerol, and 10% D2O, and spectra were recorded at 25 °C. Assigned peaks are indicated with the one-letter amino acid code and residue number: (left) interaction of L3S′ with PDK1 and (right) interaction of L3S′ with PDK2.
Isothermal Titration Calorimetric Determination of Association Constants of the C-Terminally Truncated E2·E3BP Core with PDK2
Isothermal titration calorimetry (ITC) was performed to dissect interactions between PDK2 and L2S, L1L2S, and L3S′. The binding isotherms derived from heat changes were used to calculate the standard free energy of binding (ΔG°) according to the equation ΔG° = RT ln Ka, where R is the gas constant, T the absolute temperature, and Ka the association constant. For L2S, the binding event was calculated to have an association constant Ka of 1.45 × 105 M–1 and a stoichiometry of 2.0; for L1L2S, a Ka of 1.32 × 105 M–1 with a stoichiometry of 0.83 was found, while the absence of quantifiable heat changes with L3S′ suggests an only modest strength of binding of L3S′ to PDK2. Nevertheless, the chemical shift mapping studies of L3S′ that showed the disappearance of several peaks throughout the entire lipoyl domain upon binding with PDK1 or PDK2 (Table 1 and Figures 7 and 8) affirm interaction between L3S′ and the two PDKs.
Summary
Deductions from HDX-MS Studies
(1) Given that there has been no high-resolution study of any of the 2-oxo acid dehydrogenase complexes, the HDX-MS studies accomplish a remarkable feat: they produce peptide-level resolution on both components of a complex, one partner of which, PDK1 or PDK2, has a dimer mass of 90 kDa, while the other partner, the intact E2·E3BP core of PDC, has a mass exceeding 3 MDa (as many as 60 subunits).
(2) The intact human E2·E3BP core can be effectively digested and analyzed by HDX-MS. The three different lipoyl moieties can be identified and analyzed simultaneously.
(3) Where comparable data exist from X-ray structures and kinetic and ITC studies, HDX-MS findings are in good agreement, thereby providing validation to the approach.
(4) In the complex of PDK1 and PDK2 with the E2·E3BP core, the most significant changes were detected on the N-terminal region of the L2 inner lipoyl domain; this region was more sensitive to interaction with PDK2 than with PDK1, in agreement with data in the literature.
(5) Statistically significant changes were detected in the N-terminal region of L3 (in L3S′) of E3BP upon interaction with PDK2, and to a lesser extent with PDK1. The presence of ATP enhanced the interactions for both (findings in points 4 and 5).
(6) A significant difference was observed in the behavior of binding of the E2·E3BP core to PDK1 versus PDK2.
(i) Peptide Arg51–Met59 in the N-terminal region of PDK1 revealed protection from exchange much greater than that of the corresponding peptide in PDK2. This is a major difference reported for the first time; none of the analyzed N-terminal peptides of PDK2 showed statistically significant ΔΔD values.
(ii) The HDX-MS results suggest greater ordering of the carboxyl-terminal region of the K domain in PDK1 than in PDK2 upon complexation with the E2·E3BP core (ΔΔD of ∼2.0 Da for PDK1 peptide Ile395–Asp417). According to X-ray structural data of PDK3, the C-terminal part of the K domain was suggested to be involved in the formation of the L2-binding site.
(7) The E2·E3BP core induced more changes on PDKs than any of their C-terminally truncated proteins. The effect of L1L2S on deuterium uptake by peptides from PDK1 and PDK2 was nearly as strong as that of the intact E2·E3BP core. Hence, L1L2S is an excellent candidate with which to define interaction loci with these two PDKs.
(8) Surprisingly, L3S′ induced moderate changes in many peptic peptides of PDKs, indicating that E3BP may also play an important role in PDK’s binding, activation, and regulation.
Deductions from the NMR Findings
(1) Evidence from our analysis of the interaction of the C-terminally truncated E2·E3BP proteins with PDK1 and PDK2 suggests that both L2 and L3 interact strongly with PDK1 and PDK2 (Table 1). There were fewer interaction sites with L1 than with L2 and L3. Given that the overall fold of all lipoyl domains is identical, the source of specificity of the kinases for the lipoyl domains is not obvious.
(2) Sequence comparisons of the lipoyl domains were conducted to ascertain whether amino acid differences could account for the observations (Figure S3 of the Supporting Information). The residues whose resonances disappeared in chemical shift mapping studies (upon interaction with PDK1 or PDK2) in all three lipoyl domains were correlated with the sequence. Resonances, which were too broad to be detected upon complexation, pertained to conserved or highly similar residues in the L1–L3 sequences (Figure S3 of the Supporting Information), even though the affected residues participated in different interactions. Noticeable differences were seen in the corresponding residues Gln6, Met133, and Ile4 in the L1, L2, and L3 sequences. The highly similar hydrophobic residues Met133 and Ile4 in L2 and L3 show interaction with PDK1, but residue Gln6 in L1 did not show any interaction with PDKs. Also, Ile4 (L3S′) interacts with both PDK1 and PDK2, whereas Met133 (L2S) is affected by only PDK1.
(3) A few residues in L2 and L3 exhibited unique interactions with PDKs. (a) L2 residues Leu166, Asp197, Gly201, and Leu204 interact with PDKs, whereas the corresponding residues in L3 and L1 were unaffected in the NMR studies. (b) L3 residues Leu24, Gly28, and Gly34 also display unique interaction behavior with PDKs compared with that of L1 and L2.
(4) Complexation with PDK1 or PDK2 led to more interactions with the L1L2S tridomain than with the L2S didomain. The differences in the amino acid composition of the linker residues might also play a critical role in the differential PDK regulation by the lipoyl domains. Our sequence-specific assignments identified L2 residues Ile214, Ser215, and Tyr220 to show interaction with both PDKs and residue Phe217, which was affected upon its interaction with PDK1, but not with PDK2. More complete assignments in the linker and PSBD regions might shed light on their role in the differential behavior of the lipoyl domains toward PDKs.
(5) The chemical shift perturbation maps revealed that there were some resonances corresponding to residues in each protein participating in specific interaction with PDKs. In L1L2S, two residues, Glu35 and Val198, interacting with PDK1 and two residues, Glu153 and Glu209, interacting with PDK2 displayed specificity.
(6) Analysis of the NMR data and structural information showed that the surface loop residues were consistently affected in all four lipoyl domain-containing proteins. This infers that these intrinsically flexible active site loops play multiple roles in catalysis, as well as being critical in interactions with PDKs, and further suggest that both L2 and L3 may be critical in defining PDC interactions with PDKs.
(7) Even though the ITC studies of L3S′ with PDK2 did not reveal quantifiable interaction, the NMR studies suggest that apart from L2S, the L3S′ also serves as a docking site for PDKs.
(8) The residues affected in the NMR study are mainly hydrophobic and acidic in both L2S and L3S′ proteins, in agreement with previous crystallographic studies that showed that the interfaces between the N-terminal domain of PDK3 and L2 consist of hydrophobic interactions in the lipoyl-binding pocket and its surrounding regions, as well as electrostatic interactions of the lower portion of the N-terminal domain. From our interaction studies, the “hot spots” identified in Results and Discussion and highlighted in Table 1 could serve as potential guides in a search for specific inhibition of PDK isoforms.
Advantages and Limitations of the Two Approaches
The C-terminally truncated proteins derived from the E2·E3BP core of the PDC all confirm interactions with both PDK1 and PDK2. There are also distinct hot spots suggested by the chemical shift perturbation maps, which provide suggestions for further research to be undertaken prior to inhibitor design, synthesis, and testing. Site-directed mutagenesis at the hot spots needs to be conducted, followed by NMR and ITC studies of the protein variants to rule in or out the importance of the hot spots in the interactions with the kinases. Because of the size of the PDKs (dimer of 45 kDa monomers), the NMR results inform only the interaction hot spots on the partner derived from the E2·E3BP core. The HDX-MS studies of the same C-terminally truncated proteins derived from the E2·E3BP core, and of the entire E2·E3BP core with the same two kinases, while lower in resolution than the residue-specific NMR method, add information regarding the loci of interactions on the two kinases themselves, adding to the unique capabilities of the HDX-MS method.
From these studies, it became clear that even though the L2 domain plays a major role in PDK interactions, there are other regions on E2 that interact with PDKs. According to both methods used, the larger C-terminally truncated L1L2S shows stronger, and more points of, interaction with PDKs than the didomain L2S. Because there are no X-ray structural data about these tridomain and didomain proteins, it is difficult to interpret the role of long-range interactions due to domain–domain interactions based on the chemical shift mapping studies. At the same time, it cannot be overemphasized that the HDX-MS method could use the intact E2·E3BP core (60 subunits) and provide detailed information about the effects of its interaction and/or specificity with PDK1 and PDK2. The two complementary approaches consistently suggest that the L3 domain from E3BP also interacts with PDKs. Also, chemical shift mapping identified several residues interacting uniquely with PDK1 or PDK2.
This study of E2·E3BP core–PDK1 and −PDK2 interactions could serve as a stepping stone for further research to be undertaken prior to inhibitor design for the development of isozyme-specific (and tissue-specific) inhibitors for the treatment of diabetes and cancer.
Glossary
Abbreviations
- PDC
pyruvate dehydrogenase complex
- E1
pyruvate dehydrogenase component
- E2
dihydrolipoamide acetyltransferase
- E3
dihydrolipoamide dehydrogenase
- E3BP
E3-binding protein
- ThDP
thiamin diphosphate
- L1
outer lipoyl domain
- L2
inner lipoyl domain
- L3
lipoyl domain of E3BP
- PSBD or S
peripheral subunit-binding domain on E2
- S′
E3-binding domain of E3BP
- PDK1
pyruvate dehydrogenase kinase 1
- PDK2
pyruvate dehydrogenase kinase 2
- PDP
pyruvate dehydrogenase phosphatase
- HDX-MS
hydrogen/deuterium exchange mass spectrometry
- NMR
nuclear magnetic resonance.
Supporting Information Available
Four tables on peptic peptides selected for HDX-MS analysis, one table of a summary of sequence-specific resonance assignments made, four tables of backbone resonance assignments of L1, L2S, L1L2S, and L3S′, a figure with sequence alignments for the E2 core domains of pyruvate dehydrogenase complexes, a figure showing the comparative HDX-MS analysis of the interaction of PDK2 and the E2·E3BP core, and a figure comparing the sequences of L1–L3. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
J.W. and S.K. contributed equally to this research.
The work performed by the authors and summarized here was supported, in part, by National Institutes of Health (NIH) Grants DK 20478 (M.S.P.) and GM050380 (F.J. and M.S.P.). In addition to support of this work by the NIH, research at the Rutgers laboratory is also supported by Multienzyme Druggable Targets of Chatham, NJ.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Reed L. J. (2001) A trial of research from lipoic acid to α-keto acid dehydrogenase complexes. J. Biol. Chem. 276, 38329–38336. [DOI] [PubMed] [Google Scholar]
- Perham R. N. (1991) Domains, motifs, and linkers in 2-oxo acid dehydrogenase multienzyme complexes: A paradigm in the design of a multifunctional protein. Biochemistry 30, 8501–8512. [DOI] [PubMed] [Google Scholar]
- Patel M. S.; Roche T. E. (1990) Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J. 14, 3224–3233. [DOI] [PubMed] [Google Scholar]
- Roche T. E.; Baker J. C.; Yan X.; Hiromasa Y.; Gong X.; Peng T.; Dong J.; Turkan A.; Kasten S. A. (2001) Distinct regulatory properties of pyruvate dehydrogenase kinase and phosphatase isoforms. Prog. Nucleic Acid Res. Mol. Biol. 70, 33–75. [DOI] [PubMed] [Google Scholar]
- Hiromasa Y.; Fujisawa T.; Aso Y.; Roche T. E. (2004) Organization of the cores of the mammalian pyruvate dehydrogenase complex formed by E2 and E2 plus the E3-binding protein and capacities to bind E1 and E3 components. J. Biol. Chem. 279, 6921–6933. [DOI] [PubMed] [Google Scholar]
- Roche T. E.; Hiromasa Y. (2007) Pyruvate dehydrogenase kinase regulatory mechanisms and inhibition in treating diabetes, heart ischemia, and cancer. Cell. Mol. Life Sci. 64, 830–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudi R.; Bowker-Kinley M. M.; Kedishvili N. Y.; Zhao Y.; Popov K. M. (1995) Diversity of the pyruvate dehydrogenase kinase gene family in humans. J. Biol. Chem. 270, 28989–28994. [DOI] [PubMed] [Google Scholar]
- Hiromasa Y.; Yan X.; Roche T. (2008) Specific ion influence on self-association of pyruvate dehydrogenase kinase isoform 2 (PDHK2), binding of PDHK2 to the L2 lipoyl domain, and effects of the lipoyl group-binding site inhibitor, Nov3r. Biochemistry 47, 2312–2324. [DOI] [PubMed] [Google Scholar]
- Harris R. A.; Bowker-Kenley M. M.; Huang B.; Wu P. (2002) Regulation of the activity of the pyruvate dehydrogenase complex. Adv. Enzyme Regul. 42, 249–259. [DOI] [PubMed] [Google Scholar]
- Roche T. E.; Hiromasa Y.; Turkan A.; Gong X.; Peng T.; Yan X.; Kasten S. A.; Bao H.; Dong J. (2003) Essential roles of lipoyl domains in the activated function and control of pyruvate dehydrogenase kinases and phosphatase isoform 1. Eur. J. Biochem. 270, 1050–1056. [DOI] [PubMed] [Google Scholar]
- Huang B.; Gudi R.; Wu P.; Harris R. A.; Hamilton J.; Popov K. M. (1998) Isoenzymes of pyruvate dehydrogenase phosphatase: DNA-derived amino acid sequences, expression, and regulation. J. Biol. Chem. 273, 17680–17688. [DOI] [PubMed] [Google Scholar]
- Brautigam C. A.; Wynn R. M.; Chuang J. L.; Chuang D. T. (2009) Subunit and catalytic component stoichiometries of an in vitro reconstituted human pyruvate dehydrogenase complex. J. Biol. Chem. 284, 13086–13098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciszak E. M.; Korotchkina L. G.; Dominiak P. M.; Sidhu S.; Patel M. S. (2003) Structural basis for flip-flop action of thiamin pyrophosphate-dependent enzymes revealed by human pyruvate dehydrogenase. J. Biol. Chem. 278, 21240–21246. [DOI] [PubMed] [Google Scholar]
- Brautigam C. A.; Chuang J. L.; Tomchick D. R.; Machius M.; Chuang D. T. (2005) Crystal structure of human dihydrolipoamide dehydrogenase: NAD+/NADH binding and the structural basis of disease-causing mutations. J. Mol. Biol. 350, 543–552. [DOI] [PubMed] [Google Scholar]
- Ciszak E. M.; Makal A.; Hong Y. S.; Vettaikkorumakankauv A. K.; Korotchkina L. G.; Patel M. S. (2006) How dihydrolipoamide dehydrogenase-binding protein binds dihydrolipoamide dehydrogenase in the human pyruvate dehydrogenase complex. J. Biol. Chem. 281, 648–655. [DOI] [PubMed] [Google Scholar]
- Brautigam C. A.; Wynn R. M.; Chuang J. L.; Machius M.; Tomchick D. R.; Chuang D. T. (2006) Structural insight into interactions between dihydrolipoamide dehydrogenase (E3) and E3 binding protein of human pyruvate dehydrogenase complex. Structure 14, 611–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert F.; Ciszak E.; Korotchkina L.; Golbik R.; Spinka M.; Dominiak P.; Sidhu S.; Brauer J.; Patel M. S.; Tittmann K. (2007) Phosphorylation of serine 264 impedes active site accessibility in the E1 component of the human pyruvate dehydrogenase multienzyme complex. Biochemistry 46, 6277–6287. [DOI] [PubMed] [Google Scholar]
- Kato M.; Wynn R. M.; Chuang J. L.; Tso S.-C.; Machius M.; Li J.; Chuang D. T. (2008) Structural basis for inactivation of the human pyruvate dehydrogenase complex by phosphorylation: role of disordered phosphorylation loops. Structure 16, 1849–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X.; Hiromasa Y.; Tsen H.; Stoops J. K.; Roche T. E.; Zhou Z. H. (2008) Structures of the human pyruvate dehydrogenase complex cores: A highly conserved catalytic center with flexible N-terminal domains. Structure 16, 104–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayakrishnan S.; Kelly S. M.; Gilbert R. J.; Callow P.; Bhella D.; Forsyth T.; Lindsay J. G.; Byron O. (2010) Solution structure and characterization of the human pyruvate dehydrogenase complex core assembly. J. Mol. Biol. 399, 71–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M. S.; Korotchkina L. G. (2003) The biochemistry of the pyruvate dehydrogenase Complex. Biochem. Mol. Biol. Educ. 31, 5–15. [Google Scholar]
- Patel M. S.; Korotchkina L. G. (2006) Regulation of pyruvate dehydrogenase complex. Biochem. Soc. Trans. 34, 217–222. [DOI] [PubMed] [Google Scholar]
- Kolobova E.; Tuganova A.; Boulatnikov I.; Popov K. M. (2001) Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem. J. 358, 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotchkina L. G.; Patel M. S. (2001) Probing the mechanism of inactivation of human pyruvate dehydrogenase by phosphorylation of three sites. J. Biol. Chem. 276, 5731–5738. [DOI] [PubMed] [Google Scholar]
- Korotchkina L. G.; Patel M. S. (2001) Site specificity of four pyruvate dehydrogenase kinase isoenzymes toward the three phosphorylation sites of human pyruvate dehydrogenase. J. Biol. Chem. 276, 37223–37229. [DOI] [PubMed] [Google Scholar]
- Reed L. J.; Damuni Z.; Merryfield M. L. (1985) Regulation of mammalian pyruvate α-keto acid dehydrogenase complexes by phosphorylation-dephosphorylation. Curr. Top. Cell. Regul. 27, 41–49. [DOI] [PubMed] [Google Scholar]
- Wu P.; Inskeep K.; Bowker- Kinley M. M.; Popov K. M.; Harris R. A. (1999) Mechanism responsible for inactivation of skeletal muscle pyruvate dehydrogenase complex in starvation and diabetes. Diabetes 48, 1593–1599. [DOI] [PubMed] [Google Scholar]
- Holness M. J.; Kraus A.; Harris R. A.; Sugden M. C. (2000) Targeting upregulation of pyruvate dehydrogenase kinase (PDK)-4 in slow-twitch skeletal muscle underlies the state of modification of the regulatory characteristics of PDK induced by high-fat feeding. Diabetes 49, 775–781. [DOI] [PubMed] [Google Scholar]
- Kwon H. S.; Huang B.; Unterman T. G.; Harris R. A. (2004) Protein kinase B-α inhibits human pyruvate dehydrogenase kinase-4 gene induction by dexamethasone through inactivation of FOXO transcription factors. Diabetes 53, 899–910. [DOI] [PubMed] [Google Scholar]
- Patel M. S.; Harris R. A. (1995) Mammalian α-keto acid dehydrogenase complexes gene regulation and genetic defects. FASEB J. 9, 1164–1172. [DOI] [PubMed] [Google Scholar]
- Imbard A.; Boutron A.; Vequaud C.; Zater M.; de Lonlay P.; Ogier de Baulny H.; Barnerias C.; Miné M.; Marsac C.; Saudubray J.-M.; Brivet M. (2011) Molecular characterization of 82 patients with pyruvate dehydrogenase complex deficiency. Structural implications of novel amino acid substitutions in E1 protein. Mol. Genet. Metab. 104, 507–516. [DOI] [PubMed] [Google Scholar]
- Patel K. P.; O’Brien T. W.; Subramony S. H.; Shuster J.; Stacpoole P. W. (2012) The spectrum of pyruvate dehydrogenase complex deficiency: Clinical, biochemical and genetic features in 371 patients. Mol. Genet. Metab. 106, 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeoung N. H.; Harris C. R.; Harris R. A. (2014) Regulation of pyruvate metabolism in metabolic-related diseases. Rev. Endocr. Metab. Disord. 15, 99–110. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M. G.; Cantley L. C.; Thompson C. B. (2009) Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enns L.; Ladiges W. (2012) Mitochondrial redox signaling and cancer invasiveness. J. Bioenerg. Biomembr. 44, 635–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Hernández A.; Gallardo-Pérez J. C.; Rodriguez-Enriquez S.; Moreno-Sánchez R. (2009) HIF-1α modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini-Rev. Med. Chem. 9, 1084–1101. [DOI] [PubMed] [Google Scholar]
- Schulze A.; Downward J. (2011) Flicking the Warburg switch-tyrosine phosphorylation of pyruvate dehydrogenase kinase regulates mitochondrial activity in cancer cells. Mol. Cell 44, 846–848. [DOI] [PubMed] [Google Scholar]
- Olenchock B.; Vander Heiden M. G. (2013) Pyruvate as a pivot point for oncogene-induced senescence. Cell 153, 1429–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplon J.; Zheng L.; Meissl K.; Chaneton B.; Selivanov V. A.; Mackay G.; van der Burg S. H.; Verdegaal E. M. E.; Cascante M.; Shlomi T.; Gottlieb E.; Peeper D. S. (2013) A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 498, 109–112. [DOI] [PubMed] [Google Scholar]
- Fujiwara S.; Kawano Y.; Yuki H.; Okuno Y.; Nosaka K.; Mitsuya H.; Hata H. (2013) PDK1 inhibition is a novel therapeutic target in multiple myeloma. Br. J. Cancer 108, 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur H.; Xuan Y.; Kim Y. B.; Lee G.; Shim W.; Yun J.; Ham I. H.; Han S. U. (2013) Expression of pyruvate dehydrogenase kinase-1 in gastric cancer as a potential therapeutic target. Int. J. Oncol. 42, 44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutendra G.; Michelakis E. D. (2013) Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Front. Oncol. 3, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papandreou I.; Cairns R. A.; Fontana L.; Lim A. L.; Denko N. C. (2006) HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 3, 187–197. [DOI] [PubMed] [Google Scholar]
- Kim J. W.; Tchernyshyov I.; Semenza G. L.; Dang C. V. (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177–185. [DOI] [PubMed] [Google Scholar]
- Kaplon J.; Zheng L.; Meissl K.; Chaneton B.; Selivanov V. A.; Mackay G.; van der Burg S. H.; Verdegaal E. M.; Cascante M.; Shlomi T.; Gottlieb E.; Peer D. S. (2013) A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 498, 109–112. [DOI] [PubMed] [Google Scholar]
- Michelakis E. D.; Sutendra G.; Dromparis P.; Webster L.; Haromy A.; Niven E.; Maguire C.; Gammer T. L.; Mackey J. R.; Fulton D.; Abdulkarim B.; McMurtry M. S.; Petruk K. C. (2010) Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2, 31–34. [DOI] [PubMed] [Google Scholar]
- Lu C. W.; Lin S. C.; Chen K. F.; Lai Y. Y.; Tsai S. J. (2008) Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J. Biol. Chem. 283, 28106–28114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitosugi T.; Fan J.; Chung T.-W.; Lythgoe K.; Wang X.; Xie J.; Ge Q.; Gu T.-L.; Polakiewicz R. D.; Roesel J. L.; Chen G. Z.; Boggon T. J.; Lonial S.; Fu H.; Khuri F. R.; Kang S.; Chen J. (2011) Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol. Cell 44, 864–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet S.; Archer S. L.; Allalunis-Turner J.; Haromy A.; Beaulieu C.; Thompson R.; Lee C. T.; Lopaschuk G. D.; Puttagunta L.; Bonnet S.; Harry G.; Hashimoto K.; Porter C. J.; Andrade M. A.; Thebaud B.; Michelakis E. D. (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11, 37–51. [DOI] [PubMed] [Google Scholar]
- Michelakis E. D.; Webster L.; Mackey J. R. (2008) Dichloroacetate (DCA) as a potential metabolic targeting therapy for cancer. Br. J. Cancer 99, 989–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutendra G.; Dromparis P.; Kinnaird A.; Stenson T. H.; Haromy A.; Parker J. M.; McMurtry M. S.; Michelakis E. D. (2012) Mitochondrial activation by inhibition of PDKII suppresses HIFa signaling and angiogenesis in cancer. Oncogene 32, 1638–1650. [DOI] [PubMed] [Google Scholar]
- Kato M.; Li J.; Chuang J. L.; Chuang D. T. (2007) Distinct structural mechanisms for inhibition of pyruvate dehydrogenase kinase isoforms by AZD7545, dichloroacetate, and radicicol. Structure 15, 992–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoechel T. R.; Tucker A. D.; Robinson C. M.; Phillips C.; Taylor W.; Bungay P. J.; Kasten S. A.; Roche T. E.; Brown D. G. (2006) Regulatory roles of the N-terminal domain based on crystal structures of human pyruvate dehydrogenase kinase 2 containing physiological and synthetic ligands. Biochemistry 45, 402–415. [DOI] [PubMed] [Google Scholar]
- Morrell J. A.; Orme J.; Butlin R. J.; Roche T. E.; Mayers R. M.; Kilgour E. (2003) AZD7545 is a selective inhibitor of pyruvate dehydrogenase kinase 2. Biochem. Soc. Trans. 31, 1168–1170. [DOI] [PubMed] [Google Scholar]
- Tuganova A.; Klyuyeva A.; Popov K. M. (2007) Recognition of the inner lipoyl-bearing domain of dihydrolipoyl transacetylase and of the blood glucose-lowering compound AZD7545 by pyruvate dehydrogenase kinase 2. Biochemistry 46, 8592–8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayers R. M.; Butlin R. J.; Kligour E.; Leighton B.; Martin D.; Myatt J.; Orme J. P.; Holloway B. P. (2003) AZD7545, a novel inhibitor of pyruvate dehydrogenase kinase 2 (PDK2), activates pyruvate dehydrogenase in vivo and improves blood glucose control in obese (fa/fa) Zucker rats. Biochem. Soc. Trans. 31(Part 6), 1165–1167. [DOI] [PubMed] [Google Scholar]
- Archer T. D.; Anderson R. C.; Gao J.; Shetty S. S.; Coppola G. M.; Stanton J. L.; Knorr D. C.; Sperbeck D. M.; Brand L. J.; Vinluan C. C.; Kaplan E. L.; Dragland C. J.; Tomaselli H. C.; Islam A.; Losito R. J.; Liu X.; Maniara W. M.; Fillers W. S.; DelGrande D.; Walter R. E.; Mann W. R. (2000) Secondary amides of (R)-3,3,3-trifluoro-2-hydroxy-2-methylpropionic acid as inhibitors of pyruvate dehydrogenase kinase. J. Med. Chem. 43, 236–249. [DOI] [PubMed] [Google Scholar]
- Tso S.-C.; Qi X.; Gui W.-J.; Wu C.-Y.; Chuang J. L.; Wernstedt-Asterholm I.; Morlock L. K.; Owens K. R.; Schrer P. E.; Williams N. S.; Tambar U. K.; Wynn R. M.; Chuang D. (2014) Structure-quided development of specific pyruvate dehydrogenase kinase inhibitors targeting the ATP-binding pocket. J. Biol. Chem. 289, 4432–4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steussy C. N.; Popov K. M.; Bowker-Kinley M. M.; Sloan R. B. Jr.; Harris R. A.; Hamilton J. A. (2001) Structure of pyruvate dehydrogenase kinase. Novel folding pattern for a serine protein kinase. J. Biol. Chem. 276, 37443–37450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green T.; Grigorian A.; Klyuyeva A.; Tuganova A.; Luo M.; Popov K. M. (2008) Structural and functional insights into the molecular mechanisms responsible for the regulation of pyruvate dehydrogenase kinase 2. J. Biol. Chem. 283, 15789–15798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M.; Chuang J. L.; Tso S. C.; Wynn R. M.; Chuang D. T. (2005) Crystal structure of pyruvate dehydrogenase kinase 3 bound to lipoyl domain 2 of human pyruvate dehydrogenase complex. EMBO J. 24, 1763–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devedjiev Y.; Steussy C. N.; Vassylyev D. G. (2007) Crystal structure of an asymmetric complex of pyruvate dehydrogenase kinase 3 with lipoyl domain 2 and its biological implications. J. Mol. Biol. 370, 407–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn R. M.; Kato M.; Chuang J. L.; Tso S. C.; Li J.; Chuang D. T. (2008) Pyruvate dehydrogenase kinase-4 structures reveal a metastable open conformation fostering robus core-free basal activity. J. Biol. Chem. 283, 25305–25315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowker-Kinley M. M.; Davis W. I.; Wu P.; Harris R. A.; Popov K. M. (1998) Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 329, 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P.; Blair P. V.; Sato J.; Jaskiewicz J.; Popov K. M.; Harris R. A. (2000) Starvation increases the amount of pyruvate dehydrogenase kinase in several mammalian tissues. Arch. Biochem. Biophys. 381, 1–7. [DOI] [PubMed] [Google Scholar]
- Knoechel T. R.; Tucker A. D.; Robinson C. M.; Phillips C.; Taylor W.; Bungay P. J.; Kasten S. A.; Roche T. E.; Brown D. G. (2006) Regulatory Roles of the N-Terminal Domain Based on Crystal Structures of Human Pyruvate Dehydrogenase Kinase 2 Containing Physiological and Synthetic Ligands. Biochemistry 45, 402–415. [DOI] [PubMed] [Google Scholar]
- Roche T. E.; Baker J. C.; Yan X.; Hiromasa Y.; Gong X.; Peng T.; Dong J.; Turkan A.; Kasten S. A. (2001) Distinct regulatory properties of pyruvate dehydrogenase kinase and phosphatase isoforms. Prog. Nucleic Acid Res. Mol. Biol. 70, 33–75. [DOI] [PubMed] [Google Scholar]
- Wang J.; Nemeria N. S.; Chandrasekhar K.; Kumaran S.; Arjunan P.; Reynolds S.; Calero G.; Brukh R.; Kakalis L.; Furey W.; Jordan F. (2014) Structure and Function of the Catalytic Domain of the Dihydrolipoyl Acetyltransferase Component in Escherichia coli Pyruvate Dehydrogenase Complex. J. Biol. Chem. 289, 15215–15230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekhar K.; Wang J.; Arjunan P.; Sax M.; Park Y.-H.; Nemeria N. S.; Kumaran S.; Song J.; Jordan F.; Furey W. (2013) Insight to the Interaction of the Dihydrolipoamide Acetyltransferase (E2) Core with the Peripheral Components in the Escherichia coli Pyruvate Dehydrogenase Complex via Multifaceted Structural Approaches. J. Biol. Chem. 288, 15402–15417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamuro Y.; Coales S. J.; Molnar K. S.; Tuske S. J.; Morrow J. A. (2008) Specificity of immobilized porcine pepsin in H/D exchange compatible conditions. Rapid Commun. Mass Spectrom. 22, 1041–1046. [DOI] [PubMed] [Google Scholar]
- Weis D.; Engen J.; Kass I. (2006) Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. J. Am. Soc. Mass Spectrom. 17, 1700–1703. [DOI] [PubMed] [Google Scholar]
- Kan Z. Y.; Mayne L.; Chetty P. S.; Englander S. W. (2011) ExMS: Data analysis for HX-MS experiments. J. Am. Soc. Mass Spectrom. 22, 1906–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y.; Milne J. S.; Mayne L.; Englander S. W. (1993) Primary structure effects on peptide group hydrogen exchange. Proteins 17, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T.; Lee W.; Arrowsmith C. H.; Muhandiram D. R.; Kay L. E. (1994) A Suite of Triple Resonance NMR Experiments for the Backbone Assignment of 15N, 13C, 2H Labeled Proteins with High Sensitivity. J. Am. Chem. Soc. 116, 11655–11666. [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Keller R. (2004) The Computer Aided Resonance Assignment, Cantina Verlag, Goldau, Switzerland. [Google Scholar]
- Live D. H.; Davis D. G.; Agosta W. C.; Cowburn D. (1984) Observation of 1000-fold Enhancement of 15N NMR via Proton-Detected Multiple-Quantum Coherences: Studies of Large Peptides. J. Am. Chem. Soc. 106, 6104–6105. [Google Scholar]
- Kavan D.; Man P. (2011) MSTools: Web based application for visualization and presentation of HXMS data. Int. J. Mass Spectrom. 302, 53–58. [Google Scholar]
- Ferguson N.; Sharpe T. D.; Johnson C. M.; Fersht A. R. (2006) The transition state for folding of a peripheral subunit-binding domain contains robust and ionic-strength dependent characteristics. J. Mol. Biol. 356, 1237–1247. [DOI] [PubMed] [Google Scholar]
- Ferguson N.; Sharpe T. D.; Schartau P. J.; Sato S.; Allen M. D.; Johnson C. M.; Rutherford T. J.; Fersht A. R. (2005) Ultra-fast barrier-limited folding in the peripheral subunit-binding domain family. J. Mol. Biol. 353, 427–446. [DOI] [PubMed] [Google Scholar]
- Fries M.; Stott K. M.; Reynilds S.; Perham R. N. (2007) Distinct modes of recognition of the lipoyl domain as substrate by the E1 and E3 components of the pyruvate dehydrogebase multienzyme complex. J. Mol. Biol. 366, 132–139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.