

Abstract
Poly(3-hexylthiophene) (P3HT) is one of the most extensively investigated conjugated polymers and has been employed as the active material in many devices including field-effect transistors, organic photovoltaics and sensors. As a result, methods to further tune the properties of P3HT are desirable for specific applications. Herein, we report a facile postpolymerization modification strategy to functionalize the 4-position of commercially available P3HT in two simple steps–bromination of the 4-position of P3HT (Br–P3HT) followed by lithium−bromine exchange and quenching with an electrophile. We achieved near quantitative lithium–bromine exchange with Br–P3HT, which requires over 100 thienyl lithiates to be present on a single polymer chain. The lithiated-P3HT is readily combined with functional electrophiles, resulting in P3HT derivatives with ketones, secondary alcohols, trimethylsilyl (TMS) group, fluorine, or an azide at the 4-position. We demonstrated that the azide-modified P3HT could undergo Cu-catalyzed or Cu-free click chemistry, significantly expanding the complexity of the structures that can be appended to P3HT using this method.
Introduction
Polythiophenes (PTs) are the most widely studied conjugated polymers in organic electronics and are currently employed in field-effect transistors,1,2 light-emitting diodes,3 and organic photovoltaics.4−6 Unsubstituted PT is highly crystalline and thus insoluble, hampering its processability; however, 3-hexyl substitution overcomes these limitations. The synthesis of poly(3-hexylthiophene) (P3HT) has been studied extensively over the last few decades. Grignard metathesis (GRIM) polymerization is a particularly powerful approach and allows for the large-scale preparation of highly regioregular P3HT at noncryogenic reaction temperatures.7−10
In order to enhance and fully exploit the properties of PTs, facile chemical modification of PTs is necessary. In particular, the tailoring of PT properties has largely focused on the addition of alkyl or functional side chains to the 3- and/or 4-positions in order to expand their structure–property relationships for improved utility in optoelectronic devices.11−19 For example, Ueda and co-workers reported regioregular PTs with phenyl and pyridyl side chains, which have resulted in decreased band-gaps as a result of extended π-conjugation.11 Ludwigs and co-workers reported that head-to-tail PTs having an alkylthiophene side chain display a 30% increase in open circuit voltage (Voc) in solar cells, which was attributed to a lowered highest occupied molecular orbital (HOMO).12 Our group has reported pentafluorophenoxy-containing analogues of regioregular P3HT, which behave as surfactants at the bulk heterojunction interfaces. It was found that the addition of only a small amount of these materials yielded a 30% increase in power conversion efficiency (PCE) as compared to a P3HT:PCBM standard.13 In addition to solar cells, functionalized PTs play a pivotal role in designing chemoresistive sensors when used to wrap carbon nanotubes (CNTs). Using this scheme, customized, receptor-functionalized PTs were used to create selective detection schemes for chemical warfare agents14 and were able to distinguish between structural isomers of xylene.15
Methods to prepare functionalized PTs generally integrate the side-chains or functional groups at the monomer stage. A clear advantage of this approach is that 100% of the thiophene repeating units can be reliably functionalized. However, this method is not universally applicable, and in particular, GRIM polymerization conditions are not compatible with many functional groups. As a result, postpolymerization modification20 can offer an alternative, efficient strategy to functionalize PTs. For example, current methods to modify PTs via postpolymerization modification include the GRIM polymerization of protected terminal alkyne21 or alkyl bromide thiophene monomers,22 which can be modified after polymerization through Cu-mediated Huisgen 1,3-dipolar cycloaddition23,24 or nucleophilic substitution, respectively. These approaches allow for the installation of functional groups which could not be employed in a GRIM polymerization; however, these examples still entailed preparation of specialized monomers.
A more attractive approach is the direct modification of commercially available regioregular P3HT. Previously, Holdcroft and co-workers have achieved efficient, direct modification of the 4-position of P3HT through electrophilic bromination with N-bromosuccinimide (NBS).25 The brominated P3HT (Br–P3HT) then served as a cross-coupling substrate to introduce functionality to the 4-position of P3HT26 via Suzuki–Miyaura,27 Stille,28 and Heck reactions.29 Others have extended these cross-coupling strategies to introduce fullerene derivatives,30,31 pyrene,32 perylene bisimide,33 and copolymers.34−36 Herein, we expand the utility of Br–P3HT and demonstrate that near-quantitative lithium−halogen exchange occurs at the 4-position and can be quenched with a variety of electrophiles to yield new 4-position functionalized P3HTs (Scheme 1). This methodology diversifies the portfolio of functionalized P3HTs with an efficient synthetic procedure.
Scheme 1. Postpolymerization Modification via Lithium–Bromine Exchange and Subsequent Quenching with Electrophiles.

Experimental Section
Materials and Instruments
Chemicals were purchased from Aldrich, Alfa Aesar, and TCI America without further purification unless noted otherwise. Acetic acid and methanol were purchased from Macron Fine Chemicals. Methanol-d4 “100%” (D, 99.95%) in ampules were purchased from Cambridge Isotope Laboratories, Inc. Butyraldehyde and 4-methoxybenzaldehyde were distilled before use. Regioregular P3HT was purchased from Aldrich. All reactions were carried out under argon with standard Schlenk techniques. Anhydrous tetrahydrofuran (THF) was obtained from Sigma-Aldrich dry solvent kegs and kept in a Schlenk flask with molecular sieves (3 Å). Difluorobenzocyclooctyne (20) was synthesized by the literature procedure with the use of tetrabutylammonium fluoride (TBAF), instead of CsF.37 All 1H and 19F NMR spectra were collected on a Bruker Avance-400 and data are reported in ppm. 1H NMR spectroscopy is referenced to solvent peaks, and 19F NMR spectroscopy is referenced to trifluorotoluene (δ = −62.72 ppm). The multiplicity is reported as follows: s = singlet, d = doublet, t = triplet, m = multiplet or unresolved, br = broad, bs = broad singlet, and bm = broad multiplet. Coupling constants J are reported in Hz. Elemental analyses were carried out by Robertson Microlit Laboratories, Ledgewood, NJ (USA). UV–vis spectra were recorded on an Agilent Cary 4000 spectrometer at room temperature. The solutions were prepared in dichloromethane or chloroform with concentrations between 10–4 M and 10–6 M. Thin films were prepared on glass by spin-coating 3500 rpm for 30 s from a solution of functionalized P3HT in chlorobenzene (1 mg/mL, F–P3HT is not completely soluble in this concentration, and undissolved polymers are removed by filtration). Fluorescence measurements were performed at room temperature with a Horiba Jobin Yvon SPEX Fluorolog-τ3 fluorimeter (model FL-321, 450 W xenon lamp) using right-angle conformation. Infrared (IR) spectra were measured on a Thermo Scientific Nicolet 6700 Fourier transform infrared spectrometer using the attenuated total reflectance (ATR) mode on a germanium crystal. THF gel permeation chromatography (GPC) was performed (0.5 mg/mL) on an Agilent 1260 Infinity system, calibrated with polystyrene standards. Cyclic voltammetry was carried out with an AUTOLAB PGSTAT 10 potentiostat (Eco Chemie) on a three electrode system: polymer film on indium tin oxide (ITO)-coated glass (1.2 cm × 1.2 cm) as a working electrode, Pt wire as a counter electrode, and Ag/AgCl as a reference electrode. Polymer films on ITO-coated glass were prepared by spin-coating a solution of PT in chloroform (2 mg/mL, F–P3HT was sonicated to dissolve it for about 1 h) with 2000 rpm for 30 s. The film was dried and annealed at 70 °C for 15 min. The area of 1.2 cm × 0.7 cm was immersed in the electrolyte (0.1 M tetrabutylammonium hexafluorophosphate in anhydrous acetonitrile) during measurement. The ferrocene/ferrocenium redox couple was used as an internal standard.
Procedure for Br–P3HT (1)
Br–P3HT was synthesized as previously reported.25 Briefly, to a stirring solution of commercial P3HT (300 mg, 1.80 mmol in terms of repeat unit) in chloroform (20 mL), NBS (386 mg, 2.16 mmol) was added portionwise. The reaction mixture was stirred for 12 h at room temperature. The temperature was elevated to 50 °C for 2 h, then the mixture was cooled and poured into a saturated NaHCO3 solution (50 mL). The organic layer was washed with water five times and dried over MgSO4. The mixture was precipitated in methanol. The precipitate was isolated by filtration and dried overnight under vacuum at room temperature, resulting in a yellow solid (441.6 mg, 1.80 mmol in terms of repeat unit, 99% yield based on 100% bromination).
General Procedure for the Synthesis of the Functionalized P3HT (4–15)
To a stirring solution of Br–P3HT (30 mg, 0.122 mmol in terms of repeat units) in THF (6 mL) at −78 °C, was added n-BuLi (0.612 mmol, 5 equiv, 1.6 M in hexane, 0.38 mL) dropwise. After 15 min of stirring, an electrophile (1.22 mmol, 10 equiv) was added to the mixture. Stirring continues for another 15 min. The temperature was increased to room temperature, and the resulting mixture was stirred for 4 h. Methanol (50 mL) was added and stirring continued until precipitate was generated. The precipitated solution was filtered, and the polymer was dried overnight under vacuum at 50 °C.
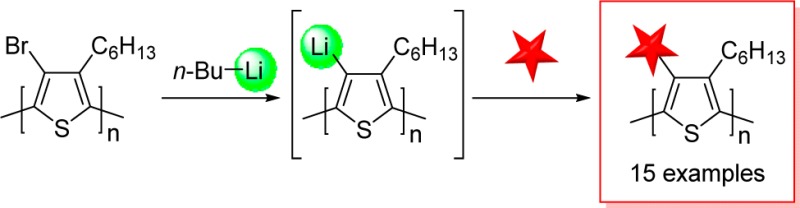
Procedure for F–P3HT (16)
To a stirring solution of Br–P3HT (30 mg, 0.122 mmol in terms of repeat units) in THF (6 mL) at −78 °C, was added n-BuLi (0.612 mmol, 5 equiv, 1.6 M in hexane, 0.38 mL) dropwise. After 20 min of stirring, N-fluorobenzenesulfonimide (1.22 mmol, 10 equiv, 0.386 mg) dissolved in THF (2 mL) was added. The mixture was stirred for 2 h, at which point the temperature was increased to room temperature and the solution was further stirred for 20 h. Methanol (50 mL) was added to precipitate the polymer. F–P3HT (16) was collected by filtration, washed with methanol and acetone, and dried under vacuum at 50 °C overnight.
Procedure for Azido-P3HT (17)
The procedure is the same as the general procedure until the quenching step (4 h stirring at room temperature). This reaction is quenched by glacial acetic acid (1.22 mmol, 10 equiv, 0.068 mL) or hydrochloric acid (1.22 mmol, 10 equiv, 3 M, 0.4 mL), and then the mixture was stirred for 30–60 min. Methanol (50 mL) was added and stirring continued until precipitate was generated. The precipitated solution was filtered, and the polymer was dried overnight under vacuum at 50 °C. Because of insolubility after precipitation, the crude mixture prior to methanol-induced precipitation was used for the next click reaction.
Procedure for Cu-Catalyzed Click Reaction (18)
To the mixture of 17 containing acetic acid were added phenyl propargyl ether (12 equiv, 0.094 mL), DIPEA (50 equiv, 0.54 mL), and CuI (14 mg, 10 mol % of the acetylene). The mixture was heated at 50 °C and stirred for 42 h. Methanol was added to precipitate the polymer, and the polymer was collected by centrifugation (11k rcf,38 10 min), washed with acetone:ethyl acetate (1:1), and dried under vacuum at 50 °C overnight.
Procedure for Cu-Free Click Reaction (19)
The mixture of 17 containing HCl was neutralized with NaOH (aq, 10%), and difluorobenzocyclooctyne (20)37 (1.47 mmol, 12 equiv, 282 mg) in THF was added. The resulting mixture was stirred at room temperature overnight. The polymer was precipitated with methanol (50 mL), collected by filtration, washed with methanol, and dried overnight under vacuum at 50 °C.
Procedure for Difluorobenzocyclooctyne (20)
This compound was synthesized by a method similar to that in the literature.37 7,7-Difluoro-5-(trimethylsilyl)-7,8,9,10-tetrahydrobenzo[8]annulen-6-yl triflate37 (700 mg, 1.69 mmol) was dissolved in THF (3 mL). To this mixture was added tetrabutylammonium fluoride solution (1 M in THF, 1.1 equiv, 1.86 mL), and the reaction mixture was stirred at room temperature for 10 min. This crude mixture was used in the synthesis of the polymer 19.
Results and Discussion
Lithium–bromine exchange is an extensively utilized chemical transformation, and most aryl or vinyl bromides react with alkyllithium reagents to form new organolithiates. These aryl- and vinyllithium species are highly reactive intermediates and readily combine with electrophiles. This strategy is widely employed in small-molecule chemistry; however, its extension to polymer chemistry has been limited, most likely as a result of concerns over polyanion formation. Specifically, it is well-accepted that generating a second anion within the same molecule is much more difficult to form than the first.39 Extrapolating this to a polymer chain, the (n + 1)th anion should be more difficult to form than the nth anion. To explore the limits of lithium–halogen exchange and its utility in polymer systems, we initiated a thorough analysis of the lithium–halogen exchange on Br–P3HT.
To test the efficiency of polyanion formation from Br–P3HT (1), we subjected 1 to varying amounts of n-butyllithium (n-BuLi), followed by quenching with excess methanol and examined the conversion back to P3HT (Scheme 2). We characterized the magnitude of the lithium–bromine exchange by comparing the NMR spectrum integration values of the thiophene 4-H (6.98 ppm, Figure 1) with the α-methylene of the hexyl chain (2.81 ppm, Figure 1) in addition to analyzing the weight percent of bromine as determined by elemental analysis (EA). The results are summarized in Table 1 and the NMR spectra are shown in Figure 1, where x and y represent monomers containing hydrogen and bromine at the 4-position, respectively. Gratifyingly, our results indicated we were indeed able to achieve near-quantitative lithium–bromine exchange on P3HT. Standard small-molecule conditions for lithium−halogen exchange often employ 1.2 equiv of butyl lithium. Using analogous conditions, we found that 73% lithiation was observed (3a). Upon increasing the amount of n-BuLi (3b–3d), more than 95% lithium–bromine exchange was achieved. The EA data for 3a–3d also display analogous trends to the NMR spectra. From Table 1, it is evident that as the amount of n-BuLi is increased, the recovered yields also increase. This relationship appears to be the result of more efficient precipitation of the polymers from methanol due to the higher salt concentration (lithium salt) in the reaction mixture. The salt weakens the solvation of the polymer by interacting with THF and thus facilitate polymer aggregation. The dispersity of 3a–3d is increased during the reaction from the corresponding Br–P3HT starting polymer (26 kDa, Mw/Mn = 1.80) to 3a (15 kDa, Mw/Mn = 3.01), 3b (28 kDa, Mw/Mn = 9.43), 3c (41 kDa, Mw/Mn = 7.39), and 3d (24 kDa, Mw/Mn = 13.4). The GPC traces of these polymers (Supporting Information) show that new peaks at higher molecular weight portions (such as 100 kDa and 550 kDa) emerged, suggesting some oligomerization and/or cross-linking. The exact nature of these high molecular weight species has not been determined. Considering the above results, we choose to develop a new PT modification methodology using 5 equiv of n-BuLi.
Scheme 2. Analysis of the Lithium–Bromine Exchange.

Figure 1.

1H NMR spectra of the polymers: (a) P3HT, (b) 1 (Br–P3HT), (c) 3c, and (d) 4. The sharp peak at 6.98 ppm from 4-position proton (●) enables the estimation of the Li–Br exchange by comparison to the peak at 2.81 ppm from α–methylene (■) of the hexyl chain. For detailed integration values and peak locations, please refer to the Supporting Information.
Table 1. Relationship between Equivalents of n-BuLi and Degree of Lithium–Bromine Exchangea.
| NMR |
EA |
|||||
|---|---|---|---|---|---|---|
| entry | n-BuLi (equiv) | x | y | x | y | % yield |
| 3a | 1.25 | 0.73 | 0.27 | 0.852 | 0.148 | 62 |
| 3b | 2.5 | 0.92 | 0.08 | 0.978 | 0.022 | 78 |
| 3c | 5 | 0.94 | 0.06 | 0.987 | 0.013 | 86 |
| 3d | 10 | >0.95 | <0.05 | 0.991 | 0.009 | 91 |
NMR indicates conversions determined by 1H NMR integration, and EA indicates conversions determined by elemental analysis.
Before proceeding with the introduction of electrophiles to the lithiated P3HT, we further investigated the origin of the 4-position proton in our methanol quenching experiments. In light of the high basicity of the multilithium intermediate 2, a portion of the recovered 4-position proton could originate from undesired proton impurities such as deprotonation of the α-methylene on the hexyl chain or the solvent, THF. To assay this, under rigorously dry conditions, Br–P3HT was combined with 5 equiv of n-BuLi and quenched with deuterated methanol (CD3OD), yielding 4 (Scheme 2). The NMR spectrum of polymer 4 (Figure 1d) showed 24% recovery of the 4-position thiophene proton at 6.98 ppm. By assuming that residual bromine content of 4 is the same as the contents of 3c (that is, 6%), 70% of the monomers contained a deuteron (z = 0.70, Scheme 2). This result demonstrates that with this methodology P3HTs containing up to 70% modification at the 4-position can be achieved. It should be noted that lower degrees of functionalization can also be achieved by controlling the levels of bromination.40 The possible avenue for introduction of the recovered 4-position proton in 4 could be E2 elimination of n-butyl bromide formed in situ by Li–Br exchange. However, the direct introduction of n-butyl bromide did not increase the recovered 4-position proton (see Supporting Information), suggesting this is not a significant source of recovered proton.
The efficient lithiation and quenching suggested that this methodology could be a versatile, efficient approach for the synthesis of 4-position functionalized P3HT by introducing lithiated intermediate 2 to a variety of electrophiles. We first applied this methodology to the synthesis of ketone-functionalized P3HTs (5–11 in Scheme 3) by the introduction of anhydrides to 2. We found that short, long, and bulky alkyl ketones (5–7), trifluoromethyl ketone (8), and aryls with different substituents (9–11) can all be appended to P3HT. The conversions (z values in Scheme 3) range from 47–65% as determined by NMR spectroscopy. Infrared (IR) spectroscopy confirms the appearance of distinct ketone peaks at around 1650–1800 cm–1 for all cases (5–11, see Supporting Information). Secondary alcohol P3HT derivatives (12–14) were obtained by quenching the intermediate 2 with the corresponding aldehydes. Despite being weaker electrophiles than anhydrides, similar conversions were achieved (51–63%). Trimethylsilyl (TMS) functionalized P3HT (15) was synthesized by quenching the lithiated P3HT with TMS chloride to result in 59% incorporation of TMS groups. Fluoride and azide moieties could also be installed at the 4-position using N-fluorobenzenesulfonimide (NFSI) and tosyl azide as electrophiles, respectively (vide infra). The dispersity of most of the synthesized polymers is increased during the chemical transformation as shown in the GPC traces (Supporting Information). Although there could be multiple interpolymer coupling in the ketone-functionalized P3HTs, it is most likely that the increased dispersity is primarily due to the lithium–bromine exchange, since 3a–3d have already possessed the increased dispersity.
Scheme 3. Scope of this Methodology: Ketone (5–11), Secondary Alcohol (12–14), TMS (15), F (16), and Azide (17) Functionalized P3HTs.

The conversion determined by 19F NMR with trifluorotoluene as an internal standard in NMR solvent. EA showed 46% conversion (See Supporting Information).
The conversion is slightly overestimated due to overlapped peaks in the aryl region of the 1H NMR spectra with CdCl3.
The conversion cannot be calculated with a single value due to the overlapped peaks of α-CH2 of hexyl chain with Ar-Me.
Slightly different reaction contions are employed. Please see below and Supporting Information for detailed conditions.
The conversion was not available due to the insolubility of the product.
Recently, Roncali and co-workers41 have proposed 4-fluoro-P3HT (F–P3HT, 16) as an advantageous material for organic photovoltaics due to the combination of its high electronegativity and small size, which will lower the HOMO and LUMO levels yet not significantly alter the crystal packing. Despite attempts to synthesize F–P3HT with a fluorine atom at each 4-position through a monomer-modification approach, Roncali’s efforts were unable to surpass 33% fluorination of PT using a monofluorinated terthiophene as the monomer unit. Our new lithium–halogen exchange methodology facilitated regioregular F–P3HT with higher percent fluorine in fewer steps. Upon subjecting lithiated P3HT to NFSI (Figure 2a), the introduction of fluorine atoms onto the 4-position was immediately evident by the broad singlet at −123.60 ppm in the 19F NMR spectrum (Supporting Information). The conversion was calculated by determining the amount of 4-position proton by 1H NMR spectroscopy and 4-position residual bromine by EA. From these values, the amount of 4-position fluorine was inferred to be 67%, twice as much as previously possible by the monomer-modification approach. We further investigated the electronic properties of the F–P3HT and compared it to P3HT and Br–P3HT through absorption spectroscopy, photoluminescence spectroscopy, and cyclic voltammetry (CV) (Figure 2 and Supporting Information). As predicted by Roncali and co-workers, the small size of the fluorine atom resulted in minimal twisting of the backbone and thus longer conjugation lengths as compared to Br–P3HT, although P3HT still appeared to have a superior conjugation length as indicated by solution spectra (Figure 2b, chloroform) and thin films (Figure 2c). The optical bandgap of F–P3HT (1.97 eV), measured from the absorption spectra of the thin film (Figure 2c), is similar to P3HT (1.90 eV), but much smaller than Br–P3HT (2.80 eV). The HOMO is measured by carrying out cyclic voltammetry in 0.1 M tetrabutylammonium hexafluorophosphate in acetonitrile as an electrolyte, along with a spin-coated polymer film on ITO-coated glass, Pt wire, and Ag/AgCl electrode as the working, counter, and reference electrode, respectively. A ferrocene/ferrocenium redox couple was used as an internal standard, assuming its redox positioned at −4.8 eV from vacuum level. CV analysis (Supporting Information) revealed that the HOMO of F–P3HT is at −5.38 eV, which is 0.51 eV lower than P3HT (−4.87 eV), indicating increased oxidative stability in the atmosphere. Br–P3HT did not show any significant current peak, indicative of its nonconductive nature and absence of π–π stacked polymer backbone. P3HT and F–P3HT thin films exhibit bathochromic shifts as compared to solution (451 to 530 nm for P3HT; 419 to 491 nm for F–P3HT) as a result of a planarization of the polymer main chain in the solid state. Thin film spectra also show shoulder signals at 600 nm for P3HT and 574 nm for F–P3HT that are well-defined vibronic transitions42,43 from the planarized π–π stacked (partially crystalline) polymer backbone. As expected in the case of Br–P3HT, no bathochromic shift or shoulder peak is observed, indicating that the steric bulk of the bromine atoms result in a twisted backbone and thus no π–π stacking is present in the Br–P3HT film. These results are consistent with the CV data.
Figure 2.
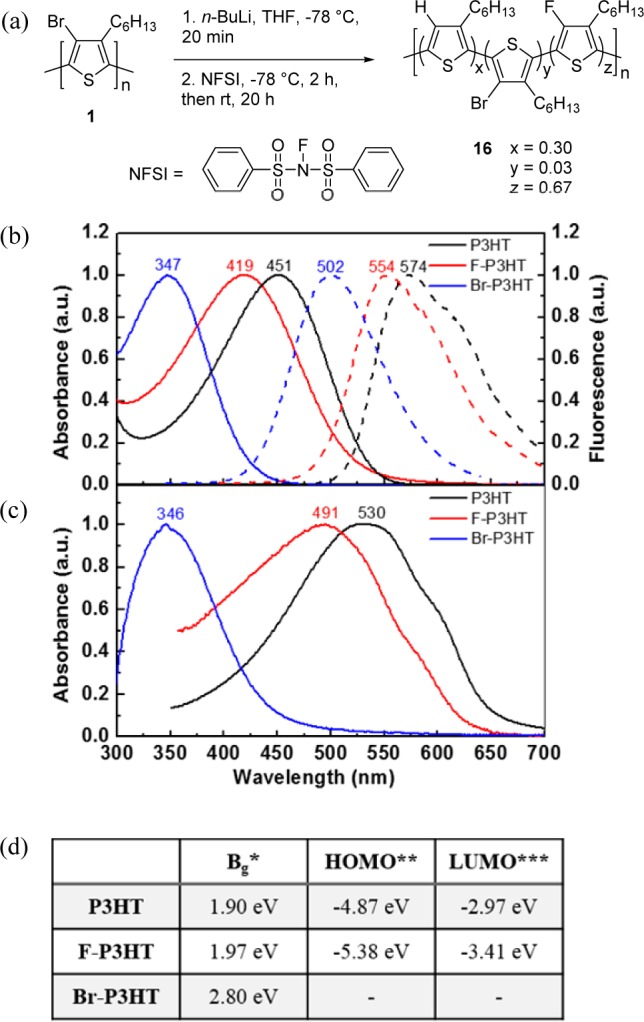
(a) Synthesis of F–P3HT (16). NFSI = N-fluorobenzenesulfonimide. (b) Absorption (solid line) and photoluminescence (dotted line) spectra of P3HT (black), F–P3HT (red), and Br–P3HT (blue) in CHCl3. Excitation at 400 nm (P3HT), 380 nm (F–P3HT), and 330 nm (Br–P3HT). (c) Absorption spectra of thin films of each polymer. The films are fabricated on glass by spin-coating each polymer from a chlorobenzene solution. (d) Electronic properties of P3HT, F–P3HT, and Br–P3HT obtained from photophysics and cyclic voltammetry measurements. (∗) Optical bandgap (Bg) was measured by onset (drawing a tangent line) of the thin film absorption spectra. (∗∗) HOMO = e(−Epolymeronset,oxidation + EFc – 4.8 V). (∗∗∗) LUMO = HOMO + Bg. For CV data, please refer to the Supporting Information.
To expand our methodology, we installed an azide group at the 4-position, which could be further modified through copper-catalyzed23,24 or strained-promoted copper-free44 cycloaddition reactions with alkynes.45 We were able to synthesize poly(3-azido-4-hexylthiophene) (N3–P3HT, 17) by the addition of tosyl azide to lithiate 2, followed by the addition of an acid, such as glacial acetic acid or hydrochloric acid (Figure 3a). The acidic work-up was employed to prevent base-mediated decomposition pathways, such as the formation of amines from 1,3-disubstituted triazenes.46 The strong peak in the IR spectrum at 2105 cm–1 (Figure 3b) indicated the presence of azide moieties. Next, we modified 17 through click reactions with alkynes. As a result of the insolubility of azido-P3HT after precipitation in methanol, we took measures to avoid precipitation (see Experimental Section) and performed an in situ click reaction on a crude solution (suspension) of 17. A Cu-catalyzed click reaction in the presence of copper iodide (CuI), diisopropylethylamine (DIPEA),21 and phenyl propargyl ether was carried out to synthesize 18. The IR spectrum of 18 (Figure 3b) reveals the disappearance of the azide peak, associated with the emergence of characteristic triazole (1599 cm–1) and phenyl (1494 cm–1) signals. We also modified N3–P3HT through a strain-promoted cycloaddition with difluorobenzocyclooctyne 20.37 The resulting triazole-containing P3HT (19) was soluble in common organic solvents, and 1H NMR spectroscopy, 19F NMR spectroscopy, as well as the disappearance of the azido group in the IR spectrum (Figure 3b) confirmed that the cycloaddition had occurred on the polymer. The analysis of 19F NMR spectrum indicated that 19 is the major regioisomer. The ability to easily synthesize N3–P3HT and its click reaction offer a plethora of opportunities for custom P3HT derivatives to be prepared from commercially available P3HT.
Figure 3.
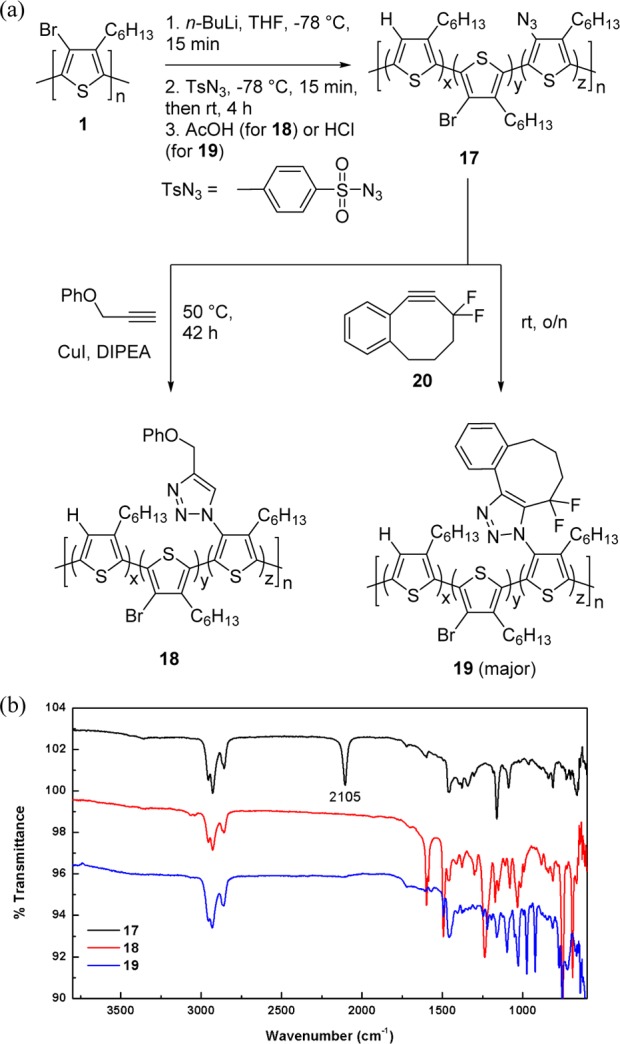
(a) Click reactions with azido-P3HT (17) in the Cu-catalyzed (18) and strain-promoted Cu-free (19) conditions. (b) Comparison of IR of 17 (black), 18 (red), and 19 (blue). 17 was prepared by quenching with HCl and resulted in a strong azide peak at 2105 cm–1. Quenching with AcOH produced similar results. In both 18 and 19, complete disappearance of the azide peak was observed.
Conclusions
A variety of functionalized P3HTs were synthesized in a simple two-step approach–bromination of P3HT25 followed by lithium–bromine exchange and quenching–from commercial P3HT. In contrast to the conventional belief that the generation of multiple reactive anions on one molecule is not favorable,39 we observed nearly quantitative lithium–bromine exchange on ca. 30 kDa Br–P3HT (more than 100 thiophene repeat units). The multilithiated P3HT is highly reactive, yet stable enough to undergo the next reaction in situ with functional electrophiles, resulting in an array of modified P3HTs. We successfully prepared ketone-, 2° alcohol-, TMS-, F-, and N3-containing P3HTs, many of which could not be synthesized by direct GRIM polymerization. The optical spectra of F–P3HT in solution and thin films as well as cyclic voltammetry were investigated to infer the electronic properties, chain configuration, and solid state behavior in comparison to P3HT and Br–P3HT. Additionally, we demonstrated that N3–P3HT could be further derivatized with alkynes using click chemistry. This novel, efficient methodology, and more generally the ability to generate polyanions through lithium−halogen exchange, will allow for the synthesis of many new functional polymers.
Acknowledgments
B.K. appreciates helpful discussions with Dr. Yanchuan Zhao. We thank Chelsea McConnell for preliminary work on the scope of this method. B.K. is grateful to Samsung Scholarship for graduate research support. E.M.S. was supported by a F32 Ruth L. Kirschstein National Research Service Award. This work was funded by grants to T.M.S. from the Institute of Soldier Nanotechnology and the NSF, Center for Energy Efficient Electronics, Award ECCS-0939514.
Supporting Information Available
Characterization of each polymer including used electrophiles, yields, absorption maxima, and emission maxima, control experiments with n-butyl bromide, cyclic voltammetry data, GPC traces, 1H NMR spectra, 19F NMR spectra, and IR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Tsumura A.; Koezuka H.; Ando T. Appl. Phys. Lett. 1986, 49, 1210–1212. [Google Scholar]
- Sirringhaus H.; Tessler N.; Friend R. H. Science 1998, 280, 1741–1744. [DOI] [PubMed] [Google Scholar]
- Ohmori Y.; Uchida M.; Muro K.; Yoshino K. Jpn. J. Appl. Phys. 2 1991, 30, L1938–L1940. [Google Scholar]
- Padinger F.; Rittberger R. S.; Sariciftci N. S. Adv. Funct. Mater. 2003, 13, 85–88. [Google Scholar]
- Yu G.; Gao J.; Hummelen J. C.; Wudl F.; Heeger A. J. Science 1995, 270, 1789–1791. [Google Scholar]
- Günes S.; Neugebauer H.; Sariciftci N. S. Chem. Rev. 2007, 107, 1324–1338. [DOI] [PubMed] [Google Scholar]
- McCullough R. D.; Lowe R. D. J. Chem. Soc., Chem. Commun. 1992, 70–72. [Google Scholar]
- Chen T. A.; Rieke R. D. J. Am. Chem. Soc. 1992, 114, 10087–10088. [Google Scholar]
- Loewe R. S.; Khersonsky S. M.; McCullough R. D. Adv. Mater. 1999, 11, 250–253. [Google Scholar]
- Loewe R. S.; Ewbank P. C.; Liu J.; Zhai L.; McCullough R. D. Macromolecules 2001, 34, 4324–4333. [Google Scholar]
- Ohshimizu K.; Takahashi A.; Rho Y.; Higashihara T.; Ree M.; Ueda M. Macromolecules 2011, 44, 719–727. [Google Scholar]
- Richter T. V.; Braun C. H.; Link S.; Scheuble M.; Crossland E. J. W.; Stelzl F.; Würfel U.; Ludwigs S. Macromolecules 2012, 45, 5782–5788. [Google Scholar]
- Lobez J. M.; Andrew T. L.; Bulović V.; Swager T. M. ACS Nano 2012, 6, 3044–3056. [DOI] [PubMed] [Google Scholar]
- Wang F.; Gu H.; Swager T. M. J. Am. Chem. Soc. 2008, 130, 5392–5393. [DOI] [PubMed] [Google Scholar]
- Wang F.; Yang Y.; Swager T. M. Angew. Chem., Int. Ed. 2008, 47, 8394–8396. [DOI] [PubMed] [Google Scholar]
- Yao K.; Chen L.; Chen Y.; Li F.; Wang P. J. Mater. Chem. 2011, 21, 13780–13784. [Google Scholar]
- Miyanishi S.; Tajima K.; Hashimoto K. Macromolecules 2009, 42, 1610–1618. [Google Scholar]
- Osaka I.; McCullough R. D. Acc. Chem. Res. 2008, 41, 1202–1214. [DOI] [PubMed] [Google Scholar]
- Holcombe T. W.; Woo C. H.; Kavulak D. F. J.; Thompson B. C.; Fréchet J. M. J. J. Am. Chem. Soc. 2009, 131, 14160–14161. [DOI] [PubMed] [Google Scholar]
- Gauthier M. A.; Gibson M. I.; Klok H.-A. Angew. Chem., Int. Ed. 2009, 48, 48–58. [DOI] [PubMed] [Google Scholar]
- Benanti T. L.; Kalaydjian A.; Venkataraman D. Macromolecules 2008, 41, 8312–8315. [Google Scholar]
- Zhai L.; Pilston R. L.; Zaiger K. L.; Stokes K. K.; McCullough R. D. Macromolecules 2003, 36, 61–64. [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. [DOI] [PubMed] [Google Scholar]
- Tornøe C. W.; Christensen C.; Meldal M. J. Org. Chem. 2002, 67, 3057–3064. [DOI] [PubMed] [Google Scholar]
- Li Y.; Vamvounis G.; Holdcroft S. Macromolecules 2001, 34, 141–143. [Google Scholar]
- Li Y.; Vamvounis G.; Yu J.; Holdcroft S. Macromolecules 2001, 34, 3130–3132. [Google Scholar]
- Miyaura N.; Suzuki A. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar]
- Stille J. K. Angew. Chem., Int. Ed. 1986, 25, 508–523. [Google Scholar]
- Beletskaya I. P.; Cheprakov A. V. Chem. Rev. 2000, 100, 3009–3066. [DOI] [PubMed] [Google Scholar]
- Li M. H.; Xu P.; Yang J. G.; Yang S. F. J. Mater. Chem. 2010, 20, 3953–3960. [Google Scholar]
- Chen M. Q.; Li M. H.; Wang H. T.; Qu S. X.; Zhao X. M.; Xie L. X.; Yang S. F. Polym. Chem. 2013, 4, 550–557. [Google Scholar]
- Li M. H.; Xu P.; Yang J. G.; Ying H.; Haubner K.; Dunsch L.; Yang S. F. J. Phys. Chem. C 2011, 115, 4584–4593. [Google Scholar]
- Zhou Z. Y.; Brusso J. L.; Holdcroft S. Chem. Mater. 2010, 22, 2287–2296. [Google Scholar]
- Chen X. W.; Gholamkhass B.; Han X.; Vamvounis G.; Holdcroft S. Macromol. Rapid Commun. 2007, 28, 1792–1797. [Google Scholar]
- Economopoulos S. P.; Chochos C. L.; Gregoriou V. G.; Kallitsis J. K.; Barrau S.; Hadziioannou G. Macromolecules 2007, 40, 921–927. [Google Scholar]
- Zhou Z. Y.; Chen X. W.; Holdcroft S. J. Am. Chem. Soc. 2008, 130, 11711–11718. [DOI] [PubMed] [Google Scholar]
- Sletten E. M.; Nakamura H.; Jewett J. C.; Bertozzi C. R. J. Am. Chem. Soc. 2010, 132, 11799–11805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- rcf = 1.1 × 10–5 × r (cm) × rpm2, where r is the rotor radius.
- Thompson C. M.; Green D. L. C. Tetrahedron 1991, 47, 4223–4285. [Google Scholar]
- With 0.1 equivalent of NBS, 10% of repeat units on P3HT were brominated, and the resulting polymer underwent Li–Br exchange followed by quenching with tosyl azide to append a few percent of azide group on P3HT. This polymer was more soluble than 17 due to fewer azide groups on P3HT.
- Gohier F.; Frère P.; Roncali J. J. Org. Chem. 2013, 78, 1497–1503. [DOI] [PubMed] [Google Scholar]
- Crossland E. J. W.; Rahimi K.; Reiter G.; Steiner U.; Ludwigs S. Adv. Funct. Mater. 2011, 21, 518–524. [Google Scholar]
- Kim J.; Swager T. M. Nature 2001, 411, 1030–1034. [DOI] [PubMed] [Google Scholar]
- Agard N. J.; Prescher J. A.; Bertozzi C. R. J. Am. Chem. Soc. 2004, 126, 15046–15047. [DOI] [PubMed] [Google Scholar]
- Palermo E. F.; Darling S. B.; McNeil A. J. J. Mater. Chem. C 2014, 2, 3401–3406. [Google Scholar]
- Smith P. A. S.; Rowe C. D.; Bruner L. B. J. Org. Chem. 1969, 34, 3430–3433. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.