

Abstract
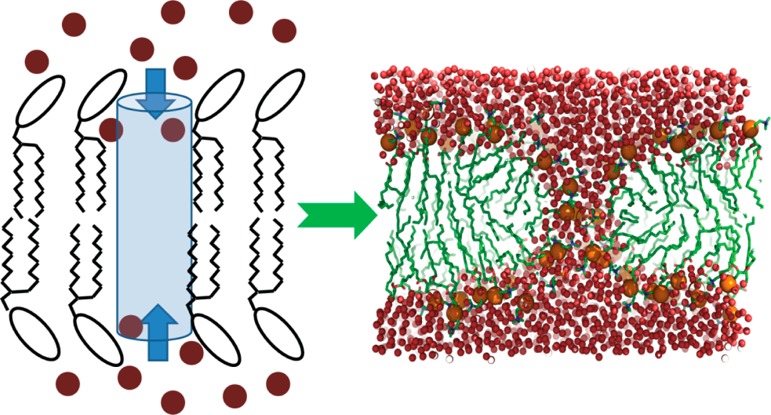
A new reaction coordinate to bias molecular dynamics simulation is described that allows enhanced sampling of density-driven processes, such as mixing and demixing two different molecular species. The methodology is validated by comparing the theoretical entropy of demixing two ideal gas species and then applied to induce deformation and pore formation in phospholipid membranes within an umbrella sampling framework. Comparison with previous biased simulations of membrane pore formation suggests overall quantitative agreement, but the density-based biasing potential results in a different, more realistic transition pathway than that in previous studies.
Introduction
Advanced computational methods have long attracted the attention of biophysicists to shed light on the behavior of biological systems. The computer simulation of proteins, membranes, and nucleic acids are a powerful technique for understanding the physical characteristics of these complex systems.1 Despite advances in computer power, the time scales required for studying many physical phenomena are still beyond the possibilities for the majority of the scientific community. However, the use of enhanced sampling methods2−6 can overcome such limitations. One example where enhanced sampling is needed is the pore formation and deformation of lipid membranes.7−16 Pore formation is involved in a variety of biological processes, such as signal transduction and small molecule transports,7−9,16 but it is also highly relevant in the context of toxins and antimicrobial peptides that induce membrane pores to cause cell leakage and ultimately kill cells.17−19
A common strategy for overcoming kinetic barriers is the use of umbrella sampling techniques,20 where a main challenge is the choice of a suitable reaction coordinate. Geometric properties such as distances, angles, or dihedrals between groups of atoms have been widely used, but some physical processes are not described well by such simple reaction coordinates. As a result, enhanced sampling simulations using such coordinates may be less effective for these systems. For example, density-driven processes may not be described well by traditional reaction coordinates. Membrane pore formation is one such process where the application of enhanced sampling methods has been challenging.21 In one previous study, the pore radius was incorporated as a reaction coordinate in a molecular dynamics framework,21,22 and the free energy cost of pore formation was measured using the potential of mean constraint field (PMCF) approach.23 Furthermore, Bennett et al.24 investigated the mechanism of pore formation initially by long equilibrium MD simulations followed by umbrella sampling where a single phosphorus atom in one of the lipids was pulled to the center. However, both choices of the reaction coordinate could be problematic as they make assumptions about how the membrane structure deforms upon pore formation.
A natural reaction coordinate for studying membrane pore formation is the density of water molecules within the membrane in the area where pore formation takes place. Using the water density instead of a structural property of the membrane avoids biasing membrane structure unnecessarily but still provides enhanced sampling across the key kinetic barrier, that is, water penetration into the membrane. Here, we are describing the development of a density-based reaction coordinate and its application in umbrella sampling simulations of membrane pore formation. The method introduced here biases the density of a group of atoms in a volume of interest, such as a cylinder. Therefore, our density biasing potential function not only can be used for studying membrane pores but also is applicable more generally for reaching a target density for a given molecular species relative to another species in any context. This methodology was implemented in the CHARMM biomolecular software package.25
In the remainder of this paper, we will provide a detailed description of the density biasing potential, followed by validation of our method by comparing entropic components of demixing free energy of two ideal gases with theoretical estimates. Then, this method is applied to a pure DPPC membrane bilayer system to demonstrate its potential for estimating free energies of membrane pore formation.
Methods
Density Biasing Potential
In this section, we provide the mathematical basis of the density biasing potential function. Given the coordinates q⃗i for atom i, the total number of atoms in any arbitrary subvolume of interest V can be calculated by integrating the product of a volume function f(r⃗) with the Dirac delta function: δ(r⃗ – q⃗i)f(r⃗) for all atoms:
| 1 |
where f(r⃗) returns one inside V while it switches smoothly to zero on the boundaries and stays zero for all the points outside the volume (see below). In general, any differentiable volume function can be used to define f(r⃗); however, simpler functions are preferred since they are easier to implement in a molecular dynamics framework.
The volume of interest in our study is a cylinder with radius Rcyl and height Zcyl with its axis aligned to the bilayer normal (Figure 1A). Therefore, we use cylindrical coordinates and decompose the volume function into radial and axial components so that
| 2 |
Choosing the switching function as a third degree polynomial used in CHARMM PBEQ26 and GBSW27,28 modules results in the following differentiable volume function:
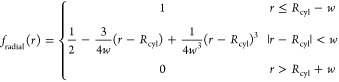 |
3 |
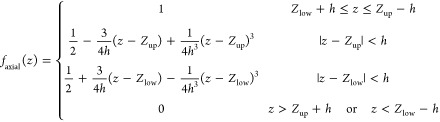 |
4 |
where w and h are the switching distances for the radial and axial terms, respectively. Figure 1B shows the shape of radial component of volume function; the axial component has a similar shape.
Figure 1.
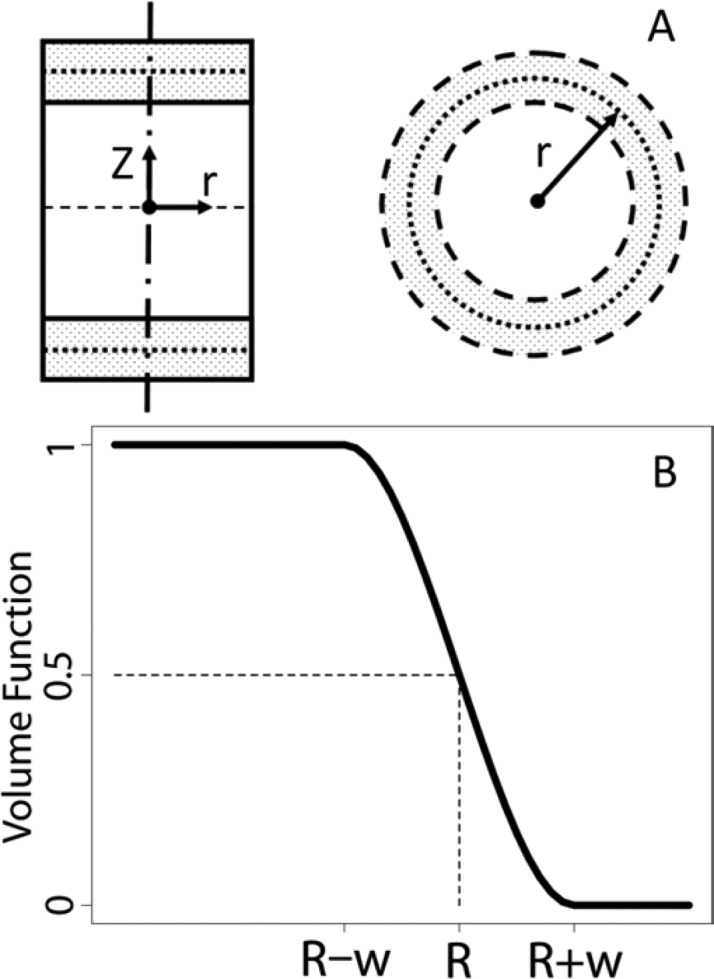
(A) Schematic representation of the biasing cylinder aligned to the bilayer normal. The center of the switching region is indicated with dashed lines. (B) Volume function used in axial and radial directions.
The number density ρV is calculated by normalizing ΓV by the cylinder volume. The potential energy is then calculated for a given value of target density ρt with the force constant k
| 5 |
The corresponding force components can be obtained from the gradient of the potential term
| 6 |
| 7 |
The details of the derivative components are provided in the Appendix.
Simulation Details
Method Validation
For validation of our computational method, we investigated the mixing entropy of two noble gas species. Helium atoms (200) were placed in a box, 40 of which were tagged to make two distinguishable species with identical parameters. The box dimensions were 200 × 200 × 50 Å3. A density biasing cylinder with a radius of 50 Å was placed in the center of the box with the cylinder axis aligned with the z axis. The switching distance in the radial direction was set to 1 Å. The cylinder height was considered to be infinite; therefore, the biasing potential did not vary along the z-axis. The number densities were normalized by the equilibrium number of particles in the cylinder volume. In order to fully separate the two molecular species, the reaction coordinate in the density biasing potential was constructed as the difference between the densities of the tagged and untagged species in the cylinder. In this case, an increase in the value of the reaction coordinate can be due to either increasing the number of tagged species or decreasing the number of untagged ones assuming that the total number of particles in the cylinder is constant on average over time.
For this system, the equilibrium value of the reaction coordinate is −6 × 10–5 Å–3 for the fully mixed state and 1 × 10–4 Å–3 for the fully separated state. Therefore, using umbrella sampling, the reaction coordinate was varied from −5.1 × 10–5 to 7.1 × 10–5 Å–3 in increments of 2.54 × 10–7 Å–3. Each umbrella window was simulated for 20 ns with a force constant of 107 kcal/mol·Å6 and a time step of 2 fs. The last 16 ns from each window were used to construct the PMF as a function of the reaction coordinate using WHAM analysis.
A theoretical estimate of the mixing entropy for two molecular gas species A and B is given by
| 8 |
where x is the mole fraction of each species, n is the total number of moles, and R is the universal gas constant. The total change in entropy is given by
| 9 |
where ntot,V1 and ntot,V2 are the average total number of atoms in volumes V1 and V2 at equilibrium, respectively. In order to compare the theoretical mixing entropy with our computational approach, we evaluated the theoretical estimate as a function of the mole fraction of species A in volume V1 in the process of going from a fully separated state (i) to a partially mixed state (ii) as shown in Figure 2. The mole fraction is then converted to the reaction coordinate (ξ) used in the umbrella sampling simulations according to
| 10 |
Figure 2.
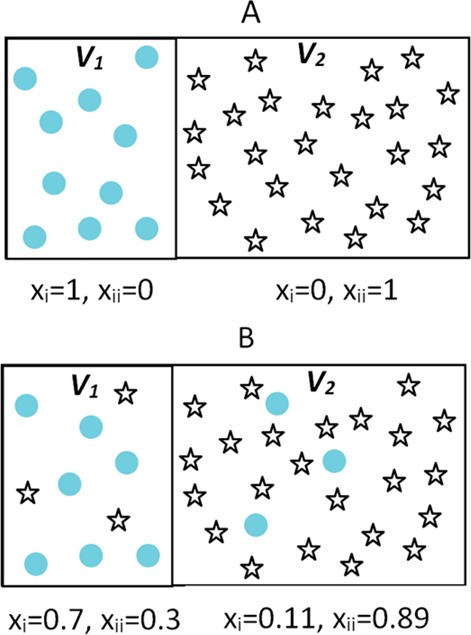
Schematic representation of the mixing process for a simple two-component noble gas mixture that is fully demixed (A) and partially mixed (B).
Membrane Simulations
A pure membrane bilayer was constructed by web-based CHARMM-GUI membrane builder,29 containing 288 dipalmitoylphosphatidylcholine (DPPC) and 8376 water molecules placed in a periodic box of size 95.2 × 95.2 × 66.6 Å3. The x–y dimensions were adjusted to match the experimental value of 63 Å2 for the area per lipid of DPPC in the fluid phase.30,31 The z dimension was chosen large enough to avoid boundary artifacts. The CHARMM36 force field32 was used along with the TIP3 water model.33 Lennard-Jones interactions were cut off at 9 Å (with a switching function beginning at 8 Å). Particle-Mesh Ewald summation34 was used for long-range electrostatic interactions with a 9 Å cutoff for the direct sum. A time step of 2 fs was used in combination with the SHAKE algorithm.35 The initially flat bilayer was heated in steps at 50, 100, 200, 250, and 323 K, each for 100 ps with a Nosé–Hoover thermostat and barostat (target pressure of 1 bar) to maintain an NPT ensemble. The center of mass of the bilayer was restrained to the plane at z = 0 with a force constant of 100 kcal/mol·Å2. The final equilibrated system was then used to study membrane deformation and pore formation with our density biased sampling method.
One-Sided Deformation of a Membrane Bilayer
The density biasing approach was applied to the DPPC membrane bilayer system. A cylinder with a radius of 8 Å was aligned to the bilayer normal (z) axis. The cylinder spanned from z = −2.5 Å to z = +15 Å, and the radial and axial switching distances were set to 1 and 5 Å, respectively. Umbrella sampling simulations were performed with 10 windows, increasing the number density of water molecules per unit area in the cylinder from 1.1 × 10–3 to 2.17 × 10–2 Å–3. A force constant of 9.2 × 105 kcal/mol·Å6 was used. To prevent deformation in the lower leaflet, a plane potential with a force constant of 100 kcal/mol·Å2 was applied to the phosphates of the lower leaflet if their distance to bilayer center was less than 8 Å. Each umbrella was simulated for 50 ns.
Pore Formation in a Membrane Bilayer
In order to create a pore in a membrane bilayer, we expanded the cylinder from the previous case to cover both leaflets, that is, from z = −18 Å to z = +18 Å. The radius of the cylinder was chosen as r = 6 Å, and the radial and axial switching distances were set to 2 and 8 Å, respectively. The parameters were adjusted based on initial trial simulations in order to achieve double-sided pore formation. Twenty umbrella windows were used to vary the number density of water molecules in the cylinder from 6.7 × 10–3 Å–3 to 2.25 × 10–2 Å–3, using a force constant of 5.18 × 106 kcal/mol·Å6. Each umbrella was simulated for 50 ns. The total simulation time for pore formation was 1 μs.
Parameter Selection
While our method can be used for a diverse set of applications, the biasing potential parameters would have to be adjusted accordingly. We will provide guidance here how to choose the two key parameters, height and radius, for the case of a cylindrical biasing volume.
Generally, the cylinder height should encompass and extend beyond the region where the density is meant to be changed. For membrane simulations, a short cylinder height would be appropriate to induce one-sided deformation while longer cylinders are necessary to induce transmembrane pores. Furthermore, for one-sided deformations, the lower bound of the cylinder was fixed at z = −2.5 Å to let water molecules reach the bilayer center without forming complete pores. In the helium gas demixing simulations, the cylinder height was chosen bigger than the box size to avoid gradients along the z axis.
The cylinder radius should be chosen large enough so that the cylinder extends beyond the pore or deformation that is meant to be formed. Otherwise, the biasing potential may affect the shape of the deformation. On the other hand, a cylinder radius that is too large may not be effective in inducing pore formation because large membrane deformation could also satisfy a bias toward increased water densities within the cylinder. Because it was not entirely clear a priori which radius and cylinder height would be optimal, we conducted a series of test simulations with varying radii and cylinder heights until pore formation was accomplished successfully.
Finally, the force constants and window spacing were optimized by trial error. We found that the final values were similar as those predicted by the criterion given by Park and Im.36
Implementation
The density biasing method using a cylinder-based volume function was implemented in the CHARMM biomolecular software package,25 version c40a1. Although not implemented so far, it would be easy to extend the method to other geometries such as a rectangular box with switching regions on each edge or a spherical geometry.
Results and Discussion
Mixing Entropy of Two-Component Gas
The free energy cost of separating two noble gas species was calculated using theoretical and computational methods. Since the two species have identical properties, there is no change in the mean of internal energy of the system upon separating the two species. Figure 3 compares theoretical estimates of −TΔS according to eqs 9 and 10 with the change in free energy computed using the density biased sampling method. The reference point in this figure is the fully mixed state that has the highest entropy. This state corresponds to a mole fraction of xA = 0.22. If the theoretical estimate assumes a perfectly uniform particle distribution to obtain the number of particles in the cylinder (Figure 3, theory A), the ΔG from the simulation underestimates the theory significantly. The agreement improves when the actual average number of particles in the umbrella windows that corresponds to the demixed states is used in the theoretical estimate (Figure 3, theory B). The remaining small discrepancy is due to a non-negligible virial term that results from a pressure difference inside and outside the cylinder during the umbrella simulations in response to the application of the biasing potential. A correction by adding −Δ(PV), calculated from the average external pressures from simulations of the fully mixed and fully demixed states as reported by CHARMM, brings the theoretical and simulation estimates in near-perfect agreement. We note that the simulated system is not an ideal gas because of weak attractive interactions and volume exclusion effects as a result of the Lennard-Jones interaction potential. This would lead to a small correction of the theoretical estimate that is expected to be smaller or on the same order as the uncertainties in the free energies obtained from the simulations. Therefore, the simple test case validates the density biasing potential introduced here.
Figure 3.
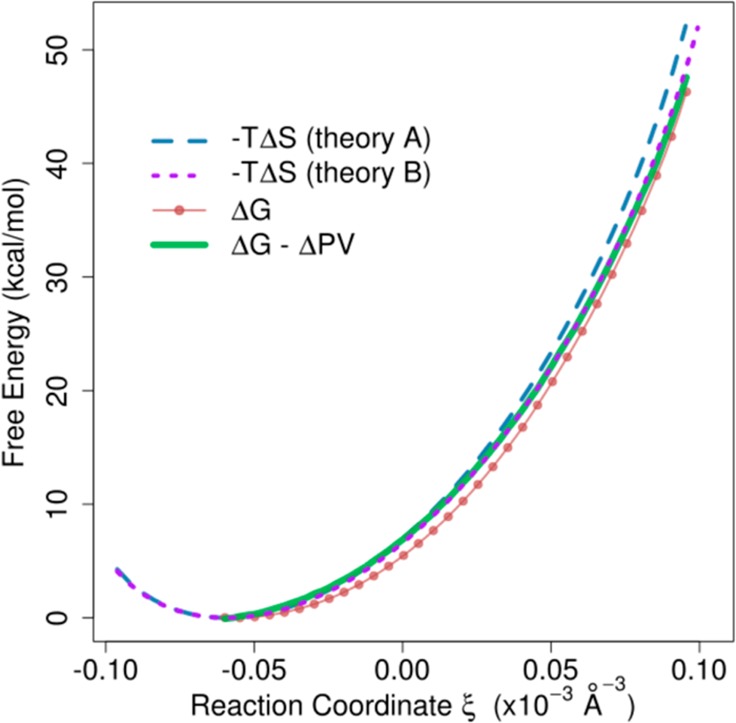
Free energy cost of mixing two noble gas species as a function of the biasing reaction coordinate ξ based on theory (mixing entropy) and simulation (free energy calculated from umbrella sampling simulations). Theory A is using uniform density to estimate total number of particle in cylinder, whereas theory B uses the empirical average number of particles observed during the simulations.
Membrane Simulations
We will now demonstrate the application of the density biasing approach to simulations of membrane bilayers. As described in detail in the Methods section, the density biasing potential was applied to water molecules within a cylinder encompassing a section of a phospholipid bilayer. Figure 4 demonstrates how local membrane thickness, calculated as the average z coordinate of the phosphorus atoms in a cylinder of radius 8 Å, responds to the water density in the cylinder when varied in umbrella sampling simulations. The strong correlation reaffirms that water density within the bilayer is a suitable reaction coordinate for inducing membrane deformations. Figure 5 shows snapshots of the membrane bilayer after 50 ns molecular dynamics simulation with the density biasing potential set to increasing target values. The increasing degree of membrane deformation is readily apparent and we note that the deformation appears to proceed with a slight bending on both leaflets (Figure 5C), presumably because this lowers the overall free energy for these intermediate states. However, a further increase in the water density results in a pronounced one-sided deformation with little apparent perturbation on the opposing leaflet. This is shown in Figure 5F. Another feature of the deformation process is that it progresses from an initially wide and shallow deformation to a narrow and deep deformation, presumably due to a balance between the elastic properties of the membrane bilayer and the free energy costs of forming water defects within the membrane. The deformation of the bilayer is further quantified in Figure 6A, where the bilayer thickness at the deformation location is shown for each umbrella window. The first umbrella is simulated with equilibrium flat bilayer conditions, therefore, no deformation is observed. However, other umbrellas increase the density of water molecules, which induces a deformation in bilayer. From the umbrella sampling, a free energy profile was obtained by weighted histogram analysis method (WHAM).37 Figure 6B shows the resulting potential of mean force (PMF) as a function of the water density in the cylinder. As would be expected, increasing the number of water molecules, and thereby deforming the bilayer, is highly unfavorable in terms of free energy with a cost exceeding 40 kcal/mol for a one-sided water defect that extends to the center of the membrane. As shown below, the cost of forming a pore is about half, so that without any restraints on the lower leaflet (see Methods section), the bilayer would not be expected to stably maintain a one-side deformation.
Figure 4.
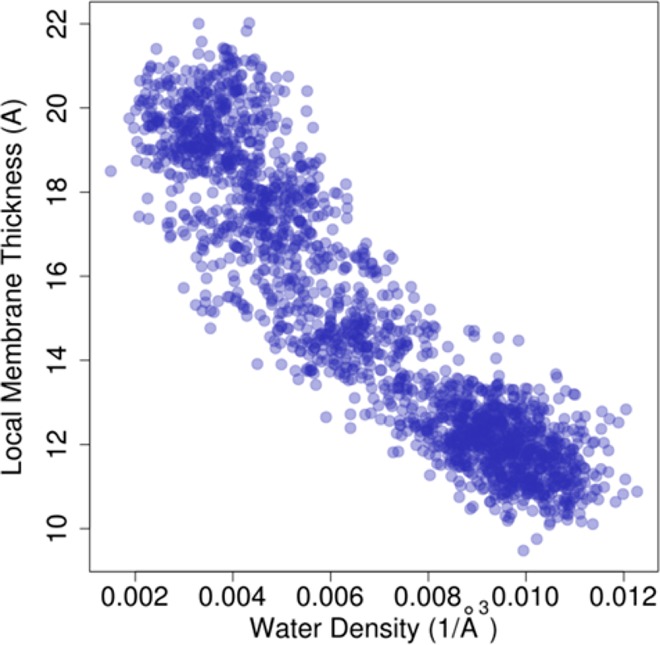
Local membrane bilayer thickness of the upper leaflet vs water density per unit volume from biased sampling of one-sided membrane deformations.
Figure 5.

Snapshots illustrating the one-sided deformation process from a flat bilayer state to a fully deformed state at water densities of 0.0016 (A), 0.0073 (B), 0.0111 (C), 0.0143 (D), 0.0167 (E), and 0.0170 Å–3 (F). Red spheres represent water molecules, brown spheres represent phosphorus atoms of the lipids, and lipid tails are shown in green.
Figure 6.
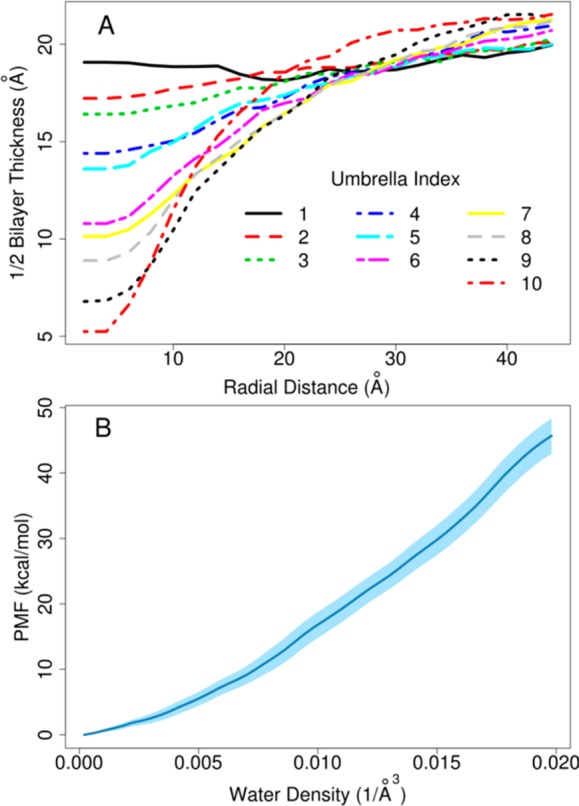
(A) Average bilayer thickness in radial slabs for each umbrella window as a function of radial distance from the pore center. (B) Free energy profile for one sided bilayer deformation as a function of water density in the cylinder. Standard error values obtained by calculating the PMF profiles over 10 2 ns subsets from the umbrella sampling simulation are shown as light blue shades.
Finally, we applied the density biasing method across the entire DPPC bilayer in order to induce pore formation. Snapshots of the bilayer after 50 ns molecular dynamics simulations with increasing water density biases are shown in Figure 7. Similar to what has been described previously,24 pore formation starts by bending both leaflets inward. A water wire forms initially (Figure 7D). The lipid head groups then rearrange and form the familiar hourglass shape of a stable pore once a critical pore radius is passed (Figure 7E). A transition involving an initial water wire is consistent with results from the equilibrium simulations by Bennett et al.24 The average number density profiles of water molecules across the bilayer normal for a flat bilayer and a bilayer with a stable pore (with average water density of 0.0216 Å–3) are compared in Figure 8. By integrating over the difference between the two curves, it is found that 148 water molecules exist in the pore. This result is comparable with the 124 water molecules obtained by Leontiadou et al.,9 in which they applied mechanical stress (surface tension) to form a pore in a DPPC bilayer.
Figure 7.
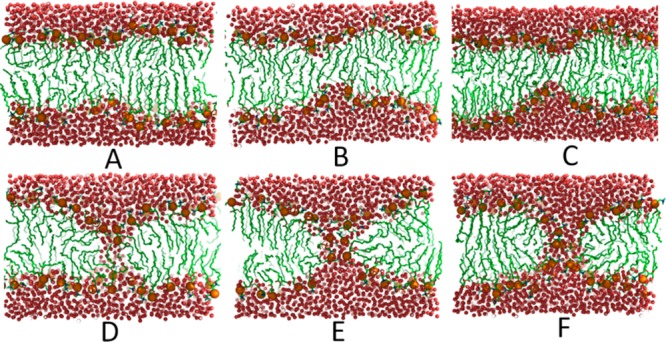
Snapshots illustrating the pore formation process from a flat bilayer state to a stable pore at water densities of 0.0067 (A), 0.0144 (B), 0.0159 (C), 0.0168 (D), 0.0196 (E), and 0.0222 Å–3 (F) with coloring as in Figure 5.
Figure 8.
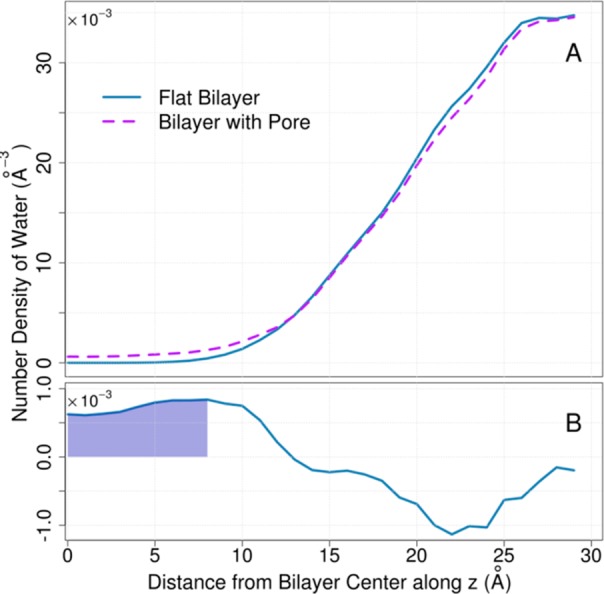
Number density of water molecules across bilayer normal compared between a flat bilayer and a bilayer with a stable pore (A) , and their differences (B).
We computed the pore size by assuming perfect cylindrical shape between z = −8 and z = 8 Å and a uniform water density in that region. The average number of water molecules in the region was found to be 117.7 in the last umbrella. The resulting pore radius is found to be 8.8 Å. Similar analyses assuming perfect cylinder for water wire result in pore radius of 4.2 Å.
Figure 9 shows the PMF of pore formation as a function of water density in the aforementioned cylinder. Again, pore formation is energetically unfavorable as expected. A plateau free energy of 22.2 (± 0.4) kcal/mol is reached at a critical water density of 0.018 Å–3 once a stable pore is formed. This result is close to the value of 19.02 kcal/mol reported by Bennett et al. for DPPC.24 The agreement is excellent, especially when considering differences in force fields. We further decomposed the free energy into enthalpic and entropic contributions. The change in enthalpy is estimated by computing the average potential energy of the system, and we found that pore formation is enthalpically favorable by 46 ± 1 kcal/mol. The simple demixing test case above suggests that there may be an additional ΔPV term but for a partially demixed system the contribution is estimated to be less than 1 kcal/mol and it is therefore neglected here. This implies an entropic cost (−TΔS) of pore formation of about 68 kcal/mol.
Figure 9.
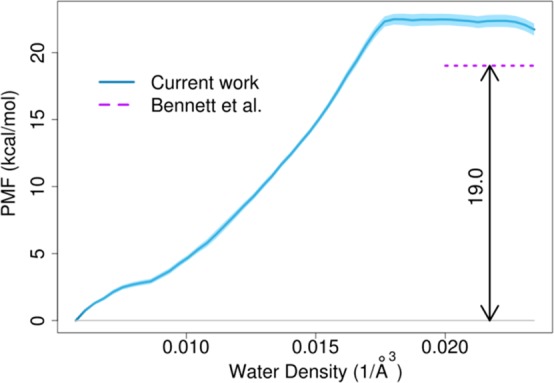
Free energy of pore formation as a function of water density in the cylinder from density-biased sampling with errors indicated as in Figure 6B. A previous result from Bennett et al. is shown for comparison.
As mentioned above, one motivation for inducing membrane pores via water density biasing rather than biasing the membrane structure directly was to avoid artifacts that could affect the pore formation pathway and thereby the energy profiles obtained from umbrella sampling. Figure 10 compares the water density (ξ), our reaction coordinate that imposes minimal bias on the membrane structure, with the average distance of the two closest phosphates from the bilayer center (λ). The latter relates to previous biased simulation studies where the distance of a single phosphate group from the bilayer center was used.24 Poor correlation between the two reaction coordinates suggests that there could be mechanistic differences when either of the two reaction coordinates is used to induce pore formation. With the density biasing term, a typical transition path (indicated in red in Figure 10) would delay a transition of phosphates to the bilayer center until a critical water density is reached at which point there is a sharp, cooperative transition that leads to formation of a full pore. On the other hand, we speculate that forming the pore by pulling down a phosphate group would follow a path indicated in green in Figure 10 where phosphates approach the center of the bilayer early and a sharp, cooperative transition could be absent. Figure 11 shows two intermediate conformations with extreme low λ values that may be intermediates on such a transition path. In these conformations, the membrane exhibits large deformations on one leaflet, and the water molecules are dragged into the center along with the lipid headgroups, as shown in Figure 11. Since free energies are state functions, overall energies of pore formation are, of course, independent of the path taken. However, the free energy profile along the transition path and any mechanistic insight obtained from such simulations does depend on the path taken as a result of the biasing potential.
Figure 10.
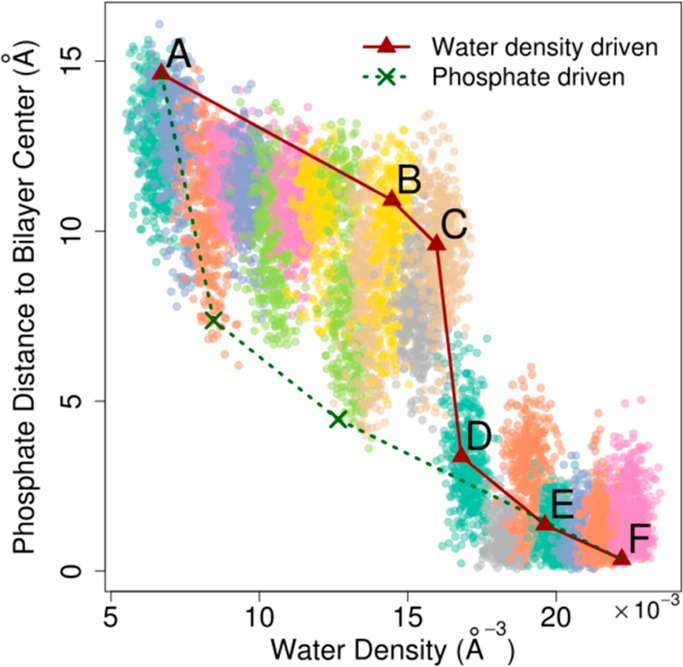
Average z coordinate of the two closest lipid phosphates from the bilayer center vs water density within pore cylinder illustrating different mechanisms between density-driven and phosphate-driven pore formation bias. Sampling from each umbrella is shown in different colors.
Figure 11.
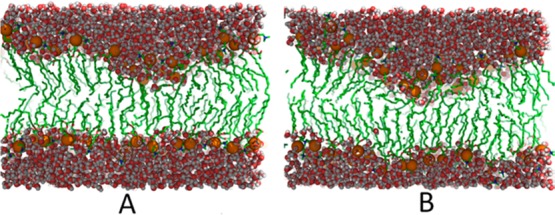
Intermediate bilayer states with low average distance of phosphates to the bilayer center.
The proposed method in this work applies a minimal bias to induce a pore in membrane. There is no assumption made about the shape of the pore or the density distribution inside the cylinder. However, the performance of this method is sensitive to the choice of cylinder parameters as described above. Therefore, we believe that this method is more universally applicable to membrane pore formation and deformations in response to interactions with other molecules, especially in cases where it is not clear a priori how exactly the membrane responds to such molecules.
The variation of the water density in our method is reminiscent of grand canonical ensemble methods38,39 that have been widely used to simulate the mixing process of model fluids.40,41 However, because demixing and bilayer pore formation processes maybe either thermodynamically unfavorable or kinetically hindered, enhanced sampling techniques such as umbrella sampling would still be required. Furthermore, a global variation of the chemical potential for water in a membrane-bilayer system may not necessarily lead to pore formation since water molecules could be added in the bulk region while a targeted change of a local chemical potential would eventually result in a method similar to ours but with the additional complications of the grand-canonical machinery.
Finally, while the method presented here focuses on overcoming the kinetic barriers in creating membrane deformations and pores, it may not fully address overcoming the slow relaxation times of lipid motions. Therefore, mechanistic studies of membrane pore formation would likely require longer simulations and/or a combination with other enhanced sampling techniques such as replica exchange sampling that can accelerate lipid motions to guarantee full convergence of deformed bilayer systems.
Conclusions
We have developed a new computational technique to bias the density of a group of molecular species, or the difference in densities of two molecular groups. The method was validated for the case of demixing two ideal gas species. Furthermore, we applied the new biasing term in the context of membrane pore formation. We believe that biasing the water density rather than structural properties of the membrane is less likely to introduce artifacts. Furthermore, the density biasing approach allows the study of one-sided deformations which has not been described with umbrella sampling techniques previously. The density biasing function is also more broadly applicable to any system involving the mixing or demixing of molecular species with respect to each other. Possible applications include lipid raft formation, cosolvent effects, and studies of concentration gradients in complex systems.
Acknowledgments
This work was supported in part by National Institute of Health Grant GM084953. Computational resources were provided by XSEDE facilities (TG-MCB090003) and High Performance Computing Center at Michigan State University.
Appendix
Derivative components of the volume function
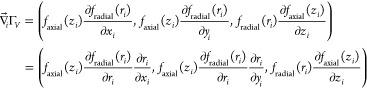 |
The radial component:
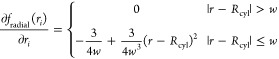 |
The axial component:
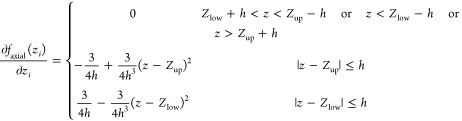 |
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Karplus M.; McCammon J. A. Molecular dynamics simulations of biomolecules. Nat. Struct. Mol. Biol. 2002, 99646–652. [DOI] [PubMed] [Google Scholar]
- Marrink S. J.; Risselada H. J.; Yefimov S.; Tieleman D. P.; de Vries A. H. The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111277812–7824. [DOI] [PubMed] [Google Scholar]
- Panahi A.; Feig M. Dynamic heterogeneous dielectric generalized Born (DHDGB): An implicit membrane model with a dynamically varying bilayer thickness. J. Chem. Theory Comput. 2013, 931709–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig M.Implicit membrane models for membrane protein simulation. In Molecular Modeling of Proteins; Kukol A., Ed.; Humana Press: Totowa, 2008; Vol. 443, pp 181–196. [DOI] [PubMed] [Google Scholar]
- Ohkubo Y. Z.; Pogorelov T. V.; Arcario M. J.; Christensen G. A.; Tajkhorshid E. Accelerating membrane insertion of peripheral proteins with a novel membrane mimetic model. Biophys. J. 2012, 10292130–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannigan G.; Philips P. F.; Brown F. L. H. Flexible lipid bilayers in implicit solvent. Phys. Rev. E 2005, 721011915. [DOI] [PubMed] [Google Scholar]
- Böckmann R. A.; de Groot B. L.; Kakorin S.; Neumann E.; Grubmüller H. Kinetics, statistics, and energetics of lipid membrane electroporation studied by molecular dynamics simulations. Biophys. J. 2008, 9541837–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtovenko A. A.; Anwar J.; Vattulainen I. Defect-mediated trafficking across cell membranes: Insights from in silico modeling. Chem. Rev. 2010, 110106077–6103. [DOI] [PubMed] [Google Scholar]
- Leontiadou H.; Mark A. E.; Marrink S. J. Molecular dynamics simulations of hydrophilic pores in lipid bilayers. Biophys. J. 2004, 8642156–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litster J. D. Stability of lipid bilayers and red blood cell membranes. Phys. Lett. A 1975, 533193–194. [Google Scholar]
- Sachs J. N.; Crozier P. S.; Woolf T. B. Atomistic simulations of biologically realistic transmembrane potential gradients. J. Chem. Phys. 2004, 1212210847. [DOI] [PubMed] [Google Scholar]
- Tieleman D. The molecular basis of electroporation. BMC Biochem. 2004, 511–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieleman D. P.; Leontiadou H.; Mark A. E.; Marrink S.-J. Simulation of pore formation in lipid bilayers by mechanical stress and electric fields. J. Am. Chem. Soc. 2003, 125216382–6383. [DOI] [PubMed] [Google Scholar]
- Weaver J. C.; Chizmadzhev Y. A. Theory of electroporation: A review. Bioelectrochem. Bioenerg. 1996, 412135–160. [Google Scholar]
- Delemotte L.; Tarek M. Molecular dynamics simulations of lipid membrane electroporation. J. Membr. Biol. 2012, 2459531–543. [DOI] [PubMed] [Google Scholar]
- Levine Z.; Vernier P. T. Life cycle of an electropore: Field-dependent and field-independent steps in pore creation and annihilation. J. Membr. Biol. 2010, 236127–36. [DOI] [PubMed] [Google Scholar]
- Arnusch C. J.; Pieters R. J.; Breukink E. Enhanced membrane pore formation through high-affinity targeted antimicrobial peptides. PLoS One 2012, 76e39768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogden K. A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria?. Nat. Rev. Microbiol. 2005, 33238–250. [DOI] [PubMed] [Google Scholar]
- Lee C.-C.; Sun Y.; Qian S.; Huang Huey W. Transmembrane pores formed by human antimicrobial peptide LL-37. Biophys. J. 2011, 10071688–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrie G. M.; Valleau J. P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 232187–199. [Google Scholar]
- Tolpekina T. V.; den Otter W. K.; Briels W. J. Nucleation free energy of pore formation in an amphiphilic bilayer studied by molecular dynamics simulations. J. Chem. Phys. 2004, 1212312060. [DOI] [PubMed] [Google Scholar]
- Wohlert J.; den Otter W. K.; Edholm O.; Briels W. J. Free energy of a trans-membrane pore calculated from atomistic molecular dynamics simulations. J. Chem. Phys. 2006, 12415154905. [DOI] [PubMed] [Google Scholar]
- Guàrdia E.; Rey R.; Padró J. A. Potential of mean force by constrained molecular dynamics: A sodium chloride ion pair in water. Chem. Phys. 1991, 1552187–195. [Google Scholar]
- Bennett W. F. D.; Sapay N.; Tieleman D. P. Atomistic simulations of pore formation and closure in lipid bilayers. Biophys. J. 2014, 1061210–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B. R.; Brooks C. L.; Mackerell A. D.; Nilsson L.; Petrella R. J.; Roux B.; Won Y.; Archontis G.; Bartels C.; Boresch S.; Caflisch A.; Caves L.; Cui Q.; Dinner A. R.; Feig M.; Fischer S.; Gao J.; Hodoscek M.; Im W.; Kuczera K.; Lazaridis T.; Ma J.; Ovchinnikov V.; Paci E.; Pastor R. W.; Post C. B.; Pu J. Z.; Schaefer M.; Tidor B.; Venable R. M.; Woodcock H. L.; Wu X.; Yang W.; York D. M.; Karplus M. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30101545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im W.; Beglov D.; Roux B. Continuum solvation model: Computation of electrostatic forces from numerical solutions to the Poisson–Boltzmann equation. Comput. Phys. Commun. 1998, 1111–359–75. [Google Scholar]
- Im W.; Lee M. S.; Brooks C. L. Generalized Born model with a simple smoothing function. J. Comput. Chem. 2003, 24141691–1702. [DOI] [PubMed] [Google Scholar]
- Im W.; Feig M.; Brooks C. L. An implicit membrane generalized Born Theory for the study of structure, stability, and interactions of membrane proteins. Biophys. J. 2003, 8552900–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Kim T.; Iyer V. G.; Im W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29111859–1865. [DOI] [PubMed] [Google Scholar]
- Kučerka N.; Tristram-Nagle S.; Nagle J. F. Closer look at structure of fully hydrated fluid phase DPPC bilayers. Biophys. J. 2006, 9011L83–L85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle J. F.; Tristram-Nagle S. Structure of lipid bilayers. Biochim. Biophys. Acta, Rev. Biomembr. 2000, 14693159–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauda J. B.; Venable R. M.; Freites J. A.; O’Connor J. W.; Tobias D. J.; Mondragon-Ramirez C.; Vorobyov I.; MacKerell A. D.; Pastor R. W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114237830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark P.; Nilsson L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105439954–9960. [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 981210089. [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. C. Numerical integration of the Cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 233327–341. [Google Scholar]
- Park S.; Im W. Two dimensional window exchange umbrella sampling for transmembrane helix assembly. J. Chem. Theory Comput. 2013, 9113–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Rosenberg J. M.; Bouzida D.; Swendsen R. H.; Kollman P. A. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 1381011–1021. [Google Scholar]
- Çagin T.; Pettitt B. M. Grand molecular dynamics: A method for open systems. Mol. Simul 1991, 61–35–26. [Google Scholar]
- Eslami H.; Müller-Plathe F. Molecular dynamics simulation in the grand canonical ensemble. J. Comput. Chem. 2007, 28101763–1773. [DOI] [PubMed] [Google Scholar]
- Wang H.; Hartmann C.; Schütte C.; Delle Site L. Grand-canonical-like molecular-dynamics simulations by using an adaptive-resolution technique. Phys. Rev. X 2013, 31011018. [Google Scholar]
- Ji J.; Çaǧin T.; Pettitt B. M. Dynamic simulations of water at constant chemical potential. J. Chem. Phys. 1992, 9621333. [Google Scholar]