

Abstract
Over the last decade, it has been discovered that the transcription factor Sox9 plays several critical roles in governing the development of the embryonic pancreas and the homeostasis of the mature organ. While analysis of pancreata from patients affected by the Sox9 haploinsufficiency syndrome campomelic dysplasia initially alluded to a functional role of Sox9 in pancreatic morphogenesis, transgenic mouse models have been instrumental in mechanistically dissecting such roles. Although initially defined as a marker and maintenance factor for pancreatic progenitors, Sox9 is now considered to fulfill additional indispensable functions during pancreogenesis and in the postnatal organ through its interactions with other transcription factors and signaling pathways such as Fgf and Notch. In addition to maintaining both multipotent and bipotent pancreatic progenitors, Sox9 is also required for initiating endocrine differentiation and maintaining pancreatic ductal identity, and it has recently been unveiled as a key player in the initiation of pancreatic cancer. These functions of Sox9 are discussed in this article, with special emphasis on the knowledge gained from various loss-of-function and lineage tracing mouse models. Also, current controversies regarding Sox9 function in healthy and injured adult pancreas and unanswered questions and avenues of future study are discussed.
Keywords: Sox9, pancreas development, beta-cell, multipotent progenitor cell, Fgf, Pdx1, Notch, Hes1, transgenic mouse, molecular marker
Abbreviations: ADPKD - autosomal dominant polycystic kidney disease; BAC - bacterial artificial chromosome; BCIP - 5-bromo-4-chloro-3-indolyl phosphate; bHLH - basic helix-loop-helix; Bmp - bone morphogenetic protein; CAII - carbonic anhydrase II; CD - campomelic dysplasia; ChIP - chromatin immunoprecipitation; CNS - central nervous system; Cpa1 - carboxypeptidase A1; DAPT - N-(N-(3,5-difluorophenacetyl)-L-alanyl)-S-phenylglycine t-butyl ester; DCC - N,N'-dicyclohexylcarbodiimide; Dll1 - delta-like ligand 1; E - embryonic day; ER - estrogen receptor; Fgf10 - fibroblast growth factor 10; Fgfr - fibroblast growth factor receptor; Foxa2 - forkhead-box protein a2; GFP - green fluorescent protein; GSI-IX - gamma-secretase inhibitor-IX; Hes1 - hairy and enhancer of split 1; hESC - human embryonic stem cell; HMG - high-mobility group; Hnf1beta - hepatocyte nuclear factor 1beta (aka Tcf2); HPD - hepatopancreatic ductal system; IAPP - islet amyloid polypeptide; IF - immunofluorescence; iPSC - induced pluripotent stem cell; IRES - internal ribosome entry site; ISH - in situ hybridization; Isl1 - ISL LIM homeobox 1; LacZ - β-galactosidase-encoding gene; Mash1 - mammalian achaete scute homolog 1; MODY - maturity-onset diabetes of the young; MPC - multipotent pancreatic progenitor cell; mRNA - messenger ribonucleic acid; NBT - nitro blue tetrazolium chloride; NeuroD - neuronal differentiation 1 (aka Beta2); Ngn3 - neurogenin 3; NICD1 - Notch 1 intracellular domain; Nkx6.1 - Nk6 homeobox protein 1; Onecut1 - one cut homeobox 1 (aka Hnf6); PanIN - pancreatic intraepithelial neoplasia; PC1/3 - proprotein convertase 1; PCFU - pancreatic colony-forming unit; PDAC - pancreatic ductal adenocarcinoma; PDL - partial duct ligation; Pdx1 - pancreatic and duodenal homeobox 1; PNS - peripheral nervous system; PP - pancreatic polypeptide; Ptf1a - pancreas transcription factor 1 subunit alpha; Rbpj - recombination signal binding protein for the immunoglobulin kappa J region; RT-PCR - reverse transcription polymerase chain reaction; Sox9 - sex-determining region Y (Sry) box 9; STZ - streptozotocin; Tcf2 - transcription factor 2 (aka Hnf1β); UTR - untranslated region ; Vegf - vascular endothelial growth factor; Wnt - wingless-type MMTV integration site family; YFP - yellow fluorescent protein
1. Introduction
Over the preceding 50 years, much effort has been expended in deciphering the mechanisms governing morphogenesis of the mammalian pancreas and, in particular, the insulin-producing β-cell, loss or dysfunction of which manifests in diabetes mellitus. Acquiring mechanistic insight into β-cell neogenesis during pancreas morphogenesis is obviously of potential benefit in the derivation of cell-based diabetes therapies. Firstly, such knowledge may be applied in the optimization of in vitro protocols for driving differentiation of functional, glucose-responsive β-cells from either human embryonic or induced pluripotent stem cells. Secondly, insight into β-cell neogenesis may be used to stimulate this process in situ in the adult diabetic pancreas to restore functional β-cell mass. Associated with this goal, recent years have seen a resurgence in the study of traditional pancreatic injury models such as partial duct ligation (PDL) [1, 2] or cerulein treatment [3], both of which induce pancreatitis.
In concert with modern inducible genetic lineage tracing of various pancreatic cell populations, these models have been employed to examine whether inflammatory stimuli are able to induce differentiation of new β-cells and to identify the source of these new β-cells, which can either be existing non-β-cell pancreatic cell types or a putative facultative adult progenitor. Efforts to mechanistically dissect β-cell neogenesis would benefit greatly from the development of molecular markers enabling identification of β-cell-competent pancreatic progenitors throughout the duration of pancreas development and, potentially, in the adult.
Since the mid 1990s, studies of knockout and mutant mice have identified crucial roles for a slew of transcription factors in pancreas and β-cell development. Strikingly, many of these factors, including Isl1, NeuroD, Nkx2.2, and Nkx6.1 are expressed during central nervous system (CNS) development [4], highlighting the conserved transcriptional mechanisms governing morphogenesis in both systems.
The sex-determining region Y (Sry)-box-containing (Sox) factors are a structurally-related family of developmentally-regulated transcription factors belonging to the high-mobility group (HMG) superfamily; members of this family are united by their highly-conserved HMG-box, a 79-amino acid DNA-binding domain [5-7]. Currently, 20 Sox factors have been identified in mammals [8]. These 20 members have each been assigned to one of eight subgroups, A-H on the basis of similarity between HMG-box sequence and protein structure; members sharing more than 80% HMG domain sequence identity occupy the same group [9], and due to biochemical similarity, often play parallel functional roles [10].
The study of Sox genes originated in 1990 with the identification of Sry, the mammalian testis-determining factor and founding member of the family [5, 6]. Subsequently, crucial roles for Sox factors were described in the maintenance of tissue-specific stem/progenitor cells in the central and peripheral nervous system [11-14]. Given the conserved roles of transcription factors regulating neuronal and pancreatic developmental programs [4], attention soon turned in the early 2000s to examining putative conserved roles of Sox transcription factors in pancreas and β-cell development. These efforts were spurred on by the report of pancreas dysmorphogenesis in human patients affected by the lethal SOX9 haploinsufficiency syndrome campomelic dysplasia [15]. This study provided the first tantalizing insight into a role for SOX9/Sox9 in governing pancreas development, and pointed to the general involvement of Sox genes in regulating the pancreatic program.
Evidence, acquired primarily from studying knockout and transgenic mouse models over the past decade, has revealed that Sox9 serves as a marker and critical maintenance factor for pancreatic progenitors [16-19]. While this role has, by popular perception, defined the function of Sox9 during pancreatic organogenesis ("pancreogenesis"), mounting evidence suggests that this factor performs additional, equally important functions in both the developing and mature pancreas. These functions include the induction of endocrine differentiation [20, 21] and maintenance of the embryonic and adult ductal state [20, 22].
While two recent reviews have covered the roles of Sox9 in regulating pancreatic as well as either hepatic [23] or duodenal [24] programs, the present review focuses exclusively on the former to enable a more comprehensive examination of the multiple roles of Sox9 in the pancreas. This review aims to describe the roles of Sox9 during pancreas development with an emphasis on mouse models, placing major findings in the context of pancreas developmental biology from a historical perspective. In the second part of this review, focusing on the postnatal pancreas, recent contradictory findings from pulse-chase labeling of Sox9+ cells in both healthy and injured conditions are discussed. Finally, recently uncovered roles for Sox9 in the initiation of pancreatic cancer are summarized.
2. Pancreas morphogenesis in the mouse
2.1 Origins of the pancreas and its differentiated cell types
In the mouse, the pancreas arises as two opposing dorsal and ventral "buds" of the foregut endoderm (Figure 1A). While these buds become morphologically discernible at around embryonic day (E) 8.75-9.0, pancreatic fate is specified in undifferentiated foregut endoderm (coined the "primary transition" [25]) several hours prior to overt bud emergence [26]. This prepancreatic endoderm is demarcated by expression of the transcription factors Pdx1 and Ptf1a ventrally from E8.5 (8-10 somites) and dorsally from E8.5-8.75 (10 somites) [27, 28]. A third pancreatic transcription factor, Nkx6.1, is expressed in prospective ventral and dorsal pancreatic endoderm from E8.75 and E9.0, respectively [27]. Pdx1, Ptf1a and Nkx6.1 continue to mark the pancreatic endoderm which, until E12.5 when the two buds fuse (Figure 1B), is comprised predominantly of early multipotent pancreatic progenitor cells (MPCs) [29, 30]. Pdx1-Cre- [29] or Ptf1aCre- [30] mediated lineage tracing have revealed that, at the population level, MPCs give rise to all mature cell types in the adult organ, including:
Figure 1. The dynamic spatiotemporal pattern of Sox9 expression in the developing and adult mouse pancreas.
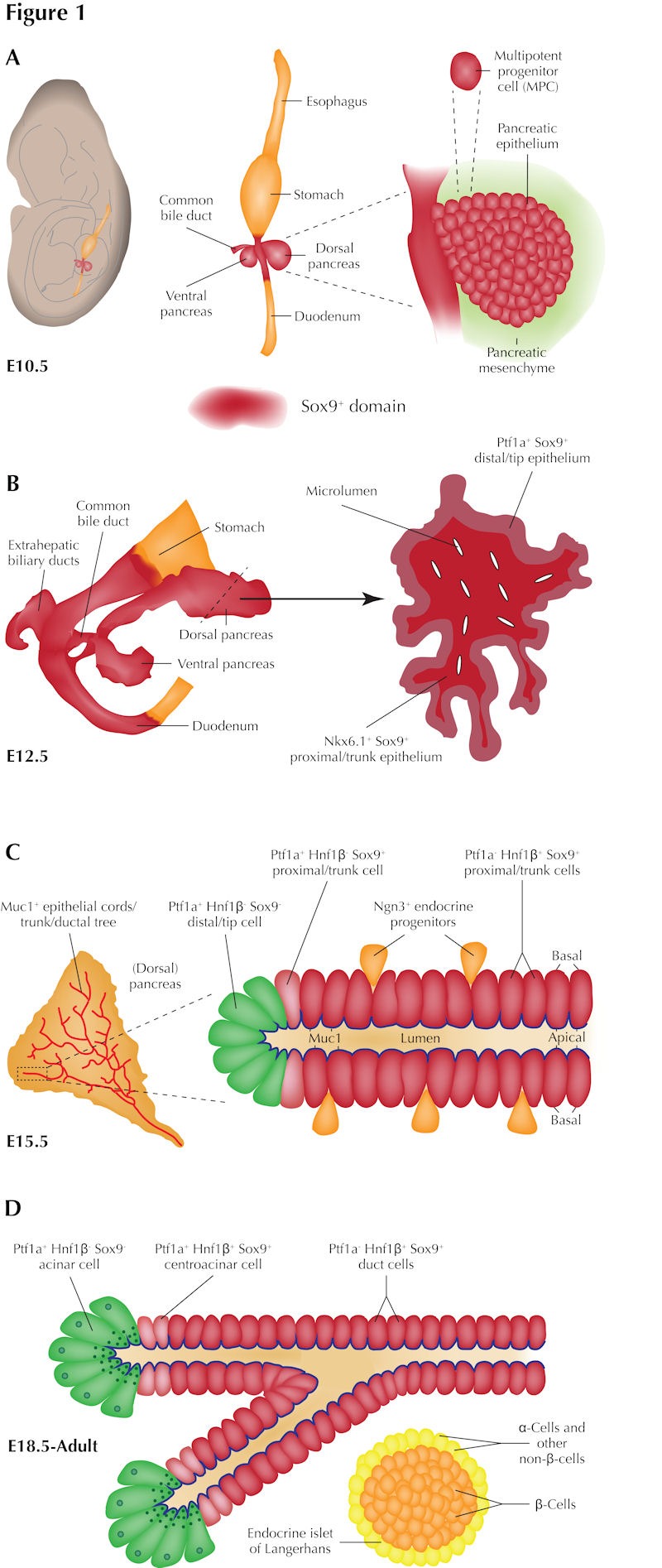
A: By E10.5, the foregut Sox9 expression domain encompasses the endoderm of the dorsal and ventral pancreatic buds as well as the antral (posterior) stomach, common bile duct (CBD), and anterior duodenum. In both pancreatic buds, Sox9 is confined to the epithelium, which at E10.5 is predominantly comprised of multipotent pancreatic progenitor cells (MPCs). B: At E12.5, Sox9 expression continues to demarcate the anterior stomach, CBD, extrahepatic biliary ducts and anterior duodenum as well as the fused dorsal and ventral pancreatic buds. By this stage, the pancreas is actively growing and is clearly lobulated; remodeling of microlumens to form a contiguous tubular plexus is also underway. These processes are concurrent with patterning of MPCs into a Ptf1a+ acinar-fated distal “tip” domain surrounding an Nkx6.1+ bipotent ductal-/endocrine-fated proximal "trunk" domain. Sox9 expression at this stage is highly dynamic: while Sox9 mRNA is receding from the tips at E12.5, Sox9 protein remains coexpressed with both Ptf1a and Nkx6.1 in the tips and the trunk, respectively. C: By E15.5, the mid-point of the secondary transition, Sox9 expression defines polarized Muc1+ epithelial cords of the proximal trunk, the predecessor of the ductal tree. Sox9 marks both Ptf1a- Hnf1β+ cells comprising the majority of the trunk as well as Ptf1a+ Hnf1β- cells at their extreme distal ends: Ptf1a+ Hnf1β- acinar cells "capping" the cords are devoid of Sox9 expression. Similarly, Ngn3+ endocrine progenitors delaminating from the Sox9+ trunk rarely coexpress high levels of Sox9 and are instead intercalated amongst Sox9+ cells. D: By late gestation (E18.5), Sox9 expression becomes confined to a subpopulation of ductal cells and centroacinar cells which are also defined by their expression of Hnf1β. Sox9 is excluded from the Ptf1a+ acinar cells as well as all endocrine cells of the islets of Langerhans.
1. Exocrine cells: the digestive enzyme-producing acinar cells and the cells of the ducts that transport these enzymes to the duodenum.
2. Endocrine cells of the islets of Langerhans (Figure 1D).
Notably, lineage tracing has shown that, in addition to Pdx1 and Ptf1a, MPCs can be similarly defined by their expression of the transcription factor Hnf1β (Tcf2) [31], the Notch downstream effector Hes1 [32], and carboxypeptidase A1 (Cpa1) [33]. Nkx6.1 undoubtedly marks this population even though this is not yet formally demonstrated because of the lack of suitable genetic tools. The five endocrine cell types derived from such progenitors comprise β-cells, α-cells, δ-cells, PP-cells, and ε-cells: respectively, they produce and secrete insulin, glucagon, somatostatin, pancreatic polypeptide (PP), or ghrelin. All endocrine cells arise in turn from a common, transient endocrine progenitor marked by expression of the transcription factor Ngn3 [29] which is both necessary [34] and sufficient [35-38] for endocrine differentiation. Ngn3 expression in MPCs results in temporally-distinct competence windows during which given endocrine cell types arise [39], commencing at E9.5 with the appearance of glucagon+ cells in the dorsal pancreas [40]. Insulin+ β-cells are not detected until E12.5; thereafter, from E14.5, their numbers expand exponentially [40] through the "secondary transition" [25], a major window of pancreatic differentiation. Somatostatin+ cells are detectable next, followed by PP-cells late in gestation [40]. Cells expressing ghrelin, the latest endocrine cell type to be discovered, can be detected as early as E10.5 and peak in number during the secondary transition [41].
2.2 Pancreatic progenitor expansion
After evaginating, the pancreatic buds rapidly expand and branch, concurrently with the synchronous extension and remodeling of a multi-lumen tubular plexus into a contiguous ductal tree [42, 43]. Expansion of MPCs is driven by both progenitor-intrinsic cues and extrinsic signals from the enveloping pancreatic mesenchyme (Figure 1A). Since final pancreas size is dictated by initial MPC pool size [44], the absence of such signals typically impacts upon progenitor proliferation, reducing progenitor numbers, manifesting in pancreatic hypoplasia. The progenitor-intrinsic transcription factors Pdx1 [45, 46] and Ptf1a [30, 47] as well as the Notch ligand Dll1 and effector Hes1 [35, 48, 49] all maintain MPC numbers, and are thus crucially required for pancreatic epithelial expansion: hypoplasia of one or both pancreatic buds ensues when MPCs are devoid of any of these factors. Likewise, deficiency for certain pancreatic mesenchyme-derived signaling cues results in hypoplasia of the pancreatic epithelium: Wnt [50, 51], Bmp [52], and, most prominently, Fgf10 [53] signaling have all been shown to stimulate proliferation of epithelial MPCs.
2.3 Proximodistal patterning
Concurrently with their expansion from E10.5 onwards, initially multipotent pancreatic progenitors become physically segregated by the secondary transition in terms of their developmental competency. MPCs in the distal “tip” domain marked by Pdx1, Ptf1a, and Cpa1 [33], gradually become acinar-fated while those progenitors left in their wake in the proximal "trunk", expressing Pdx1, Nkx6.1 (and its paralog, Nkx6.2), and Hnf1β [54], form both the ductal tree and endocrine cells and are thus "bipotent progenitors" [33] (Figure 1B, C). This compartmentalization is established through the cross-antagonism between Ptf1a and Nkx6.1/Nkx6.2; while initially co-expressed in early progenitors, by E14.5 they resolve into distinct Ptf1a+ distal tip and Nkx6.1+ proximal trunk domains [55]. More recently, a crucial requirement for Notch signaling has been demonstrated in the patterning of the trunk domain [56, 57].
2.4 The secondary transition and pancreatic differentiation
During the secondary transition, Ngn3+ endocrine progenitors appear in an intercalated pattern between the cells of the trunk, which comprises a primitive ductal epithelium marked by Hnf1β [54, 58], as well as the transcription factors Onecut1 (previously Hnf6) [59, 60] and Foxa2 (formerly Hnf3β) [61]. It has long been assumed that endocrine progenitors originate from the secondary transition ductal epithelium [54]. However, the relative paucity of exclusive markers of this domain and, hence, suitable genetic tools has meant that it is only quite recently, with their advent, that it has become feasible to test this hypothesis formally. The strictly mutually-exclusive expression domains of Ngn3 and Hes1 within the trunk have led to the widely-held notion that endocrine progenitors are specified through the process of Notch-mediated lateral inhibition [35, 62] (Figure 1C). As endocrine progenitors differentiate into the five endocrine cell types, they coalesce to form polyclonal islets [63]. In the mouse, islets display a highly stereotyped cytoarchitecture comprising a central core of insulin+ β-cells and a peripheral mantle composed predominantly of glucagon+ α-cells as well as the other three scarcer endocrine cell types (Figure 1D). It is only with the onset of postnatal life that the β-cells become fully-functional, capable of sensing glucose and modulating their secretion of insulin accordingly [64]. Concomitantly and during the preceding period, the pancreas grows rapidly, principally through the proliferation of acinar cells [65].
3. Expression of Sox factors in the developing and adult pancreas
The first comprehensive survey of Sox family gene expression in the mammalian pancreas [66] revealed that transcripts for a dozen Sox genes are detectable in the developing mouse pancreas: Sox11, Sox4, Sox13, Sox5, Sox9, Sox8, Sox10, Sox7, Sox17, Sox18, Sox15, and Sox30, three of which (Sox4, Sox9, and Sox13) were reported to be additionally detected in mature islets (Table 1). Notwithstanding the additional detection of Sox2 and Sox12 in developing mouse pancreas, findings of a similar analysis were in close concordance with this outcome [67] (Table 1). Of these dozen Sox genes expressed during mouse pancreogenesis, orthologs of five (SOX4, SOX9, SOX10, SOX11, and SOX12) have been shown to be enriched in the developing human pancreas versus adult islets [68], testament to the conserved nature of the pancreatic programs in mouse and man.
Table 1. Qualitative relative expression of Sox factors in developing pancreas and adult islets of the mouse.
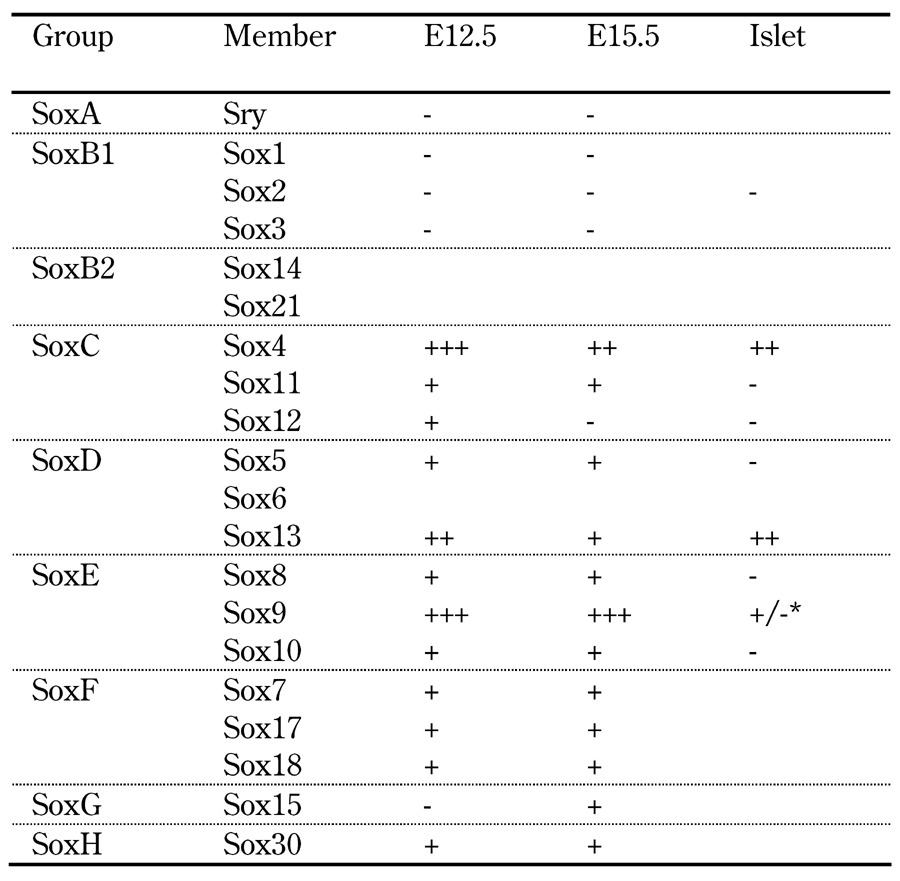
Legend: Data indicate the qualitative representation of relative abundance of Sox family member expression in whole embryonic mouse E12.5 and E15.5 pancreata and isolated adult islets, as surveyed by RT-PCR [66, 67]. Symbols: -: not detected; +, ++, +++: detected in increasing abundance, respectively. Absence of a symbol indicates expression not examined [66, 67]. *+: Sox9 transcripts initially detected in isolated adult islets [66], but (-) not in a subsequent study [16].
Via a more in-depth in situ hybridization (ISH) expression survey by Sox group, the Sander group [66] showed group C member Sox11 to be expressed predominantly in early pancreatic mesenchyme, then exclusively within some epithelial cells by E12.5, while Sox4 (also group C) expression is widespread in pancreatic epithelium from E12.5; both factors become islet-localized by birth with Sox4 being expressed in all mature islet cell types [67]. Corresponding with a pro-endocrine role for Sox4, explanted E11.5 Sox4-/- pancreata exhibit reduced numbers of insulin+ and glucagon+ cells [67]. Given the lack of quantification of Ngn3+ endocrine progenitors in Sox4-deficient explants coupled with no detectable effect of Sox4 deletion on either proliferation or survival of insulin+ cells [67], the underlying cause of endocrine hypoplasia in Sox4-/- mice remains obscure. Zebrafish studies, however, provide some insight. The Sox4 ortholog, sox4b, is expressed in endocrine precursors and glucagon+ cells in the developing zebrafish pancreas; when its expression is downregulated, glucagon+ cells are specifically lost, revealing a role in α-cell differentiation [69]. While the absence of an effect on insulin+ cell numbers suggests their differentiation to be sox4b-independent in zebrafish, specific effects on a single endocrine cell type suggest sox4b to either function downstream of a common endocrine precursor or to impact endocrine precursor numbers specifically during the window of α-cell neogenesis.
Of the group D factors, Sox13 is expressed at E10.5 in some, then by E14.5, in almost all pancreatic epithelial cells [66]. Likewise, L-Sox5 is first detectable at E12.5 in some pancreatic epithelial cells and is broadly epithelially expressed by the secondary transition. After this point, both factors are rapidly downregulated, becoming undetectable by birth. In contrast, Sox8 and Sox10, two of the three group E factors, are expressed in dispersed glucagon-negative cells at the epithelial periphery by E10.5 through E12.5, paralleling the pattern of Sox8 expression during chick pancreogenesis [70]. By birth, expression of both factors is confined to cells enveloping the islets [66]. Given that in the peripheral nervous system (PNS), both Sox factors are expressed in glial cells with Sox10 being required for their development [71, 72] and glia have been reported [73, 74] to ensheath the pancreatic islets, it was posited [66] that these Sox8+/Sox10+ cells are islet glial cells. Despite the unique peri-islet expression patterns of Sox8 and Sox10, analysis of pancreata from Sox8-/- or Sox10-/- mice revealed no gross dysgenesis of either the endocrine or exocrine compartments, indicating these factors to be dispensable for normal pancreatic morphogenesis [66]. While the precise function of peri-islet glia remains unknown, it has been proposed that they may provide neurotropic support to islet-innervating nerves and endocrine cells, and also play a role in neuroendocrine regulation [75]. While the possible function of Sox10 and peri-islet glia in adult endocrine function warrants closer examination, the perinatal lethality of Sox10-/- mice will dictate the requirement for a conditional Sox10 ablation model.
4. Sox9 expression in the developing pancreas
Alongside Sox8 and Sox10, Sox9 completes the Sox group E trifecta. Of the seven Sox factors, whose pancreatic expression patterns were comprehensively assessed by Lioubinski et al. [66], Sox9 proved the most intriguing. Sox9 was found to be highly expressed in both emerging pancreatic buds at E9.5 when they comprise almost exclusively of multipotent progenitors, hinting at a role in this population [66]. While Sox9 ISH signal remains elevated throughout E12.5 pancreatic epithelium, some diminution is evident at the periphery, denoting Sox9 withdrawal from the distal tips: concordantly, Sox9 becomes confined to trunk epithelium by the secondary transition. In line with this proximal expression pattern, the distribution of Sox9 ISH signal at birth was interpreted as marking a subset of ductal cells, islets, and a few acinar cells [66]. At the time of publication a decade ago, proximodistal patterning and secondary transition bipotent ductal/endocrine progenitors had yet to be conceptualized and thus, their markers identified. Notwithstanding, embryonic ductal Sox9 expression was deemed significant, given the assumption that endocrine progenitors arise from this domain during the secondary transition [36].
To better characterize dynamic spatiotemporal Sox9 expression during mouse pancreogenesis and to circumvent limited ISH resolution, co-immunofluorescence (IF) analysis was performed with a well-characterized Sox9-specific antiserum [76]. While colocalizing with the MPC marker Pdx1 in early (by E9.0) dorsal and ventral pancreatic endoderm (Figure 1A), Sox9 was excluded from glucagon+ endocrine cells [16, 77]. Strikingly, while at E12.5 Sox9 mRNA has receded from the epithelial tips [66], Sox9 protein is retained throughout the entire E12.5 pancreatic epithelium (excluding endocrine cells) [16], a finding validated by later studies [19] (Figure 1B). While implying Sox9 to be a novel pancreatic progenitor marker, these analyses also indicated that concomitant with proximodistal patterning, Sox9 expression is highly dynamic. Notably, hinting at evolutionarily-conserved roles for Sox9 in vertebrate pancreatic morphogenesis, this pattern of Sox9 expression in the early pancreatic rudiment is recapitulated in the frog Xenopus laevis, zebrafish, and human. Sox9 expression, within a broader Pdx1+ endodermal domain, specifically demarcates dorsal and ventral prepancreatic endoderm in X. laevis from its initial specification and continues to define the pancreatic buds through the point at which they fuse [78]. Similarly, the Sox9 paralog sox9b is expressed in the early zebrafish hepatopancreatic endoderm, in the anterior Pdx1+ domain, marking both the base of the dorsal pancreas and the prospective ventral pancreas [79]. A very recent immunohistochemistry-based expression analysis [80] has provided unprecedented (and beautiful) insight into the expression patterns of a number of pancreatic transcription factors (including SOX9) during the earliest steps of human pancreatic morphogenesis. This study shows that in humans, SOX9 is weakly coexpressed with PDX1 in prospective duodenal-pancreatic endoderm by Carnegie Stage (CS) 12 (29-31 days post-conception, dpc), equivalent to ~E9-9.5 in mouse embryogenesis [80]. By CS13 (30-33 dpc ≈E9.5-10 in mouse), SOX9 is strongly expressed in the readily-discernible dorsal and ventral pancreatic buds and is maintained through CS16 (37-40 dpc ≈E12.25-12.75) in the branched pancreatic epithelium [80], in agreement with earlier work [15].
Consistent with withdrawal of Sox9 from the murine pancreatic tip domain after E12.5, IF showed Sox9 to be restricted by E15.5 to a subpopulation of Pdx1+ cells comprising secondary transition pancreatic "epithelial cords" [16, 77] (Figure 1C). Such cords, historically posited to give rise to all differentiated cell types arising during the secondary transition [81] are now known as the proximal trunk domain. Notably, the process of segregating SOX9 to the proximal trunk appears to occur relatively late during the human pancreatic program, between 10-14 weeks post-conception (wpc) [80].
Supporting a continuing progenitor role through the secondary transition, a large proportion (~40%) of Sox9+ cells in the E15.5 mouse pancreas are both proliferative and Notch-responsive (Hes1+) [16]. In contrast, little overlap is evident between strongly Sox9-stained (Sox9Hi) cells and those expressing the endocrine progenitor marker Ngn3; Ngn3+ cells tend instead to be intercalated between Sox9Hi trunk cells [16, 21, 77] (Figure 1C). Similarly, in the developing human pancreas, cells expressing high levels of the Ngn3 ortholog NEUROG3 occur in juxtaposition with SOX9+ cells, concurrently with endocrine differentiation at 10 wpc [80]. Discordance between the reported extent of Sox9/Ngn3 colocalization in the embryonic mouse pancreas [16, 77] triggered a more comprehensive examination. This study concluded that, while only a small proportion (13.1%) of Ngn3+ cells are Sox9Hi, the vast majority (74.1%) are weakly Sox9-immunoreactive (Sox9Lo), with the remaining 12.8% of Ngn3+ cells being Sox9-, consistent with Sox9+ cells initiating Ngn3 coexpression then extinguishing Sox9 followed by Ngn3 expression before commencing endocrine hormone production [21]. In accordance with Ngn3+ cells arising from the Sox9+ trunk then downregulating Sox9 expression, Sox9 rarely colocalizes with late endocrine progenitor markers and is excluded from all differentiated endocrine and acinar cells [16] (Figure 1D). This is consistent with Sox9 being enriched in pancreatic progenitors while being largely excluded from endocrine progenitors and differentiated endocrine and acinar cells. Confirming this notion, pancreatic Sox9 expression is unaffected in embryos lacking the endocrine differentiation/maturation factors Ngn3 or Nkx6.1, showing Sox9 to be upstream of both [16]. Cementing its characterization as a pancreatic progenitor marker, expression of Sox9 is maintained throughout pancreata from Pdx1-Fgf10 mice in which the MPC mitogen Fgf10 is forcibly expressed throughout the pancreatic epithelium under the governance of the Pdx1 promoter, arresting pancreatic cells in the progenitor state and abrogating differentiation [16, 82].
5. Lineage tracing Sox9+ cells during pancreas development
5.1 Sox9 marks MPCs during the primary transition
While Sox9 coexpression with known MPC markers and regulation consistent with being a progenitor-expressed factor both infer such a role, these lines of evidence by themselves are insufficient to prove this. Such incontrovertible evidence for Sox9 defining MPCs can only be provided by genetic lineage tracing of the pancreatic Sox9+ population. Using this approach, the de Crombrugghe group [18] was the first to show indisputably that expression of Sox9 defines multipotent progenitors not only for the pancreas but other organs as well. The authors generated a Sox9IRES-Cre knock-in line through the ingenious stratagem of inserting the Cre transgene into the 3'-UTR of mouse Sox9; in theory, Cre expression faithfully recapitulates endogenous Sox9 expression while maintaining Sox9 dosage integrity [18]. Notably, such recapitulation of whole-organism expression domains is of particular relevance to Sox9, given that three separate long-range (251 kb 5' to 95 kb 3') upstream and downstream enhancers have been demonstrated to govern distinct organ-specific subsets of the complex spatiotemporal Sox9 expression domain [83]. Analysis of embryos carrying both Sox9IRES-Cre and the Rosa26RlacZ Cre reporter allele [84] revealed that by either E17.0 [18] or postnatal day (P)1 [17], the entire pancreatic (exocrine and endocrine) epithelium comprised descendants of Sox9+ cells as marked by heritable β-gal expression resulting from Sox9IRES-Cre-mediated Rosa26RlacZ recombination (Figure 2A). This genetic lineage tracing study showed Sox9 to mark progenitors not only of the pancreas, but also those of testis, intestine and spinal cord which were all similarly uniformly β-gal-labeled in E17.0 Rosa26RlacZ; Sox9IRES-Cre embryos [18].
Figure 2. Developmental potency of Sox9+ cells becomes progressively restricted during pancreogenesis.
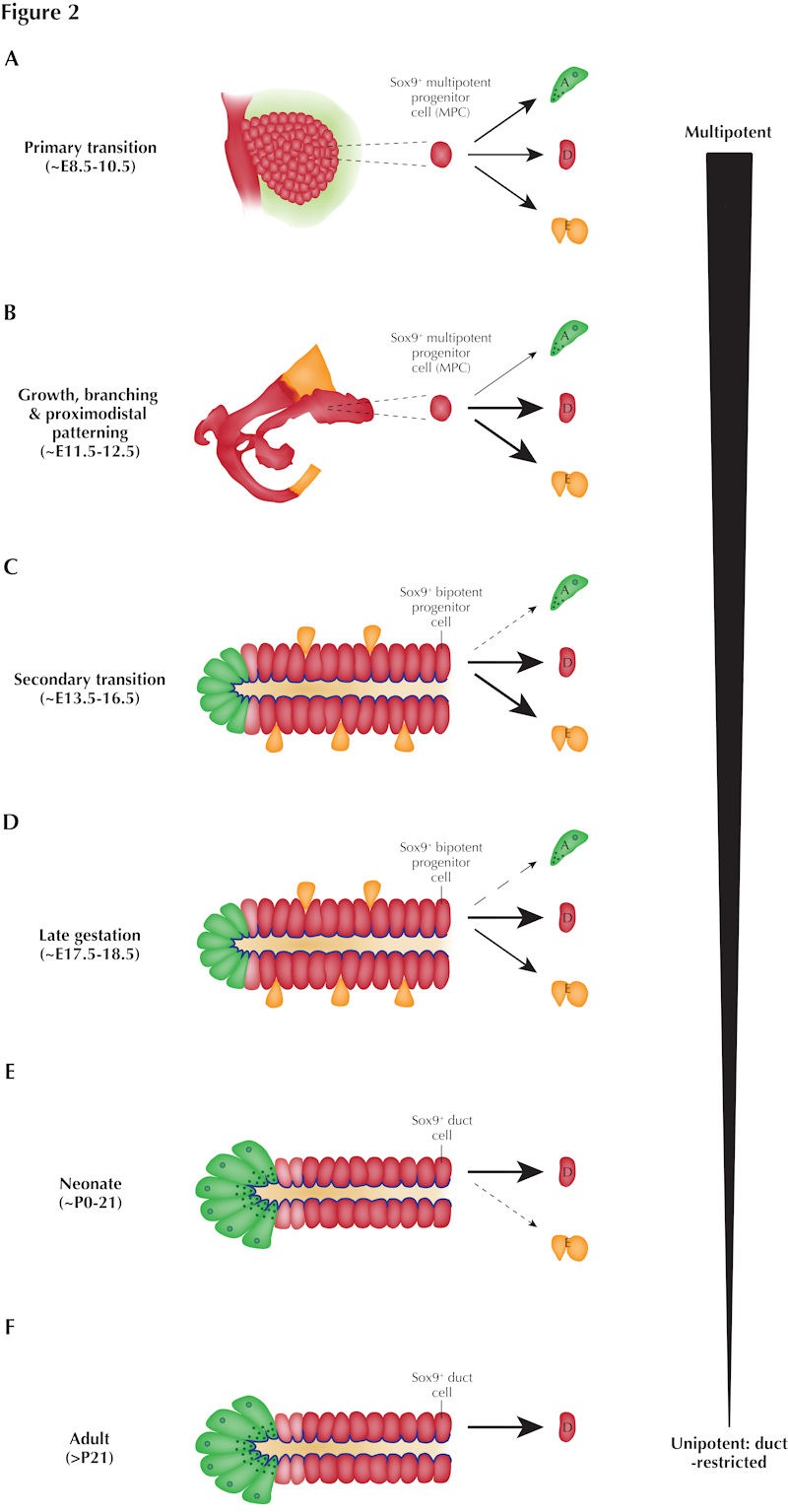
A: Genetic lineage tracing has shown that during the primary transition, Sox9 defines multipotent MPCs (also marked by Pdx1 and Ptf1a) comprising the pancreatic epithelium which give rise to all three pancreatic compartments: exocrine acinar (A), ductal (D), and endocrine (E) cells (via an intermediate Ngn3+ endocrine progenitor cell). B: Concurrently with branching and proximodistal patterning of the pancreatic epithelium between approximately E11.5-12.5, Sox9 continues to mark MPCs with an increased propensity to give rise to ductal and endocrine cells at the expense of acinar cells. C: During the secondary transition, Sox9 expression defines bipotent progenitor cells which almost exclusively yield ductal or endocrine cells; Sox9+ cells at the extreme distal ends of the trunk can, however, yield acinar cells. D: By late gestation, Sox9+ cells predominantly contribute to the ductal compartment; they can, however, still yield endocrine cells and, very rarely, acinar cells. E: During the first three weeks of life, Sox9+ cells almost exclusively yield ductal cells; endocrine neogenesis can, however, still rarely occur from the Sox9+ compartment. F: In contrast, in later adulthood, Sox9+ cells are unipotent, giving rise only to ductal cells. Thickness of arrows denotes relative propensity to yield cells of each of the three pancreatic compartments.
5.2 Sox9 continues to define multipotent progenitors throughout pancreogenesis
Since in Rosa26RlacZ; Sox9IRES-Cre embryos, Sox9+ cells (and their progeny) are labeled from the onset of pancreatic Sox9 expression, this model cannot be used to trace the descendants of Sox9+ cells at later time-points. To circumvent this issue in the absence of purpose-made genetic tools (namely, a Sox9-CreER line), "pseudo short-term" lineage tracing of Sox9+ cells was performed by exploiting the perdurance of Sox9 promoter-driven eGFP over endogenous Sox9 in the Sox9-eGFP BAC transgenic line [85]. As a consequence of the ~26-hour half-life of eGFP [86], retention of eGFP label for 1-2 days past the point of Sox9 promoter inactivity allows descendants of Sox9+ cells to be followed through this window. Consistent with Sox9 marking early pancreatic progenitors, Sox9-eGFP colocalized with Sox9 in almost all cells of the early (E10.5) pancreatic epithelium, including Sox9+ progenitor-derived glucagon+ endocrine cells [21]. Concordantly, it has recently been demonstrated that ex vivo, when diluted to clonal density, isolated Sox9-eGFP+ cells from E11.5 pancreata are capable of undergoing an estimated >8-9 doublings and consequently expand to form clonal epithelial "pancreatic spheres" containing several endocrine cell types, including glucose-responsive insulin-secreting cells [87]. Thus, primary Sox9+ cells from early pancreatic rudiments are capable of self-renewal and multilineage differentiation, hallmarks of multipotent progenitor cells. Later in pancreogenesis, during the secondary transition (E15.5), short-term lineage tracing showed Sox9-eGFP, mirroring Sox9, to be restricted to the epithelial cords/trunk [21]. Strikingly, until E17.5, eGFP was also detected in a proportion of Sox9- Ngn3+ endocrine progenitors, lending further credence to the notion that endocrine progenitors arise from Sox9+ cells. Consistent with both this and the transient ~18-hour existence of Ngn3+ cells [88], eGFP was detected in some insulin+ and glucagon+ cells at E15.5 and birth. Together, these findings imply that endocrine cells arise from Sox9+ progenitors through ~E14-18 (Figure 2C, D). Like endocrine cells, at E15.5, acinar cells abutting the distal tips of the Sox9+ trunk also bore eGFP label, inferring that they also arise from Sox9+ cells (Figure 2C). However, since no eGFP label was evident in acinar cells at birth, this suggests that acinar neogenesis ceases in late embryogenesis (Figure 2D) and proliferation becomes the predominant method of acinar expansion from then onwards. In sum, the results of this "pseudo short-term" lineage tracing study imply that at the population level, Sox9+ cells are multipotent during the secondary transition, capable of giving rise to both endocrine and exocrine cells. Unarguably, colocalization upon which this approach is based cannot substitute fully for genetic lineage tracing, since the identity of labeled and traced or "chased" cells cannot be fully defined and to this end has (justifiably) invoked criticism [31]. Notwithstanding its caveats however, subsequent tamoxifen-inducible genetic lineage tracing of Sox9+ cells during pancreogenesis using BAC transgenic Sox9-CreERT2 mice [19] bore out the main conclusions of this earlier study, validating the efficacy of this approach. In these mice, administration of tamoxifen results in nuclear translocation of Sox9 promoter-driven CreERT2, which remains active and capable of recombination ~12-36 hours following tamoxifen injection [33]. Thus, in mice carrying both Sox9-CreERT2 and Rosa26RlacZ alleles, a single injection of tamoxifen will result in a "pulse" of CreERT2-mediated β-gal labeling of all cells in which Sox9 is active during this 12-36-hour post-injection window such that these cells and their progeny can then be traced or "chased".
Replicating the results of labeling Sox9+ cells from the onset of pancreogenesis [18], administering tamoxifen to Sox9-CreERT2; Rosa26RlacZ embryos at E8.5 resulted in β-gal labeling of all three pancreatic compartments―acinar, ductal, and endocrine―by three weeks of age [19] (Figure 2A). Strikingly, and largely corresponding with the preceding Sox9-eGFP analysis, exposure to tamoxifen between E12.5-18.5 similarly resulted in Sox9+ cells contributing to all three compartments [19], showing that the pancreatic Sox9+ population serves as progenitors, giving rise to ductal, endocrine, and acinar cells, throughout embryogenesis (Figure 2B-D). Quantification of the relative distributions of β-gal-labeled cells between ducts, islets, and acini revealed that with progressively later tamoxifen administration, relative contribution to the ducts increased at the expense of the acini [19]. However, as stressed by Kopp and colleagues [19], since mitotic indices differ between endocrine, ductal, and acinar cells, quantification of such genetic pulse-chase labeling cannot provide a direct index of their relative rates of neogenesis. Notwithstanding this caveat, the decline in acinar labeling with progressively later pulses implies that the competence of Sox9+ progenitors to give rise to acinar cells decreases as pancreogenesis proceeds (Figure 2A-D). This caveat might also conceivably account for the discrepancy in late acinar cell labeling between the pseudo short-term and genetic lineage-tracing studies (Figure 2D): similar rates of acinar neogenesis from Sox9+ cells during late embryogenesis might result in different labeling outcomes in both. While Sox9-eGFP in a small proportion of progenitor-derived acinar cells might be diluted out by subsequent cell division, the extent of heritable Sox9-CreERT2-mediated β-gal labeling in the same proportion of acinar cells might be amplified by rapid proliferative expansion of acini in the immediate postnatal period [65] comprising the three-week chase.
Findings from an additional genetic lineage-tracing study [17], while generally in agreement with Sox9-eGFP- and Sox9-CreERT2-mediated tracing, further emphasize how varying proliferative rates of the pancreatic compartments confound interpretation of pulse-chase labeling data. Using an analogous approach to their earlier generation of Sox9IRES-Cre knock-in mice, Furuyama and colleagues [17] generated a Sox9IRES-CreERT2 knock-in line by inserting CreERT2 into the 3'-UTR of mouse Sox9. Analysis of Sox9IRES-CreERT2; Rosa26RlacZ mice at eight weeks of age revealed that following tamoxifen administration at E16.5, β-gal labeling was observed in ducts and islets, although the largest proportion of all β-gal-labeled cells (73%) were acinar cells [17]. While qualitatively this study validates that of Kopp et al. [19] in showing that in late embryogenesis, Sox9+ cells continue to beget endocrine and acinar cells, the protracted chase period confounds interpretation of labeling patterns. Following tamoxifen administration at E16.5, Sox9-CreERT2-mediated labeling (as a proportion of total labeled cells) of ducts at three weeks of age is just over twice that of acini [19] while tamoxifen exposure at the same stage in embryos carrying Sox9IRES-CreERT2 results in duct labeling being a little over three-fold less than in acinar cells when analyzed at eight weeks of age [17]. Given the irrelevance of tamoxifen dosage on such relative quantifications and recombination of the same Rosa26RlacZ allele being examined, other factors must account for this large discrepancy in the ratio of ductal:acinar labeling between the two studies. The rapid proliferative expansion of the acini in the immediate postnatal period [65] renders it impossible to distinguish relative proportions of acinar cells arising through either: 1) neogenesis from Sox9+ progenitors or 2) proliferation of labeled acinar cells. Hence, the longer chase period in the study by Furuyama and coworkers [17] might conceivably account for a greater degree of "amplification" of initial numbers of β-gal-labeled acinar cells through their proliferation, skewing the labeling ratio towards such cells. While plausible, other factors might equally account for the discrepancy between lineage tracing data generated using the two CreERT2 lines, which will be speculated upon later.
While the findings of these Sox9+ cell lineage tracing analyses differ in their details, their main conclusions are unified in showing that Sox9 expression in the developing pancreas defines a multipotent progenitor population which gives rise to ductal, endocrine, and acinar cells throughout the duration of pancreogenesis (Figure 2A-D). The multipotency of Sox9+ progenitors declines progressively however: while at the population level early MPCs are "tripotent", capable of giving rise to endocrine, acinar and ductal cells (Figure 2A-C), perinatal Sox9+ progenitors are bipotent, giving rise to mostly ductal, and a few endocrine, cells (Figure 2D). By adulthood, Sox9+ cells become "locked" into a ductal identity (Figure 2F). It should be noted that, while such "tripotency" of secondary transition trunk Sox9+ cells holds true at the population level, it most likely does not apply to individual cells. Despite its caveats, short-term Sox9-eGFP tracing yielded insight at single-cell resolution into lineage events immediately preceding examination. Such analysis revealed that Sox9-eGFP-labeled acinar cells arise from only the (relatively sparse) cells at the extreme distal tips of the Sox9+ trunk, while the remaining majority of Sox9+ trunk cells give rise to cells of the ductal tree or endocrine cells (via an Ngn3+ intermediate) [21]. It remains unknown whether this acinar-endocrine differentiation bias is governed by a proximal-distal distribution of fate determinants within the trunk or instead is dictated by relative proximity to the surrounding stroma and associated signaling cues. Comparison of the transcriptional signatures of proximal versus distal Sox9+ trunk cells will help resolve this question. Since the majority of cells marked by Sox9 in the secondary transition pancreas are capable of giving rise either to duct or endocrine cells, at this stage Sox9+ cells are commonly referred to as "bipotent progenitors". Several groups in the field are currently working on deciphering the molecular mechanisms underlying the endocrine-versus-duct fate decision at the level of a single bipotent progenitor cell.
Strikingly, parallel lineage tracing studies using Hnf1β-CreERT2 mice have revealed that mirroring the Sox9+ domain, Hnf1β+ cells exhibit a remarkably similar pattern of diminishing potency during pancreogenesis: Hnf1β marks MPCs during early organogenesis (E11.5-13.5), duct-endocrine bipotent progenitors during the secondary transition (E13.5-16.5), and then duct (including terminal duct or centroacinar) cells from late gestation (E16.5) onwards [31]. This is not unexpected given that Sox9 and Hnf1β expression is almost perfectly colocalized throughout pancreas development and beyond, in the adult organ [19], the possible functional significance of which is worthy of speculation. Intriguingly however, the Sox9+ and Hnf1β+ domains diverge during the secondary transition: while Sox9 expression extends the entire length of the Nkx6.1+ Pdx1+ epithelial trunk to the most distal trunk cells at the tip-trunk interface which are, in addition, marked by Ptf1a, Hnf1β expression is extinguished in these distal-most trunk cells [19] (Figure 1C). Thus, since exclusion of Ptf1a from bipotent progenitors is associated with their loss of acinar competence, and MPCs are marked by expression of Sox9, Nkx6.1, and Ptf1a [55], the retention of Ptf1a in Nkx6.1+ Sox9+ distal trunk cells suggests that they retain multipotency and the ability to give rise to acinar as well as duct and endocrine cells. This notion has been borne out by a recent Ptf1aCreERTM-mediated lineage tracing study, showing that rare multipotent progenitors persist late into the secondary transition [89]. Expression of Sox9 but not Hnf1β in rare distal trunk Ptf1a+ Nkx6.1+ "late MPCs" is consistent with the observation of Sox9-eGFP-labeled acinar cells at the very distal tips of the secondary transition trunk [21]. This finding also explains why lineage tracing reveals Sox9+ cells to give rise to all three pancreatic cell types, including acinar cells, throughout pancreogenesis [19], while Hnf1β+ cells are unable to yield acinar cells beyond E13.5 [31]: Hnf1β+ cells lose their multipotency prior to Sox9+ cells during pancreogenesis. On account of their highly defined tip-trunk junction localization and unique transcriptional signature, it has been proposed [90] that these rare Ptf1a+ Sox9+ Hnf1β- distal tip cells represent the earliest precursors of terminal duct/centroacinar cells which reside at the duct-acinus interface (Figure 1C, D). However, for lack of centroacinar cell-specific markers, this remains to be formally tested through genetic lineage tracing. Partly on account of their retention of a multipotent transcriptional signature (Sox9+ Hnf1β+ Ptf1a+ Hes1+), such centroacinar cells have been proposed to serve as multipotent progenitors in the adult pancreas (reviewed in [90]). Concordantly, isolated centroacinar cells are able to function as progenitors ex vivo, giving rise to self-renewing "pancreatospheres" capable of endocrine and exocrine differentiation [91]. It remains to be seen whether centroacinar cells can similarly function as progenitors in vivo in the adult pancreas.
6. Sox9 expression in adult pancreas
Following the secondary transition, Sox9 expression in embryonic mouse pancreas is progressively down-regulated, becoming restricted to a subpopulation of ductal cells and centroacinar cells in the adult [16] (Figure 1D). Mirroring this, SOX9 is restricted to duct-like cells by 14 wpc in the developing human pancreas and by birth is similarly restricted to only ductal and centroacinar cells [15, 16, 80]. This is particularly poignant since, as mentioned above, it has been widely posited that a subset of pancreatic ductal cells, including centroacinar cells, might function as facultative progenitor cells in adulthood [90, 92]. It is equally intriguing since it shows Sox9, like the other early MPC markers Pdx1 and Ptf1a, to become confined in adulthood to a single compartment. While Sox9 becomes postnatally restricted to ducts, Pdx1 becomes confined to endocrine (β- and δ-) cells [93] and Ptf1a becomes restricted to the acini [94]. It is quite worthy of speculation why these three MPC markers become exclusively confined to distinct pancreatic compartments.
Notably, in contrast to previous reports of Sox9 expression in mouse islets (as detected by ISH or RT-PCR), it was subsequently reported that Sox9, as in embryonic endocrine cells, was not immunodetectable in the adult islet [16] (Figure 1D). In conjunction with the failure to amplify Sox9 mRNA from isolated islets [16], this constitutes clear evidence that neither Sox9 RNA nor protein is expressed in endocrine cells. Leaching of BCIP/NBT used to visualize Sox9 ISH probe from ducts to adjacent islets is likely to have caused an apparent Sox9 mRNA signal in E18.5 islets [66]. Similarly, detection of Sox9 transcripts in adult islets in the same study can be attributed to residual genomic DNA or ductal/centroacinar cell contamination of isolated islet preparations. A factor that further confounds IHC detection of Sox9 in endocrine cells and one that we have experienced first-hand is the variable pancreatic Sox9 staining patterns resulting from IHC using a range of Sox9-specific antisera (personal communication). In addition to the bona fide ductal and centroacinar cell Sox9-specific staining typically observed in adult mouse pancreas, several commonly-used anti-Sox9 antisera yield either α-cell-like cytoplasmic staining or pan-endocrine perinuclear staining patterns. Such non-specific signal is not to be confused with that which might result from sera cross-reactivity with Sox8 and/or Sox10 in the peripheral islet glial sheath outside the islet cells proper. However, both staining patterns are consistent with the notoriously immunogenic nature of pancreatic islets, α-cells in particular [95]. Accordingly, preadsorption of such Sox9 antisera with the immunizing peptide typically abolishes ductal and centroacinar IHC signal while the non-specific islet staining persists (personal communication). Thus, while it is now clearly apparent that Sox9 is not expressed in endocrine cells in the developing or adult pancreas, this notion continues to be perpetuated in the literature.
7. Functional roles of Sox9 in the developing pancreas
7.1 Sox9 maintains pancreatic progenitors through Notch-dependent and -independent mechanisms
While its expression defines pancreatic progenitors, it does not necessarily follow that Sox9 serves a functional role in this population. However, amassing evidence prior to 2007 increasingly hinted at this being likely given the requirement for Sox9 in maintaining progenitors in the neural crest [13], intestinal epithelium [96] and hair bulge [97] during embryogenesis. Since neonatal lethality of Sox9+/- mice [98] precludes breeding Sox9-null embryos, Pdx1-Cre-mediated conditional Sox9 ablation in the pancreas was used to test for a role of this factor in pancreas development. Deletion of Sox9 in Sox9fl/fl; Pdx1-Cre pancreata, while rapid and efficient, only occurred once specification and outgrowth of the pancreatic buds was complete, precluding testing a requirement for Sox9 in these processes [16]. Dramatic hypoplasia of both pancreatic buds became apparent almost immediately after Sox9 deletion due to the loss of Pdx1+ MPCs, revealing a requirement for Sox9 in early pancreatic growth [16]. This strong phenotype indicates an apparent lack of functional redundancy in pancreogenesis between Sox9 and the additional SoxE subgroup members Sox8 and Sox10. This is consistent with the fact that, while members of the same Sox subgroup often play parallel functional roles [10], the pancreatic expression domains of Sox8 and Sox10 do not overlap with that of Sox9 [66]. Moreover, given such a crucial role for Sox9 in maintaining the early pancreatic endoderm, it is tempting to speculate whether Sox9 is required for its initial specification and, if so, how this is achieved. This will require the generation and analysis of mice in which Sox9 is ablated in the gut endoderm preceding the initiation of pancreogenesis.
Consistent with final pancreas size being constrained by the initial number of MPCs specified between ~E8.5-12.5 [44], the pancreas remained hypoplastic at birth in Sox9 mutants, causing perinatally-lethal hyperglycemia due to a near-absence of all endocrine cells. Lineage tracing at birth revealed an over-representation of the progeny of unrecombined (Sox9+) progenitors, suggesting that the mutant pancreas becomes repopulated by unrecombined MPCs. Thus, Sox9-deleted progenitors bear a reduced capacity to contribute to the growing organ. A mechanistic dissection attributed this to a 50% reduction in proliferation, increased apoptosis, and precocious differentiation of Sox9-deleted progenitors. Notably, subsequent examination of Sox9fl/+; Pdx1-Cre embryos revealed a 25% reduction in progenitor proliferation in the Sox9-haploinsufficient pancreas [21]. Thus, MPC proliferation is exquisitely Sox9 dosage-sensitive with loss of each functional Sox9 allele conferring a 25% decrease in mitotic index. Previous reports of similar pancreas hypoplasia in mice lacking the Notch effector Hes1 due to both precocious progenitor differentiation [48] and reduced MPC proliferation [49, 99] prompted an analysis of Hes1 expression in Sox9-deleted pancreata. This revealed an almost two-fold reduction in Hes1+ progenitors at E10.5. Sox9 thus plays a crucial role in driving early pancreatic growth by maintaining MPC pool size through promoting the survival, proliferation, and undifferentiated status of progenitors (Figure 3A). Given that Hes1-deficient pancreatic progenitors exhibit reduced proliferation and precocious differentiation but not decreased survival, it is highly plausible that Sox9 maintains the MPC pool through both Notch-dependent and -independent mechanisms (Figure 3A).
Figure 3. Sox9 serves multiple crucial roles in the developing and adult pancreas.
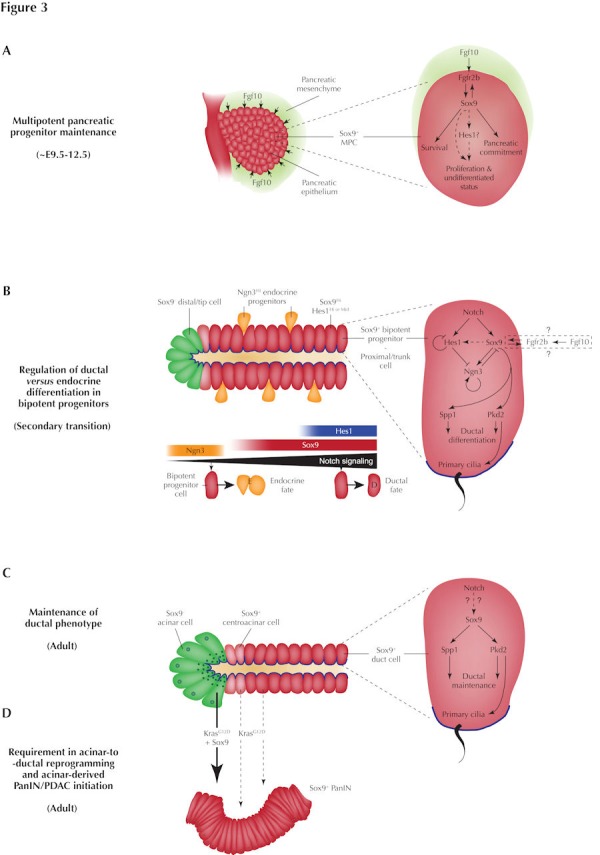
A: Fgf10 from the pancreatic mesenchyme is transduced by Fgfr2b to maintain Sox9 expression in early (E9.5-12.5) MPCs comprising the pancreatic epithelium. Sox9 in turn maintains their Fgfr2b expression. This Sox9/Fgf10/Fgfr2b feed-forward loop maintains both proliferation and pancreatic fate commitment of MPCs. In addition, Sox9 is also required for maintaining both MPC survival and their undifferentiated state. Sox9 acts as a modulator of Hes1 expression in MPCs and possibly regulates MPC proliferation and differentiation status in a Hes1-dependent manner. B: In ductal-/endocrine-fated bipotent progenitors comprising the majority of the proximal trunk in the secondary transition pancreas, graded Notch signaling regulates ductal versus endocrine fate commitment by controlling the cellular equilibrium between Sox9 and Hes1. Notch signaling promotes expression of Sox9 which cell-autonomously activates Ngn3 which positively autoregulates itself, enabling an Ngn3Hi state to be attained. However, high-level Notch activity induces expression of both Sox9 and the Ngn3 repressor Hes1, preventing Ngn3 induction and endocrine differentiation. At intermediate levels of Notch signaling, only Sox9 is induced, initiating Ngn3 expression; endocrine differentiation then requires cell-autonomous repression of Sox9 by Ngn3 to repress the ductal program. Bipotent progenitors remaining Sox9+ Hes1+ adopt a ductal fate by default as Sox9 positively regulates expression of ductal genes including Spp1 and Pkd2. Therefore, Pkd2 and Sox9 are required for maintaining primary cilia. Continued Fgfr2b expression in secondary transition Sox9+ cells raises the possibility of a Sox9/Fgf feed-forward loop functioning in bipotent progenitors as in MPCs. C: Analogously to its role in embryonic bipotent progenitors, Sox9 maintains ductal identity in the adult by maintaining expression of both Spp1 and Pkd2 in mature ductal cells and is therefore also required for maintaining primary cilia on ductal cells. Whether Sox9 expression is positively regulated by Notch activity in adult duct cells as in the embryo remains to be ascertained. D: Genetic lineage tracing following induction of the oncogenic constitutively active KrasG12D allele reveals ductal and centroacinar cells to be relatively refractory to oncogenic transformation into PDAC precursor lesions called PanINs (dashed arrows). In contrast, acinar cells readily form duct-like premalignant lesions following KrasG12D expression; while ectopic induction of Sox9 is required for this reprogramming, coexpression of Sox9 with KrasG12D accelerates PanIn formation (broad, solid arrow).
7.2 Sox9 interacts with the Fgf pathway to reinforce pancreatic fate commitment
Recently, mouse genetic studies provided deeper mechanistic insight into how Sox9 governs MPC proliferation, and in so doing, revealed that in addition to Notch, Sox9 interacts with the Fgf signaling pathway to maintain both expansion and organ identity of early MPCs [100]. Transcriptional profiling of early Sox9fl/fl; Pdx1-Cre pancreata uncovered upregulation of liver markers such as AFP and albumin, borne out by IF studies confirming their widespread expression in Pdx1+ Sox9-deleted MPCs by E11.5 [100]. When deprived of Sox9 up until ~E12.5, pancreas-fated progenitors adopt an alternative hepatic program. MPCs are labile in their organ commitment through this window, requiring Sox9 for reinforcing their pancreatic commitment [100]. Such aberrant hepatic differentiation in the early pancreas mirrors that in fgf10 mutant zebrafish [101]. In such mutants, the hepatopancreatic ductal system (HPD), which connects the ducts of the proximal pancreas and liver to the intestine, is dysmorphic, with some HPD cells aberrantly adopting pancreatic and hepatic fates; similarly, some cells in the proximal pancreas and liver misdifferentiate into hepatic and pancreatic cells, respectively [101].
As mentioned, in mice, Fgf10 from the pancreatic mesenchyme serves a crucial mitogenic role in driving pancreatic progenitor proliferation [53] (Figure 3A). Growth arrest of MPCs therefore results in mice lacking either Fgf10 [53] or its receptor Fgfr2b [102-104] expressed in early pancreatic epithelium [53, 105, 106], resulting in a mirroring of the pancreatic hypoplasia seen in Sox9-deficient mice. Fgfr2b and Sox9 almost perfectly colocalize within MPCs between E9.0-12.5, concurrently with mesenchymal Fgf10 expression and ablation of Sox9 results in an almost immediate extinguishing of Fgfr2b with only unrecombined Sox9+ progenitors retaining its expression [100]. Sox9 therefore cell-autonomously maintains Fgfr2b expression in MPCs and thus their Fgf10 receptivity (Figure 3A). While Pdx1 also regulates Fgfr2b expression in MPCs [107], the synchronous downregulation of Sox9 and Fgfr2b in Pdx1-deficient pancreata suggests that loss of Fgfr2b in Pdx1-/- mice is Sox9-dependent [100]. As predicted by their downregulation of Sox9, MPCs in Pdx1-null embryos also undergo hepatic fate conversion [100], inferring Sox9 to be dominant over Pdx1 in maintaining MPC pancreatic commitment. Together with the finding that SU5402-mediated Fgfr inhibition in E9.5 foregut explants downregulates Sox9 and induces AFP in the pancreas, these data support a model in which Fgfr-transduced Fgf10 signaling maintains Sox9 and so Fgfr2b expression in MPCs [100]. Completing this model, Sox9 and Fgfr2b are synchronously downregulated in MPCs of E9.5 Fgf10-/- embryos [100], which is consistent with Fgf10 maintaining Sox9 expression in progenitors, as seen in Pdx1-Fgf10 pancreata [16, 82]. As predicted by their losing Sox9 and Fgfr2b expression, Fgf10-deficient MPCs also undergo hepatic reprogramming [100]. This study therefore supports the operation of an Fgf10/Fgfr2b/Sox9 feed-forward loop in early pancreatic progenitors that maintains both their proliferation and pancreatic commitment (Figure 3A).
Mesenchymal Fgf10 maintains Sox9 expression in epithelial progenitors, which in turn, maintains their expression of Fgfr2b, and thus, their receptivity to mitogenic, pro-pancreatic Fgf10 signaling. Abrogation of any component of this loop manifests in MPC growth-arrest and pancreatic hypoplasia, as well as loss of pancreatic commitment and the adoption of an alternative hepatic program. This mechanism unifies the pancreatic hypoplasia observed in mice lacking Sox9, Pdx1, Fgf10 or Fgfr2b [16, 45, 46, 53, 103, 104, 108]. Significantly, Sox9 positively regulates Fgfr2 expression in other developmental contexts such as testes [109] and ventral prostate [110]. Whether this regulation is direct remains unknown and will require chromatin immunoprecipitation (ChIP) studies to elucidate. Similarly, while Fgf10-Fgfr2 signaling promotes Sox9 expression in developing lung [111], this study is the first demonstration of the complete Fgf10/Fgfr2b/Sox9 loop in the same context. It is highly likely that such a loop maintains the expansion and/or fate commitment of progenitors in other developmental and regenerative contexts. In the case of early pancreogenesis, further work will be required to mechanistically dissect how the activity of this loop maintains MPC proliferation while repressing the hepatic differentiation program. Such studies will undoubtedly provide insight into downstream effects of Fgf-Sox9 signaling in other tissue progenitors.
Strikingly, recent genetic studies indicate the Sox9-Fgf relationship to be evolutionarily conserved in zebrafish hepatopancreatic patterning, although its pro-pancreatic influence applies only to the pancreas. During zebrafish development, the paralog sox9b is expressed in the hepatopancreatic endoderm and subsequently in developing intrapancreatic and intrahepatic ducts as well as in the HPD connecting them to the intestine [79, 112]. In sox9b mutants, these structures are dysmorphic with ectopic hepatic and pancreatic cells appearing within the HPD, proximal pancreas (hepatic), and liver (pancreatic) [79, 112], phenocopying zebrafish fgf10 mutants [101]. Consistent with this, early expression of sox9b in the hepatopancreatic endoderm is lost in compound mutants lacking both fgf10 and fgf24, although not in single fgf10 mutants [79], which is consistent with fgf10 acting redundantly with fgf24 in specification of the zebrafish pancreas [113]. These findings imply that sox9b functions downstream of Fgf signaling to pattern the zebrafish gut. Intriguingly, in the same study [79], loss of sox9b expression was observed in hepatopancreatic endoderm following morpholino-mediated bmp2a knockdown. Thus, multiple cues undoubtedly govern the induction and/or maintenance of sox9b/Sox9 expression in the gut endoderm.
While there are parallels in Sox9/Fgf-governance of gut endoderm patterning between mouse and fish, it is unclear whether a similar feed-forward loop functions in the latter. Although fgfr2 is expressed in zebrafish anterior embryonic endoderm concurrently with hepatopancreatic patterning [101], its expression has not been examined at single organ resolution. Furthermore, fgfr2 expression has yet to be assayed in sox9b mutants. It thus remains to be seen whether sox9b likewise maintains expression of fgfr2 in zebrafish gut endoderm cells, completing such a regulatory loop.
Finally, given the central role of Sox9 in early MPCs in modulating the Notch effector Hes1 and maintaining receptivity to Fgf10, Sox9 appears to provide a nexus between the Fgf and Notch pathways during early pancreas development (Figure 3A). Several studies have unambiguously shown that persistent Fgf10 expression during pancreogenesis increases proliferation and arrests pancreatic cells in a progenitor state and is associated with their maintained Notch activation, as indicated by maintained Notch1, Notch2 and Hes1 expression [82, 114]. While not demonstrated incontrovertibly, two facts strongly implicate this progenitor arrest occurring within MPC-stage progenitors: Fgf10 expression being Pdx1 promoter-driven (so MPC-expressed) and maintained Notch1/Notch2 co-expression with the Notch ligands Jagged1 and Jagged2 in later ectopic Fgf10-expressing cells as in normal (E12.5) MPCs [82]. In agreement, abrogation of Notch signaling in explanted E10.5 pancreatic epithelium is able to suppress the proliferative and progenitor-maintaining effects of Fgf10 treatment [115]. Together, these studies imply that Notch signaling is an obligate downstream effector of Fgf10 signaling in MPCs. However, exactly how Fgf10 modulates Notch activity in progenitors remains obscure. As Sox9 serves both as a downstream effector of Fgf10 signaling and as an upstream modulator of Hes1 in MPCs, it is conceivable that Sox9 acts as the Fgf10-Hes1/Notch conduit (Figure 3A). This notion is made even more plausible by the recent finding that another pancreatic progenitor maintenance factor, Ptf1a, maintains Hes1 expression in MPCs via the Notch ligand Dll1 [49]. Given the central role that modulation of Fgf and Notch signaling pathways plays in protocols for differentiating hESC/iPS cells into insulin+ cells in vitro [116], establishing whether Sox9 is the point of Fgf10-Hes1/Notch cross-talk is likely to provide insights to aid their optimization. Dissecting this regulatory network should drive the development of protocols aimed at specifically expanding hESC/iPS-derived cells at the pancreatic progenitor stage.
7.3 Sox9 is required in a dosage-dependent manner for endocrine differentiation
At birth, the hypoplastic pancreas in Sox9fl/fl; Pdx1-Cre mice is cystic, comprising dispersed acini, a poorly-branched ductal tree, and almost no endocrine cells [16]. Thus, while Sox9 is required for pancreatic growth, the greater perturbation of endocrine than exocrine morphogenesis reveals that differentiation of endocrine cells is more Sox9 dosage-sensitive than that of exocrine cells. Accordantly, islet-specific defects occur in Sox9fl/+; Pdx1-Cre mice deficient for one functional copy of Sox9 in the pancreas [21]. Size and gross morphology of the pancreas are unperturbed by such pancreatic Sox9 haploinsufficiency [21], in a way consistent with no detectable impact on the morphogenesis of exocrine tissue which comprises 98-99% of the adult mouse pancreas [26]. However, while exocrine morphogenesis is insensitive to a halving of Sox9 gene dosage, the number of endocrine cells (and both α- and β-cell mass) is halved at birth, with all islet cell types being equally affected [21]. As both pro rata β-cell insulin content and expression of mature β-cell markers (such as Glut2, PC1/3 and islet amyloid polypeptide - IAPP) are unperturbed in Sox9fl/+; Pdx1-Cre mice [21], this suggests that terminal differentiation of β-cells is unhampered by Sox9 haploinsufficiency. This endocrine hypoplasia phenocopies that reported by the Hanley group [15] in human patients affected by the lethal SOX9 haploinsufficiency syndrome campomelic dysplasia (CD; OMIM 114290) characterized by severe skeletal dysplasia and variable autosomal XY sex reversal [117, 118]. Notably, this study was the first to hint at a potential functional role for Sox9/SOX9 in pancreatic morphogenesis. Histological examination revealed two of three perinatal pancreata from severe, perinatally-lethal CD cases to display hypoplastic islets with variable hormone expression as well as reduced expression of PC1/3 and IAPP [15]. While this discrepancy in marker expression between β-cells of Sox9-haploinsufficient mouse and human pancreata might reflect minor deviations in their pancreatic programs, it is more likely to be attributable to divergent tissue fixation given the more challenging nature of preserving human autopsy-derived tissues.
Given that in Sox9-haploinsufficient mice endocrine differentiation is unperturbed and islet hypoplasia affects all cell types equally, this suggests that such hypoplasia results from an upstream defect impacting endocrine progenitor specification. Confirming this notion, Ngn3+ endocrine progenitor cell numbers are halved in Sox9fl/+; Pdx1-Cre embryos [21]. In the light of lineage tracing data that support one Ngn3+ progenitor giving rise to one endocrine cell [119], this halving of Ngn3+ cells is entirely consistent with the halving of islet cell numbers. Therefore, islet hypoplasia in Sox9-haploinsufficient mice and, presumably, humans, occurs through impaired endocrine neogenesis, revealing Sox9 to directly govern the initiation of endocrine differentiation. Further supporting this role, while inducible deletion of Sox9 immediately prior to the secondary transition only negligibly impacts pancreatic growth and exocrine morphogenesis, Ngn3+ endocrine progenitors and, therefore, endocrine cells are almost completely ablated [20]. Lineage tracing reveals that Sox9-deleted progenitors exhibit a reduced capacity to give rise to Ngn3+ progenitors and endocrine cells but their probability of yielding acinar or ductal cells is unaffected [20]. Thus, Sox9 is required cell-autonomously for initiating the endocrine program. ChIP studies have provided mechanistic insight into the molecular basis underlying this cell-autonomous role. ChIP performed both in primary embryonic (E15.5) mouse pancreata [21] and the mouse pancreatic duct cell line mPAC L20 [77, 120] has revealed Sox9 occupancy on the Ngn3 promoter. Furthermore, Sox9 has been demonstrated to directly stimulate Ngn3 promoter activity by the finding that it can bind to the promoter of human NEUROG3 and increase its expression [77]. Knockdown of Sox9 expression in mPAC L20 cells in the same study was demonstrated to abrogate expression of Ngn3 induced by adenoviral expression of the bHLH protein Mash1 [77]. Together, these studies show that, while exocrine differentiation is Sox9-independent, Sox9 is absolutely required for initiating endocrinogenesis by inducing Ngn3 expression (Figure 3B) and furthermore, Sox9 regulates this process in a dosage-sensitive, cell-autonomous manner.
7.4 Discrepancies between in vitro- and in vivo-identified progenitor transcriptional networks
Having shown that Sox9 regulates Ngn3 expression in vitro, Lynn et al. [77] sought to understand how Sox9 induces Ngn3 expression in a portion of progenitor cells only. Using mPac L20 duct cells, they attempted to characterize the position of Sox9 in the transcriptional network governing progenitor maintenance versus differentiation. Sox9 was found to bind to the promoters of the pancreatic transcription factors Hnf1β, Onecut1, and Foxa2 [77], all previously implicated in governing Ngn3 expression and coexpressed with Sox9 in MPCs and bipotent progenitors [121]. Furthermore, partial (∼50%) knockdown of Sox9 expression was demonstrated to result in a 20-30% decrease in expression of Onecut1 and Foxa2 and a 50% increase in Hnf1β expression [77].
A parallel approach saw Hnf1β knockdown modestly reduce expression of Foxa2 without affecting that of Onecut1 while knockdown of Foxa2 diminished expression of Sox9 without affecting that of either Hnf1β or Onecut1 [77]. It was concluded from these results that Foxa2 is required for maintaining the expression of Sox9 in a transcriptional network of endocrine differentiation genes including Hnf1β, Onecut1, and Foxa2, which is required for proper pancreatic morphogenesis. However, as this in vitro study is based upon the mPAC L20 duct cell line, it is unclear whether the proposed transcriptional network is conserved in progenitors of the developing pancreas in vivo and, if so, given the coexpression of these factors in MPCs and later bipotent progenitors, to which progenitor population(s) it is applicable. To resolve this issue, Dubois and colleagues [121] assessed the expression of the same endocrine differentiation genes in Sox9-deficient pancreata. Sox9 ablation by E10.5 in Sox9fl/fl; Pdx1-Cre embryos, or at E13 or E15 in Sox9fl/fl; Rosa26-CreER embryos exposed to tamoxifen at E12.5 or E14.5 respectively failed to perturb pancreatic expression of Hnf1β, Onecut1 or Foxa2 [121].
Thus, contrary to in vitro studies, Sox9 is dispensable for the expression of all three factors during pancreatic organogenesis. In contrast however, Pdx1 expression was strongly downregulated following Sox9 ablation [121], uncovering a requirement for Sox9 in maintenance of Pdx1 expression in pancreatic progenitors. As Pdx1 is crucially required for endocrinogenesis and regulates expression of Ngn3 [122], this would argue that Sox9-dependent regulation of Pdx1 expression in secondary transition bipotent progenitors is required for the major wave of β-cell neogenesis from the secondary transition until birth. This is consistent with the loss of β-cells seen following Sox9 deletion at E13 or E15 [121]. While Sox9 therefore induces expression of Ngn3 both directly as well as indirectly through Pdx1, it is arguably loss of the former which predominantly accounts for endocrine hypoplasia in Sox9-haploinsufficient mice as their levels of pancreatic Pdx1 expression remain unperturbed. Further work will be required to resolve whether or not Sox9 regulates Pdx1 expression directly in bipotent progenitors. The fact that Pdx1 expression is maintained in MPCs immediately following Pdx1-Cre-mediated Sox9 ablation [16] and is only downregulated at E12.5 [121] suggests that Sox9 does not directly regulate Pdx1 during early pancreogenesis and/or that Sox9 regulates Pdx1 via distinct mechanisms in MPCs versus bipotent progenitors. Given that in humans mutations in Pdx1 manifest in MODY (maturity-onset diabetes of the young) 4 [123], the maintenance of Pdx1 by Sox9 during the major wave of β-cell neogenesis implicates SOX9 loss-of-function mutations being a putative causative factor of diabetes in humans. While SOX9-haploinsufficiency is associated with lethal CD as mentioned above, mutations affecting the long-range E1 enhancer driving pancreas-specific SOX9 expression [83] might conceivably manifest in human diabetes. Although the majority of MODY gene products function in β-cells, Hnf1β/Tcf2―mutations of which are associated with MODY5 [124]―is excluded from such cells, only being expressed like Sox9 in the MPCs and bipotent progenitors from which they arise [31, 54]. This sets a precedent for dysfunction of pancreatic progenitor genes to manifest in diabetes independently of β-cell dysfunction, presumably as a consequence of endocrine dysgenesis instead. Thus, mutations reducing Sox9/SOX9 dosage might conceivably manifest in MODY, consistent with glucose intolerance in Sox9-haploinsufficient adult mice [121] as mentioned below. Whether SOX9 mutations are associated with human diabetes remains to be seen.
7.5 Notch activity governs ductal-versus-endocrine fate choice through Sox9 and Hes1
While Sox9 is required to induce pro-endocrine Ngn3 expression in secondary transition bipotent progenitors, this contradicts with the Sox9+ population being Notch-active at this time, as evidenced by widespread Sox9/Hes1 colocalization at E15.5 [16] (Figure 3B). Sox9 inducing Ngn3 is discordant with it also positively regulating Hes1 expression [16] since to activate Ngn3, Sox9+ progenitors must escape from the Ngn3-repressing influence of Hes1 [125]. Recently, an attractive model was posited to account for how such "escape" might occur, reconciling these apparently contradictory Sox9 functions [20]. Confirming secondary transition Sox9+ bipotent progenitors to be Notch-active [16], Shih and colleagues [20] showed a subpopulation of E15.5 Sox9+ progenitors to express high levels of the Notch target Hes1 and Notch receptor Notch2 as well as lower levels of Notch1. Concurrently, these cells also expressed NICD1 and NICD2. This was further supported by expression of the Notch ligands Dll1 and Jag1 and the Notch mediator Rbpj in the Sox9+ trunk. Thus, Sox9+ progenitors appear to be Notch-regulated during pancreogenesis as they are in the embryonic liver [112, 126].
In confirmation of previous findings [16, 21, 127], while elevated Ngn3 expression was seen in a small proportion of Sox9+ cells, it was never detected in Hes1+ cells (Figure 3B). Combined with previous reports of NICD1 and Sox9 downregulation in Ngn3+ cells [128], this indicates that Ngn3 induction and endocrine progenitor delamination is associated with diminished Notch signaling [20]. Apparent Notch governance of Sox9+ bipotent progenitors coupled with Hes1 being expressed in only a proportion of them led to the hypothesis that Sox9 and Hes1 are Notch-responsive at distinct thresholds. By culturing pancreatic explants in graded concentrations of the Notch inhibitor γ-secretase inhibitor-IX (GSI-IX), Shih et al. [20] found that at low (1 μM) inhibitor concentrations, expression of Hes1 is reduced while that of Sox9 is unaffected while at higher (5 or 10 μM) concentrations, expression of both factors is abrogated. Thus, Sox9, like Hes1, is a Notch target: it remains to be resolved however whether this regulation is direct as during liver development when NICD1 directly binds the Sox9 promoter [126]. While both are Notch targets, Hes1 and Sox9 respond to Notch at distinct thresholds: both are expressed in NotchHi progenitors, but "intermediate" Notch activity results in Sox9 expression only, accounting for the fact that only a subset of Sox9+ progenitors is Hes1+ (Figure 3B). The resulting prediction that Sox9 should be more sensitive to increased Notch activity was proved by showing that Pdx1-Cre-driven NICD misexpression induced a far greater upregulation of Sox9 than Hes1 in bipotent progenitors [20]. As Notch inhibition induces both Ngn3 expression and endocrine differentiation [128, 129], the authors tested whether this induction is Sox9-dependent. As GSI-IX fails to induce Ngn3 expression in Sox9-deleted pancreatic explants, this suggests that Sox9 is an obligatory inducer of Ngn3 [20]. It would appear therefore that Notch signaling induces both activators and inhibitors of endocrine differentiation, and competition between the two maintains the undifferentiated status quo (Figure 3B).
To better investigate the interaction between Sox9 and Ngn3, Shih and colleagues [20] misexpressed Ngn3 throughout MPCs. The resulting repression of Sox9 in progenitors shows Sox9 and Ngn3 to participate in a cell-autonomous negative-feedback loop (Figure 3B). As Sox9 loss in Notch-inhibited Ngn3-null pancreatic explants excludes Ngn3 repression of Sox9 accounting for Sox9 loss following Notch inhibition, this shows that via Sox9, Notch signaling induces expression of Ngn3, which, at high levels, then represses Sox9. This finding is consistent with the earlier demonstration of persistent Sox9 mRNA [130] and protein [128] in Ngn3-deficient pancreatic cells activating the Ngn3 promoter. Contradicting reports of Ngn3 repressing itself [131], Shih et al. [20] then went on to show that Ngn3+ (eGFP+) cells in the pancreata of Ngn3eGFP/eGFP (functionally Ngn3-nullizygous) embryos display reduced eGFP intensity compared with those in Ngn3eGFP/+ (Ngn3-haploinsufficient) siblings, a result which is conserved following Notch inhibition of explanted pancreata. Taken together, these findings suggest that during pancreogenesis, Ngn3 is positively autoregulated in a Notch-independent fashion so that once Sox9-induced Ngn3 expression is initiated, a positive feedback loop enables an Ngn3Hi state to be achieved, ensuring endocrine commitment (Figure 3B). If this state is not attained, a bipotent progenitor adopts a ductal fate, as illustrated by the persistent expression of bipotent progenitor/ductal genes (such as Sox9, Hes1 and Hnf1β) in eGFP+ cells of Ngn3eGFP/eGFP embryos [20]. While this suggests that Ngn3 represses the ductal program, Sox9 in contradiction is directly required for differentiation and maintenance of pancreatic ducts. Inducible Sox9 deletion immediately prior to the secondary transition manifests in dysgenic ducts with dilated lumina at E15.5 and a cystic phenotype after birth which is mirrored by conditional Sox9 ablation in adulthood [20]. Expression profiling revealed ductal dysgenesis to be associated with downregulation of ductal genes including Spp1 [132], a known Sox9 target [133] and polycystin 2 (Pkd2), mutations of which are associated with autosomal dominant polycystic kidney disease (ADPKD), an inherited systemic disorder characterized by the development of renal, hepatic, and pancreatic cysts [134, 135]. Further supporting a requirement for Sox9 in maintaining ductal Pkd2 expression, primary cilia, which Pkd2 maintains [136], are lost from the apical membrane of duct cells in embryonic and adult pancreas following Sox9 ablation, showing Sox9 to be required for primary cilia formation in the pancreas [20] (Figure 3B).
Finally, Shih et al. examined in more detail the relationship between Notch and Sox9 in regulating the ductal program [20]. Not normally expressed in endocrine progenitors, Ngn3-Cre-driven expression of NICD in such cells induced expression of Sox9 and Hes1 as well as ductal markers such as Spp1 and Pkd2 and, as shown [137, 138], perturbed endocrine differentiation [20]. On a Sox9-deficient background, Hes1 expression was maintained in NICD-expressing Ngn3+ cells, confirming that Sox9 serves as a modulator of Hes1 expression [20]. Mirroring Ngn3+ cells expressing both NICD and Sox9, endocrine differentiation was still abrogated in Sox9-deleted mice [20]. This failure of high Notch activity to rescue endocrinogenesis in the absence of Sox9 is consistent with an absolute requirement for Sox9 in this process. Likewise, in the Sox9-deficient state, NICD misexpression was insufficient to induce a ductal program in bipotent progenitors, showing Sox9 to be an obligatory effector of Notch-induced ductal differentiation [20].
Together, the findings of this intricate study by Shih and colleagues point to the fate of secondary transition bipotent duct/endocrine progenitors being governed by a Notch-dependent molecular network [20] (Figure 3B). According to their model, given that Hes1 expression in bipotent progenitors represses expression of Ngn3 and Sox9 induces it, intermediate levels of Notch activity favor Sox9 over Hes1 expression, promoting Ngn3 induction. Positive autoregulation of Ngn3 ensures that once induced, progenitors are able to rapidly attain an Ngn3Hi state, committing them to the endocrine program. Ngn3 then cell-autonomously represses Sox9, and thus, ductal development, enabling endocrine differentiation to occur. Thus, different gradations of Notch activity are able to specify distinct endocrine or ductal fates within the same bipotent progenitor population.
Studies in zebrafish point towards this Notch-Sox9 interaction being conserved in mid-pancreogenesis. Treatment of late zebrafish embryos with the γ-secretase inhibitor DAPT between three and four days post-fertilization results in both loss of sox9b expression and excess endocrine differentiation in the intrapancreatic ducts [79], showing Notch activity to be required for maintaining ductal sox9b expression as it is in the mouse. Notably, this effect is recapitulated in the extrapancreatic duct by treatment with the Fgf receptor inhibitor SU5402 between two and three days post-fertilization [79]. Thus, Fgf, like Notch signaling, is required to maintain ductal sox9b expression in zebrafish. The conservation of relationships between Fgf, Notch and Sox9 during mouse and zebrafish pancreogenesis invites speculation as to whether a Sox9-Fgf loop is active in bipotent progenitors in the same way as it is in MPCs (Figure 3B). In support of this contention, as well as being expressed in MPCs, Fgfr2b expression persists in Sox9+ bipotent trunk progenitors during the secondary transition [100], revealing that this population remains Fgf10-receptive. While Fgf10 is downregulated in mouse pancreatic mesenchyme after E12.5 [53], it is purportedly weakly expressed in the ductal tree of the secondary transition pancreas [114, 139]. Fgf10 is also detectable in the pancreas by RT-PCR throughout embryogenesis [140].
Additional studies will be required to ascertain whether physiological levels of Fgf10 are expressed in the secondary transition pancreas and if so, whether Fgf10 interacts with Fgfr2b and Sox9 to maintain trunk progenitors during this window as it does MPCs in earlier development. If so, this immediately invokes the question of how Notch and Fgf cues interact to govern the adoption of ductal versus endocrine fates in bipotent progenitor cells. As mentioned above, on account of the integral role of Fgf and Notch modulation in hESC/iPS cell to insulin+ cell differentiation protocols [116], the molecular dissection of such putative Fgf-Notch cross-talk is likely to prove instrumental in guiding the optimization of such protocols.
8. Roles of Sox9 in the postnatal pancreas
As mentioned above, by birth, expression of Sox9 in both mouse and human pancreas is confined to ductal and centroacinar cells [15, 16, 80] (Figure 1D). While Sox9 expression is required in adult as well as in developing ductal cells to maintain ductal integrity and primary cilia formation [20] (Figure 3C), this requirement - unlike that for endocrine differentiation - does not appear to be dosage-sensitive, as a halved complement of pancreatic Sox9 gene dosage does not manifest in detectable ductal dysmorphogenesis [121]. In fact, despite two-fold reduced islet size and β-cell mass at birth [21], while Sox9-haploinsufficient (Sox9fl/+; Pdx1-Cre) mice are glucose-intolerant, this fails to progress to overt diabetes, most likely as a consequence of a compensatory increase in β-cell proliferation, and hence, mass, in the early postnatal period [121]. Superficially, this would seem to imply that homeostasis of the adult pancreas is relatively insensitive to changes in Sox9 dosage. However, genetic lineage tracing studies have revealed that this is not necessarily the case, as will be expanded upon below.
Given the role of Sox9 as a maintenance factor for progenitors in the developing pancreas [16] and the commonly held belief that in adulthood facultative progenitors reside in the pancreatic ducts [92], an additional role for Sox9 in tissue homeostasis in the healthy and/or injured pancreas was heavily anticipated. Several studies over the last few years have begun to offer insight into the functions of this factor in a number of pathological states including neonatal growth, pancreatitis-induced regeneration, and pancreatic cancer.
8.1 Sox9+ cells retain some bipotency in the immediate postnatal period
Given that Sox9+ cells retain the potential to give rise to duct, acinar, and endocrine cells as late as E18.5 in embryogenesis [19], genetic lineage tracing was conducted neonatally to ascertain whether they retain such multipotency after birth. While tamoxifen administration to five-day-old, Sox9-CreERT2-expressing mice did not result in any acinar labeling, scarce labeling was detected in small endocrine cell clusters, with non-β-cells labeling more frequently than insulin+ cells [19]. While inappropriate transgene activation cannot be fully excluded, such labeling implies that endocrine neogenesis continues, albeit at a negligible rate, from the Sox9+ domain in the immediate postnatal period (Figure 2E). However, despite intensive regimens of tamoxifen administration at three weeks of age, no evidence was obtained in this analysis [19] to support endocrine (or acinar) neogenesis from Sox9+ cells beyond the immediate postnatal stage, implying that Sox9+ cells become progressively "locked in" to a ductal fate beyond birth (Figure 2F).
8.2 Neogenic potential of Sox9+ cells in the mature pancreas is contentious
In stark contrast to the mature pancreatic Sox9+ cell pulse-chase findings of Kopp and coworkers [19], tamoxifen-induced marking of Sox9+ duct and centroacinar cells in two-month-old Sox9IRES-CreERT2; Rosa26RlacZ mice surprisingly resulted in progressively increased labeling of the acinar compartment over the following months such that almost all exocrine (duct, centroacinar, and acinar), but no endocrine, cells were labeled after a 12-month chase [17]. This pronounced acinar labeling was recapitulated following a tamoxifen pulse to E16.5 Sox9IRES-CreERT2; Rosa26RlacZ embryos [17] but was not evident in Sox9-CreERT2; Rosa26RlacZ mice pulsed at the same stage [19]. This dramatic, postnatal acinar repopulation by descendants of labeled Sox9+ ductal and/or centroacinar cells is consistent with continuous acinar replenishment by Sox9+ progenitors during normal homeostasis. In attempting to resolve these conflicting studies, it is conceivable that this discordance with Sox9-CreERT2-mediated tracing stems from an acinus-supplying centroacinar sub-population being differentially-labeled by the two different CreERT2 lines. A side-by-side survey of the exact adult ductal sub-populations targeted by the two lines will be required to test this hypothesis.
A second, alternative explanation for these discordant findings is, however, equally plausible. While Sox9-CreERT2 mice were derived through BAC transgenesis, the Sox9IRES-CreERT2 line was generated by knock-in of IRES-CreERT2 into the Sox9 3'-UTR. Such regions modulate eukaryotic gene expression by governing mRNA translation, stability, and subcellular localization [141] and, in accordance with such crucial roles, comprise some of the most conserved elements within the mammalian genome [142]. As 3'-UTR-mediated post-transcriptional regulation is necessary for the appropriate spatial and temporal expression of the mRNA-encoded protein [143], knock-in into the 3'-UTR is sometimes known to disrupt expression levels of the targeted gene, often in quite unpredictable fashion. In illustration, knock-in of lacZ into the 3'-UTR of mouse Vegf (encoding vascular endothelial growth factor A) has been reported to result in either increased [144] or decreased [145] Vegf expression levels, contingent upon the exact insertion site. Given the exquisite Sox9 dosage-sensitivity of pancreatic morphogenesis, it is plausible that even slight perturbations in Sox9 expression level caused by disruption of the Sox9 3'-UTR might impact Sox9+ cell behavior. Indeed, lending credence to this notion, deletion of a recently-discovered enhancer located 70 kb upstream of mouse Sox9, produces an 18-37% decrease in Sox9 expression levels in a number of organs including pancreas, phenocopying (albeit to a lesser degree) islet hypoplasia resulting from Sox9-haploinsufficiency [146]. This complex transcriptional regulation of Sox9 is further compounded by its positive autoregulation as implied by a previous study [77]: Sox9 dimers bind multiple recognition sites within the enhancer region and are both necessary and sufficient for its activity [146]. Thus, together, even relatively minor reductions in Sox9 expression levels are sufficient to affect the neogenic potential of Sox9+ pancreatic cells.
Confirming the notion that IRES-CreERT2 insertion into its 3'-UTR impacts Sox9 dosage, while the level of endogenous pancreatic (and hepatic) Sox9 expression is unaffected in Sox9IRES-CreERT2 neonates, it is decreased in adult mice [23], implying divergent transcriptional mechanisms regulating Sox9 expression in development versus adulthood. Such postnatal Sox9 hypomorphism is likely to affect the neogenic competence of Sox9+ ductal (including centroacinar) cells, rendering them more plastic [23] and able to give rise to acinar cells which repopulate the acinar compartment through self-duplication [147]. This hypothesis might be easily tested by pulse-chasing Sox9+ cells in adult Sox9-CreERT2; Rosa26RlacZ mice on Sox9flox/+ (Sox9-haploinsufficient) versus Sox9+/+ backgrounds. While reduced Sox9 expression in adult pancreatic cells might conceivably result in them yielding acinar cells, how such labeled acinar cells populate the entire acinar compartment is a matter of contention. This phenomenon might result from labeled acinar cells having some selective advantage and "outcompeting" unlabeled cells in the same acinus. However, it may also result from previously unappreciated mechanisms of cell renewal in pancreas homeostasis such that acinar cells juxtaposing terminal duct/centroacinar cells are predisposed to repopulate the entire acinus at intervals, consistent with individual pancreatic acini being clonal [148]. While unintentional, this tracing of Sox9-hypomorphic adult duct cells [17] has deftly revealed that Sox9 dosage regulates neogenic potential of Sox9+ cells in adult as well as embryonic pancreas. It is clear that further work will be required to determine how exactly Sox9 dosage modulates adult cell plasticity and how this finding might be exploited for therapeutic gain.
8.3 Pancreatic injury invokes incomplete endocrine differentiation of Sox9+ cells
Despite their discrepancies, the aforementioned pulse-chase studies [17, 19] are unanimous in presenting no evidence for endocrine neogenesis from Sox9+ cells in the mature pancreas during normal tissue homeostasis. However, injury to the adult organ might invoke the necessary stimuli required to induce endocrine differentiation from a putative facultative adult ductal progenitor [92]. Over the last decade, the study of pancreatic regeneration initiated in the nineteenth century has undergone a major revival, which is in large part due to the advent of modern molecular techniques and suitable transgenic tools. Historically, a range of insults has been exploited experimentally to induce pancreatic regeneration including partial pancreatectomy [149, 150], cerulein-induced pancreatitis [3, 151] and β-cell ablation - either pharmacologically using streptozotocin (STZ) or alloxan [152] or genetically in conjunction with diphtheria toxin [153, 154]. However, the partial duct ligation (PDL) model [1, 2] has garnered most attention in recent years, largely owing to the publication of a high-profile study by the Heimberg group demonstrating that PDL stimulates Ngn3-dependent β-cell neogenesis from adult pancreatic ducts [155].
Following ligation of the duct leading to the "tail" (the splenic portion) of the pancreas, pancreatitis leads to rapid acinar apoptosis in the tail while the "head" (the duodenal portion) of the pancreas remains unaffected. β-cell mass in the tail was reported to more than double one week post-PDL, concomitantly with strong, tail-specific upregulation of Ngn3 expression [155]. Abrogation of this Ngn3 induction via lentiviral knockdown significantly diminished the PDL-induced increase in β-cell mass showing at least a portion of this increase to be Ngn3-dependent. Following PDL in mice expressing Ngn3 promoter-driven lacZ, β-gal+ cells were detected specifically in the ligated tail, many residing within ducts. Akin to Sox9-eGFP pseudo short-term tracing, β-gal perdurance [156] allowed Ngn3+ cells to be traced and, consistent with an endocrine progenitor identity, a third of β-gal+ cells expressed islet hormones one week post-PDL. As well as being ultrastructurally similar to embryonic Ngn3+ endocrine progenitors, when PDL-induced adult Ngn3+ cells were grafted into Ngn3-null embryonic pancreas explants, they could be induced to differentiate into endocrine cells, including glucose-responsive β-cells. Together, these observations infer activation of β-cell neogenesis from Ngn3+ endocrine progenitors induced in ducts by PDL-mediated adult pancreatic injury.
This contention is one of the most controversial topics in current diabetes biology. Several groups have argued that, while PDL does induce ductal Ngn3 expression, absolute β-cell number in the ligated tail remains unchanged, although β-cell mass (i.e. calculated morphometrically as proportional area of insulin-immuno-positive pancreas tissue x pancreatic wet weight) increased as an artifact of the ~70% decrease in total pancreatic tail area seen one week after PDL due to extensive acinar apoptosis [19, 32, 157]. While pancreatic edema, fatty degeneration, and fibrosis resulting from ligation-induced pancreatitis contribute to wet weight, such tissue regions will be excluded from morphometric area measurements, artificially inflating calculated β-cell mass in ligated pancreata [19]. However, in conflict with this and in validation of their initial findings, Heimberg and colleagues recently demonstrated a post-PDL doubling of total β-cell volume and islet number in the tail, the latter being especially attributable to an increase in small β-cell clusters [158].
Furthermore, pulse-chase labeling of Ngn3+ cells in Ngn3CreERT; Rosa26RYFP mice following PDL revealed YFP label in duct-associated insulin+ cell clusters as well as islets, which is consistent with induction of ductal Ngn3+ endocrine progenitors, β-cell neogenesis, and their contribution to existing islets [158]. Interpretation of Ngn3+ cell lineage tracing is, however, confounded by persistent low-level endogenous Ngn3 expression in mature adult endocrine cells, including those in duct-ligated pancreata [19, 159]. Thus, genetic lineage tracing of β-cells from pancreatic ducts post-PDL would constitute the most incontrovertible evidence to support β-cell neogenesis in this injury model. To this end, pulse-chase duct cell labeling has been undertaken following PDL in several transgenic lines in which CreER expression is duct-specific in the adult pancreas, including CAII (carbonic anhydrase II)-CreERTM [160], Hnf1β-CreERT2 [31] and Hes1CreERT2 [32] mice. The outcomes of these studies are, however, conflicting as only the first study shows labeling in insulin+ cells following ligation [160]. While such β-cell labeling is consistent with β-cell neogenesis from ducts following PDL, it might conceivably also arise from non-ductal CreER expression, given the low-frequency endocrine and acinar labeling seen in control animals [160]. Equally, the lack of contribution of labeled cells to islets following PDL in Hnf1β-CreERT2 or Hes1CreERT2 mice might reflect heterogeneity in labeling ductal subpopulations so that a rare putative adult progenitor population expresses neither transgene. Unlike Hnf1β-CreERT2 [31], CK(cytokeratin)19CreERT [161] and Muc1IRES-CreERT2 [162], Sox9-CreERT2 labels a ductal population during the immediate postnatal period which gives rise to endocrine cells [19]. Thus, it is plausible that this line is more likely to target a putative adult progenitor population than other ductally expressed CreERs.
To test whether β-cell neogenesis is induced from Sox9+ cells following ligation injury, Kopp et al. performed PDL or sham surgery on seven-week-old Sox9-CreERT2; Rosa26RYFP mice four weeks after they were pulse-labeled via a high-dose tamoxifen regimen, resulting in two-thirds of Sox9+ cells being YFP-labeled [19]. Despite a 40-fold upregulation of Ngn3 expression in the ligated tail, which was consistent with the detection of Ngn3+ hormone- cells within Sox9+ lineage-labeled ducts of ligated pancreata, labeling was seen in virtually no (fewer than 1 in 1,000) insulin+ cells at either one or eight weeks post-PDL [19]. Accordingly, no increase in duct-adjacent small β-cell clusters was evident seven days after surgery [19]. Mirroring these findings, PDL of Sox9IRES-CreERT2; Rosa26RlacZ mice pulsed with a high-dose tamoxifen regimen at 4 or 8 weeks of age similarly failed to elicit any appreciable β-cell labeling [17]. Independent of the possible failure of both Sox9-CreERT2 and Sox9IRES-CreERT to mark a scarce Sox9+ progenitor population, it appears that PDL-induced injury does not induce neogenesis of bona fide β-cells from the Sox9+ duct compartment. Indeed, given the replication of this outcome in transgenic lines, in which CreER is expressed in other ductal subpopulations [31, 32], it is becoming improbable that neogenesis of mature insulin+ cells from ducts occurs to any appreciable extent in the PDL model. This, however, does not preclude PDL injury stimulating at least a partial program of endocrine differentiation in injured adult duct cells, as suggested by activation of ductal Ngn3 expression post-PDL. Presumably, however, the duct-ligated adult pancreas lacks the vital cues required for the continued differentiation of endocrine progenitors into hormone+ endocrine cells. This is consistent with Ngn3+ cells in ligated adult pancreata being able to give rise to insulin+ cells in embryonic pancreas explants [155]. Contingent upon feasibility, identifying the molecular cues required for terminal β-cell differentiation from ductal Ngn3+ cells in the PDL model will facilitate the development of novel therapies for stimulating β-cell regeneration in situ in the diabetic pancreas.
Notably, confirming the inability of Sox9+ cells to yield β-cells following PDL-induced injury, alternative pancreatic injury models including partial pancreatectomy, cerulein-induced pancreatitis or STZ-induced β-cell ablation all similarly failed to label β-cells in Sox9IRES-CreERT2; Rosa26RlacZ mice [17]. Together, these observations unanimously argue that Sox9+ cells in the adult mouse pancreas do not serve as facultative β-cell progenitors following injury. In apparent contrast, regeneration of β-cells following their specific ablation at the end of embryogenesis, is compromised in sox9b mutant zebrafish larvae [79]. While this result would ostensibly implicate a requirement for sox9b in β-cell regeneration in zebrafish, it is important to consider the challenges encountered in attempting to identify comparable stages in the mouse and zebrafish pancreatic programs. Thus, while conceivable that β-cell regeneration occurs via distinct mechanisms in the mature rodent versus zebrafish pancreas, it is more plausible that β-cell neogenesis during the early post-embryogenesis stage in zebrafish corresponds with the mouse secondary transition. Thus, in zebrafish, sox9b appears to be required for pancreatic ductal cells to generate secondary transition endocrine cells. Given that zebrafish sox9b mutants are capable of surviving to adulthood [79, 112], it is tempting to speculate whether β-cell regeneration following ablation will be similarly compromised in mature adult sox9b mutants.
8.4 Adult pancreatic Sox9+ cells can be induced to form insulin+ cells ex vivo
Whereas Sox9+ cells in the intact adult mouse pancreas can only be stimulated to undergo partial differentiation toward an endocrine fate, they can seemingly be induced to form endocrine cells when cultured ex vivo. Where in vivo pulse-chase studies have failed, a recent ex vivo hematopoietic colony assay study [163] identified a putative adult pancreatic progenitor cell expressing Sox9-eGFP [85] as well as the ductal marker CD133 [164]. Constituting ~1% of total adult pancreatic cells, such so-called "pancreatic colony-forming units" (PCFUs) form ductal-like colonies in Matrigel-containing semisolid culture media which are capable of >100,000-fold expansion following treatment with the Wnt agonist R-spondin 1 [163]. Moreover, when transferred to a Matrigel-free laminin-based semisolid medium, some cells give rise to endocrine/acinar colonies containing glucose-responsive β-cell-like cells [163]. This result apparently confirms the notion that mature pancreatic Sox9+ cells can be induced to form β-cells when provided with the requisite signaling milieu. It is also particularly striking when it is considered that the same BAC clone was used to generate both Sox9-eGFP and Sox9-CreERT2 mice [19, 85]. Thus, the same Sox9+ pancreatic population should theoretically be targeted in studies using either mouse, notwithstanding integration site-specific effects. Given the relatively young age (2-4 months) of the mice from which such PCFUs are isolated, only a few weeks after Sox9+ cells remain duct/endocrine-bipotent [19], it will be interesting to examine whether equally multipotent PCFUs can be isolated from more mature animals. Furthermore, it is interesting to speculate whether the dissociation process itself elicits a regenerative stimulus provoking a facultative progenitor response to injury. Since the Sox9-eGFP+ CD133+ ductal population comprises between 2-6% of total pancreatic cells while only ~1% of such cells exhibit colony-forming ability, further efforts should be expended on characterizing the adult ductal population to gain insight into this heterogeneity. Undoubtedly, this ex vivo study will redouble current efforts aimed at identifying adult pancreatic progenitor cells corresponding to PCFUs in situ.
8.5 Sox9+ cells in adult liver are competent to initiate a pancreatic program
Intriguingly, while in vivo, Sox9+ duct cells in mature adult pancreas apparently cannot be coaxed into giving rise to insulin+ cells, those in the liver can. In a recent study by Slack and colleagues STZ-induced diabetes in NOD-SCID mice was rescued by adenoviral administration of a transcription factor cocktail comprising Pdx1, Ngn3, and the β-cell maturation factor MafA [165, 166]. This was associated with a 25-fold increase in hepatic insulin content consistent with the presence of glucose-responsive insulin-immunopositive CK19+ ectopic duct-like structures in the livers of infected mice. While insulin+, such cells appear to be polyhormonal, additionally expressing glucagon and somatostatin, consistent with the expression of the pan-endocrine markers Isl1 and Rfx6 [165, 167, 168]. Using Sox9-CreERT2-mediated lineage tracing, such insulin+ cells were demonstrated to arise from Sox9+ hepatic cells, proposed by the authors to constitute:
1. Small bile ducts,
2. Hepatoblast-like progenitors assumed to reside in the adjoining periportal zone, or
3. Peribiliary glands associated with larger bile ducts.
Supporting the second hypothesis, the Chan group convincingly showed that helper-dependent adenoviral delivery of Ngn3 and the growth factor betacellulin in STZ-treated diabetic mice induced the appearance of periportal "neo-islets" comprising the four principal endocrine cell types, including functional insulin-immunopositive β-cell-like cells capable of restoring euglycemia in infected mice [169].
Several strands of evidence including marker expression, location, morphology, and lineage tracing supported the cell of origin being hepatic oval cells, bipotent hepatic progenitors able to give rise to hepatocytes or biliary cells [170]. Thus, it is plausible that in both studies, insulin+ cells in the adult liver arise from the same progenitor population in which Sox9 expression affords endocrine competence. Supporting this contention, in both healthy adult mice and those subjected to an oval cell-inducing injury (treatment with the chemical DCC), Sox9 marks a clonal liver progenitor population residing within biliary ducts that is capable of in vitro bilineage (hepatocyte and biliary duct cell) colony formation [171]. This differentiation potential of Sox9+ cells in adult liver is conserved in vivo: genetic pulse-chase labeling using either Sox9IRES-CreERT2 [17] or Sox9-CreERT2 [171] mice reveals that Sox9+ cells contribute to both hepatocyte and bile duct compartments in the uninjured adult liver during normal tissue homeostasis. Hepatic injury through treatment with either DCC [171] or a methionine- and choline-deficient/ethionine-supplemented diet [17] to induce an oval cell response, dramatically augmented the contribution to both lineages from Sox9+ progenitors, as did acute liver insult through carbon tetrachloride administration or bile duct ligation [17].
These studies thus show that Sox9 marks a bipotential adult hepatic progenitor which gives rise to both hepatocytes and biliary duct cells even in the absence of a regenerative stimulus. Therefore, while Sox9+ ductal cells in the adult pancreas do not appear to serve an obvious progenitor role, at least during normal organ homeostasis, they evidently do so in the mature liver. The ability to reprogram adult hepatic Sox9+ progenitors into insulin+ cells when provided with the appropriate cues suggests that transcriptional programs are at least partially conserved between Sox9+ progenitors in adult liver and those in the embryonic pancreas. By extension, it will be fascinating to test whether their expression of Sox9 provides hepatic progenitors the competence to give rise to insulin+ cells. This might be examined by testing whether the adenoviruses mentioned above are still able to induce hepatic insulin expression following conditional hepatic Sox9 ablation.
8.6 Sox9 is a key player in the initiation of pancreatic ductal adenocarcinoma
In light of many tumors originating from adult organ stem cells [172], it was almost inevitable that a potential role for Sox9 in pancreatic cancer pathogenesis would be examined. Recent studies have revealed that Sox9 does indeed serve a crucial role in the initiation of pancreatic ductal adenocarcinoma (PDAC). In the US, PDAC is the fourth leading cause of cancer-associated mortality, with a five-year survival rate of less than 5%, which is in large part due to the advanced and/or metastatic state of neoplastic lesions at the time of diagnosis [173, 174]. The development of both early detection assays and novel therapies will require an understanding of the mechanisms underpinning PDAC initiation. PDAC is a malignant neoplasm which arises from normal pancreatic tissue through a series of histologically well-defined preinvasive ductal lesions (termed pancreatic intraepithelial neoplasias or PanINs) to invasive carcinoma [175, 176]. PDAC is widely regarded as a "genetic disease" with genetic mutations accumulating in lesions during PDAC progression, the first of these being activating mutations of KRAS [177, 178]. Seminal studies in the field of pancreatic cancer biology revealed that expression of constitutively active KrasG12D in mice induces PDAC several months after PanIN induction [179], roughly at the time when acinar-to-ductal metaplasia (ADM) occurs [180, 181]. Despite PDAC progression being histologically well-characterized, the cellular origin of PDAC has remained unproven: tumor histology can quite often be deceptive in terms of ascertaining tumor origin. While centroacinar cells have been widely implicated by several studies [182, 183], the lack of transgenic lines expressing CreER exclusively in centroacinar and ductal cells until recently has precluded testing this hypothesis. Activation of oncogenic Kras in cells of the larger ducts has revealed a low propensity for PanIN induction from ductal cells [184, 185], while its activation in acinar cells has been shown to convert them into duct-like cells and PanINs [180, 186-189], implicating ADM as a putative cause of PanIN formation.
In an effort to dissect the mechanisms inducing PanIN and PDAC formation, Kopp and colleagues demonstrated that activation of oncogenic KrasG12D in acinar cells using the Ptf1aCreERTM line readily induces PanINs while Sox9-CreERT2-driven KrasG12D activation in ductal/centroacinar cells does not (Figure 3D) [22, 89]. Consistent with this, acute cerulein-induced pancreatitis, previously shown to exacerbate KrasG12D-mediated PanIN and PDAC formation [180, 188, 190, 191], potentiated KrasG12D-mediated PanIN formation from acinar, but not ductal, cells [22]. Thus, the metaplasia of acinar to ductal cells associated with PanIN induction has served as a red herring, hampering the identification of the acinar cell as the true cell of origin of pancreatic cancer. Kopp et al. went on to show that aberrant induction of Sox9 expression in KrasG12D-expressing acinar cells precedes ADM and PanIN initiation [22]. Another recent study suggests that this event is conserved in human PDAC etiology: Sox9 and a second pancreatic ductal factor, Onecut1 [59, 60], are ectopically expressed in metaplastic acinar cells in close proximity to PDAC lesions in sections of resected human pancreas [192]. If this Sox9 induction is prevented in mice by simultaneous acinar-specific KrasG12D expression and Sox9 deletion, PanIN formation is blocked, revealing Sox9 induction in Kras-active acinar cells to be necessary for PanIN initiation [22]. In support of this result, Prévot et al. showed that ADM induced by PDL-mediated pancreatitis is blunted by acinar-specific Sox9 deletion, revealing Sox9 to be required in acinar cells for their metaplasia [192]. The converse is also true in that forced expression of Sox9 in KrasG12D-expressing acinar cells accelerates both ADM and PanIN formation [22] (Figure 3D). The expression of ductal markers in Sox9-expressing acinar cells in the absence of oncogenic Kras suggests that this is associated with a "destabilization" of acinar identity, increasing the potential for ADM given the appropriate inflammatory cues [22, 192]. Consistent with this, forced acinar Sox9 expression, mirroring oncogenic Kras, promotes ADM following cerulein-induced pancreatitis [22].
Finally, Kopp and coworkers showed that, while acinar-specific Sox9 ablation is able to block KrasG12D-mediated ADM and PanIN initiation, it is insufficient to prevent ADM following cerulein-induced pancreatitis [22], which suggested that Sox9 is dispensable for ADM in this pancreatic injury model. While this is the case, Sox9 expression is obligatory for the further progression of ADM into PanINs since cerulein-treated mice subjected to acinar-specific KrasG12D expression and Sox9 deletion develop virtually no PanINs [22]. Together, these studies show that Sox9 is crucially required for the conversion of acinar cells into PanINs by conferring them with ductal characteristics [22, 192], which is consistent with the aforementioned role of Sox9 in regulating duct-specific genes [20, 79]. Kopp and colleagues showed that, while mature acinar cells are readily PanIN-competent following KrasG12D-induced Sox9 activation, Sox9-expressing ductal and centroacinar cells remain highly refractory to PanIN initiation [22] (Figure 3D). The relatively low efficiency of Sox9-CreERT2-mediated recombination (in ∼12% of Sox9+ cells) does not, however, preclude the existence of scarce (either Sox9+ or Sox9-) PanIN-initiating cells in the ductal tree which were not recombined in this analysis. Notwithstanding this caveat, while tumor histology has long implicated ductal/centroacinar cells as the originator of PDAC, they paradoxically represent the only pancreatic lineage so far examined that does not ostensibly undergo neoplastic transformation readily. Contingent upon these pathophysiological findings in mouse translating to human PDAC etiology, they suggest that: 1) Sox9 (and Onecut1) constitute novel biomarkers of ADM and 2) prophylactic therapeutic strategies for PDAC should focus on preventing PanIN initiation from the acinar compartment by preventing acinar Sox9 induction. Thus, befitting the intimate relationship between Sox9 and Notch signaling in the pancreas, Sox9 now joins the Notch pathway as a therapeutic target in pancreatic cancer prevention.
9. Summary and future perspectives
As well as being required for maintaining progenitors in the developing neural crest [13], intestinal epithelium [96], hair bulge/follicle [97, 193], and pancreas [16], Sox9 has been subsequently shown to maintain the neural stem cells of the CNS [194] and mammary stem cells [195] and mark progenitors of the developing lung [196, 197] and retina [198] and adult liver [17, 171] and intestine [17]. Undoubtedly, this roster of organ-specific stem/progenitor cells marked and/or maintained by Sox9 will be added to by future studies. While this progenitor marker and maintenance function has seemingly defined its role in the pancreatic program, the multiple indispensable tasks performed by Sox9 in the developing and adult pancreas outlined here show Sox9 to be far more multi-functional than generally perceived. In sum, during the morphogenesis and postnatal maintenance of the pancreas:
1. Sox9 marks and maintains multipotent pancreatic progenitors by promoting their survival and proliferation as well as retention of their undifferentiated status. Sox9+ cells retain their multipotency throughout gestation, including the secondary transition, but become progressively "locked in" to a ductal fate as birth approaches. Maintenance of the MPC pool is likely to be achieved in part through modulation of Hes1/Notch signaling. Furthermore, by maintaining MPC Fgfr2b expression and receptivity to mitogenic mesenchymal Fgf10, Sox9 participates in a feed-forward Fgf10/Fgfr2b/Sox9 loop to maintain MPC proliferation and pancreatic commitment. Consequently, pancreas-specific Sox9 ablation produces pancreatic hypoplasia and a pancreas-to-liver fate switch.
2. Sox9 marks secondary transition duct-endocrine bipotent pancreatic progenitors and is absolutely required for their endocrine differentiation. Like Hes1, Sox9 is a Notch target in bipotent progenitors which lay the foundations of the presumptive ductal tree. While pro-endocrine Ngn3 is inhibited by Hes1, it is directly activated by Sox9. As a consequence of Sox9 being more sensitive than Hes1 to increased Notch activity: 1) bipotent progenitors with high Notch activity express both Hes1 and Sox9 and remain undifferentiated while 2) those with intermediate Notch activity induce only Sox9. Because of a positive autoregulatory feedback loop, once expression of Ngn3 is initiated, an Ngn3Hi state is rapidly attained, ensuring endocrine commitment. Cell-autonomous repression of Sox9 by Ngn3 is then required for endocrine differentiation. Sox9+ bipotent progenitors which fail to initiate an endocrine program adopt the alternative ductal fate instead. Consistent with Sox9 being an obligatory effector of Ngn3 induction and endocrine commitment, morphogenesis of the endocrine compartment is more susceptible to Sox9 dosage than that of the exocrine tissue such that Sox9-haploinsufficiency manifests in islet-specific hypoplasia, phenocopying the islet defects seen in human CD cases.
3. Sox9 is directly required for differentiation and maintenance of pancreatic ducts and their cilia, in part by maintaining expression of ductal genes including Spp1 and Pkd2, which maintains primary cilia. Concordantly, Sox9 ablation either prior to the secondary transition or in adulthood results in duct dysgenesis and cysts as well as loss of primary cilia.
4. Sox9 marks duct and centroacinar cells in the postnatal pancreas which retain some neogenic potential. In vivo, the Sox9+ pancreatic compartment is able to give rise to small numbers of endocrine cells in the immediate neonatal period, but not beyond. However, given the requisite culture conditions, isolated Sox9+ adult ductal cells are able to expand and give rise to acinar and endocrine cells, including β-cell-like cells ex vivo. While pancreatic injury such as PDL is ostensibly able to initiate an endocrine program (as indicated by Ngn3 induction) in adult pancreatic Sox9+ cells, endocrine differentiation is incomplete, presumably owing to the absence of the necessary cues. Reduction of Sox9 dosage in adult pancreas appears to invoke their acinar (but not endocrine) differentiation, revealing plasticity of mature Sox9+ duct/centroacinar cells.
5. Ectopic activation of Sox9 expression in acinar cells is integral for their metaplasia to ductal cells and subsequent PanIN/PDAC progression, unveiling Sox9 as a key player in the initiation of pancreatic cancer.
These divergent roles are clearly dependent upon the interaction of Sox9 with various transcription factors and signaling pathways, foremost Fgf and Notch. It is highly probable that this milieu of interacting partners is spatiotemporally dynamic during the course of pancreatic organogenesis, changing with both developmental stage and cellular context. It is envisaged that characterizing the Sox9 interactome throughout pancreas organogenesis and in both the healthy and injured pancreas will provide great insight into the mechanisms through which Sox9 regulates pancreogenesis and adult pancreas homeostasis. This will require ChIP-Seq of various Sox9+ pancreatic cell populations to identify Sox9 pancreatic targets on a genome-wide scale. Such analyses are of course contingent upon the future development of necessary ChIP-grade reagents. Dissecting Sox9-mediated governance of the pancreatic program will undoubtedly provide unprecedented insight into the transcriptional mechanisms regulating the differentiation of pancreatic endoderm into β-cells. Such knowledge will be highly beneficial in optimizing current protocols for differentiating hESC/iPS cells into functionally-mature insulin+ cells. Elucidation of the changing pancreatic Sox9 interactome is in turn likely to lead to the identification of further roles for Sox9 in the pancreas, assuring its position as a multi-tasking master regulator of the pancreatic program during pancreas development and beyond.
Disclosures: The author reports no conflict of interests.
Acknowledgments
I would like to thank Drs. Mette C. Jørgensen and Palle Serup for their constructive comments on the manuscript. I also owe a debt of gratitude to Palle Serup for providing me the latitude to prepare this work. Apologies are offered in advance to colleagues whose work could not be cited here because of editorial constraints.
References
- 1.Goss RJ. The physiology of growth. Academic Press; 1978. chapters 13 and 19. [Google Scholar]
- 2.Hughes H. An experimental study of regeneration in the islets of Langerhans with reference to the theory of balance. Acta Anat (Basel) 1956;27(1-2):1–61. doi: 10.1159/000141111. [DOI] [PubMed] [Google Scholar]
- 3.Adler G, Hupp T, Kern HF. Course and spontaneous regression of acute pancreatitis in the rat. Virchows Arch A Pathol Anat Histol. 1979;382(1):31–47. doi: 10.1007/BF01102739. [DOI] [PubMed] [Google Scholar]
- 4.Edlund H. Pancreatic organogenesis - developmental mechanisms and implications for therapy. Nat Rev Genet. 2002;3(7):524–532. doi: 10.1038/nrg841. [DOI] [PubMed] [Google Scholar]
- 5.Gubbay J, Collignon J, Koopman P, Capel B, Economou A, Munsterberg A, Vivian N, Goodfellow P, Lovell-Badge R. A gene mapping to the sex-determining region of the mouse Y chromosome is a member of a novel family of embryonically expressed genes. Nature. 1990;346(6281):245–250. doi: 10.1038/346245a0. [DOI] [PubMed] [Google Scholar]
- 6.Sinclair AH, Berta P, Palmer MS, Hawkins JR, Griffiths BL, Smith MJ, Foster JW, Frischauf AM, Lovell-Badge R, Goodfellow PN. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature. 1990;346(6281):240–244. doi: 10.1038/346240a0. [DOI] [PubMed] [Google Scholar]
- 7.Soullier S, Jay P, Poulat F, Vanacker JM, Berta P, Laudet V. Diversification pattern of the HMG and SOX family members during evolution. J Mol Evol. 1999;48(5):517–527. doi: 10.1007/pl00006495. [DOI] [PubMed] [Google Scholar]
- 8.Schepers GE, Teasdale RD, Koopman P. Twenty pairs of sox: extent, homology, and nomenclature of the mouse and human sox transcription factor gene families. Dev Cell. 2002;3(2):167–170. doi: 10.1016/s1534-5807(02)00223-x. [DOI] [PubMed] [Google Scholar]
- 9.Sarkar A, Hochedlinger K. The sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell. 2013;12(1):15–30. doi: 10.1016/j.stem.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wegner M. All purpose Sox: The many roles of Sox proteins in gene expression. Int J Biochem Cell Biol. 2010;42(3):381–390. doi: 10.1016/j.biocel.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 11.Bylund M, Andersson E, Novitch BG, Muhr J. Vertebrate neurogenesis is counteracted by Sox1-3 activity. Nat Neurosci. 2003;6(11):1162–1168. doi: 10.1038/nn1131. [DOI] [PubMed] [Google Scholar]
- 12.Graham V, Khudyakov J, Ellis P, Pevny L. SOX2 functions to maintain neural progenitor identity. Neuron. 2003;39(5):749–765. doi: 10.1016/s0896-6273(03)00497-5. [DOI] [PubMed] [Google Scholar]
- 13.Cheung M, Briscoe J. Neural crest development is regulated by the transcription factor Sox9. Development. 2003;130(23):5681–5693. doi: 10.1242/dev.00808. [DOI] [PubMed] [Google Scholar]
- 14.Kim J, Lo L, Dormand E, Anderson DJ. SOX10 maintains multipotency and inhibits neuronal differentiation of neural crest stem cells. Neuron. 2003;38(1):17–31. doi: 10.1016/s0896-6273(03)00163-6. [DOI] [PubMed] [Google Scholar]
- 15.Piper K, Ball SG, Keeling JW, Mansoor S, Wilson DI, Hanley NA. Novel SOX9 expression during human pancreas development correlates to abnormalities in Campomelic dysplasia. Mech Dev. 2002;116(1-2):223–226. doi: 10.1016/s0925-4773(02)00145-4. [DOI] [PubMed] [Google Scholar]
- 16.Seymour PA, Freude KK, Tran MN, Mayes EE, Jensen J, Kist R, Scherer G, Sander M. SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc Natl Acad Sci U S A. 2007;104(6):1865–1870. doi: 10.1073/pnas.0609217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Furuyama K, Kawaguchi Y, Akiyama H, Horiguchi M, Kodama S, Kuhara T, Hosokawa S, Elbahrawy A, Soeda T, Koizumi M. et al. Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat Genet. 2011;43(1):34–41. doi: 10.1038/ng.722. [DOI] [PubMed] [Google Scholar]
- 18.Akiyama H, Kim JE, Nakashima K, Balmes G, Iwai N, Deng JM, Zhang Z, Martin JF, Behringer RR, Nakamura T, de Crombrugghe B. Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. Proc Natl Acad Sci U S A. 2005;102(41):14665–14670. doi: 10.1073/pnas.0504750102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kopp JL, Dubois CL, Schaffer AE, Hao E, Shih HP, Seymour PA, Ma J, Sander M. Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development. 2011;138(4):653–665. doi: 10.1242/dev.056499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shih HP, Kopp JL, Sandhu M, Dubois CL, Seymour PA, Grapin-Botton A, Sander M. A Notch-dependent molecular circuitry initiates pancreatic endocrine and ductal cell differentiation. Development. 2012;139(14):2488–2499. doi: 10.1242/dev.078634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seymour PA, Freude KK, Dubois CL, Shih HP, Patel NA, Sander M. A dosage-dependent requirement for Sox9 in pancreatic endocrine cell formation. Dev Biol. 2008;323(1):19–30. doi: 10.1016/j.ydbio.2008.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kopp JL, von Figura G, Mayes E, Liu FF, Dubois CL, Morris JP 4th, Pan FC, Akiyama H, Wright CV, Jensen K. et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22(6):737–750. doi: 10.1016/j.ccr.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawaguchi Y. Sox9 and programming of liver and pancreatic progenitors. J Clin Invest. 2013;123(5):1881–1886. doi: 10.1172/JCI66022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Belo J, Krishnamurthy M, Oakie A, Wang R. The role of SOX9 transcription factor in pancreatic and duodenal development. Stem Cells Dev. 2013;22(22):2935–2943. doi: 10.1089/scd.2013.0106. [DOI] [PubMed] [Google Scholar]
- 25.Pictet RL, Clark WR, Williams RH, Rutter WJ. An ultrastructural analysis of the developing embryonic pancreas. Dev Biol. 1972;29(4):436–467. doi: 10.1016/0012-1606(72)90083-8. [DOI] [PubMed] [Google Scholar]
- 26.Slack JM. Developmental biology of the pancreas. Development. 1995;121(6):1569–1580. doi: 10.1242/dev.121.6.1569. [DOI] [PubMed] [Google Scholar]
- 27.Jorgensen MC, Ahnfelt-Ronne J, Hald J, Madsen OD, Serup P, Hecksher-Sorensen J. An illustrated review of early pancreas development in the mouse. Endocr Rev. 2007;28(6):685–705. doi: 10.1210/er.2007-0016. [DOI] [PubMed] [Google Scholar]
- 28.Hald J, Sprinkel AE, Ray M, Serup P, Wright C, Madsen OD. Generation and characterization of Ptf1a antiserum and localization of Ptf1a in relation to Nkx6.1 and Pdx1 during the earliest stages of mouse pancreas development. J Histochem Cytochem. 2008;56(6):587–595. doi: 10.1369/jhc.2008.950675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129(10):2447–2457. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- 30.Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, Wright CV. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet. 2002;32(1):128–134. doi: 10.1038/ng959. [DOI] [PubMed] [Google Scholar]
- 31.Solar M, Cardalda C, Houbracken I, Martin M, Maestro MA, De Medts N, Xu X, Grau V, Heimberg H, Bouwens L, Ferrer J. Pancreatic exocrine duct cells give rise to insulin-producing beta cells during embryogenesis but not after birth. Dev Cell. 2009;17(6):849–860. doi: 10.1016/j.devcel.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 32.Kopinke D, Brailsford M, Shea JE, Leavitt R, Scaife CL, Murtaugh LC. Lineage tracing reveals the dynamic contribution of Hes1+ cells to the developing and adult pancreas. Development. 2011;138(3):431–441. doi: 10.1242/dev.053843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou Q, Law AC, Rajagopal J, Anderson WJ, Gray PA, Melton DA. A multipotent progenitor domain guides pancreatic organogenesis. Dev Cell. 2007;13(1):103–114. doi: 10.1016/j.devcel.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 34.Gradwohl G, Dierich A, LeMeur M, Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A. 2000;97(4):1607–1611. doi: 10.1073/pnas.97.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Apelqvist A, Li H, Sommer L, Beatus P, Anderson DJ, Honjo T, Hrabe de Angelis M, Lendahl U, Edlund H. Notch signalling controls pancreatic cell differentiation. Nature. 1999;400(6747):877–881. doi: 10.1038/23716. [DOI] [PubMed] [Google Scholar]
- 36.Schwitzgebel VM, Scheel DW, Conners JR, Kalamaras J, Lee JE, Anderson DJ, Sussel L, Johnson JD, German MS. Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development. 2000;127(16):3533–3542. doi: 10.1242/dev.127.16.3533. [DOI] [PubMed] [Google Scholar]
- 37.Grapin-Botton A, Majithia AR, Melton DA. Key events of pancreas formation are triggered in gut endoderm by ectopic expression of pancreatic regulatory genes. Genes Dev. 2001;15(4):444–454. doi: 10.1101/gad.846001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heremans Y, Van De Casteele M, In't Veld P, Gradwohl G, Serup P, Madsen O, Pipeleers D, Heimberg H. Recapitulation of embryonic neuroendocrine differentiation in adult human pancreatic duct cells expressing neurogenin 3. J Cell Biol. 2002;159(2):303–312. doi: 10.1083/jcb.200203074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johansson KA, Dursun U, Jordan N, Gu G, Beermann F, Gradwohl G, Grapin-Botton A. Temporal control of neurogenin3 activity in pancreas progenitors reveals competence windows for the generation of different endocrine cell types. Dev Cell. 2007;12(3):457–465. doi: 10.1016/j.devcel.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 40.Herrera PL, Huarte J, Sanvito F, Meda P, Orci L, Vassalli JD. Embryogenesis of the murine endocrine pancreas; early expression of pancreatic polypeptide gene. Development. 1991;113(4):1257–1265. doi: 10.1242/dev.113.4.1257. [DOI] [PubMed] [Google Scholar]
- 41.Prado CL, Pugh-Bernard AE, Elghazi L, Sosa-Pineda B, Sussel L. Ghrelin cells replace insulin-producing beta cells in two mouse models of pancreas development. Proc Natl Acad Sci U S A. 2004;101(9):2924–2929. doi: 10.1073/pnas.0308604100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kesavan G, Sand FW, Greiner TU, Johansson JK, Kobberup S, Wu X, Brakebusch C, Semb H. Cdc42-mediated tubulogenesis controls cell specification. Cell. 2009;139(4):791–801. doi: 10.1016/j.cell.2009.08.049. [DOI] [PubMed] [Google Scholar]
- 43.Villasenor A, Chong DC, Henkemeyer M, Cleaver O. Epithelial dynamics of pancreatic branching morphogenesis. Development. 2010;137(24):4295–4305. doi: 10.1242/dev.052993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stanger BZ, Tanaka AJ, Melton DA. Organ size is limited by the number of embryonic progenitor cells in the pancreas but not the liver. Nature. 2007;445(7130):886–891. doi: 10.1038/nature05537. [DOI] [PubMed] [Google Scholar]
- 45.Ahlgren U, Jonsson J, Edlund H. The morphogenesis of the pancreatic mesenchyme is uncoupled from that of the pancreatic epithelium in IPF1/PDX1-deficient mice. Development. 1996;122(5):1409–1416. doi: 10.1242/dev.122.5.1409. [DOI] [PubMed] [Google Scholar]
- 46.Offield MF, Jetton TL, Labosky PA, Ray M, Stein RW, Magnuson MA, Hogan BL, Wright CV. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 1996;122(3):983–995. doi: 10.1242/dev.122.3.983. [DOI] [PubMed] [Google Scholar]
- 47.Krapp A, Knofler M, Ledermann B, Burki K, Berney C, Zoerkler N, Hagenbuchle O, Wellauer PK. The bHLH protein PTF1-p48 is essential for the formation of the exocrine and the correct spatial organization of the endocrine pancreas. Genes Dev. 1998;12(23):3752–3763. doi: 10.1101/gad.12.23.3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jensen J, Pedersen EE, Galante P, Hald J, Heller RS, Ishibashi M, Kageyama R, Guillemot F, Serup P, Madsen OD. Control of endodermal endocrine development by Hes-1. Nat Genet. 2000;24(1):36–44. doi: 10.1038/71657. [DOI] [PubMed] [Google Scholar]
- 49.Ahnfelt-Ronne J, Jorgensen MC, Klinck R, Jensen JN, Fuchtbauer EM, Deering T, MacDonald RJ, Wright CV, Madsen OD, Serup P. Ptf1a-mediated control of Dll1 reveals an alternative to the lateral inhibition mechanism. Development. 2012;139(1):33–45. doi: 10.1242/dev.071761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jonckheere N, Mayes E, Shih HP, Li B, Lioubinski O, Dai X, Sander M. Analysis of mPygo2 mutant mice suggests a requirement for mesenchymal Wnt signaling in pancreatic growth and differentiation. Dev Biol. 2008;318(2):224–235. doi: 10.1016/j.ydbio.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Landsman L, Nijagal A, Whitchurch TJ, Vanderlaan RL, Zimmer WE, Mackenzie TC, Hebrok M. Pancreatic mesenchyme regulates epithelial organogenesis throughout development. Plos Biol. 2011;9(9):e1001143. doi: 10.1371/journal.pbio.1001143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ahnfelt-Rønne J, Ravassard P, Pardanaud-Galvieux C, Scharfmann R, Serup P. Mesenchymal bone morphogenetic protein signaling is required for normal pancreas development. Diabetes. 2010;59:1948–1956. doi: 10.2337/db09-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bhushan A, Itoh N, Kato S, Thiery JP, Czernichow P, Bellusci S, Scharfmann R. Fgf10 is essential for maintaining the proliferative capacity of epithelial progenitor cells during early pancreatic organogenesis. Development. 2001;128(24):5109–5117. doi: 10.1242/dev.128.24.5109. [DOI] [PubMed] [Google Scholar]
- 54.Maestro MA, Boj SF, Luco RF, Pierreux CE, Cabedo J, Servitja JM, German MS, Rousseau GG, Lemaigre FP, Ferrer J. Hnf6 and Tcf2 (MODY5) are linked in a gene network operating in a precursor cell domain of the embryonic pancreas. Hum Mol Genet. 2003;12(24):3307–3314. doi: 10.1093/hmg/ddg355. [DOI] [PubMed] [Google Scholar]
- 55.Schaffer AE, Freude KK, Nelson SB, Sander M. Nkx6 transcription factors and Ptf1a function as antagonistic lineage determinants in multipotent pancreatic progenitors. Dev Cell. 2010;18(6):1022–1029. doi: 10.1016/j.devcel.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Afelik S, Qu X, Hasrouni E, Bukys MA, Deering T, Nieuwoudt S, Rogers W, Macdonald RJ, Jensen J. Notch-mediated patterning and cell fate allocation of pancreatic progenitor cells. Development. 2012;139(10):1744–1753. doi: 10.1242/dev.075804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Horn S, Kobberup S, Jorgensen MC, Kalisz M, Klein T, Kageyama R, Gegg M, Lickert H, Lindner J, Magnuson MA. et al. Mind bomb 1 is required for pancreatic beta-cell formation. Proc Natl Acad Sci U S A. 2012;109(19):7356–7361. doi: 10.1073/pnas.1203605109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Coffinier C, Barra J, Babinet C, Yaniv M. Expression of the vHNF1/HNF1beta homeoprotein gene during mouse organogenesis. Mech Dev. 1999;89(1-2):211–213. doi: 10.1016/s0925-4773(99)00221-x. [DOI] [PubMed] [Google Scholar]
- 59.Jacquemin P, Lemaigre FP, Rousseau GG. The Onecut transcription factor HNF-6 (OC-1) is required for timely specification of the pancreas and acts upstream of Pdx-1 in the specification cascade. Dev Biol. 2003;258(1):105–116. doi: 10.1016/s0012-1606(03)00115-5. [DOI] [PubMed] [Google Scholar]
- 60.Jacquemin P, Durviaux SM, Jensen J, Godfraind C, Gradwohl G, Guillemot F, Madsen OD, Carmeliet P, Dewerchin M, Collen D. et al. Transcription factor hepatocyte nuclear factor 6 regulates pancreatic endocrine cell differentiation and controls expression of the proendocrine gene ngn3. Mol Cell Biol. 2000;20(12):4445–4454. doi: 10.1128/mcb.20.12.4445-4454.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee CS, Sund NJ, Behr R, Herrera PL, Kaestner KH. Foxa2 is required for the differentiation of pancreatic alpha-cells. Dev Biol. 2005;278(2):484–495. doi: 10.1016/j.ydbio.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 62.Jensen J, Heller RS, Funder-Nielsen T, Pedersen EE, Lindsell C, Weinmaster G, Madsen OD, Serup P. Independent development of pancreatic alpha- and beta-cells from neurogenin3-expressing precursors: a role for the notch pathway in repression of premature differentiation. Diabetes. 2000;49(2):163–176. doi: 10.2337/diabetes.49.2.163. [DOI] [PubMed] [Google Scholar]
- 63.Deltour L, Leduque P, Paldi A, Ripoche MA, Dubois P, Jami J. Polyclonal origin of pancreatic islets in aggregation mouse chimaeras. Development. 1991;112(4):1115–1121. doi: 10.1242/dev.112.4.1115. [DOI] [PubMed] [Google Scholar]
- 64.Rozzo A, Meneghel-Rozzo T, Delakorda SL, Yang SB, Rupnik M. Exocytosis of insulin: in vivo maturation of mouse endocrine pancreas. Ann N Y Acad Sci. 2009;1152:53–62. doi: 10.1111/j.1749-6632.2008.04003.x. [DOI] [PubMed] [Google Scholar]
- 65.Dutta S, Gannon M, Peers B, Wright C, Bonner-Weir S, Montminy M. PDX:PBX complexes are required for normal proliferation of pancreatic cells during development. Proc Natl Acad Sci U S A. 2001;98(3):1065–1070. doi: 10.1073/pnas.031561298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lioubinski O, Muller M, Wegner M, Sander M. Expression of Sox transcription factors in the developing mouse pancreas. Dev Dyn. 2003;227(3):402–408. doi: 10.1002/dvdy.10311. [DOI] [PubMed] [Google Scholar]
- 67.Wilson ME, Yang KY, Kalousova A, Lau J, Kosaka Y, Lynn FC, Wang J, Mrejen C, Episkopou V, Clevers HC, German MS. The HMG Box Transcription Factor Sox4 Contributes to the Development of the Endocrine Pancreas. Diabetes. 2005;54(12):3402–3409. doi: 10.2337/diabetes.54.12.3402. [DOI] [PubMed] [Google Scholar]
- 68.McDonald E, Krishnamurthy M, Goodyer CG, Wang R. The emerging role of SOX transcription factors in pancreatic endocrine cell development and function. Stem Cells Dev. 2009;18(10):1379–1388. doi: 10.1089/scd.2009.0240. [DOI] [PubMed] [Google Scholar]
- 69.Mavropoulos A, Devos N, Biemar F, Zecchin E, Argenton F, Edlund H, Motte P, Martial JA, Peers B. sox4b is a key player of pancreatic alpha cell differentiation in zebrafish. Dev Biol. 2005;285(1):211–223. doi: 10.1016/j.ydbio.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 70.Bell KM, Western PS, Sinclair AH. SOX8 expression during chick embryogenesis. Mech Dev. 2000;94(1-2):257–260. doi: 10.1016/s0925-4773(00)00296-3. [DOI] [PubMed] [Google Scholar]
- 71.Britsch S, Goerich DE, Riethmacher D, Peirano RI, Rossner M, Nave KA, Birchmeier C, Wegner M. The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev. 2001;15(1):66–78. doi: 10.1101/gad.186601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sock E, Schmidt K, Hermanns-Borgmeyer I, Bosl MR, Wegner M. Idiopathic weight reduction in mice deficient in the high-mobility-group transcription factor Sox8. Mol Cell Biol. 2001;21(20):6951–6959. doi: 10.1128/MCB.21.20.6951-6959.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Teitelman G, Guz Y, Ivkovic S, Ehrlich M. Islet injury induces neurotrophin expression in pancreatic cells and reactive gliosis of peri-islet Schwann cells. J Neurobiol. 1998;34(4):304–318. doi: 10.1002/(sici)1097-4695(199803)34:4<304::aid-neu2>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 74.Ushiki T, Watanabe S. Distribution and ultrastructure of the autonomic nerves in the mouse pancreas. Microsc Res Tech. 1997;37(5-6):399–406. doi: 10.1002/(SICI)1097-0029(19970601)37:5/6<399::AID-JEMT4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 75.Tsui H, Winer S, Chan Y, Truong D, Tang L, Yantha J, Paltser G, Dosch HM. Islet glia, neurons, and beta cells. Ann N Y Acad Sci. 2008;1150:32–42. doi: 10.1196/annals.1447.033. [DOI] [PubMed] [Google Scholar]
- 76.Stolt CC, Lommes P, Sock E, Chaboissier MC, Schedl A, Wegner M. The Sox9 transcription factor determines glial fate choice in the developing spinal cord. Genes Dev. 2003;17(13):1677–1689. doi: 10.1101/gad.259003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lynn FC, Smith SB, Wilson ME, Yang KY, Nekrep N, German MS. Sox9 coordinates a transcriptional network in pancreatic progenitor cells. Proc Natl Acad Sci U S A. 2007;104(25):10500–10505. doi: 10.1073/pnas.0704054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee YH, Saint-Jeannet JP. Sox9, a novel pancreatic marker in Xenopus. Int J Dev Biol. 2003;47(6):459–462. [PubMed] [Google Scholar]
- 79.Manfroid I, Ghaye A, Naye F, Detry N, Palm S, Pan L, Ma TP, Huang W, Rovira M, Martial JA. et al. Zebrafish sox9b is crucial for hepatopancreatic duct development and pancreatic endocrine cell regeneration. Dev Biol. 2012;366(2):268–278. doi: 10.1016/j.ydbio.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jennings RE, Berry AA, Kirkwood-Wilson R, Roberts NA, Hearn T, Salisbury RJ, Blaylock J, Hanley KP, Hanley NA. Development of the human pancreas from foregut to endocrine commitment. Diabetes. 2013;62(10):3514–3522. doi: 10.2337/db12-1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fujitani Y, Fujitani S, Boyer DF, Gannon M, Kawaguchi Y, Ray M, Shiota M, Stein RW, Magnuson MA, Wright CV. Targeted deletion of a cis-regulatory region reveals differential gene dosage requirements for Pdx1 in foregut organ differentiation and pancreas formation. Genes Dev. 2006;20(2):253–266. doi: 10.1101/gad.1360106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Norgaard GA, Jensen JN, Jensen J. FGF10 signaling maintains the pancreatic progenitor cell state revealing a novel role of Notch in organ development. Dev Biol. 2003;264(2):323–338. doi: 10.1016/j.ydbio.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 83.Bagheri-Fam S, Barrionuevo F, Dohrmann U, Gunther T, Schule R, Kemler R, Mallo M, Kanzler B, Scherer G. Long-range upstream and downstream enhancers control distinct subsets of the complex spatiotemporal Sox9 expression pattern. Dev Biol. 2006;291(2):382–397. doi: 10.1016/j.ydbio.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 84.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21(1):70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 85.Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425(6961):917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- 86.Corish P, Tyler-Smith C. Attenuation of green fluorescent protein half-life in mammalian cells. Protein Eng. 1999;12(12):1035–1040. doi: 10.1093/protein/12.12.1035. [DOI] [PubMed] [Google Scholar]
- 87.Sugiyama T, Benitez CM, Ghodasara A, Liu L, McLean GW, Lee J, Blauwkamp TA, Nusse R, Wright CV, Gu G, Kim SK. Reconstituting pancreas development from purified progenitor cells reveals genes essential for islet differentiation. Proc Natl Acad Sci U S A. 2013;110(31):12691–12696. doi: 10.1073/pnas.1304507110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Miyatsuka T, Li Z, German MS. Chronology of islet differentiation revealed by temporal cell labeling. Diabetes. 2009;58(8):1863–1868. doi: 10.2337/db09-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pan FC, Bankaitis ED, Boyer D, Xu X, Van de Casteele M, Magnuson MA, Heimberg H, Wright CV. Spatiotemporal patterns of multipotentiality in Ptf1a-expressing cells during pancreas organogenesis and injury-induced facultative restoration. Development. 2013;140(4):751–764. doi: 10.1242/dev.090159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cleveland MH, Sawyer JM, Afelik S, Jensen J, Leach SD. Exocrine ontogenies: on the development of pancreatic acinar, ductal and centroacinar cells. Semin Cell Dev Biol. 2012;23(6):711–719. doi: 10.1016/j.semcdb.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rovira M, Scott SG, Liss AS, Jensen J, Thayer SP, Leach SD. Isolation and characterization of centroacinar/terminal ductal progenitor cells in adult mouse pancreas. Proc Natl Acad Sci U S A. 2010;107(1):75–80. doi: 10.1073/pnas.0912589107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sharma A, Zangen DH, Reitz P, Taneja M, Lissauer ME, Miller CP, Weir GC, Habener JF, Bonner-Weir S. The homeodomain protein IDX-1 increases after an early burst of proliferation during pancreatic regeneration. Diabetes. 1999;48(3):507–513. doi: 10.2337/diabetes.48.3.507. [DOI] [PubMed] [Google Scholar]
- 93.Guz Y, Montminy MR, Stein R, Leonard J, Gamer LW, Wright CV, Teitelman G. Expression of murine STF-1, a putative insulin gene transcription factor, in beta cells of pancreas, duodenal epithelium and pancreatic exocrine and endocrine progenitors during ontogeny. Development. 1995;121(1):11–18. doi: 10.1242/dev.121.1.11. [DOI] [PubMed] [Google Scholar]
- 94.Krapp A, Knofler M, Frutiger S, Hughes GJ, Hagenbuchle O, Wellauer PK. The p48 DNA-binding subunit of transcription factor PTF1 is a new exocrine pancreas-specific basic helix-loop-helix protein. Embo J. 1996;15(16):4317–4329. [PMC free article] [PubMed] [Google Scholar]
- 95.Scopsi L, Wang BL, Larsson LI. Nonspecific immunocytochemical reactions with certain neurohormonal peptides and basic peptide sequences. J Histochem Cytochem. 1986;34(11):1469–1475. doi: 10.1177/34.11.2877024. [DOI] [PubMed] [Google Scholar]
- 96.Blache P, van de Wetering M, Duluc I, Domon C, Berta P, Freund JN, Clevers H, Jay P. SOX9 is an intestine crypt transcription factor, is regulated by the Wnt pathway, and represses the CDX2 and MUC2 genes. J Cell Biol. 2004;166(1):37–47. doi: 10.1083/jcb.200311021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vidal VP, Chaboissier MC, Lutzkendorf S, Cotsarelis G, Mill P, Hui CC, Ortonne N, Ortonne JP, Schedl A. Sox9 is essential for outer root sheath differentiation and the formation of the hair stem cell compartment. Curr Biol. 2005;15(15):1340–1351. doi: 10.1016/j.cub.2005.06.064. [DOI] [PubMed] [Google Scholar]
- 98.Bi W, Huang W, Whitworth DJ, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc Natl Acad Sci U S A. 2001;98(12):6698–6703. doi: 10.1073/pnas.111092198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Georgia S, Soliz R, Li M, Zhang P, Bhushan A. p57 and Hes1 coordinate cell cycle exit with self-renewal of pancreatic progenitors. Dev Biol. 2006;298(1):22–31. doi: 10.1016/j.ydbio.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 100.Seymour PA, Shih HP, Patel NA, Freude KK, Xie R, Lim CJ, Sander M. A Sox9/Fgf feed-forward loop maintains pancreatic organ identity. Development. 2012;139(18):3363–3372. doi: 10.1242/dev.078733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dong PD, Munson CA, Norton W, Crosnier C, Pan X, Gong Z, Neumann CJ, Stainier DY. Fgf10 regulates hepatopancreatic ductal system patterning and differentiation. Nat Genet. 2007;39(3):397–402. doi: 10.1038/ng1961. [DOI] [PubMed] [Google Scholar]
- 102.Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F, Gao G, Goldfarb M. Receptor specificity of the fibroblast growth factor family. J Biol Chem. 1996;271(25):15292–15297. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- 103.Pulkkinen MA, Spencer-Dene B, Dickson C, Otonkoski T. The IIIb isoform of fibroblast growth factor receptor 2 is required for proper growth and branching of pancreatic ductal epithelium but not for differentiation of exocrine or endocrine cells. Mech Dev. 2003;120(2):167–175. doi: 10.1016/s0925-4773(02)00440-9. [DOI] [PubMed] [Google Scholar]
- 104.Revest JM, Spencer-Dene B, Kerr K, De Moerlooze L, Rosewell I, Dickson C. Fibroblast growth factor receptor 2-IIIb acts upstream of Shh and Fgf4 and is required for limb bud maintenance but not for the induction of Fgf8, Fgf10, Msx1, or Bmp4. Dev Biol. 2001;231(1):47–62. doi: 10.1006/dbio.2000.0144. [DOI] [PubMed] [Google Scholar]
- 105.Elghazi L, Cras-Meneur C, Czernichow P, Scharfmann R. Role for FGFR2IIIb-mediated signals in controlling pancreatic endocrine progenitor cell proliferation. Proc Natl Acad Sci U S A. 2002;99(6):3884–3889. doi: 10.1073/pnas.062321799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sylvestersen KB, Herrera PL, Serup P, Rescan C. Fgf9 signalling stimulates Spred and Sprouty expression in embryonic mouse pancreas mesenchyme. Gene Expr Patterns. 2011;11:105–111. doi: 10.1016/j.gep.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 107.Svensson P, Williams C, Lundeberg J, Ryden P, Bergqvist I, Edlund H. Gene array identification of Ipf1/Pdx1-/- regulated genes in pancreatic progenitor cells. BMC Dev Biol. 2007;7:129. doi: 10.1186/1471-213X-7-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 1994;371(6498):606–609. doi: 10.1038/371606a0. [DOI] [PubMed] [Google Scholar]
- 109.Bagheri-Fam S, Sim H, Bernard P, Jayakody I, Taketo MM, Scherer G, Harley VR. Loss of Fgfr2 leads to partial XY sex reversal. Dev Biol. 2008;314(1):71–83. doi: 10.1016/j.ydbio.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 110.Thomsen MK, Butler CM, Shen MM, Swain A. Sox9 is required for prostate development. Dev Biol. 2008;316(2):302–311. doi: 10.1016/j.ydbio.2008.01.030. [DOI] [PubMed] [Google Scholar]
- 111.Abler LL, Mansour SL, Sun X. Conditional gene inactivation reveals roles for Fgf10 and Fgfr2 in establishing a normal pattern of epithelial branching in the mouse lung. Dev Dyn. 2009;238(8):1999–2013. doi: 10.1002/dvdy.22032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Delous M, Yin C, Shin D, Ninov N, Debrito Carten J, Pan L, Ma TP, Farber SA, Moens CB, Stainier DY. Sox9b is a key regulator of pancreaticobiliary ductal system development. Plos Genet. 2012;8(6):e1002754. doi: 10.1371/journal.pgen.1002754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Manfroid I, Delporte F, Baudhuin A, Motte P, Neumann CJ, Voz ML, Martial JA, Peers B. Reciprocal endoderm-mesoderm interactions mediated by fgf24 and fgf10 govern pancreas development. Development. 2007;134(22):4011–4021. doi: 10.1242/dev.007823. [DOI] [PubMed] [Google Scholar]
- 114.Hart A, Papadopoulou S, Edlund H. Fgf10 maintains notch activation, stimulates proliferation, and blocks differentiation of pancreatic epithelial cells. Dev Dyn. 2003;228(2):185–193. doi: 10.1002/dvdy.10368. [DOI] [PubMed] [Google Scholar]
- 115.Miralles F, Lamotte L, Couton D, Joshi RL. Interplay between FGF10 and Notch signalling is required for the self-renewal of pancreatic progenitors. Int J Dev Biol. 2006;50(1):17–26. doi: 10.1387/ijdb.052080fm. [DOI] [PubMed] [Google Scholar]
- 116.D'Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, Smart NG, Moorman MA, Kroon E, Carpenter MK, Baetge EE. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol. 2006;24(11):1392–1401. doi: 10.1038/nbt1259. [DOI] [PubMed] [Google Scholar]
- 117.Foster JW, Dominguez-Steglich MA, Guioli S, Kowk G, Weller PA, Stevanovic M, Weissenbach J, Mansour S, Young ID, Goodfellow PN. et al. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature. 1994;372(6506):525–530. doi: 10.1038/372525a0. [DOI] [PubMed] [Google Scholar]
- 118.Wagner T, Wirth J, Meyer J, Zabel B, Held M, Zimmer J, Pasantes J, Bricarelli FD, Keutel J, Hustert E. et al. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell. 1994;79(6):1111–1120. doi: 10.1016/0092-8674(94)90041-8. [DOI] [PubMed] [Google Scholar]
- 119.Desgraz R, Herrera PL. Pancreatic neurogenin 3-expressing cells are unipotent islet precursors. Development. 2009;136(21):3567–3574. doi: 10.1242/dev.039214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yoshida T, Hanahan D. Murine pancreatic ductal adenocarcinoma produced by in vitro transduction of polyoma middle T oncogene into the islets of Langerhans. Am J Pathol. 1994;145(3):671–684. [PMC free article] [PubMed] [Google Scholar]
- 121.Dubois CL, Shih HP, Seymour PA, Patel NA, Behrmann JM, Ngo V, Sander M. Sox9-haploinsufficiency causes glucose intolerance in mice. Plos One. 2011;6(8):e23131. doi: 10.1371/journal.pone.0023131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Oliver-Krasinski JM, Kasner MT, Yang J, Crutchlow MF, Rustgi AK, Kaestner KH, Stoffers DA. The diabetes gene Pdx1 regulates the transcriptional network of pancreatic endocrine progenitor cells in mice. J Clin Invest. 2009;119(7):1888–1898. doi: 10.1172/JCI37028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Stoffers DA, Ferrer J, Clarke WL, Habener JF. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat Genet. 1997;17(2):138–139. doi: 10.1038/ng1097-138. [DOI] [PubMed] [Google Scholar]
- 124.Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, Lindner T, Yamagata K, Ogata M, Tomonaga O. et al. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat Genet. 1997;17(4):384–385. doi: 10.1038/ng1297-384. [DOI] [PubMed] [Google Scholar]
- 125.Lee JC, Smith SB, Watada H, Lin J, Scheel D, Wang J, Mirmira RG, German MS. Regulation of the pancreatic pro-endocrine gene neurogenin3. Diabetes. 2001;50(5):928–936. doi: 10.2337/diabetes.50.5.928. [DOI] [PubMed] [Google Scholar]
- 126.Zong Y, Panikkar A, Xu J, Antoniou A, Raynaud P, Lemaigre F, Stanger BZ. Notch signaling controls liver development by regulating biliary differentiation. Development. 2009;136(10):1727–1739. doi: 10.1242/dev.029140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Esni F, Ghosh B, Biankin AV, Lin JW, Albert MA, Yu X, MacDonald RJ, Civin CI, Real FX, Pack MA. et al. Notch inhibits Ptf1 function and acinar cell differentiation in developing mouse and zebrafish pancreas. Development. 2004;131(17):4213–4224. doi: 10.1242/dev.01280. [DOI] [PubMed] [Google Scholar]
- 128.Magenheim J, Klein AM, Stanger BZ, Ashery-Padan R, Sosa-Pineda B, Gu G, Dor Y. Ngn3(+) endocrine progenitor cells control the fate and morphogenesis of pancreatic ductal epithelium. Dev Biol. 2011;359(1):26–36. doi: 10.1016/j.ydbio.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Duvillie B, Attali M, Bounacer A, Ravassard P, Basmaciogullari A, Scharfmann R. The mesenchyme controls the timing of pancreatic beta-cell differentiation. Diabetes. 2006;55(3):582–589. doi: 10.2337/diabetes.55.03.06.db05-0839. [DOI] [PubMed] [Google Scholar]
- 130.Wang S, Yan J, Anderson DA, Xu Y, Kanal MC, Cao Z, Wright CV, Gu G. Neurog3 gene dosage regulates allocation of endocrine and exocrine cell fates in the developing mouse pancreas. Dev Biol. 2010;339(1):26–37. doi: 10.1016/j.ydbio.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Smith SB, Watada H, German MS. Neurogenin3 activates the islet differentiation program while repressing its own expression. Mol Endocrinol. 2004;18(1):142–149. doi: 10.1210/me.2003-0037. [DOI] [PubMed] [Google Scholar]
- 132.Kilic G, Wang J, Sosa-Pineda B. Osteopontin is a novel marker of pancreatic ductal tissues and of undifferentiated pancreatic precursors in mice. Dev Dyn. 2006;235(6):1659–1667. doi: 10.1002/dvdy.20729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Peacock JD, Huk DJ, Ediriweera HN, Lincoln J. Sox9 transcriptionally represses Spp1 to prevent matrix mineralization in maturing heart valves and chondrocytes. Plos One. 2011;6(10):e26769. doi: 10.1371/journal.pone.0026769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wu G, D'Agati V, Cai Y, Markowitz G, Park JH, Reynolds DM, Maeda Y, Le TC, Hou H Jr, Kucherlapati R. et al. Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell. 1998;93(2):177–188. doi: 10.1016/s0092-8674(00)81570-6. [DOI] [PubMed] [Google Scholar]
- 135.Wu G, Markowitz GS, Li L, D'Agati VD, Factor SM, Geng L, Tibara S, Tuchman J, Cai Y, Park JH. et al. Cardiac defects and renal failure in mice with targeted mutations in Pkd2. Nat Genet. 2000;24(1):75–78. doi: 10.1038/71724. [DOI] [PubMed] [Google Scholar]
- 136.Kim I, Ding T, Fu Y, Li C, Cui L, Li A, Lian P, Liang D, Wang DW, Guo C. et al. Conditional mutation of Pkd2 causes cystogenesis and upregulates beta-catenin. J Am Soc Nephrol. 2009;20(12):2556–2569. doi: 10.1681/ASN.2009030271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Greenwood AL, Li S, Jones K, Melton DA. Notch signaling reveals developmental plasticity of Pax4(+) pancreatic endocrine progenitors and shunts them to a duct fate. Mech Dev. 2007;124(2):97–107. doi: 10.1016/j.mod.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 138.Murtaugh LC, Stanger BZ, Kwan KM, Melton DA. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci U S A. 2003;100(25):14920–14925. doi: 10.1073/pnas.2436557100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kayali AG, Stotland A, Gunst KV, Kritzik M, Liu G, Dabernat S, Zhang YQ, Wu W, Sarvetnick N. Growth factor-induced signaling of the pancreatic epithelium. J Endocrinol. 2005;185(1):45–56. doi: 10.1677/joe.1.05949. [DOI] [PubMed] [Google Scholar]
- 140.Dichmann DS, Miller CP, Jensen J, Heller RS, Serup P. Expression and misexpression of members of the FGF and TGFbeta families of growth factors in the developing mouse pancreas. Dev Dyn. 2003;226(4):663–674. doi: 10.1002/dvdy.10270. [DOI] [PubMed] [Google Scholar]
- 141.Kuersten S, Goodwin EB. The power of the 3' UTR: translational control and development. Nat Rev Genet. 2003;4(8):626–637. doi: 10.1038/nrg1125. [DOI] [PubMed] [Google Scholar]
- 142.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S. et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15(8):1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mercer TR, Wilhelm D, Dinger ME, Solda G, Korbie DJ, Glazov EA, Truong V, Schwenke M, Simons C, Matthaei KI. et al. Expression of distinct RNAs from 3' untranslated regions. Nucleic Acids Res. 2011;39(6):2393–2403. doi: 10.1093/nar/gkq1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Miquerol L, Langille BL, Nagy A. Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development. 2000;127(18):3941–3946. doi: 10.1242/dev.127.18.3941. [DOI] [PubMed] [Google Scholar]
- 145.Damert A, Miquerol L, Gertsenstein M, Risau W, Nagy A. Insufficient VEGFA activity in yolk sac endoderm compromises haematopoietic and endothelial differentiation. Development. 2002;129(8):1881–1892. doi: 10.1242/dev.129.8.1881. [DOI] [PubMed] [Google Scholar]
- 146.Mead TJ, Wang Q, Bhattaram P, Dy P, Afelik S, Jensen J, Lefebvre V. A far-upstream (-70 kb) enhancer mediates Sox9 auto-regulation in somatic tissues during development and adult regeneration. Nucleic Acids Res. 2013;41(8):4459–4469. doi: 10.1093/nar/gkt140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Desai BM, Oliver-Krasinski J, De Leon DD, Farzad C, Hong N, Leach SD, Stoffers DA. Preexisting pancreatic acinar cells contribute to acinar cell, but not islet beta cell, regeneration. J Clin Invest. 2007;117(4):971–977. doi: 10.1172/JCI29988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Swenson ES, Xanthopoulos J, Nottoli T, McGrath J, Theise ND, Krause DS. Chimeric mice reveal clonal development of pancreatic acini, but not islets. Biochem Biophys Res Commun. 2009;379(2):526–531. doi: 10.1016/j.bbrc.2008.12.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Friedmann NB, Marble A. Island hyperplasia in the partially depancreatized rat. Endocrinology. 1941;29:577–582. [Google Scholar]
- 150.Lehv M, Fitzgerald PJ. Pancreatic acinar cell regeneration. IV. Regeneration after resection. Am J Pathol. 1968;53(4):513–535. [PMC free article] [PubMed] [Google Scholar]
- 151.Elsasser HP, Adler G, Kern HF. Time course and cellular source of pancreatic regeneration following acute pancreatitis in the rat. Pancreas. 1986;1(5):421–429. doi: 10.1097/00006676-198609000-00006. [DOI] [PubMed] [Google Scholar]
- 152.Chang AY, Diani AR. Chemically and hormonally induced diabetes mellitus. In: Volk BW, Arquilla ER, editors. The diabetic pancreas. Plenum; 1985. pp. 415–438. [Google Scholar]
- 153.Palmiter RD, Behringer RR, Quaife CJ, Maxwell F, Maxwell IH, Brinster RL. Cell lineage ablation in transgenic mice by cell-specific expression of a toxin gene. Cell. 1987;50(3):435–443. doi: 10.1016/0092-8674(87)90497-1. [DOI] [PubMed] [Google Scholar]
- 154.Herrera PL, Huarte J, Zufferey R, Nichols A, Mermillod B, Philippe J, Muniesa P, Sanvito F, Orci L, Vassalli JD. Ablation of islet endocrine cells by targeted expression of hormone-promoter-driven toxigenes. Proc Natl Acad Sci U S A. 1994;91(26):12999–13003. doi: 10.1073/pnas.91.26.12999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Xu X, D'Hoker J, Stange G, Bonne S, De Leu N, Xiao X, Van de Casteele M, Mellitzer G, Ling Z, Pipeleers D. et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell. 2008;132(2):197–207. doi: 10.1016/j.cell.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 156.Gonda DK, Bachmair A, Wunning I, Tobias JW, Lane WS, Varshavsky A. Universality and structure of the N-end rule. J Biol Chem. 1989;264(28):16700–16712. [PubMed] [Google Scholar]
- 157.Rankin MM, Wilbur CJ, Rak K, Shields EJ, Granger A, Kushner JA. Beta-cells are not generated in pancreatic duct ligation-induced injury in adult mice. Diabetes. 2013;62(5):1634–1645. doi: 10.2337/db12-0848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Van de Casteele M, Leuckx G, Baeyens L, Cai Y, Yuchi Y, Coppens V, De Groef S, Eriksson M, Svensson C, Ahlgren U. et al. Neurogenin 3+ cells contribute to beta-cell neogenesis and proliferation in injured adult mouse pancreas. Cell Death Dis. 2013;4:e523. doi: 10.1038/cddis.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wang S, Jensen JN, Seymour PA, Hsu W, Dor Y, Sander M, Magnuson MA, Serup P, Gu G. Sustained Neurog3 expression in hormone-expressing islet cells is required for endocrine maturation and function. Proc Natl Acad Sci U S A. 2009;106(24):9715–9720. doi: 10.1073/pnas.0904247106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Inada A, Nienaber C, Katsuta H, Fujitani Y, Levine J, Morita R, Sharma A, Bonner-Weir S. Carbonic anhydrase II-positive pancreatic cells are progenitors for both endocrine and exocrine pancreas after birth. Proc Natl Acad Sci U S A. 2008;105(50):19915–19919. doi: 10.1073/pnas.0805803105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Means AL, Xu Y, Zhao A, Ray KC, Gu G. A CK19(CreERT) knockin mouse line allows for conditional DNA recombination in epithelial cells in multiple endodermal organs. Genesis. 2008;46(6):318–323. doi: 10.1002/dvg.20397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kopinke D, Murtaugh LC. Exocrine-to-endocrine differentiation is detectable only prior to birth in the uninjured mouse pancreas. BMC Dev Biol. 2010;10:38. doi: 10.1186/1471-213X-10-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Jin L, Feng T, Shih HP, Zerda R, Luo A, Hsu J, Mahdavi A, Sander M, Tirrell DA, Riggs AD, Ku HT. Colony-forming cells in the adult mouse pancreas are expandable in Matrigel and form endocrine/acinar colonies in laminin hydrogel. Proc Natl Acad Sci U S A. 2013;110(10):3907–3912. doi: 10.1073/pnas.1301889110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sugiyama T, Rodriguez RT, McLean GW, Kim SK. Conserved markers of fetal pancreatic epithelium permit prospective isolation of islet progenitor cells by FACS. Proc Natl Acad Sci U S A. 2007;104(1):175–180. doi: 10.1073/pnas.0609490104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Banga A, Akinci E, Greder LV, Dutton JR, Slack JM. In vivo reprogramming of Sox9+ cells in the liver to insulin-secreting ducts. Proc Natl Acad Sci U S A. 2012;109(38):15336–15341. doi: 10.1073/pnas.1201701109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Artner I, Hang Y, Mazur M, Yamamoto T, Guo M, Lindner J, Magnuson MA, Stein R. MafA and MafB regulate genes critical to beta-cells in a unique temporal manner. Diabetes. 2010;59(10):2530–2539. doi: 10.2337/db10-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ahlgren U, Pfaff SL, Jessell TM, Edlund T, Edlund H. Independent requirement for ISL1 in formation of pancreatic mesenchyme and islet cells. Nature. 1997;385(6613):257–260. doi: 10.1038/385257a0. [DOI] [PubMed] [Google Scholar]
- 168.Smith SB, Qu HQ, Taleb N, Kishimoto NY, Scheel DW, Lu Y, Patch AM, Grabs R, Wang J, Lynn FC. et al. Rfx6 directs islet formation and insulin production in mice and humans. Nature. 2010;463(7282):775–780. doi: 10.1038/nature08748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yechoor V, Liu V, Espiritu C, Paul A, Oka K, Kojima H, Chan L. Neurogenin3 is sufficient for transdetermination of hepatic progenitor cells into neo-islets in vivo but not transdifferentiation of hepatocytes. Dev Cell. 2009;16(3):358–373. doi: 10.1016/j.devcel.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zheng YW, Taniguchi H. Diversity of hepatic stem cells in the fetal and adult liver. Semin Liver Dis. 2003;23(4):337–348. doi: 10.1055/s-2004-815557. [DOI] [PubMed] [Google Scholar]
- 171.Dorrell C, Erker L, Schug J, Kopp JL, Canaday PS, Fox AJ, Smirnova O, Duncan AW, Finegold MJ, Sander M. et al. Prospective isolation of a bipotential clonogenic liver progenitor cell in adult mice. Genes Dev. 2011;25(11):1193–1203. doi: 10.1101/gad.2029411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Visvader JE. Cells of origin in cancer. Nature. 2011;469(7330):314–322. doi: 10.1038/nature09781. [DOI] [PubMed] [Google Scholar]
- 173.Sener SF, Fremgen A, Menck HR, Winchester DP. Pancreatic cancer: a report of treatment and survival trends for 100,313 patients diagnosed from 1985-1995, using the National Cancer Database. J Am Coll Surg. 1999;189(1):1–7. doi: 10.1016/s1072-7515(99)00075-7. [DOI] [PubMed] [Google Scholar]
- 174.Lillemoe KD, Yeo CJ, Cameron JL. Pancreatic cancer: state-of-the-art care. CA Cancer J Clin. 2000;50(4):241–268. doi: 10.3322/canjclin.50.4.241. [DOI] [PubMed] [Google Scholar]
- 175.Hruban RH, Adsay NV, Albores-Saavedra J, Compton C, Garrett ES, Goodman SN, Kern SE, Klimstra DS, Kloppel G, Longnecker DS. et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25(5):579–586. doi: 10.1097/00000478-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 176.Giovinazzo F, Turri G, Zanini S, Butturini G, Scarpa A, Bassi C. Clinical implications of biological markers in Pancreatic Ductal Adenocarcinoma. Surg Oncol. 2012;21(4):e171–e182. doi: 10.1016/j.suronc.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 177.Feldmann G, Beaty R, Hruban RH, Maitra A. Molecular genetics of pancreatic intraepithelial neoplasia. J Hepatobiliary Pancreat Surg. 2007;14(3):224–232. doi: 10.1007/s00534-006-1166-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kanda M, Matthaei H, Wu J, Hong SM, Yu J, Borges M, Hruban RH, Maitra A, Kinzler K, Vogelstein B, Goggins M. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. 2012;142(4):730–733. doi: 10.1053/j.gastro.2011.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA. et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4(6):437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 180.Morris JP, Cano DA, Sekine S, Wang SC, Hebrok M. Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J Clin Invest. 2010;120(2):508–520. doi: 10.1172/JCI40045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zhu L, Shi G, Schmidt CM, Hruban RH, Konieczny SF. Acinar cells contribute to the molecular heterogeneity of pancreatic intraepithelial neoplasia. Am J Pathol. 2007;171(1):263–273. doi: 10.2353/ajpath.2007.061176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Miyamoto Y, Maitra A, Ghosh B, Zechner U, Argani P, Iacobuzio-Donahue CA, Sriuranpong V, Iso T, Meszoely IM, Wolfe MS. et al. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell. 2003;3(6):565–576. doi: 10.1016/s1535-6108(03)00140-5. [DOI] [PubMed] [Google Scholar]
- 183.Stanger BZ, Stiles B, Lauwers GY, Bardeesy N, Mendoza M, Wang Y, Greenwood A, Cheng KH, McLaughlin M, Brown D. et al. Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell. 2005;8(3):185–195. doi: 10.1016/j.ccr.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 184.Brembeck FH, Schreiber FS, Deramaudt TB, Craig L, Rhoades B, Swain G, Grippo P, Stoffers DA, Silberg DG, Rustgi AK. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res. 2003;63(9):2005–2009. [PubMed] [Google Scholar]
- 185.Ray KC, Bell KM, Yan J, Gu G, Chung CH, Washington MK, Means AL. Epithelial tissues have varying degrees of susceptibility to Kras(G12D)-initiated tumorigenesis in a mouse model. Plos One. 2011;6(2):e16786. doi: 10.1371/journal.pone.0016786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Carriere C, Seeley ES, Goetze T, Longnecker DS, Korc M. The Nestin progenitor lineage is the compartment of origin for pancreatic intraepithelial neoplasia. Proc Natl Acad Sci U S A. 2007;104(11):4437–4442. doi: 10.1073/pnas.0701117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.De La OJ, Emerson LL, Goodman JL, Froebe SC, Illum BE, Curtis AB, Murtaugh LC. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci U S A. 2008;105(48):18907–18912. doi: 10.1073/pnas.0810111105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, Dubus P, Sandgren EP, Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11(3):291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 189.Habbe N, Shi G, Meguid RA, Fendrich V, Esni F, Chen H, Feldmann G, Stoffers DA, Konieczny SF, Leach SD, Maitra A. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A. 2008;105(48):18913–18918. doi: 10.1073/pnas.0810097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Carriere C, Young AL, Gunn JR, Longnecker DS, Korc M. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem Biophys Res Commun. 2009;382(3):561–565. doi: 10.1016/j.bbrc.2009.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Gidekel Friedlander SY, Chu GC, Snyder EL, Girnius N, Dibelius G, Crowley D, Vasile E, DePinho RA, Jacks T. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell. 2009;16(5):379–389. doi: 10.1016/j.ccr.2009.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Prevot PP, Simion A, Grimont A, Colletti M, Khalaileh A, Van den Steen G, Sempoux C, Xu X, Roelants V, Hald J. et al. Role of the ductal transcription factors HNF6 and Sox9 in pancreatic acinar-to-ductal metaplasia. Gut. 2012;61(12):1723–1732. doi: 10.1136/gutjnl-2011-300266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Nowak JA, Polak L, Pasolli HA, Fuchs E. Hair follicle stem cells are specified and function in early skin morphogenesis. Cell Stem Cell. 2008;3(1):33–43. doi: 10.1016/j.stem.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Scott CE, Wynn SL, Sesay A, Cruz C, Cheung M, Gomez Gaviro MV, Booth S, Gao B, Cheah KS, Lovell-Badge R, Briscoe J. SOX9 induces and maintains neural stem cells. Nat Neurosci. 2010;13(10):1181–1189. doi: 10.1038/nn.2646. [DOI] [PubMed] [Google Scholar]
- 195.Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, Itzkovitz S, Noske A, Zurrer-Hardi U, Bell G. et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell. 2012;148(5):1015–1028. doi: 10.1016/j.cell.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Liu Y, Hogan BL. Differential gene expression in the distal tip endoderm of the embryonic mouse lung. Gene Expr Patterns. 2002;2(3-4):229–233. doi: 10.1016/s1567-133x(02)00057-1. [DOI] [PubMed] [Google Scholar]
- 197.Perl AK, Kist R, Shan Z, Scherer G, Whitsett JA. Normal lung development and function after Sox9 inactivation in the respiratory epithelium. Genesis. 2005;41(1):23–32. doi: 10.1002/gene.20093. [DOI] [PubMed] [Google Scholar]
- 198.Poche RA, Furuta Y, Chaboissier MC, Schedl A, Behringer RR. Sox9 is expressed in mouse multipotent retinal progenitor cells and functions in Muller glial cell development. J Comp Neurol. 2008;510(3):237–250. doi: 10.1002/cne.21746. [DOI] [PMC free article] [PubMed] [Google Scholar]