

Abstract
The brain vasculature has been increasingly recognized as a key player that directs brain development, regulates homeostasis, and contributes to pathological processes. Following ischemic stroke, the reduction of blood flow elicits a cascade of changes and leads to vascular remodeling. However, the temporal profile of vascular changes after stroke is not well understood. Growing evidence suggests that the early phase of cerebral blood volume (CBV) increase is likely due to the improvement in collateral flow, also known as arteriogenesis, whereas the late phase of CBV increase is attributed to the surge of angiogenesis. Arteriogenesis is triggered by shear fluid stress followed by activation of endothelium and inflammatory processes, while angiogenesis induces a number of pro-angiogenic factors and circulating endothelial progenitor cells (EPCs). The status of collaterals in acute stroke has been shown to have several prognostic implications, while the causal relationship between angiogenesis and improved functional recovery has yet to be established in patients. A number of interventions aimed at enhancing cerebral blood flow including increasing collateral recruitment are under clinical investigation. Transplantation of EPCs to improve angiogenesis is also underway. Knowledge in the underlying physiological mechanisms for improved arteriogenesis and angiogenesis shall lead to more effective therapies for ischemic stroke.
Keywords: arteriogenesis, angiogenesis, collateral flow, fluid shear stress, pro-angiogenic factors, inflammation, macrophage polarization
1. Collateral circulation and arteriogenesis as the new therapeutic targets of cerebral ischemia
Stroke or cerebral ischemia leads to cerebrovascular adaptations both acutely and chronically. During acute flow obstruction, some parts of the brain tissue suffer from ischemia while others are sustained by collateral flow through pre-existing anastomoses. Collateral circulation refers to the supplemental network of vessels that compensate blood flow and offset the adverse effect of ischemia (Kucinski et al., 2003, Liebeskind, 2003, 2005b, a, Shuaib et al., 2011b, Liebeskind, 2012). Collateral flow recruitment plays a critical role in maintaining regional cerebral blood flow upon reduction of cerebral perfusion pressure or failure of cerebral autoregulation (Ozgur et al., 2001, Liebeskind, 2003). Mounting clinical evidence suggests that the collateral status is an independent predictor of outcome and response to recanalization therapies in patients with ischemic stroke.
Arteriogenesis, the development of functional collateral flow from pre-existing arterial anastomoses, is a process distinct from angiogenesis (Schaper and Buschmann, 1999, Schaper, 2009). Arteriogenesis is induced by mechanical forces such as shear stress and circumferential wall stress, and its induction is independent of a hypoxia state. By contrast, angiogenesis involves the proliferation of endothelial cells and formation of new vessels, and can potentially increase the total resistance of the vascular bed of the afflicted artery (Troidl and Schaper, 2012), and thus may be of limited value for the acute functional replacement of occluded arteries. As substrates of arteriogenesis, collateral circulation plays a crucial role in determining the outcome and severity of ischemic injury. From this perspective, understanding of collateral anatomy and regulation is essential for the development of effective stroke interventions. Due to recent progress in neuroimaging, we now have a fair understanding of the anatomy of human cerebral collateral circulation and its role in mediating pathophysiology as well as treatment efficacy in cerebrovascular diseases (Liebeskind, 2003, Shuaib et al., 2011b, McVerry et al., 2012). Highlights of experimental studies including those in non-cerebral collaterals will also be discussed to complement findings in humans, as the former are particularly instrumental in revealing the molecular mechanisms underlying collateral flow development and regulation and their potential application to the latter (Fung and Helisch, 2012, la Sala et al., 2012, Troidl and Schaper, 2012). Figures of mouse intracranial arterial collateral circulation are shown for comparison (Figure 1). However, due to physiological differences between species, care should be taken when extrapolating data between humans and animals (Liebeskind, 2008).
Figure 1. Murine intracranial arterial collateral circulation in ventral (A) and dorsal (B) views.

A), A number of major cerebral arteries are interconnected to form the Circle of Willis, as the primary collateral circulation in the mouse brain. In addition, posterior communicating artery (PcomA) forms anastomosis between superior cerebellar artery (SCA) and posterior cerebral artery (PCA), connecting the anterior and posterior cerebral circulation. Similar to the function of anterior communicating artery in humans, the Azygos anterior cerebral artery (Azygos ACA) bridges two ACAs in C57BL/6 mice, providing collateral circulation between two cerebral hemispheres. B), Leptomeningeal anastomoses between ACA and middle cerebral artery (MCA) (red), between ACA and PCA (green) and between PCA and MCA (blue). ICA: internal carotid artery; BA: basilar artery; VA: vertebral artery; OA: olfactory artery. The axes indicate the anterior (A) and posterior (P) directions.
1.1 The anatomy of collateral circulation
A number of collateral circuits exist between the extracranial and intracranial circulations, making the external carotid artery (ECA) a potential source of collateral flow when the internal carotid artery (ICA) experiences chronic stenosis or occlusion. The more proximal connecting collaterals from ECA branches to the brain include connections through the ophthalmic and superficial temporal arteries to the intracranial vessels. Distal anastomoses form at the cervical level between the vertebral arteries and muscular branches of the ECA, and between the spinal arteries and branches of the proximal intracranial arteries (Liebeskind, 2003, 2005b, Krishnaswamy et al., 2010).
In humans, it is generally recognized that the circle of Willis may be the primary source of intracranial collateral pathway. Secondary collaterals can be accessed through the ophthalmic artery and leptomeningeal vessels when collateral flow through the circle of Willis is inadequate. Apart from distributing blood to the main branches of the ICA, the circle of Willis also provides connecting collaterals among the branching arteries (Alpers et al., 1959). For example, the anterior communicating artery (AComA) bridges the two anterior cerebral arteries (ACAs) from the two hemispheres, while the posterior communicating artery (PComA) connects the anterior and posterior circulations via the ICA and posterior cerebral artery (PCA). The retrograde flow of the proximal anterior cerebral artery supports the anterior portion of the circle of Willis, while the proximal posterior cerebral arteries compensate flow between hemispheres at the posterior portion of the circle of Willis (Liebeskind, 2003, Shuaib et al., 2011b).
Retrograde flow via the ophthalmic artery constitutes an important source of collateral flow second to the Willisian collaterals. The leptomeningeal anastomoses potentially provide bidirectional arterial blood to the cortical surface, connecting between ACA and middle cerebral artery (MCA), MCA and PCA, and to a lesser extent, PCA and ACA (Brozici et al., 2003, Krishnaswamy et al., 2010). The small arteriolar connections within the leptomeningeal anastomoses linking distal parts of the major cerebral arteries are important routes for retrograde collateral flow during vessel occlusion. Thus, the collateral status in the leptomeningeal anastomoses is a strong predictor of outcome in stroke patients (Lima et al., 2010).
The anatomy of the collateral circulation varies between individuals suggesting a contribution of genetic elements. A significant portion of the human population possesses an incomplete configuration of the circle of Willis, including the most common abnormalities involving the absence or hypoplasia of either PComA or proximal ACA in 30% and 10% of the human population, respectively (Liebeskind, 2003). Substantial variability in size, number, and location of connecting collaterals were also observed in the leptomeningeal anastomoses between individuals (Vander Eecken, 1954). The contribution of genetic factors in the development of collateral vessels and stroke outcome is well supported by comparing mice of various strains. For example, the relatively poor patency of the PComA in the C57Bl/6 mice was associated with a larger infarct size and hypoperfusion of the PCA territory following MCA occlusion (Akamatsu et al., 2012). However, the C57BL/6 strain has a more robust collateral connection between the ACA and MCA compared to Balb/C strain as evidenced by vessel anatomy and retrograde recruitment of the collateral flow (Figure 2) (Defazio et al., 2011). Interestingly, genetic factors are not the only determinants for collateral status. A recent case suggests that at least PComA hypoplasia is not a static feature, as the flow conductance improved over the course of chronic stenosis (Hartkamp et al., 1999). In addition, PComA dilatation occurring as a result of moyamoya disease may indicate disease progression and portend hemorrhagic sequelae (Morioka et al., 2003). Lastly, although some collaterals such as leptomeningeal anastomoses may be anatomically present, the recruitment and conductance of these vessels are influenced by pathophysiological factors (Menon et al., 2013), which can only be appreciated following a thorough understanding of the mechanisms regulating normal collateral flow under physiological state.
Figure 2. Strain differences in leptomeningeal collateral circulation.

A). The number of connecting collaterals between ACA and MCA is significantly less in the Balb/C compared to C57BL/6 mice (p<0.0001). B). Representative DiI labeling images of Balb/C and C57BL/6 strains at baseline. Magnified views inside of the yellow grids are shown in the right panel. Contrary to the abundant connecting collaterals between MCA and ACA networks seen in the C57BL/6 mice, very few connecting collaterals were found in the Balb/C strain. C). Representative Doppler Optical Coherence Tomography (DOCT) images of MCA and ACA branches at baseline, one hour after MCAO and one hour after reperfusion in C57BL/6 and Balb/C mice. The direction of cerebral blood flow is color-coded, with the blood flowing towards the DOCT probe beam coded as red, and the opposite direction as green. At baseline, the flow direction of MCA and ACA was coded as red and green, respectively. During MCAO, some branches of MCA were filled by ACA retrograde flow via the tortuous anastomoses in the C57BL/6 mouse only, with a reversal of flow direction from red (baseline) to green (occlusion and reperfusion). ACA flow (shown as green) in the Balb/c mice never reached MCA territory during occlusion and reperfusion due to a paucity of the connecting collaterals. The axes indicate the rostral (R) and lateral (L) directions.
1. 2. Imaging collateral flow
1.2.1 Clinical methods in imaging collateral flow
Advanced neuroimaging modalities that are capable of providing both angiographic and perfusion information may help to identify regions of vulnerability following stroke and thus guide therapeutic interventions. Currently, there is no ideal imaging modality for an accurate measurement of the collateral flow and structure, as each method possesses various advantages and limitations. Among the four common clinical methods for imaging collateral status, conventional digital subtraction angiography (DSA) is considered the gold standard (Liebeskind, 2009). It allows the measurement of collateral extent and number in all collateral circulations with excellent spatial and temporal resolution. However, the invasive nature of contrast dye injection into femoral artery precludes it to be used routinely, particularly under the acute setting when patients are at risk for complications. CT angiography provides good inter-observer reliability (McVerry et al., 2012), thus also a good choice for grading collateral status in multicenter studies. In addition, the non-invasive nature and rapid availability makes it ideal for assessing the collateral status in patients with acute stroke. MR angiography has been used to grade collateral status and its relation to outcome (Lee et al., 2009). Like CT angiography, the advantage of MR angiography also lies in the non-invasive nature and the increased diagnostic power when combined with other MR imaging modalities. However, no inter-observer reliability is possible when using different MR imaging characteristics to infer the presence of collaterals (Chalela et al., 2000, Hermier et al., 2005, Lee et al., 2009). Transcranial Doppler (TCD) is the least used method to investigate collateral flow by using relative blood flow velocity and vessel pulsatility as surrogate markers, despite its non-invasive advantage. Due to differences in methods and scoring criteria, a systematic review of methods used for assessing the leptomeningeal collateral flow was recently completed, and found a sizeable inconsistency in the grading of LMF among clinical literature (McVerry et al., 2012).
1.2.2 Pre-clinical methods in imaging collateral flow
Owing to the relatively small sizes of rodent brains, conventional MR or PET techniques suffering from poor spatial resolution are not ideal to image regional flow in preclinical studies. A number of optical based techniques have been invented to circumvent this limit, enabling the measurement of regional blood flow changes with a spatial resolution <100 μm. Among these techniques, laser speckle contrast imaging (LSCI) is the most cost effective method to image collateral flow in laboratory animals. LSCI creates high-resolution maps of blood flow on the cortical surface based on the blurring of the laser speckle patters by the motion of blood cells (Dunn et al., 2001). Using repeated LSCI, new patterns of collateral flow between the ACA and MCA were detected soon after MCAO, and persisted 24 hours after stroke (Armitage et al., 2010). In addition, increased venous flow following reperfusion was detected within the territory of MCA. In another recent study evaluating the effect of G-CSF and GM-CSF on arteriogenesis using LSCI, the authors found that both growth factors promoted leptomeningeal collateral growth after CCA occlusion, which coincided with an increase in circulating monocytes (Sugiyama et al., 2011). In addition to the sensitivity and non-invasiveness, LSCI offers nearly real time imaging of blood flow. However, the quantitative relationship between speckle contrast and blood flow velocity remains undefined, and thus LSCI can not provide absolute value in blood flow velocity (Ayata et al., 2004b, Parthasarathy et al., 2008), limiting its utility in comparing between animals and between time points.
Two-photon laser scanning microscopy (TPLSM) allows absolute speed and direct measurement of individual vessels (Schaffer et al., 2006). This technique was elegantly applied in a series of studies investigating hemodynamic consequences of an occlusion in cortical surface arterioles and penetrating arterioles following vessel occlusion. In contrast to the poor perfusion status extending at least 7 branches downstream from the occlusions in penetrating arterioles (Nishimura et al., 2007), there was a robust redistribution in blood flow after occlusion of cortical surface arterioles via reversal of flow from downstream branches (Schaffer et al., 2006), providing evidences of functional collateral flow in the surface artery network. Due to its high resolution, the greatest advantage of TPLSM is the ability to quantify blood flow in small vessels including capillaries. However, it can only measure flow in vessels that are oriented parallel to the imaging plane, and in one vessel at a time. Other limitations include a relatively shallow tissue penetration due to light scattering and requirement for a cranial window that reduces the frequency of repeated imaging.
Doppler optical coherence tomography (DOCT) has emerged as a new method to determine cerebral blood flow. It measures the Doppler shift of light scattered off from moving red blood cells to quantify flow velocity with 3-D spatial resolution. Unlike TPLSM, DOCT is capable of quantifying blood flow across many individual vessels simultaneously at a relatively high speed in the brain of a living animal (Wang and An, 2009, Srinivasan et al., 2011). Absolute blood perfusion measured using DOCT yields a vastly improved spatial resolution over previous methods (Santisakultarm and Schaffer, 2011). As DOCT is best suited to measure the top 150 μm of the brain in relatively larger vessels, it thus is an ideal imaging modality for studying dynamic flow changes in leptomeningeal anastomoses in rodents after MCA stroke. Using this technique, we compared the collateral flow recruitment from ACA to MCA following distal MCA occlusion in C57BL/6 and Balb/C mice (Figure 2). Retrograde flow from the ACA to MCA during MCA occlusion and reperfusion in the Balb/C mice was nearly undetectable due to very few existing connecting collaterals between the two vascular networks. However, the robustness of collateral flow after ischemic stroke is not entirely dependent on the number of existing collateral connections. For example, despite the similar numbers of connecting collateral vessels between the ACA and the MCA of diabetic and normoglycemic db mice, insulin resistant db/db mice exhibited impairment in retrograde recruitment of collateral flow from ACA to MCA immediately following MCA occlusion compared to their normoglycemic counterpart db/+ (Akamatsu et al., 2013). This suggests that multiple vascular and intravascular factors contribute to a functional collateral circulation.
1.3 Regulation of arteriogenesis and collateral flow
Arteriogenesis is triggered by fluid shear stress (FSS) and increased circumferential wall stress following a sudden occlusion or a chronically progressing stenosis in the artery, leading to the activation of endothelium, monocyte invasion, secretion of growth factors and cytokines, followed by matrix digestion and ultimately proliferation of smooth muscle cells for the development of collateral flow conductance and the remodeling of collateral vessels (Heil and Schaper, 2004, van Oostrom et al., 2008, Fung and Helisch, 2012, la Sala et al., 2012, Troidl and Schaper, 2012). The molecular mechanisms involved in this complex process are discussed below.
1.3.1 Mechanical force as the initial trigger
Arterial occlusion lowers the pressure in the distal vasculature, and thus increases flow through pre-existing collaterals due to the resulting pressure gradient. The initial increase of FSS exerted by the blood on the endothelium as it flows by activates the endothelium and stimulates a cascade of signaling events (Pipp et al., 2004, Schierling et al., 2009a, Schierling et al., 2011), leading to the conductance of the collateral vessels. As the collateral vessels grow in diameter, fluid shear stress falls and collateral flow decreases, providing a self-regulating mechanism. The collateral flow developed proximal to the occlusion does not require the local expression of either vascular endothelial growth factor (VEGF) or hypoxia-inducible factor 1 (HIF1) (Lee et al., 2004). It is this mechanosensitive feature of arteriogenesis that prominently differentiates itself from other types of vascular growth such as angiogenesis. By maximizing FSS via an artificial arteriovenous (AV) shunt, collateral artery growth can be induced beyond the physiological extent and can overcome anatomical restrictions (Eitenmuller et al., 2006). FSS leads to both peripheral and cerebral arteriogenesis. Using a ligature-shunt model by bilateral ligation of the common carotid arteries and creation of an AV fistula between the distal stump of the ligated carotid artery and the adjacent jugular vein, Schierling and colleagues observed considerable arteriogenesis and vessel growth in the FSS stimulated arteries (Schierling et al., 2009a).
1.3.2 Activation of endothelium
Microarray data identified a potential role for the shear stress sensitive gene transient receptor potential cation channel subfamily V member 4 (Trpv4), a Ca2+ channel in endothelial cells, in transducing the stimuli induced by FSS. Pharmacological activation of Trpv4 strongly increased cerebral arteriogenesis and collateral flow (Schierling et al., 2011), while Trpv-channel blocker reduced collateral flow (Troidl et al., 2009a). Complementary to these findings, arteriogenesis was found impaired in mice lacking Trpv4 (Troidl et al., 2009a). Additional evidence suggested that actin-binding Rho activating protein may also be involved in FSS-induced arteriogenesis (Troidl et al., 2009b). Although the precise downstream signaling of Trpv4 has yet to be identified, Ca2+-dependent mechanisms including the activation of endothelial-derived hyperpolarization factor (EDHF), cyclooxygenase, as well as NOS are potential candidates (Troidl et al., 2009a, Schierling et al., 2011). Among these, the most likely molecule transmitting the signal triggered by FSS is NO, which can diffuse through basement membrane that separates the endothelial cells and smooth muscle cells. Targeted deletion of the eNOS gene led to a loss of vasodilation but not of arteriogenesis, while targeted deletion of iNOS led to partial but not complete impairment of arteriogenesis. Only the deletion of both eNOS and iNOS resulted in a complete loss of collateral vessel remodeling during arterial occlusion (Troidl et al., 2010b). In addition to the above-mentioned proteins, FSS also upregulates endothelial microRNA-21 which suppresses its target gene phosphatase and tensin homolog (PTEN), contributing to increased eNOS phosphorylation and NO production (Weber et al., 2010).
1.3.3 Infiltration of inflammatory cells and subsequent inflammatory response
In addition to alterations in intracellular signaling and gene expression, recent studies have identified functional roles for various immune cell subsets as regulators of arteriogenesis, including monocytes/macrophages, T helper 17 (Th17) cells, regulatory T lymphocytes (Tregs), and natural killer (NK) cells (la Sala et al., 2012). Several lines of evidence support the critical role of monocytes and monocyte signaling in arteriogenesis. Pharmacological depletion of monocytes impairs arteriogenesis in rabbit and mouse models of hindlimb ischemia (Heil et al., 2002). Consistent results were also obtained in osteopetrotic mice with natural monocytopenia using a hindlimb ischemia model (Bergmann et al., 2006). In addition, adoptive transfer of wild type bone marrow mononuclear cells enhanced reperfusion following hindlimb ischemia (Capoccia et al., 2008). The improved reperfusion following wild type adoptive transfer was absent in mice defective in MCP-1/CCL2, suggesting that the recruitment of the inflammatory subset of monocytes to sites of ischemia is a critical step in collateral growth (Capoccia et al., 2008). Finally, deletion of one allele of the prolyl hydroxylase domain-containing protein 2 (PHD2) gene not only polarized macrophages toward a M2 phenotype but also promoted arteriogenesis, which was associated with an increased production of SDF-1 and PDGF-B (Takeda et al., 2011). A recent study by Troidl and colleagues determined the temporal and spatial distribution of macrophage subpopulation during arteriogenesis in a rat model of chronic FSS (Troidl et al., 2013). In this model, M2 macrophages appeared early following reperfusion and maintained up to 28d, concentrated particularly in the region of collateral growth. M1 macrophages were also present, but to less of an extent. In keeping with animal data, impaired chemotaxis of monocytes appeared to account for the impaired formation of collaterals in patients with diabetes (Waltenberger et al., 2000, Tchaikovski et al., 2009).
In many ways, arteriogenesis shares similarities with an active inflammatory process. For example, growing collaterals are characterized by the presence of infiltrating monocytes/macrophages in the lumen and in the perivascular space (Arras et al., 1998). Gene expression profiling during collateral development in a mouse model of hindlimb ischemia indicated a prominent role for inflammatory response-related genes, including CXCL5, MCP-1, CXCL9, and CXCL10. Furthermore, the nature of a coordinated appearance of proinflammatory genes followed by anti-inflammatory genes suggests that inflammation contributes to the initiation of collateral development (Meisner and Price, 2010). Lastly, infiltrating macrophages – more commonly associated with inflammatory response - also secrete a number of angiogenic growth factors including MCP-1, VEGF, FGF, GM-CSF, HGF, TNF-α, TGF-β and PDGF (Schierling et al., 2009b). The infusion of some of the most potent angiogenic factors at high pharmacological doses achieved only a fraction of the maximum conductance obtained by high FSS in a rabbit model of femoral artery occlusion, suggesting that these factors alone cannot fully account for the arteriogenic effects. Nonetheless, the findings implicate potential in using these cytokines or growth factors to promote arteriogenesis.
1.3.4 Structural remodeling via the activation of proteases, basal membrane degradation and changes of the extracellular matrix
Following the infiltration of monocytes and macrophages into the sites of collateral vessel, a structural remodeling process occurs that leads to the maturation and enlargement of collateral arteries. The orchestrated production of cytokines and growth factors induces the degradation of the extracellular matrix and stimulates the proliferation of endothelial (EC) and smooth muscle cells (SMC) in preparation for the remodeling of extracellular matrix and the expansion of the collateral diameter. However, the mechanisms leading to the production of cytokines and growth factors by monocytes and macrophages at sites of collateral growth are not well understood.
The turnover and remodeling of the extracellular matrix is carried out by the matrix metalloproteinases (MMPs) and their inhibitors, namely tissue inhibitor of metalloproteinases (TIMPs). During arteriogenesis, the external elastic lamina and elastin are broken down by MMPs and plasmin, creating space for the expanding vessel (Fung and Helisch, 2012, la Sala et al., 2012). Earlier studies indicated that MMP-2, MMP-9 and TIMP-1 were up-regulated in the intima during collateral remodeling (Kadoglou et al., 2005), suggesting that it is the balance between MMPs and TIMPs which is critical for the maintenance and remodeling of the vessel wall. Metabolic disorders such as diabetes disrupts this balance during arteriogenesis (Schatteman et al., 2000). For example, Lepr-db/db mutation blunted the ischemia-induced up-regulation of MMP-2, MMP- 12 and MMP-16 in the murine hindlimb ischemia model (Schiekofer et al., 2005).
During the late phase of arteriogenesis, ECs and SMCs proliferate and migrate (Scholz et al., 2000). SMCs account for a large part of the production of new tissue, changing their phenotype from contractile to a synthetic and proliferative phenotype (Schaper, 2009). As the collateral vessels grow in diameter with cell proliferation, FSS falls and collateral flow decreases as a feedback autoregulatory mechanism.
1.4 Clinical significance of collateral circulation
As noted previously, both extracranial sources of cerebral blood flow (CBF) and intracranial routes of ancillary perfusion contribute to collateral circulation (Liebeskind, 2003). These ancillary perfusion sources are divided into primary (e.g., circle of Willis) or secondary (i.e., ophthalmic artery and leptomeningeal vessels) collateral pathways. In general, poor collateral status not only increases the risk for stroke, but also serves as an independent predictor of poorer outcomes in patients with ischemic stroke (Henderson et al., 2000, Bang et al., 2008, Maas et al., 2009, Lima et al., 2010, Liebeskind et al., 2011b). Conversely, patients with good collateral flow not only experience more favorable stroke outcomes, but also show better clinical response to recanalization therapy even under conditions wherein treatment was initiated at delayed hours after the onset of stroke (Kucinski et al., 2003, Bang et al., 2011a, Bang et al., 2011b, Ribo et al., 2011). Metabolic risk factors such as type II diabetes mellitus (T2DM) are associated with poor leptomeningeal collateral status (Menon et al., 2013). Details of how the primary and secondary collateral flow contribute to the outcome of cerebrovascular occlusive diseases are discussed below.
1.4.1 Effect of primary collaterals
Willisian collaterals provide rapid routing of CBF to ischemic regions via existing anastomoses, critical under conditions involving occlusion or stenosis of the major feeding arteries (i.e., ICAs and the vertebrobasilar arteries) in the brain (Liebeskind, 2003). Unilateral ICA occlusion led to augmented collateral flow through the anterior Circle of Willis as well as an increase in the diameter of the anterior communicating arteries (Hartkamp et al., 1999). In contrast, bilateral ICA occlusion led to both enhanced collateral flow through the posterior circle of Willis as well as an increase in the diameter of the posterior communicating artery (Hartkamp et al., 1999). Furthermore, in patients with ICA occlusion, vertebrobasilar arteries appeared to be responsible for blood flow in the territory of the MCA ipsilateral to the occluded ICA, whereas the contralateral ICA provided collateral flow to the ACA territory of the ischemic side (van Laar et al., 2007). Enhanced collateral flow following ICA obstruction has been correlated with both increased vessel diameter (Hartkamp et al., 1999) as well as patient recovery, including an improved survival rate and decreased risk of ischemic events in patients with severe stenosis of the ICA (Henderson et al., 2000, Lima et al., 2010).
1.4.2 Effect of secondary collaterals in acute ischemia
The development of secondary collateral pathways appears to be initiated once primary collaterals at the circle of Willis become insufficient. In patients with complete occlusion of the intracranial ICA and/or the MCA (M1 or M2 segments), enhancement of leptomeningeal collaterals on CT angiography was correlated with lower post-stroke mortality (Lima et al., 2010). Patients with both MCA occlusion and rapid recruitment of secondary collateral pathways followed a similar symptomatic trajectory compared to ischemic patients without occlusion, whereas patients with MCA occlusion in the absence or reduction of collaterals fared significantly worse in the progression of symptoms post-stroke (Maas et al., 2009). Although the determinants of variability in collateral abundance in patients with acute ischemic stroke are largely unknown, a recent study reported that age and metabolic syndromes including hyperuricemia were associated with poor leptomeningeal collateral status in patients with acute ischemic stroke (Menon et al., 2013).
With respect to stroke therapy, although rapid recanalization has a strong association with clinical improvement, it remains inaccessible to many patients due to its limited time window and potential for hemorrhagic complication. In patients treated by local intra-arterial or systemic thrombolysis within 6 hours of the onset, adequate collateral supply and successful recanalization with thrombolytic treatment had a better clinical outcome (Kucinski et al., 2003). In the patients who received endovascular treatment, angiographic collateral grade determined the recanalization rate and clinical outcome after endovascular revascularization therapy (Bang et al., 2008, Bang et al., 2011a, Bang et al., 2011b, Liebeskind, 2013). Additionally, good collateral blood flow is linked to lower rates of hemorrhagic transformation after thrombolytic therapy and endovascular therapy (Bang et al., 2008, Christoforidis et al., 2009, Bang et al., 2011a, Bang et al., 2011b). Therefore, collateral status readily available from baseline angiography may refine therapeutic decision-making and prognostic assessments in the setting of acute cerebral ischemia. Good collateral status predicts favorable clinical response to systemic or intra-arterial treatment given even beyond 5–6 hours from stroke onset (Kucinski et al., 2003, Ribo et al., 2011), and may encourage further recanalization efforts. Inadequate information regarding collateral status might account for the unsuccessful clinical recovery in patients received endovascular therapy despite high recanalization rates (Penumbra Pivotal Stroke Trial, 2009). Thus, the state of collateral flow may be necessary to consider in the development of protocols for clinical trials.
However, the development of collaterals does not necessarily guarantee post-stroke sustained flow conductance. Hemodynamic fluctuations during the occlusion or following recanalization may disrupt collateral circulation, possibly threatening steady cerebral blood flow, and thrombolysis within the parent vessel may lead to clot fragments that travel and occlude collateral branches supplying retrograde flow from cortical arteries (Liebeskind, 2003). Furthermore, it is still not well understood how genetic and environmental factors influence acute collateral hemodynamic change after abrupt occlusion. A thorough understanding of these factors may better explain differences in clinical outcome and enable risk stratification for individual subjects.
1.4.3 Effect of secondary collaterals in chronic hypoperfusion (ischemia)
Although enhanced leptomeningeal collaterals are seen as a positive indicator of acute post stroke outcome, the presence of leptomeningeal collaterals prior to stroke onset can indicate cerebrovascular impairment in the patients with chronic hypoperfusion. In contrast to acute embolic or thrombotic occlusion, the development of collaterals over time may occur due to progressive atherosclerotic stenosis of intracranial segments. Increased compensation via collaterals correlated with severity of chronic stenosis and downstream perfusion (Liebeskind et al., 2011a, Liebeskind et al., 2011b), suggesting that enhancement of collaterals may be a characteristic of the underlying chronic pathophysiology and thus may be a marker of increased risk of stroke in intracranial atherosclerosis patients (Liebeskind et al., 2011a, Liebeskind et al., 2011b). However, various factors such as time course, extent of stenosis as well as collateral circulation likely impact the clinical presentation and progression of intracranial steno-occlusive arterial disease. The complex interplay of these and other factors (including differences in methodology) could explain the conflicting results described in clinical studies which attempt to correlate collateral patterns with hemodynamic and metabolic parameters (Muller and Schimrigk, 1996, Derdeyn et al., 1999a, Derdeyn et al., 1999b). In addition, limitations in the angiographic assessment of all potential collateral routes may have contributed to the discrepancy between clinical studies. In addition, the causes of variability in collateral robustness in patients with acute ischemic stroke are not well understood. A recent study suggests that metabolic syndrome, hyperuricemia and age are associated with poor leptomeningeal collateral status in patients with M1 segment middle cerebral artery with or without intracranial ICA occlusions (Menon et al., 2013). Furthermore, commonly used pharmacological treatments for co-morbidities may impact collateral flow. For example, a relationship between pre-stroke statin use and enhanced angiographic collateral grade was observed among acute ischemic stroke patients with occlusion of a major cerebral artery (Ovbiagele et al., 2007).
Unlike acute ischemic stroke, Moyamoya disease is a chronic, occlusive cerebrovascular disease characterized by bilateral steno-occlusive changes at the terminal portion of the ICA and an abnormal vascular network at the base of the brain (Suzuki and Takaku, 1969). In these patients, developments of leptomeningeal collaterals (termed “Moyamoya-like vessels”) compensate for insufficient blood flow, and are thus characterized as a type of collateral vessels, although pathological (Hinshaw et al., 1976, Fukui et al., 2000). The development of these vessels is negatively correlated with the development of leptomeningeal collaterals from the PCA, and thus are positively correlated with the severity of the stenotic lesions in the major arterial trunks (Yamada et al., 1995). Despite the pathological effects of the development of Moyamoya-like vessels, they may exist as an attempt to compensate the insufficient collateral networks in the ipsilateral cortical area (Kato et al., 2008).
1.4.4 Summary of clinical perspectives
Collateral flow in acute stroke may have several prognostic implications. Because collateral supply helps to limit the extent of infarction prior to the restoration of reperfusion, collateral perfusion in an ischemic region of the brain is an important determinant of clinical recovery with or without recanalization therapy. Aging and metabolic factors may impair collateral development in the setting of intracranial steno-occlusive disease and therefore affect ischemic stroke risk and outcome. Rapid clot lysis has an established strong association with clinical improvement, and reperfusion treatments must be provided as fast as possible in patients most likely to benefit. Patients who fail to rapidly reperfuse may benefit from other strategies that increase collateral flow as adjunctive and alternative approach. Thus, therapeutic strategies geared toward the enhancement of leptomeningeal collaterals or augmentation of their function could be an effective focus for clinical trials, either preventively before stroke, or in the acute phase after stroke onset.
1.5 Therapies to improve collateral flow in ischemic stroke
As experimental and clinical data have demonstrated the importance of collateral circulation and the benefit of spontaneous or therapeutically induced reperfusion (Shuaib et al., 2011b, Liebeskind, 2012, 2013), interventions aimed at augmenting and stabilizing collateral recruitment might be efficacious in treating acute ischemic stroke. In this section, we will provide an overview of a few notable approaches towards collateral blood flow augmentation and the observed efficacy in patients of acute ischemic stroke and prestroke.
1.5.1 Volume expansion or hemodilution
Increased cerebral perfusion or oxygen delivery can be achieved by decreasing blood viscosity using various hemodiluting agents (Ginsberg, 2008). Interestingly, hemodilutants, including dextran 40, hydroxyethyl starch (HES) and human albumin, have differential effects towards blood cells versus plasma. For example, decreased whole blood viscosity and lowered hematocrit are effectively induced by both dextran 40 and hydroxyethyl starch (HES), However, dextran 40 led to an increase in plasma viscosity and red-cell aggregation. Conversely, HES decreased plasma viscosity and red-cell aggregation (Haass et al., 1986, Kroemer et al., 1987), underscoring a distinction between the two hemodiluting agents in conditions where plasma volume expansion may be desirable. Human albumin is also a hemodiluting agent and, in moderate- to high-doses, has been demonstrated to exert neuroprotective effects in animal models of temporary and permanent focal cerebral ischemia (Belayev et al., 1997, Belayev et al., 1998, Liu et al., 2001) as well as in global cerebral ischemia (Belayev et al., 1999). Neuroprotection afforded by albumin has been attributed at least in part to its non-hemodiluting effect, including decreased oxidative stress (Wayner et al., 1985, Halliwell, 1988) and induction of endothelium-derived relaxing factor-like properties by reacting with nitric oxide (Keaney et al., 1993a, Keaney et al., 1993b). However, albumin also has multiple roles in the vasculature that likely play a role in ischemic neuroprotection, including the maintenance of normal microvascular permeability via hemodilution (He and Curry, 1993), decreased red blood cell sedimentation under low-flow conditions (Reinhart and Nagy, 1995), improved blood flow (Huh et al., 1998), increased microvascular flow velocity within the ischemic cortex (Nimmagadda et al., 2008) and reduced post-ischemic thrombosis and blood-element adhesion within the brain’s microvasculature (Belayev et al., 2002). Due to its multiple salutary actions, high-dose human albumin therapy is now under investigation in an international phase III multicenter clinical efficacy trial for stroke (Ginsberg et al., 2006, Hill et al., 2007) (Albumin in Acute Stroke, abbreviated as ALIAS) (http://www.NIHClinicalTrials.gov, Identifier NCT00235495).
1.5.2 Vasodilation
Because arteriogenesis involves NO release and vasodilation (Schaper, 2009, Troidl et al., 2010a), it is intuitive to develop therapies that enhance collateral flow by inducing vasodilation. However, small clinical trials have not shown any consistent benefit in neurological outcome (Rordorf et al., 1997, Rordorf et al., 2001). One explanation could be that because ischemic vasculature is already fully dilated during stroke, thus any vasodilation intervention is effective only in non-ischemic vessels and may lead to vascular stealing that is deleterious to ischemic tissue (Bremer et al., 1980, Kuwabara et al., 1995). If this explanation is correct, then effective collateral therapies should target vasodilation of the collateral vessels specifically. Unfortunately, no therapeutic strategy yet developed has achieved this level of specificity.
1.5.3 Induced hypertension
In the ischemic cortex, the normal autoregulatory mechanisms of cerebral blood flow are impaired, leading to a passive perfusion dependent on blood pressure variations. Although deleterious in nature, an induced rise in systemic blood pressure via pharmacotherapeutic intervention should improve blood flow to the brain, possibly bypassing occluded regions through increased collateral flow (Bogoslovsky et al., 2006, Wityk, 2007). The beneficial effects of induced mild hypertension have been confirmed in animal models of stroke. Induction of mild hypertension with phenylephrine increased blood pressure by 65 mm Hg and had a dramatic effect on infarct reduction in rabbits, with 97% and 45% reduction after one and two hours of MCAO, respectively (Smrcka et al., 1998). Intravenous infusion of phenylephrine in rats beginning as late as 60 minutes after the onset of distal MCAO increased blood pressure by 30% for the remaining duration of ischemia, and was associated with reduced infarct volume, improved CBF and CMRO2 in the penumbra, as well as increased leptomeningeal collateral blood flow (Shin et al., 2008). Angiotensin was also used to increase the mean arterial pressure by 40–60% and reduced infarct volume after transient MCAO in rats (Chileuitt et al., 1996). Although mild hypertension induced during acute stroke appears to be neuroprotective, chronic hypertension paradoxically worsens stroke outcome (Aslanyan et al., 2003, Koga et al., 2009, Geeganage et al., 2011), possibly due to inhibition of collateral blood flow. Consistent with this notion, compensatory growth of leptomeningeal collaterals in response to chronic cerebral hypoperfusion was impaired in spontaneously hypertensive rats compared to normotensive rats; however, anti-hypertensive drugs restored the collateral growth (Omura-Matsuoka et al., 2011). In support of experimental data, some clinical studies have suggested benefit in tissue perfusion or functional improvement with induced hypertension (Rordorf et al., 2001, Hillis et al., 2003, Chalela et al., 2005, Bogoslovsky et al., 2006). However, it is unclear whether these results could be attributed to improved collateral flow recruitment, as collateral flow was not directly assessed in these studies. Despite the encouraging results and feasibility of inducing mild hypertension, there are many potential risks associated with hypertensive paradigms in patients with acute stroke, including the side effect of increased intracranial pressure. One study found that norepinephrine improved perfusion pressure and flow velocity in patients with large MCA stroke with increased the mean arterial pressure by 10 mmHg and only a slight increase in intracranial pressure (Schwarz et al., 2002), supporting the benevolent nature of this approach.
1.5.4 Collateral flow improvement by growth factors
Arteriogenesis and angiogenesis can be stimulated by various chemokines and growth factors secreted from circulating inflammatory cells, including MCP-1, fibroblast growth factor (FGF)-2, TGF-b, VEGF and granulocyte-macrophage colony-stimulating factor (GM-CSF) (Buschmann et al., 2003, Schirmer et al., 2009). Addition of many of these chemokines and growth factors can modulate the rate of arteriogenesis. Indeed, collateral conductance was enhanced by the infusion of CC chemokines (CCL2), FGF, or GM-CSF into the peripheral or coronary collateral circulation (Asahara et al., 1995, Ito et al., 1997, Zbinden et al., 2004). The therapeutic potential of these substances has been demonstrated in pre-clinical animal models by improving collateral conductance, extending neo-vascularization in the collateral dependent tissue regions, decreasing infarct area after hemodynamic stroke and improving functional parameters in myocardial ischemia (Erdo and Buschmann, 2007).
G-CSF and GM-CSF in particular have been fairly well investigated for treating cerebral ischemia (Buschmann et al., 2001). Administration of G-CSF and GM-CSF before or after ischemia was neuroprotective in rodent models (Nakagawa et al., 2006, Schabitz et al., 2008, Todo et al., 2008), likely in part due to its upregulation in the collateral vascular channels. Prophylactic injection of GM-CSF in rats for 6 weeks prior to bilateral common carotid artery occlusion attenuated functional impairment of cerebrovascular reserve capacity and increased leptomeningeal collateral density (Schneider et al., 2007). A recent study demonstrated that five days of daily GM-CSF or G-CSF administered to C57/BL6 mice after unilateral occlusion of the common carotid artery promoted leptomeningeal collateral growth and decreased infarct volume (Sugiyama et al., 2011). When a subset of these animals were subjected to MCAO seven days after common carotid occlusion and treated with G-CSF or GM-CSF, infarct size was reduced and cerebral perfusion improved compared to those with MCAO without growth factor treatment.
In addition to GM-CSF, other prophylactic therapies to improve collateral circulation should be developed to improve outcomes after stroke. As a previous clinical study has demonstrated, metabolic syndrome, hyperuricemia and age may affect collateral status (Menon et al., 2013), although the underlying mechanism is not yet understood. With the exception of aging, these risk factors are all modifiable. Therefore, the role in improving native collateral status warrants further studies in future. Ovbiagele and colleagues reported that prestroke statin use is associated with better cerebral collateral supply (Ovbiagele et al., 2007). Chronic angiogenic and more immediate arteriogenic effects of statins after arterial occlusion support the use of these agents in prevention and acute treatment of stroke.
1.5.5 Emerging technologies to improve cerebral blood flow in stroke patients
A number of emerging therapeutic techniques have shown promise in improving perfusion in stroke patients, including sphenopalatine ganglion stimulation, partial aortic occlusion and external counterpulsation, although their specific effects on increasing collateral flow have not been demonstrated. The principle underlying each strategy and the respective stage of investigation are discussed below.
Stimulation of the sphenopalatine ganglion increases intracranial blood flow and induces vasodilation through the parasympathetic innervation in the intracranial blood vessels (Suzuki et al., 1991, Tomita et al., 2000, Ayajiki et al., 2005, Yarnitsky et al., 2005). Data in experimental models of stroke have demonstrated that stimulation of this ganglion increased blood flow, reduced infarct volume and improved functional outcomes (Henninger and Fisher, 2007, Bar-Shir et al., 2010). Conversely, severing the nerves emanating from the sphenopalatine ganglion increased infarct volume after MCAO (Diansan et al., 2010). Based on encouraging preclinical data, the safety of sphenopalatine ganglion stimulation is currently being evaluated in human stroke patients (Khurana et al., 2009).
Temporary occlusion of the abdominal aorta at the suprarenal and infrarenal levels was tested to augment cerebral blood flow to the brain. This novel approach to increase blood flow in the upper body appears safe and feasible in stroke patients eight to 24 hours after symptom onset (Hammer et al., 2012). Although the efficacy of NeuroFlo in ischemic stroke was not proven in the Safety and Efficacy of NeuroFlo in Acute Ischemic Stroke (SENTIS) trial (Shuaib et al., 2011a), the technique reduced mortality without an increase in severe disability (Schellinger et al., 2013). It still remains possible that this approach is effective as an adjunct to thrombolysis, due to its proven safety and feasibility within 30 days of partial aortic occlusion immediately after thrombolytic treatment (Emery et al., 2011).
External counterpulsation (ECP) is a novel method used to improve the perfusion of vital organs, and may serve to benefit ischemic stroke patients. Unlike intra-aortic counterpulsation using intra-aortic balloon inflation (Wesley and Morgan, 1990), blood flow can be diverted from the capacitance vessels in the lower limbs by non-invasive means of external compression via antigravity suits or leg air cuffs. Antigravity suits induced a transient increase in cardiac output and a marked and sustained decrease in plasma norepinephrine and plasma renin activity (Kravik et al., 1986, Geelen et al., 1992), which reflect a decrease in sympathetic activity. During ECP, middle cerebral artery mean flow velocities increased in stroke patients by elevating blood pressure (Lin et al., 2012). However, the study failed to correlate improved functional outcomes and cerebral blood flow. Further randomized controlled trials with conventional medical treatment of stroke patients used as controls to investigate the effects of ECP on ischemic stroke patients are warranted.
2. The role of angiogenesis in cerebrovascular diseases and therapy
Angiogenesis is a tightly controlled process in the central nervous system (CNS). In cancer, inhibition of angiogenesis is an effective therapeutic strategy for treating solid tumors. Conversely, promoting focal angiogenesis could provide an opportunity for improving outcomes of ischemia-related diseases. Understanding the molecular and cellular mechanisms by which angiogenesis occurs appears promising to yield potential therapeutic targets for ischemic brain diseases. However, numerous questions remain unanswered. Which molecules or stem cells are crucial for triggering angiogenesis in the adult brain? How do these molecules or stem cells respond to ischemic episodes? What brain-specific pro-angiogenic factors are involved during brain repair and what are their roles in promoting angiogenesis? How do vascular and neural cells interact to promote angiogenesis? Answering these questions would help to develop new therapeutics to promote functional angiogenesis after ischemia.
2.1 Vessel development in the normal brain
Development of vessels in the adult brain uses many similar molecules and signaling pathways as in other parts of the body, which involves an orchestrated interplay of growth factors and regulators. However, the vascularization of the developmental CNS and the maintenance of brain vascular function also require several special structures, including the blood-brain barrier (BBB) and neurovascular unit (NVU). The structural uniqueness of the vasculature in the brain and molecular mechanisms that regulate the dynamic interactions of the neural and vascular system give rise to a highly complex system that may be a key target for therapeutic interventions.
2.1.1 Modes of vessel growth
Vessels can grow in the body via several modes, which include vasculogenesis, angiogenesis, arteriogenesis and collateral vessel growth (Carmeliet and Jain, 2011). Vasculogenesis is defined as a formation of blood vessels by progenitor cells, which is most common during embryogenesis but also occurs in adult vessel generation. Angiogenesis refers to new microvessel formation via sprouting or splitting from pre-existing vessels and is mainly responsible for the development of blood vessels after birth. As described above, arteriogenesis typically involves vessel maintenance and flow stabilization. Specifically, collateral growth refers to the formation of interconnected bridges of arterial systems. It is noted that specific angiogenic factor signaling system corresponds to each mode of vessel formation and growth.
During brain development, both vasculogenesis and angiogenesis occur. Mesodermal angioblasts form an initial vascular network around the neural tube via vasculogenesis. Further branching of newly formed microvessels into neural tube is accomplished through angiogenesis. Upon exposure to pro-angiogenic signals, endothelial cells (ECs) become mobile and protrude filopodia, forming tip cells (Jakobsson et al., 2010). Tip cells initiate new sprouts and sense guidance cues from focal environment. Stalk cells follow tip cells, proliferate to support sprout elongation, and establish a vessel lumen. Microvessel loops are then formed when tip cells anastomose with neighboring sprouts (Eilken and Adams, 2010, Potente et al., 2011). After the microvessel forms a lumen, mural cells such as pericytes and vascular smooth muscle cells (VSMCs) stabilize the capillaries and small arterioles (Quaegebeur et al., 2011). Focal cerebral angiogenesis is closely regulated by angiogenic factors, mediators, and local circumstances. These factors work together to ensure angiogenesis with correct induction of tip cells, maintenance of tip cell function and stalk cell identity, and recruiting of pericytes to the nascent capillary. In recent years, evidence indicates that several regulatory factors are shared by vascular system and the nervous system (Larrivee et al., 2009). These factors play important roles in aligning the vessels and nerves. Alignment of blood vessels and nerves is commonly observed and has been postulated to be mutually dependent on each other.
Vascular recruitment and neurogenesis are often associated. Studies of adult hippocampal neurogenesis revealed that newly formed microvessels are also used as migratory conduits for neural progenitor cells (Palmer et al., 2000). Within the neural stem cell niche in the brain, vascular components regulate neurogenesis through paracrine signaling as well as direct contact with neural progenitor cells(Goldberg and Hirschi, 2009). Understanding the interdependent relationship between the vascular and nervous system would help the development of therapeutics for the CNS.
2.1.2 Vascular cells
Even more so than in other organs, the relationship between the vascular and nervous systems in the CNS are tightly interwoven. They form intimately connected and highly-coordinated neurovascular unit composed of ECs, pericytes, VSMCs, astrocytes, microglia and neurons (Winkler et al., 2011a). Lining the interior of the vessel lumen are ECs, which originate from endothelial progenitor cells (EPCs). At the capillary level, pericytes attach to ECs and project elongated, finger-like processes that sheath the capillary wall. At the arterial level, VSMCs stabilize vessel wall and deposit extracellular matrix. CNS homeostasis depends upon the proper communication and functional interaction of these diverse cell types, and in turn, the mature vascular system requires innervation for vasoconstriction and vasodilation for proper function.
Perivascular signals are important for regulating both EC proliferation and neuronal development. Signaling pathways that promote neurogenesis, neural stem cell expansion, differentiation, migration and homing have been extensively studied. Interestingly, CNS pericytes possess both mesodermal and neuroectodermal origins (Winkler et al., 2011a). Mature pericytes play critical roles in regulating focal cerebral blood flow (CBF) (Peppiatt et al., 2006, Bell et al., 2010), controlling BBB maturation and integrity (Armulik et al., 2010, Bell et al., 2010, Daneman et al., 2010), supporting cerebrovascular system stability (Winkler et al., 2011a) and regulating angiogenesis (Stratman et al., 2009). Several CNS disorders, including diabetic retinopathy and some neurodegenerative states, have been associated with abnormal pericyte function (Hammes et al., 2004, Bell et al., 2010).
Located directly on the capillary wall, pericytes share a common basement membrane with ECs, attaching by different integrins to extracellular matrix (ECM) proteins (Diaz-Flores et al., 2009, Stratman et al., 2009). In areas that lack a basement membrane, pericytes and ECs make direct cell-to-cell contacts via junction proteins such as N-cadherin (Gerhardt et al., 2000, Li et al., 2011, Winkler et al., 2011a) and connexin-43 hemichannels, which form gap junctions between the two cell types and allow the transfer of nutrients, metabolites, secondary messengers and ions (Bobbie et al., 2010).
Normal function of neuronal circuits and synaptic transmission require an optimal neuronal microenvironment that relies on successful BBB maturation and maintenance. BBB is formed by continuous EC membrane characterized by the presence of tight junctions and adherens junctions (Hawkins and Davis, 2005), and acts as a regulated interface between the peripheral blood circulation and the CNS environment. Pericytes play a role in forming and maintaining BBB integrity in both the developing and adult brain (Armulik et al., 2010, Bell et al., 2010, Daneman et al., 2010, Quaegebeur et al., 2010, Li et al., 2011).
2.1.3 Angiogenic molecules
Angiogenesis in the brain is regulated by both general angiogenic factors and brain-specific angiogenic factors. These factors regulate EC migration, maintain cell identity, guide cell growth and elongation, and regulate BBB formation. Recent evidence suggests that specific factors are also involved in ensuring nerve-vessel alignment and nerve-artery alignment in the brain.
2.1.3.1 Vascular endothelial growth factor (VEGF)
VEGF is a critical stimulator of angiogenesis. VEGF-A is the main component of the VEGF family, and stimulates angiogenesis through VEGF receptor-2 (VEGFR-2) (Nagy et al., 2007, Ferrara, 2009). VEGFR-2 deficiency and/or loss of VEGF cause defects in vascular development (Carmeliet, 2003). VEGF is presented in both soluble and matrix-bound isoforms. Soluble VEGF mainly promotes vessel enlargement while matrix-bound isoforms stimulates more branching (Carmeliet, 2003). The release of soluble VEGF into the extracellular space leads to a VEGF gradient. This gradient signals tip cells to up-regulate DLL4 expression and subsequent activation of NOTCH signaling in adjacent stalk cells. The activation of NOTCH in stalk cells down-regulates VEGFR-2 expression, leading to a cessation of response to VEGF and thereby ensuring that the tip cell takes the lead (Phng and Gerhardt, 2009). The biological effect of VEGFR-2 signaling can vary greatly based on its subcellular localization. For example, during VEGF induced arterial morphogenesis, VEGFR-2 must signal from intracellular compartments (Lanahan et al., 2010). Paracrine VEGF, released by tumor, myeloid or other stromal cells, increase vessel branching and render tumor vessels abnormal (Stockmann et al., 2008), whereas autocrine VEGF, released by ECs, maintains vascular homeostasis (Lee et al., 2007).
2.1.3.2 Netrins (Nishiyama et al., 2003)
Concurrent with neural growth, vasculature formation occurs in the same direction in brain tissue. The similarities between these two systems in terms of their developmental timing and migration raise the possibility that ECs use an analogous mechanism similar to axonal growth cones in order to migrate toward their proper targets, and may even overlap in the response to the same extracellular molecules. Growth cones are specialized structure formed at the tip of a developing axon that function in axonal guidance by sensing guidance molecules in the extracellular environment (Carmeliet and Tessier-Lavigne, 2005). Indeed, the morphology of endothelial tip cells are strikingly similar to axonal growth cones and even express receptors for axonal guidance cues (Gerhardt et al., 2003), providing evidence that ECs and neurons may use similar mechanisms, and even overlapping cues, to correctly reach their appropriate targets.
Interactions between four classes of guidance cues (specifically, ephrins, semaphorins, netrins and slits) and their specific receptors guide axonal formation during neuronal development. Interestingly, many of these molecules appear to also be involved in vascular morphogenesis (Suchting et al., 2006). Guidance cues can function either as an attractive signal to guide the axon (or developing vessel) toward a specific region, or as a repulsive signal to repel formation in an area. Of the primary classes of axonal guidance cues, slits, semaphorins, and ephrins act primarily as repellents, although in some situations, they can exert an attractive signal. Netrins have been widely studied, and have been found to act as either attractants or repellents, depending on the specific cell type and expressed receptors.
Netrins are a family of evolutionarily conserved secreted molecules (Huber et al., 2003), three members of which (netrin-1, netrin-3 and netrin-4) have been identified in mammals (Moore et al., 2007). The common structural elements of members of the netrin family include a laminin VI domain, three EGF-like repeats similar to the laminin V domain, and a heparin-binding carboxyl-terminal domain. Functionally, netrins act as bifunctional guidance cues, exerting attractant or repellent modalities depending on the receptors expressed by various cell phenotypes. For example, Netrin-1 functions as an attractant for dorsal commissural neurons, but as a repellent for certain classes of motor neurons (Culotti and Merz, 1998). Attraction to netrin-1 is mediated by deleted–in-colorectal-carcinoma (DCC) receptors (Keino-Masu et al., 1996). Consistently, mice deficient in DCC receptors or in netrin-1 retard commissural axonal extension and result in a failure of these projections to cross the midline (Fazeli et al., 1997). On the other hand, netrin-1-mediated repulsion is mediated via UNC5H2 receptor homodimers or UNC5-DCC receptor heterodimers (Leonardo et al., 1997, Hong et al., 1999). In addition to the expression of specific receptors that influence the attractant or repellent modalities of netrin, the level of intracellular cyclic nucleotides may also switch the cellular response to netrin from attractant to repellent (Nishiyama et al., 2003). To support the concept that netrin guidance cues may impact vascular formation, navigating ECs express netrin-related receptors, including DCC, UNC5 and ephrin receptors. Overexpression of netrin-1 in the mouse brain after ischemia not only improved neurological function, but also promoted functional angiogenesis (Lu et al., 2012), providing a correlation between netrin-1, angiogenesis and functional improvement.
2.1.3.3 Angiopoietins (Angs) and Tie
The Ang/Tie system is involved in vessel maintenance, growth and stabilization. An important function of Ang/Tie system is allowing vessels to maintain quiescence while remain responsive to angiogenic stimuli. In humans, Ang/Tie family is composed of three ligands, Ang-1, Ang-2 and Ang-4 and two receptors, Tie-1 and Tie-2. Ang-1 is expressed by mural cells, and Ang-2 is expressed by angiogenic tip cells. Ang-1 activates Tie-2 reduces EC permeability, enhances vascular stabilization and suppresses thrombin-induced calcium influx and inflammation (Koh, 2013). Ang-1 induces Tie-2 clustering at cell–cell junctions to maintain EC quiescence (Saharinen et al., 2008). Ang-1 also promotes vessel thickening by stimulating mural coverage and basement membrane deposition. Conversely, Ang-2 induces EC destabilization and may compete with Ang-1 at the EC-EC junctions, leading to the decrease of vascular stability, which is an important step at the beginning of angiogenesis (Saharinen et al., 2008). In the presence of angiogenic stimulators, sprouting ECs release Ang-2, which antagonizes Ang-1 and Tie-2 signaling to enhance mural cell detachment, vascular permeability and EC sprouting (Augustin et al., 2009).
2.1.3.4 Fibroblast growth factor (FGF) family
The FGF super-family has many members with high degrees of redundancy (Beenken and Mohammadi, 2009). bFGF, FGF1, FGF2, and FGF9 all have angiogenic properties (Cao et al., 2003, Carmeliet and Jain, 2011), via directly activating receptors on ECs or indirectly via inducing the release of angiogenic factors (Beenken and Mohammadi, 2009). FGF signaling has been demonstrated to be important for the maintenance of vascular integrity, as inhibition of FGF signaling caused disassembly of adherens and tight junctions, and led to disintegration of the vasculature (Murakami et al., 2008).
2.1.3.5 Platelet-derived growth factor (PDGF)
Mural cell coverage is crucial for vascular maturation and proper vessel function. PDGF-β recruits mural cells through its receptor PDGFR-β (Gaengel et al., 2009). PDGF-β released by ECs stimulates pericytes proliferation and migration via cell surface PDGFR-β (Hellberg et al., 2010). Inactivation of either PDGF-B or PDGFR-β is associated with hallmarks of vascular breakdown, including pericyte deficiency, vascular dysfunction, and micro-aneurysm formation (Gaengel et al., 2009). Similarly, PDGFR-β hypomorphic mice exhibit BBB defects and neurodegenerative damage due to insufficient pericytes around brain vessels (Quaegebeur et al., 2010).
2.1.3.6 Transforming growth factor-beta (TGF-β)
TGF-β is a multifunctional cytokine that exerts context-dependent pro- and anti-angiogenic effects (Sieczkiewicz and Herman, 2003, Lebrin et al., 2005, Walshe et al., 2009). A variety of cell types secrete TGF-β in the brain, including ECs, pericytes, neurons and glia. Secreted TGF-β is an inactive protein-bound form that can be activated by thrombospondin or integrin (Lebrin et al., 2005, Gaengel et al., 2009). When activated, TGF-β binds to TGF-β receptor R2, which initiates the recruitment and phosphorylation of TGF-β receptor R1 and activin-like kinase 1 or 5 (ALK1 or ALK5), leading to transcriptional changes via the Smad signaling pathway (Lebrin et al., 2005). TGF-β can also regulate pericyte differentiation and adhesion (Gaengel et al., 2009). Genetic mutations of TGF-β receptors, such as endoglin (Eng) or ALK1, led to arteriovenous malformation (AVM) (Pardali et al., 2010).
2.1.3.7 Chemokines
Recent evidence suggests that specific chemokines are capable of regulating angiogenesis by recruiting leukocytes and EPCs (Duda et al., 2011, Wang et al., 2012). During hypoxia, chemokine CXCL12 (also known as stromal derived factor-1, SDF-1) is upregulated by hypoxia-induced factor-1α (HIF-1α) and binds to its receptor CXCR4 on target cells such as peripheral EPCs to promote angiogenesis. Neural progenitor cells (NPCs) also express CXCR4 and therefore could be attracted by CXCL12 (Robin et al., 2006). More evidence to support a role for CXCL12 in angiogenesis is derived by the observation that peripheral neurons express CXCL12 to control nerve-vessel alignment over a distance (Scalzo et al., 2013). In zebra fish, CXCL12/CXCR4 signaling controls the formation of arteriovenous connections in the developing CNS (Bussmann et al., 2011, Fujita et al., 2011).
2.1.3.8 Inhibitors of angiogenesis
As angiogenic processes fuel and sustain cancerous tumor growth, studies of angiogenesis inhibitors have largely been powered by the desire to control cancer progression. Over the years, many naturally occurring angiogenesis inhibitors are identified. Angiostatin was first identified in the serum and urine of tumor-bearing mice (Chen et al., 1995). As a proteolytic fragment of plasminogen, angiostatin inhibits the growth of many experimental tumors propagated from human or mouse tumor cell lines (Lannutti et al., 1997, Gorski et al., 1998, Kirsch et al., 1998, Redlitz et al., 1999). Shortly after the discovery of angiostatin, endostatin was identified as a proteolytic fragment of collagen type XVIII and a potent inhibitor of angiogenesis (O’Reilly et al., 1997). Recombinant endostatin inhibits tumor growth (Boehm et al., 1997) and causes transplanted tumor regression (Boehm et al., 1997). In addition to angiostatin and endostatin, a number of other endogenous proteins show anti-angiogenic activity. Many of these proteins are products of proteinase cleavage. These include ECM/basement membrane components (e.g. Arresten, Canstatin and Tumstatin from collagen IV; Vastatin from collagen VIII; Restin from collagen XV; Endostatin from collagen XVIII), proteinases or enzymes (e.g. PEX from MMP2; Mini-TrpRS from tryptophanyl-tRNA synthetase) or plasma proteins (e.g. Angiostatin from plasminogen; 16K Prolactin from prolactin; fragments of several serpins) (D’Amore and Ng, 2002). Further exploration of the physiological and pathological functions of these inhibitors could yield potential tumor treatments, as well as serve as tools for proof-of-concept in models of cerebrovascular disease.
2.2 Aberrant angiogenesis in cerebrovascular diseases
2.2.1 Arteriovenous malformations (AVMs)
Abnormal molecular regulation during vessel quiescence could lead to abnormal growth of vessels and even vascular malformations. Brain AVMs can cause an array of neurological symptoms including headache, focal neurological deficits, seizures, and even hemorrhagic stroke. The formation of AVMs leads to a direct linkage between arterial and venous circulation, bypassing the normally interposed capillary bed. The occurrence of such brain AVMs can be either sporadic or in the context of well-defined genetic disorders. Human hereditary telangiectasia (HHT) is an autosomal-dominant inherited vascular dysplasia that causes AVMs in the brain as well as other organs (Shovlin, 2010). HHT is caused by loss-of-function mutations of Endoglin or ALK1 gene, both of which are involved in TGF-β signaling pathway (Leblanc et al., 2009, Dupuis-Girod et al., 2010, Pardali et al., 2010). Emerging evidence suggests that AVM result from defective vessel maturation. However, mechanisms by which Endoglin or ALK1 could cause AVMs remain largely unknown. It is likely that both SMCs and ECs are potentially affect by mutations and thus both contribute to the defect (Ricard et al., 2010, Carmeliet and Jain, 2011, Storkebaum et al., 2011).
2.2.2 Moyamoya syndrome
Moyamoya syndrome is a cerebrovascular condition with progressive stenosis of the intracranial internal carotid arteries (ICA) and their proximal branches (Scott and Smith, 2009). Although Moyamoya is associated with 6% of childhood strokes (Smith and Scott, 2005), it is increasingly recognized cause of stroke in both children and adults. The stenosis characteristic of Moyamoya syndrome reduces blood flow in the major vessels of the anterior circulation of the brain. As discussed above, chronic hypoperfusion can lead to compensatory development of collateral vasculature, in this case by small vessels near the apex of the carotid, on the cortical surface, leptomeninges, and branches of the external carotid artery (ECA). In Japanese, Moyamoya means “like a puff of smoke”, descriptive of the characteristic angiography image of the associated network of abnormally dilated collateral vessels (Takeuchi and Shimizu, 1957). Genetic factors appear to play a major role in Moyamoya syndrome (Ikeda et al., 1999, Nanba et al., 2005), as a single nucleotide polymorphism in the promoter region of tissue inhibitor of matrix metalloproteinase type 2 (TIMP-2) gene has been associated with Moyamoya syndrome (Kang et al., 2006). However, genetic links are likely not the only contributing factor of Moyamoya syndrome, as epidemiological studies of identical twins suggest that environmental factors may precipitate the clinical emergence of the condition in susceptible individuals (Tanghetti et al., 1983, Scott et al., 2004).
2.2.3 Neonatal intraventricular hemorrhage
Intraventricular hemorrhage is a substantial cause of morbidity and mortality in premature infants (Ballabh, 2010). The shortage of pericytes specifically in the germinal matrix vasculature is thought to contribute to the hemorrhagic state, as this region has a high degree of angiogenesis (Braun et al., 2007). The vascular fragility caused by reduced pericytes combined with altered hemodynamics likely promotes vessel rupture and hemorrhage (Ballabh, 2010). The underlying cause of the pericyte shortage and resulting hemorrhage is still unclear, but has been linked with a region-specific deficiency in TGF-β signaling (Braun et al., 2007, Li et al., 2011, Winkler et al., 2011b). Prenatal glucocorticoid, which increased TGF-β, protected against the disease, leading to both enhanced pericyte coverage and suppressed VEGF-driven angiogenesis in a rabbit model (Vinukonda et al., 2010).
2.2.4 Ischemic Stroke
Although a host of risk factors is linked to the development of ischemic stroke in adult patients, the contribution of aberrant vessel development is not evident (with the exception of Moyamoya syndrome). However, abnormal vascular changes have been widely observed in preclinical stroke models. For example, a prolonged increase in vascular permeability is detected in the reperfused outer cortical layers and leptomeninges in rats (Schierling et al., 2009a).
2.3 Angiogenesis after ischemic stroke
2.3.1 Evidences of post stroke angiogenesis
During chronic phase of stroke, the formation of new vessels by angiogenesis, is hypothesized to participate in brain plasticity and functional recovery after stroke (Arai et al., 2009). Data from MR imaging of laboratory animals suggest that the early phase of cerebral blood volume (CBV) increase may be a result of improved collateral flow, and the late phase of CBV increase due to resulting angiogenesis (Lin et al., 2008). Angiogenesis is a key restorative mechanism in response to ischemia. Ischemia triggers endogenous angiogenesis in brain, heart and hind limb, and interventions that promote angiogenesis are correlated with reduced injury in animal models (Banai et al., 1994, Takeshita et al., 1994, Fan and Yang, 2007). In humans, post-mortem analysis of brain tissue from ischemic stroke patients demonstrates that stroke tissue isolated from patients contains angiogenic activity and angiogenesis seems more developed in the penumbra (Krupinski et al., 1993a, 1994). In ischemic stroke patients, a correlation was found between the numbers of new vessels in the ischemic penumbral regions with prolonged survival, suggesting that activated angiogenesis could be beneficial for the ischemic brain (Krupinski et al., 1993b, Krupinski et al., 1994). It should be noted that angiogenesis is not always detected in peri-infarct area. Aging is a complex and significant factor that impacts post-ischemic angiogenesis (Petcu et al., 2010). Hypoxia-inducible angiogenesis in rodent hippocampus decreases with aging (Ingraham et al., 2008). Since ischemic stroke is a disease that occurs more often in seniors, the age criterion should be taken into consideration in experimental design.
The three-dimensional structure and survival of newly formed blood vessels can be assessed using scanning electron microscopy (SEM) of vascular corrosion casts from ex-vivo brain samples. Using this technique, Krupinski and colleagues found that neocortical arterioles and venues lost their radial patterns within one day of occlusion, quickly reforming into a highly dense network of anastomosing microvessels. Interestingly, vascular budding and microvessel connections of the newly formed vessels were similar to normal rat brain, but distinct to those formed in growing tumors (Krupinski et al., 2003).
2.3.2 Imaging of post stroke angiogenesis
Post-ischemic angiogenesis is associated with an increase in cerebral blood flow (CBF) and cerebral blood volume (CBV) (Lin et al., 2002, Schierling et al., 2009a). Using noninvasive magnetic resonance imaging (MRI), monitoring of vascular remodeling as well as quantitative assessment of tissue perfusion and microvascular characteristics, including CBF and CBV, vascular density, size and integrity, can be obtained after stroke (Ayata et al., 2004a, Boas and Dunn, 2010). CBF and CBV are directly affected by angiogenesis and can be measured with perfusion MRI techniques such as dynamic susceptibility contrast-enhanced (DSC) MRI, steady state contrast-enhanced (ssCE) MRI and arterial spin labeling (ASL). Vessel size index (VSI) and microvessel density (MVD) can be assessed with ssCE-MRI. These techniques have been the subject of several recent and thorough reviews (Ayata et al., 2004a, Chen et al., 2013). In addition, blood oxygenation level-dependent (BOLD) MRI and positron emission tomography (PET) are useful for the detection of increased oxygenated blood flow or metabolism as indirect evidence for angiogenesis (Marchal et al., 1993, Ding et al., 2008). All of these methods have inherent limitations and pitfalls. An increase in CBF and CBV may not be exclusively specific to neovascularization, unless corroborated with data from perfusion imaging and histology. For example, vasodilatation of existing vessels and collateral flow could contribute to increases of CBF and CBV. BBB disruption could also complicate the computation and interpretation of the results. Increased Ki (i.e. contrast agent leakage) and shortened of T2* (i.e. more deoxygenated blood) could occur as results of BBB disruption and hemorrhage in response to vascular injury and limit the analysis of MRI data (Ayata et al., 2004a). In another example, despite of the proliferation of ECs in the ischemic striatum, ssCE MRI detected a significant decrease in rCBV in small vessels, leading to a decrease in calculated vessel density (Boas and Dunn, 2010). The authors attributed the discrepancy from previous reports of increased vessel density to cystic transformation.
The spatial resolution of MR angiography is limited to approximately 50 micron for small animal MR imaging, and to approximately 250 micron for clinical MRI scanners, which do not allow direct visualization of newly formed vasculature. Histological staining of sectioned brain thus is often used in experimental studies of post-ischemic angiogenesis in combination with imaging tools. Double staining of EC marker CD31 and proliferation marker proliferating cell nuclear antigen (PCNA) or pre-injected Bromodeoxyuridine (BrdU) is used to detect newly formed vessels. Recently, high-resolution synchrotron radiation X-ray angiography has been developed to image microvessels with diameters in the range of several microns to tens of microns (Yuan et al., 2013). This technique allows direct comparison of microvessel density pre- and post- ischemia and may be used to evaluate the efficacy of pro-angiogenesis interventions (Lu et al., 2012).
2.3.3 Angiogenesis and post stroke neuroplasticity/functional recovery
Angiogenesis and tissue remodeling is highly active following stroke, particularly in penumbral regions. The penumbra is extremely resilient and quickly mounts and active process to recover from the ischemic event, rather than slowly succumbing passively. In the penumbra, partially depolarized tissue adjacent to the infract core sustain reduced cerebral blood flow and increased oxygen extraction rate, and thus struggle for survival in a very limited time-window. A major part of the recovery process is the stimulation of angiogenesis, and the initial struggle to survive contributes towards later neurogenesis, vascular remodeling and final recovery (Figure 2). Indeed, EC proliferation appears to begin as early as 12–24 hours after focal cerebral ischemia and to persist for up to several weeks thereafter (Marti et al., 2000, Hayashi et al., 2003). Microvascular ECs secrete growth factors and chemokines, which in turn support the survival of newly formed neurons.
Angiogenesis, neurogenesis and synaptic plasticity are endogenous processes in under both normal and pathological conditions. However, these processes can also be stimulated by pharmacological treatments. Administration of human cord blood-derived CD34+ cells following stroke induced revascularization in the peri-infarct cortex and increased neuroblast migration towards the damaged tissue (Taguchi et al., 2004). Transplantation of EPCs promoted angiogenesis and also improved long-term stroke outcomes in mice (Fan et al., 2010). Interestingly, many common pathways are triggered after stroke over different recovery periods (Lo et al., 2003), and may possess a biphasic nature in the penumbra in terms of injury versus repair (Lo, 2008). The metalloproteinase (MMP) family and possibly chemokine stromal derived factor-1 (SDF-1, also known as CXCL12) exhibit such biphasic natures (Figure 3). Both of these pathways are associated with detrimental effects in the early phase after stroke, but during the later remodeling phase, both MMPs and CXCL12 appear to be crucial for neuroblast migration and neurorepair (Zhao et al., 2006, Huang et al., 2013). Further understanding of the role of these factors could aid the development of new therapeutics to treat ischemic stroke.
Figure 3. Cerebral Ischemia induces angiogenesis.
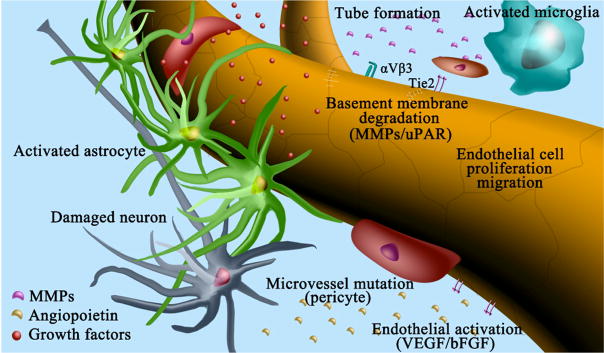
Schematic representation shows the process of angiogenesis in the brain during cerebral ischemia. 1) Damaged neurons, activated endothelial cells and astrocytes release growth factors such as VEGF and bFGF, etc. 2) At the same time, matrix metalloproteinases (MMPs), urokinase-type receptor (uPAR) and inflammatory mediators are highly expressed in the focal environments, which trigger the degradation of basement membrane. 3) Activated endothelial cells proliferate, migrate and sprout into new vessels locally. 4) Activated microglia express many angiogenic and inflammatory factors to promote the formation of new tubes by endothelial cells, endothelial progenitor cells, and mesenchymal stem cells. 5) Upon the activation of Ang/Tie2 signaling, smooth muscle cells and pericytes are attracted to surrounding newly formed microvessels and promote vessel maturation.
 Growth factors,
Growth factors,
 MMPs,
MMPs,
 Angiopoietins
Angiopoietins
2.4 Angiogenesis as targets for therapy in CVD
2.4.1 Arteriovenous malformations
Treatment of AVMs has largely been restricted to micro-neurosurgery, stereotactic radiotherapy or endovascular embolization. Understanding the molecular mechanism of AVMs is crucial for designing less invasive therapeutic approaches. Thalidomide, which has antiangiogenic effects, improved the outcome of HHT in experimental model and small group of patients by augmenting PDGF-βexpression in ECs and stimulating mural cell activation (Lebrin et al., 2010). Additionally, treatment with bevacizumab, and angiogenesis inhibitor, improved clinical symptoms in AVM patients (Bose et al., 2009). Recently, a miR-132 blocker delivered using nanoparticles was found to suppress pathological angiogenesis, which suggests its potential for the treatment of cerebrovascular malformations (Anand et al., 2010).
2.4.2 Ischemic stroke
Very limited progress has been made in developing therapies to effectively treat ischemic stroke. Currently the only FDA approved treatment is thrombolysis with tissue plasminogen activator (tPA). Unfortunately, less than 5% of patients are treated with tPA due to its narrow therapeutic time window and safety concern. Thus, most stroke victims receive only supportive care (Fonarow et al., 2011). Acute carotid endocardectomy and endovascular reperfusion strategies appeared beneficial to selected patients (Liebeskind, 2010), but is contraindicated in most of the stroke population. Therefore, the development of other therapeutic strategies is critical for patients who are ineligible for tPA or interventional approaches (Fonarow et al., 2011). Many promising therapeutic approaches have been developed in the preclinical stage, focusing on neuroprotection by preventing excitotoxicity, oxidative stress, inflammation or apoptosis in rodent models of cerebral ischemia (Moskowitz et al., 2010). However, these experimental designs have failed in large clinical trials, perhaps due to the weighted focus on neuroprotection rather than to include support processes (Moskowitz, 2010). It is now widely accepted that the brain should be treated as an entity event that requires synergistic interaction of the neural and vascular systems (Quaegebeur et al., 2011). Moreover, understanding how the brain mounts an endogenous response to protect itself under various settings may also reveal better therapeutic targets (Iadecola and Anrather, 2011).
Targeting individual pathogenic components of the ischemic cascade has been proven to be an insufficient strategy. Coordinated and multifunctional therapeutic approaches are needed to treat acute stroke efficiently; to this end, progress has been made with several multifunctional therapeutic candidates. Minocycline, an agent with anti-apoptotic, anti-inflammatory, and anti-excitotoxic properties, protects the animal brain with a wide therapeutic window (Fagan et al., 2011), and is currently being tested in a large phase III trial after promising results in smaller clinical trials. Ischemic preconditioning induces protection in both early and delayed time windows (Iadecola and Anrather, 2011). Additionally, brain protection is observed even when the procedure was applied during (pro-conditioning) or after the ischemic event (post conditioning) (Dirnagl et al., 2009), indicating that paradigms to establish ischemic tolerance could be used not only as a preventive strategy in individuals at high risk but also as a treatment following acute stroke (Iadecola and Anrather, 2011).
Promoting angiogenesis and subsequent improvement of cerebral blood flow appears to an important treatment strategy for ischemic stroke on the basis of promising results from pre-clinical studies. VEGF overexpression promoted focal angiogenesis following focal cerebral ischemia in mice (Shen et al., 2008). Adeno-associated virus (AAV) mediated netrin-1 overexpression promoted both angiogenesis and long-term neurological recovery after transient focal ischemia in mice (Lu et al., 2012). However, one challenge for developing pro-angiogenic therapy is the promotion of functional angiogenesis without causing uncontrolled vessel growth. It is also critical to ensure the integrity of BBB while promoting new vessel growth. There is a balance between BBB leakage and vessel growth. If vessels grow too fast and pericytes and other mural cells do not have sufficient time to stabilize the vessels, BBB leakage could occur(Navaratna et al., 2009). It is therefore important to develop methods that can monitor functional vessel growth and perfusion in experimental animal models. Recent development of real-time high-resolution angiography for rodents provides a unique tool for such evaluation (Guan et al., 2012, Yuan et al., 2013). A further improvement in our understanding of the negative regulation of angiogenesis is also critical.
Stem cell based therapy for ischemic stroke has been a major focus in recent years. Transplantation of EPCs promoted angiogenesis and improved long-term stroke outcomes in mice (Fan et al., 2010). Potential sources of EPC for vascular remodeling include bone marrow and peripheral blood for adults and umbilical cord blood of newborns (George et al., 2011). Several clinical trials are currently underway to evaluate the feasibility and safety of EPCs for the treatment of patients with ischemic stroke (ClinicalTrials.gov Identifiers: NCT01468064, NCT01289795). Recent discoveries of new angiogenesis regulators also bring new promises of stroke treatment. microRNAs (mirRs) are essential determinants of EC biology and contributors to stroke pathogenesis. Angiogenesis-regulating mirRs are potential therapeutic targets for ischemic stroke (Yin et al., 2013).
We previously mentioned that MMP exhibits biphasic nature in terms of injury versus repair (Rosell and Lo, 2008). Although MMPs mediate acute neurovascular disruption and parenchymal destruction, their activity is required for angiogenesis and vasculogenesis in the recovery phase. Similarly, other acute neurovascular events classically considered neurotoxic may represent an endogenous attempt by the brain to initiate angiogenic recovery (Arai et al., 2009). Altered calcium signaling between astrocytes and the corresponding ECs may function as signals for vascular remodeling. Disruption of tight junctions may be indicative of ECs disengagement in preparation to move. The concept that these acute neurovascular events may have a beneficial aspect as well as a detrimental effect necessitate that interventions must be carefully balanced so that delayed neuronal, glial and endothelial recovery is not impaired.
3. Conclusions and perspectives
Both arteriogenesis and angiogenesis are tightly controlled processes under physiological and pathological conditions. Arteriogenesis is triggered by mechanical force, while angiogenesis is induced by a hypoxic environment. Strategies to increase collateral flow have been shown to hold promise in improving the outcome of acute stroke and recanalization therapies in stroke patients. Experimental studies suggest that enhancing angiogenesis is linked to an improved recovery of function and brain plasticity, although the efficacy of angiogenesis therapy has yet to be established in stroke patients. A better understanding of collateral flow recruitment and angiogenesis can not only improve our knowledge in vascular remodeling and brain plasticity after stroke, but also enable us to harness the therapeutic potential of each process to promote vascularization and enhance cerebral blood flow, either as adjunct therapies or interventions by themselves.
Figure 4. Angiogenic molecules involve in focal angiogenesis.
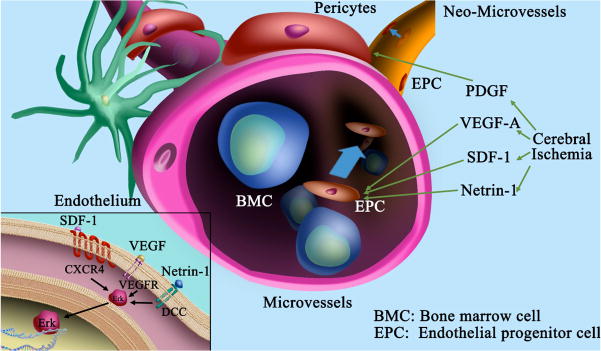
Schematic representation shows the function of angiogenic molecules during cerebral ischemia. Injured neurons and activated astrocytes and endothelial cells express growth factors such as VEGF, PDGF, MMPs and etc., which degrade basement membrane, activate endothelial cells and induce sprout. Bone marrow cells (BMCs) and endothelial progenitor cells (EPCs) are homing towards the sprout regions via chemokines CXCL12/SDF-1 and netrin-1, leading to tube formation. Lower left inset indicates that VEGF signals through VEGF-R1 or VEGF-R2, CXCL12/SDF-1 through CXCR4 or CXCR7, and Netrin-1 through DCC or UNC5H2 receptors, all of which further activate ERK signaling pathway and induce downstream cascade activation.
Highlights.
Distinct mechanisms underlying arteriogenesis and angiogenesis are delineated
Technology in detecting each vascular remodeling process
Clinical and preclinical implications of arteriogenesis and angiogenesis
Potential therapies in promoting vascular remodeling including those in trials
Acknowledgments
This work was supported by NIH grant R01 NS071050 (JL), AHA EIA 0940065N (JL), China 973 Program 2010CB834306 and 2011CB504405 (GYY, WY), NSFC 81070939 (GYY) and U1232205 (GYY).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akamatsu Y, Lee CC, Liu J. Impaired acute collateral flow dynamics following experimental stroke in Type 2 diabetic mice. Stroke. 2013;44:ATP146. [Google Scholar]
- Akamatsu Y, Shimizu H, Saito A, Fujimura M, Tominaga T. Consistent focal cerebral ischemia without posterior cerebral artery occlusion and its real-time monitoring in an intraluminal suture model in mice. J Neurosurg. 2012;116:657–664. doi: 10.3171/2011.11.JNS111167. [DOI] [PubMed] [Google Scholar]
- Alpers BJ, Berry RG, Paddison RM. Anatomical studies of the circle of Willis in normal brain. AMA Arch Neurol Psychiatry. 1959;81:409–418. doi: 10.1001/archneurpsyc.1959.02340160007002. [DOI] [PubMed] [Google Scholar]
- Anand S, Majeti BK, Acevedo LM, Murphy EA, Mukthavaram R, Scheppke L, Huang M, Shields DJ, Lindquist JN, Lapinski PE, King PD, Weis SM, Cheresh DA. MicroRNA-132-mediated loss of p120RasGAP activates the endothelium to facilitate pathological angiogenesis. Nat Med. 2010;16:909–914. doi: 10.1038/nm.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai K, Jin G, Navaratna D, Lo EH. Brain angiogenesis in developmental and pathological processes: neurovascular injury and angiogenic recovery after stroke. FEBS J. 2009;276:4644–4652. doi: 10.1111/j.1742-4658.2009.07176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armitage GA, Todd KG, Shuaib A, Winship IR. Laser speckle contrast imaging of collateral blood flow during acute ischemic stroke. J Cereb Blood Flow Metab. 2010;30:1432–1436. doi: 10.1038/jcbfm.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, Johansson BR, Betsholtz C. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- Arras M, Ito WD, Scholz D, Winkler B, Schaper J, Schaper W. Monocyte activation in angiogenesis and collateral growth in the rabbit hindlimb. J Clin Invest. 1998;101:40–50. doi: 10.1172/JCI119877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahara T, Bauters C, Zheng LP, Takeshita S, Bunting S, Ferrara N, Symes JF, Isner JM. Synergistic effect of vascular endothelial growth factor and basic fibroblast growth factor on angiogenesis in vivo. Circulation. 1995;92:II365–371. doi: 10.1161/01.cir.92.9.365. [DOI] [PubMed] [Google Scholar]
- Aslanyan S, Fazekas F, Weir CJ, Horner S, Lees KR Committee, GIS investigator . Effect of blood pressure during the acute period of ischemic stroke on stroke outcome: a tertiary analysis of the GAIN International Trial. Stroke. 2003;34:2420–2425. doi: 10.1161/01.STR.0000091233.04524.0C. [DOI] [PubMed] [Google Scholar]
- Augustin HG, Koh GY, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol. 2009;10:165–177. doi: 10.1038/nrm2639. [DOI] [PubMed] [Google Scholar]
- Ayajiki K, Fujioka H, Shinozaki K, Okamura T. Effects of capsaicin and nitric oxide synthase inhibitor on increase in cerebral blood flow induced by sensory and parasympathetic nerve stimulation in the rat. J Appl Physiol. 2005;98:1792–1798. doi: 10.1152/japplphysiol.00690.2004. [DOI] [PubMed] [Google Scholar]
- Ayata C, Dunn AK, Gursoy OY, Huang Z, Boas DA, Moskowitz MA. Laser speckle flowmetry for the study of cerebrovascular physiology in normal and ischemic mouse cortex. J Cereb Blood Flow Metab. 2004a;24:744–755. doi: 10.1097/01.WCB.0000122745.72175.D5. [DOI] [PubMed] [Google Scholar]
- Ayata C, Shin HK, Salomone S, Ozdemir-Gursoy Y, Boas DA, Dunn AK, Moskowitz MA. Pronounced hypoperfusion during spreading depression in mouse cortex. J Cereb Blood Flow Metab. 2004b;24:1172–1182. doi: 10.1097/01.WCB.0000137057.92786.F3. [DOI] [PubMed] [Google Scholar]
- Ballabh P. Intraventricular hemorrhage in premature infants: mechanism of disease. Pediatr Res. 2010;67:1–8. doi: 10.1203/PDR.0b013e3181c1b176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banai S, Jaklitsch MT, Shou M, Lazarous DF, Scheinowitz M, Biro S, Epstein SE, Unger EF. Angiogenic-induced enhancement of collateral blood flow to ischemic myocardium by vascular endothelial growth factor in dogs. Circulation. 1994;89:2183–2189. doi: 10.1161/01.cir.89.5.2183. [DOI] [PubMed] [Google Scholar]
- Bang OY, Saver JL, Buck BH, Alger JR, Starkman S, Ovbiagele B, Kim D, Jahan R, Duckwiler GR, Yoon SR, Vinuela F, Liebeskind DS, Investigators UC. Impact of collateral flow on tissue fate in acute ischaemic stroke. J Neurol Neurosurg Psychiatry. 2008;79:625–629. doi: 10.1136/jnnp.2007.132100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang OY, Saver JL, Kim SJ, Kim GM, Chung CS, Ovbiagele B, Lee KH, Liebeskind DS. Collateral flow predicts response to endovascular therapy for acute ischemic stroke. Stroke. 2011a;42:693–699. doi: 10.1161/STROKEAHA.110.595256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang OY, Saver JL, Kim SJ, Kim GM, Chung CS, Ovbiagele B, Lee KH, Liebeskind DS Collaborators UC-SS. Collateral flow averts hemorrhagic transformation after endovascular therapy for acute ischemic stroke. Stroke. 2011b;42:2235–2239. doi: 10.1161/STROKEAHA.110.604603. [DOI] [PubMed] [Google Scholar]
- Bar-Shir A, Shemesh N, Nossin-Manor R, Cohen Y. Late stimulation of the sphenopalatine-ganglion in ischemic rats: improvement in N-acetyl-aspartate levels and diffusion weighted imaging characteristics as seen by MR. J Magn Reson Imaging. 2010;31:1355–1363. doi: 10.1002/jmri.22110. [DOI] [PubMed] [Google Scholar]
- Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8:235–253. doi: 10.1038/nrd2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belayev L, Busto R, Zhao W, Clemens JA, Ginsberg MD. Effect of delayed albumin hemodilution on infarction volume and brain edema after transient middle cerebral artery occlusion in rats. J Neurosurg. 1997;87:595–601. doi: 10.3171/jns.1997.87.4.0595. [DOI] [PubMed] [Google Scholar]
- Belayev L, Pinard E, Nallet H, Seylaz J, Liu Y, Riyamongkol P, Zhao W, Busto R, Ginsberg MD. Albumin therapy of transient focal cerebral ischemia: in vivo analysis of dynamic microvascular responses. Stroke. 2002;33:1077–1084. doi: 10.1161/hs0402.105555. [DOI] [PubMed] [Google Scholar]
- Belayev L, Saul I, Huh PW, Finotti N, Zhao W, Busto R, Ginsberg MD. Neuroprotective effect of high-dose albumin therapy against global ischemic brain injury in rats. Brain Res. 1999;845:107–111. doi: 10.1016/s0006-8993(99)01952-6. [DOI] [PubMed] [Google Scholar]
- Belayev L, Zhao W, Pattany PM, Weaver RG, Huh PW, Lin B, Busto R, Ginsberg MD. Diffusion-weighted magnetic resonance imaging confirms marked neuroprotective efficacy of albumin therapy in focal cerebral ischemia. Stroke. 1998;29:2587–2599. doi: 10.1161/01.str.29.12.2587. [DOI] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic BV. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68:409–427. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann CE, Hoefer IE, Meder B, Roth H, van Royen N, Breit SM, Jost MM, Aharinejad S, Hartmann S, Buschmann IR. Arteriogenesis depends on circulating monocytes and macrophage accumulation and is severely depressed in op/op mice. J Leukoc Biol. 2006;80:59–65. doi: 10.1189/jlb.0206087. [DOI] [PubMed] [Google Scholar]
- Boas DA, Dunn AK. Laser speckle contrast imaging in biomedical optics. J Biomed Opt. 2010;15:011109. doi: 10.1117/1.3285504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobbie MW, Roy S, Trudeau K, Munger SJ, Simon AM, Roy S. Reduced connexin 43 expression and its effect on the development of vascular lesions in retinas of diabetic mice. Invest Ophthalmol Vis Sci. 2010;51:3758–3763. doi: 10.1167/iovs.09-4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm T, Folkman J, Browder T, O’Reilly MS. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature. 1997;390:404–407. doi: 10.1038/37126. [DOI] [PubMed] [Google Scholar]
- Bogoslovsky T, Happola O, Salonen O, Lindsberg PJ. Induced hypertension for the treatment of acute MCA occlusion beyond the thrombolysis window: case report. BMC Neurol. 2006;6:46. doi: 10.1186/1471-2377-6-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose P, Holter JL, Selby GB. Bevacizumab in hereditary hemorrhagic telangiectasia. N Engl J Med. 2009;360:2143–2144. doi: 10.1056/NEJMc0901421. [DOI] [PubMed] [Google Scholar]
- Braun A, Xu H, Hu F, Kocherlakota P, Siegel D, Chander P, Ungvari Z, Csiszar A, Nedergaard M, Ballabh P. Paucity of pericytes in germinal matrix vasculature of premature infants. J Neurosci. 2007;27:12012–12024. doi: 10.1523/JNEUROSCI.3281-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremer AM, Yamada K, West CR. Ischemic cerebral edema in primates: effects of acetazolamide, phenytoin, sorbitol, dexamethasone, and methylprednisolone on brain water and electrolytes. Neurosurgery. 1980;6:149–154. doi: 10.1227/00006123-198002000-00006. [DOI] [PubMed] [Google Scholar]
- Brozici M, van der Zwan A, Hillen B. Anatomy and functionality of leptomeningeal anastomoses: a review. Stroke. 2003;34:2750–2762. doi: 10.1161/01.STR.0000095791.85737.65. [DOI] [PubMed] [Google Scholar]
- Buschmann I, Heil M, Jost M, Schaper W. Influence of inflammatory cytokines on arteriogenesis. Microcirculation. 2003;10:371–379. doi: 10.1038/sj.mn.7800199. [DOI] [PubMed] [Google Scholar]
- Buschmann IR, Hoefer IE, van Royen N, Katzer E, Braun-Dulleaus R, Heil M, Kostin S, Bode C, Schaper W. GM-CSF: a strong arteriogenic factor acting by amplification of monocyte function. Atherosclerosis. 2001;159:343–356. doi: 10.1016/s0021-9150(01)00637-2. [DOI] [PubMed] [Google Scholar]
- Bussmann J, Wolfe SA, Siekmann AF. Arterial-venous network formation during brain vascularization involves hemodynamic regulation of chemokine signaling. Development. 2011;138:1717–1726. doi: 10.1242/dev.059881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Brakenhielm E, Pawliuk R, Wariaro D, Post MJ, Wahlberg E, Leboulch P, Cao Y. Angiogenic synergism, vascular stability and improvement of hind-limb ischemia by a combination of PDGF-BB and FGF-2. Nat Med. 2003;9:604–613. doi: 10.1038/nm848. [DOI] [PubMed] [Google Scholar]
- Capoccia BJ, Gregory AD, Link DC. Recruitment of the inflammatory subset of monocytes to sites of ischemia induces angiogenesis in a monocyte chemoattractant protein-1-dependent fashion. J Leukoc Biol. 2008;84:760–768. doi: 10.1189/jlb.1107756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Tessier-Lavigne M. Common mechanisms of nerve and blood vessel wiring. Nature. 2005;436:193–200. doi: 10.1038/nature03875. [DOI] [PubMed] [Google Scholar]
- Chalela JA, Alsop DC, Gonzalez-Atavales JB, Maldjian JA, Kasner SE, Detre JA. Magnetic resonance perfusion imaging in acute ischemic stroke using continuous arterial spin labeling. Stroke. 2000;31:680–687. doi: 10.1161/01.str.31.3.680. [DOI] [PubMed] [Google Scholar]
- Chalela JA, Dunn B, Todd JW, Warach S. Induced hypertension improves cerebral blood flow in acute ischemic stroke. Neurology. 2005;64:1979. doi: 10.1212/01.WNL.0000156360.70336.18. [DOI] [PubMed] [Google Scholar]
- Chen C, Parangi S, Tolentino MJ, Folkman J. A strategy to discover circulating angiogenesis inhibitors generated by human tumors. Cancer Res. 1995;55:4230–4233. [PubMed] [Google Scholar]
- Chen CC, Chen YC, Hsiao HY, Chang C, Chern Y. Neurovascular abnormalities in brain disorders: highlights with angiogenesis and magnetic resonance imaging studies. J Biomed Sci. 2013;20:47. doi: 10.1186/1423-0127-20-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chileuitt L, Leber K, McCalden T, Weinstein PR. Induced hypertension during ischemia reduces infarct area after temporary middle cerebral artery occlusion in rats. Surg Neurol. 1996;46:229–234. doi: 10.1016/0090-3019(95)00453-x. [DOI] [PubMed] [Google Scholar]
- Christoforidis GA, Karakasis C, Mohammad Y, Caragine LP, Yang M, Slivka AP. Predictors of hemorrhage following intra-arterial thrombolysis for acute ischemic stroke: the role of pial collateral formation. AJNR Am J Neuroradiol. 2009;30:165–170. doi: 10.3174/ajnr.A1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culotti JG, Merz DC. DCC and netrins. Curr Opin Cell Biol. 1998;10:609–613. doi: 10.1016/s0955-0674(98)80036-7. [DOI] [PubMed] [Google Scholar]
- D’Amore PA, Ng YS. Tales of the cryptic: unveiling more angiogenesis inhibitors. Trends Mol Med. 2002;8:313–315. doi: 10.1016/s1471-4914(02)02367-5. [DOI] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defazio RA, Levy S, Morales CL, Levy RV, Dave KR, Lin HW, Abaffy T, Watson BD, Perez-Pinzon MA, Ohanna V. A protocol for characterizing the impact of collateral flow after distal middle cerebral artery occlusion. Transl Stroke Res. 2011;2:112–127. doi: 10.1007/s12975-010-0044-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derdeyn CP, Shaibani A, Moran CJ, Cross DT, 3rd, Grubb RL, Jr, Powers WJ. Lack of correlation between pattern of collateralization and misery perfusion in patients with carotid occlusion. Stroke. 1999a;30:1025–1032. doi: 10.1161/01.str.30.5.1025. [DOI] [PubMed] [Google Scholar]
- Derdeyn CP, Videen TO, Fritsch SM, Carpenter DA, Grubb RL, Jr, Powers WJ. Compensatory mechanisms for chronic cerebral hypoperfusion in patients with carotid occlusion. Stroke. 1999b;30:1019–1024. doi: 10.1161/01.str.30.5.1019. [DOI] [PubMed] [Google Scholar]
- Diansan S, Shifen Z, Zhen G, Heming W, Xiangrui W. Resection of the nerves bundle from the sphenopalatine ganglia tend to increase the infarction volume following middle cerebral artery occlusion. Neurol Sci. 2010;31:431–435. doi: 10.1007/s10072-010-0238-0. [DOI] [PubMed] [Google Scholar]
- Diaz-Flores L, Gutierrez R, Madrid JF, Varela H, Valladares F, Acosta E, Martin-Vasallo P, Diaz-Flores L., Jr Pericytes. Morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histol Histopathol. 2009;24:909–969. doi: 10.14670/HH-24.909. [DOI] [PubMed] [Google Scholar]
- Ding G, Jiang Q, Li L, Zhang L, Zhang ZG, Ledbetter KA, Gollapalli L, Panda S, Li Q, Ewing JR, Chopp M. Angiogenesis detected after embolic stroke in rat brain using magnetic resonance T2*WI. Stroke. 2008;39:1563–1568. doi: 10.1161/STROKEAHA.107.502146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol. 2009;8:398–412. doi: 10.1016/S1474-4422(09)70054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda DG, Kozin SV, Kirkpatrick ND, Xu L, Fukumura D, Jain RK. CXCL12 (SDF1alpha)-CXCR4/CXCR7 pathway inhibition: an emerging sensitizer for anticancer therapies? Clin Cancer Res. 2011;17:2074–2080. doi: 10.1158/1078-0432.CCR-10-2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn AK, Bolay H, Moskowitz MA, Boas DA. Dynamic imaging of cerebral blood flow using laser speckle. J Cereb Blood Flow Metab. 2001;21:195–201. doi: 10.1097/00004647-200103000-00002. [DOI] [PubMed] [Google Scholar]
- Dupuis-Girod S, Bailly S, Plauchu H. Hereditary hemorrhagic telangiectasia: from molecular biology to patient care. J Thromb Haemost. 2010;8:1447–1456. doi: 10.1111/j.1538-7836.2010.03860.x. [DOI] [PubMed] [Google Scholar]
- Eilken HM, Adams RH. Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr Opin Cell Biol. 2010;22:617–625. doi: 10.1016/j.ceb.2010.08.010. [DOI] [PubMed] [Google Scholar]
- Eitenmuller I, Volger O, Kluge A, Troidl K, Barancik M, Cai WJ, Heil M, Pipp F, Fischer S, Horrevoets AJ, Schmitz-Rixen T, Schaper W. The range of adaptation by collateral vessels after femoral artery occlusion. Circ Res. 2006;99:656–662. doi: 10.1161/01.RES.0000242560.77512.dd. [DOI] [PubMed] [Google Scholar]
- Emery DJ, Schellinger PD, Selchen D, Douen AG, Chan R, Shuaib A, Butcher KS. Safety and feasibility of collateral blood flow augmentation after intravenous thrombolysis. Stroke. 2011;42:1135–1137. doi: 10.1161/STROKEAHA.110.607846. [DOI] [PubMed] [Google Scholar]
- Erdo F, Buschmann IR. Arteriogenesis: a new strategy of therapeutic intervention in chronic arterial disorders. Cellular mechanism and experimental models. Orv Hetil. 2007;148:633–642. doi: 10.1556/OH.2007.27916. [DOI] [PubMed] [Google Scholar]
- Fagan SC, Cronic LE, Hess DC. Minocycline Development for Acute Ischemic Stroke. Transl Stroke Res. 2011;2:202–208. doi: 10.1007/s12975-011-0072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Shen F, Frenzel T, Zhu W, Ye J, Liu J, Chen Y, Su H, Young WL, Yang GY. Endothelial progenitor cell transplantation improves long-term stroke outcome in mice. Ann Neurol. 2010;67:488–497. doi: 10.1002/ana.21919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Yang GY. Therapeutic angiogenesis for brain ischemia: a brief review. J Neuroimmune Pharmacol. 2007;2:284–289. doi: 10.1007/s11481-007-9073-3. [DOI] [PubMed] [Google Scholar]
- Fazeli A, Dickinson SL, Hermiston ML, Tighe RV, Steen RG, Small CG, Stoeckli ET, Keino-Masu K, Masu M, Rayburn H, Simons J, Bronson RT, Gordon JI, Tessier-Lavigne M, Weinberg RA. Phenotype of mice lacking functional Deleted in colorectal cancer (Dcc) gene. Nature. 1997;386:796–804. doi: 10.1038/386796a0. [DOI] [PubMed] [Google Scholar]
- Ferrara N. VEGF-A: a critical regulator of blood vessel growth. Eur Cytokine Netw. 2009;20:158–163. doi: 10.1684/ecn.2009.0170. [DOI] [PubMed] [Google Scholar]
- Fonarow GC, Smith EE, Saver JL, Reeves MJ, Bhatt DL, Grau-Sepulveda MV, Olson DM, Hernandez AF, Peterson ED, Schwamm LH. Timeliness of tissue-type plasminogen activator therapy in acute ischemic stroke: patient characteristics, hospital factors, and outcomes associated with door-to-needle times within 60 minutes. Circulation. 2011;123:750–758. doi: 10.1161/CIRCULATIONAHA.110.974675. [DOI] [PubMed] [Google Scholar]
- Fujita M, Cha YR, Pham VN, Sakurai A, Roman BL, Gutkind JS, Weinstein BM. Assembly and patterning of the vascular network of the vertebrate hindbrain. Development. 2011;138:1705–1715. doi: 10.1242/dev.058776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui M, Kono S, Sueishi K, Ikezaki K. Moyamoya disease. Neuropathology. 2000;20(Suppl):S61–64. doi: 10.1046/j.1440-1789.2000.00300.x. [DOI] [PubMed] [Google Scholar]
- Fung E, Helisch A. Macrophages in collateral arteriogenesis. Front Physiol. 2012;3:353. doi: 10.3389/fphys.2012.00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaengel K, Genove G, Armulik A, Betsholtz C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol. 2009;29:630–638. doi: 10.1161/ATVBAHA.107.161521. [DOI] [PubMed] [Google Scholar]
- Geeganage C, Tracy M, England T, Sare G, Moulin T, Woimant F, Christensen H, De Deyn PP, Leys D, O’Neill D, Ringelstein EB, Bath PM for TI. Relationship between baseline blood pressure parameters (including mean pressure, pulse pressure, and variability) and early outcome after stroke: data from the Tinzaparin in Acute Ischaemic Stroke Trial (TAIST) Stroke. 2011;42:491–493. doi: 10.1161/STROKEAHA.110.596163. [DOI] [PubMed] [Google Scholar]
- Geelen G, Arbeille P, Saumet JL, Cottet-Emard JM, Patat F, Vincent M. Hemodynamic and hormonal effects of prolonged anti-G suit inflation in humans. J Appl Physiol (1985) 1992;72:977–984. doi: 10.1152/jappl.1992.72.3.977. [DOI] [PubMed] [Google Scholar]
- George AL, Bangalore-Prakash P, Rajoria S, Suriano R, Shanmugam A, Mittelman A, Tiwari RK. Endothelial progenitor cell biology in disease and tissue regeneration. J Hematol Oncol. 2011;4:24. doi: 10.1186/1756-8722-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C, Alitalo K, Shima D, Betsholtz C. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. 2003;161:1163–1177. doi: 10.1083/jcb.200302047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt H, Wolburg H, Redies C. N-cadherin mediates pericytic-endothelial interaction during brain angiogenesis in the chicken. Dev Dyn. 2000;218:472–479. doi: 10.1002/1097-0177(200007)218:3<472::AID-DVDY1008>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Ginsberg MD. Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology. 2008;55:363–389. doi: 10.1016/j.neuropharm.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg MD, Palesch YY, Hill MD. The ALIAS (ALbumin In Acute Stroke) Phase III randomized multicentre clinical trial: design and progress report. Biochem Soc Trans. 2006;34:1323–1326. doi: 10.1042/BST0341323. [DOI] [PubMed] [Google Scholar]
- Goldberg JS, Hirschi KK. Diverse roles of the vasculature within the neural stem cell niche. Regen Med. 2009;4:879–897. doi: 10.2217/rme.09.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski DH, Mauceri HJ, Salloum RM, Gately S, Hellman S, Beckett MA, Sukhatme VP, Soff GA, Kufe DW, Weichselbaum RR. Potentiation of the antitumor effect of ionizing radiation by brief concomitant exposures to angiostatin. Cancer Res. 1998;58:5686–5689. [PubMed] [Google Scholar]
- Guan Y, Wang Y, Yuan F, Lu H, Ren Y, Xiao T, Chen K, Greenberg DA, Jin K, Yang GY. Effect of suture properties on stability of middle cerebral artery occlusion evaluated by synchrotron radiation angiography. Stroke. 2012;43:888–891. doi: 10.1161/STROKEAHA.111.636456. [DOI] [PubMed] [Google Scholar]
- Haass A, Kroemer H, Jager H, Muller K, Decker I, Wagner EM, Schimrigk K. Dextran 40 or HES 200/0.5? Hemorheology of the long-term treatment of ischemic cerebral attacks. Dtsch Med Wochenschr. 1986;111:1681–1686. doi: 10.1055/s-2008-1068692. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Albumin--an important extracellular antioxidant? Biochem Pharmacol. 1988;37:569–571. doi: 10.1016/0006-2952(88)90126-8. [DOI] [PubMed] [Google Scholar]
- Hammer MD, Schwamm L, Starkman S, Schellinger PD, Jovin T, Nogueira R, Burgin WS, Sen S, Diener HC, Watson T, Michel P, Shuaib A, Dillon W, Liebeskind DS. Safety and feasibility of NeuroFlo use in eight- to 24-hour ischemic stroke patients. Int J Stroke. 2012;7:655–661. doi: 10.1111/j.1747-4949.2011.00719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes HP, Lin J, Wagner P, Feng Y, Vom Hagen F, Krzizok T, Renner O, Breier G, Brownlee M, Deutsch U. Angiopoietin-2 causes pericyte dropout in the normal retina: evidence for involvement in diabetic retinopathy. Diabetes. 2004;53:1104–1110. doi: 10.2337/diabetes.53.4.1104. [DOI] [PubMed] [Google Scholar]
- Hartkamp MJ, van Der Grond J, van Everdingen KJ, Hillen B, Mali WP. Circle of Willis collateral flow investigated by magnetic resonance angiography. Stroke. 1999;30:2671–2678. doi: 10.1161/01.str.30.12.2671. [DOI] [PubMed] [Google Scholar]
- Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Noshita N, Sugawara T, Chan PH. Temporal profile of angiogenesis and expression of related genes in the brain after ischemia. J Cereb Blood Flow Metab. 2003;23:166–180. doi: 10.1097/01.WCB.0000041283.53351.CB. [DOI] [PubMed] [Google Scholar]
- He P, Curry FE. Albumin modulation of capillary permeability: role of endothelial cell [Ca2+]i. Am J Physiol. 1993;265:H74–82. doi: 10.1152/ajpheart.1993.265.1.H74. [DOI] [PubMed] [Google Scholar]
- Heil M, Schaper W. Influence of mechanical, cellular, and molecular factors on collateral artery growth (arteriogenesis) Circ Res. 2004;95:449–458. doi: 10.1161/01.RES.0000141145.78900.44. [DOI] [PubMed] [Google Scholar]
- Heil M, Ziegelhoeffer T, Pipp F, Kostin S, Martin S, Clauss M, Schaper W. Blood monocyte concentration is critical for enhancement of collateral artery growth. Am J Physiol Heart Circ Physiol. 2002;283:H2411–2419. doi: 10.1152/ajpheart.01098.2001. [DOI] [PubMed] [Google Scholar]
- Hellberg C, Ostman A, Heldin CH. PDGF and vessel maturation. Recent Results Cancer Res. 2010;180:103–114. doi: 10.1007/978-3-540-78281-0_7. [DOI] [PubMed] [Google Scholar]
- Henderson RD, Eliasziw M, Fox AJ, Rothwell PM, Barnett HJ. Angiographically defined collateral circulation and risk of stroke in patients with severe carotid artery stenosis. North American Symptomatic Carotid Endarterectomy Trial (NASCET) Group. Stroke. 2000;31:128–132. doi: 10.1161/01.str.31.1.128. [DOI] [PubMed] [Google Scholar]
- Henninger N, Fisher M. Stimulating circle of Willis nerve fibers preserves the diffusion-perfusion mismatch in experimental stroke. Stroke. 2007;38:2779–2786. doi: 10.1161/STROKEAHA.107.485581. [DOI] [PubMed] [Google Scholar]
- Hermier M, Nighoghossian N, Derex L, Wiart M, Nemoz C, Berthezene Y, Froment JC. Hypointense leptomeningeal vessels at T2*-weighted MRI in acute ischemic stroke. Neurology. 2005;65:652–653. doi: 10.1212/01.wnl.0000173036.95976.46. [DOI] [PubMed] [Google Scholar]
- Hill MD, Moy CS, Palesch YY, Martin R, Dillon CR, Waldman BD, Patterson L, Mendez IM, Ryckborst KJ, Tamariz D, Ginsberg MD, Investigators A. The albumin in acute stroke trial (ALIAS); design and methodology. Int J Stroke. 2007;2:214–219. doi: 10.1111/j.1747-4949.2007.00143.x. [DOI] [PubMed] [Google Scholar]
- Hillis AE, Wityk RJ, Barker PB, Ulatowski JA, Jacobs MA. Change in perfusion in acute nondominant hemisphere stroke may be better estimated by tests of hemispatial neglect than by the National Institutes of Health Stroke Scale. Stroke. 2003;34:2392–2396. doi: 10.1161/01.STR.0000089681.84041.69. [DOI] [PubMed] [Google Scholar]
- Hinshaw DB, Jr, Thompson JR, Hasso AN. Adult arteriosclerotic moyamoya. Radiology. 1976;118:633–636. doi: 10.1148/118.3.633. [DOI] [PubMed] [Google Scholar]
- Hong K, Hinck L, Nishiyama M, Poo MM, Tessier-Lavigne M, Stein E. A ligand-gated association between cytoplasmic domains of UNC5 and DCC family receptors converts netrin-induced growth cone attraction to repulsion. Cell. 1999;97:927–941. doi: 10.1016/s0092-8674(00)80804-1. [DOI] [PubMed] [Google Scholar]
- Huang J, Li Y, Tang Y, Tang G, Yang GY, Wang Y. CXCR4 antagonist AMD3100 protects blood-brain barrier integrity and reduces inflammatory response after focal ischemia in mice. Stroke. 2013;44:190–197. doi: 10.1161/STROKEAHA.112.670299. [DOI] [PubMed] [Google Scholar]
- Huber AB, Kolodkin AL, Ginty DD, Cloutier JF. Signaling at the growth cone: ligand-receptor complexes and the control of axon growth and guidance. Annu Rev Neurosci. 2003;26:509–563. doi: 10.1146/annurev.neuro.26.010302.081139. [DOI] [PubMed] [Google Scholar]
- Huh PW, Belayev L, Zhao W, Busto R, Saul I, Ginsberg MD. The effect of high-dose albumin therapy on local cerebral perfusion after transient focal cerebral ischemia in rats. Brain Res. 1998;804:105–113. doi: 10.1016/s0006-8993(98)00674-x. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Anrather J. Stroke research at a crossroad: asking the brain for directions. Nat Neurosci. 2011;14:1363–1368. doi: 10.1038/nn.2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda H, Sasaki T, Yoshimoto T, Fukui M, Arinami T. Mapping of a familial moyamoya disease gene to chromosome 3p24.2–p26. Am J Hum Genet. 1999;64:533–537. doi: 10.1086/302243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingraham JP, Forbes ME, Riddle DR, Sonntag WE. Aging reduces hypoxia-induced microvascular growth in the rodent hippocampus. J Gerontol A Biol Sci Med Sci. 2008;63:12–20. doi: 10.1093/gerona/63.1.12. [DOI] [PubMed] [Google Scholar]
- Ito WD, Arras M, Winkler B, Scholz D, Schaper J, Schaper W. Monocyte chemotactic protein-1 increases collateral and peripheral conductance after femoral artery occlusion. Circ Res. 1997;80:829–837. doi: 10.1161/01.res.80.6.829. [DOI] [PubMed] [Google Scholar]
- Jakobsson L, Franco CA, Bentley K, Collins RT, Ponsioen B, Aspalter IM, Rosewell I, Busse M, Thurston G, Medvinsky A, Schulte-Merker S, Gerhardt H. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol. 2010;12:943–953. doi: 10.1038/ncb2103. [DOI] [PubMed] [Google Scholar]
- Kadoglou NP, Daskalopoulou SS, Perrea D, Liapis CD. Matrix metalloproteinases and diabetic vascular complications. Angiology. 2005;56:173–189. doi: 10.1177/000331970505600208. [DOI] [PubMed] [Google Scholar]
- Kang HS, Kim SK, Cho BK, Kim YY, Hwang YS, Wang KC. Single nucleotide polymorphisms of tissue inhibitor of metalloproteinase genes in familial moyamoya disease. Neurosurgery. 2006;58:1074–1080. doi: 10.1227/01.NEU.0000215854.66011.4F. discussion 1074–1080. [DOI] [PubMed] [Google Scholar]
- Kato H, Shimosegawa E, Oku N, Kimura Y, Kajimoto K, Tanaka M, Hori M, Kitagawa K, Hatazawa J. Cerebral hemodynamics and oxygen metabolism in patients with moyamoya syndrome associated with atherosclerotic steno-occlusive arterial lesions. Cerebrovasc Dis. 2008;26:9–15. doi: 10.1159/000135647. [DOI] [PubMed] [Google Scholar]
- Keaney JF, Jr, Gaziano JM, Xu A, Frei B, Curran-Celentano J, Shwaery GT, Loscalzo J, Vita JA. Dietary antioxidants preserve endothelium-dependent vessel relaxation in cholesterol-fed rabbits. Proc Natl Acad Sci U S A. 1993a;90:11880–11884. doi: 10.1073/pnas.90.24.11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keaney JF, Jr, Simon DI, Stamler JS, Jaraki O, Scharfstein J, Vita JA, Loscalzo J. NO forms an adduct with serum albumin that has endothelium-derived relaxing factor-like properties. J Clin Invest. 1993b;91:1582–1589. doi: 10.1172/JCI116364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keino-Masu K, Masu M, Hinck L, Leonardo ED, Chan SS, Culotti JG, Tessier-Lavigne M. Deleted in Colorectal Cancer (DCC) encodes a netrin receptor. Cell. 1996;87:175–185. doi: 10.1016/s0092-8674(00)81336-7. [DOI] [PubMed] [Google Scholar]
- Khurana D, Kaul S, Bornstein NM Imp ACTSG. Implant for augmentation of cerebral blood flow trial 1: a pilot study evaluating the safety and effectiveness of the Ischaemic Stroke System for treatment of acute ischaemic stroke. Int J Stroke. 2009;4:480–485. doi: 10.1111/j.1747-4949.2009.00385.x. [DOI] [PubMed] [Google Scholar]
- Kirsch M, Strasser J, Allende R, Bello L, Zhang J, Black PM. Angiostatin suppresses malignant glioma growth in vivo. Cancer Res. 1998;58:4654–4659. [PubMed] [Google Scholar]
- Koga M, Toyoda K, Naganuma M, Kario K, Nakagawara J, Furui E, Shiokawa Y, Hasegawa Y, Okuda S, Yamagami H, Kimura K, Okada Y, Minematsu K Stroke Acute Management with Urgent Risk-factor A, Improvement Study I. Nationwide survey of antihypertensive treatment for acute intracerebral hemorrhage in Japan. Hypertens Res. 2009;32:759–764. doi: 10.1038/hr.2009.93. [DOI] [PubMed] [Google Scholar]
- Koh GY. Orchestral actions of angiopoietin-1 in vascular regeneration. Trends Mol Med. 2013;19:31–39. doi: 10.1016/j.molmed.2012.10.010. [DOI] [PubMed] [Google Scholar]
- Kravik SE, Keil LC, Geelen G, Wade CE, Barnes PR, Spaul WA, Elder CA, Greenleaf JE. Effect of antigravity suit inflation on cardiovascular, PRA, and PVP responses in humans. J Appl Physiol (1985) 1986;61:766–774. doi: 10.1152/jappl.1986.61.2.766. [DOI] [PubMed] [Google Scholar]
- Krishnaswamy A, Klein JP, Kapadia SR. Clinical cerebrovascular anatomy. Catheter Cardiovasc Interv. 2010;75:530–539. doi: 10.1002/ccd.22299. [DOI] [PubMed] [Google Scholar]
- Kroemer H, Haass A, Muller K, Jager H, Wagner EM, Heimburg P, Klotz U. Haemodilution therapy in ischaemic stroke: plasma concentrations and plasma viscosity during long-term infusion of dextran 40 or hydroxyethyl starch 200/0.5. Eur J Clin Pharmacol. 1987;31:705–710. doi: 10.1007/BF00541299. [DOI] [PubMed] [Google Scholar]
- Krupinski J, Kaluza J, Kumar P, Kumar S, Wang JM. Some remarks on the growth-rate and angiogenesis of microvessels in ischemic stroke. Morphometric and immunocytochemical studies. Patologia polska. 1993a;44:203–209. [PubMed] [Google Scholar]
- Krupinski J, Kaluza J, Kumar P, Kumar S, Wang JM. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke. 1994;25:1794–1798. doi: 10.1161/01.str.25.9.1794. [DOI] [PubMed] [Google Scholar]
- Krupinski J, Kaluza J, Kumar P, Wang M, Kumar S. Prognostic value of blood vessel density in ischaemic stroke. Lancet. 1993b;342:742. doi: 10.1016/0140-6736(93)91734-4. [DOI] [PubMed] [Google Scholar]
- Krupinski J, Stroemer P, Slevin M, Marti E, Kumar P, Rubio F. Three-dimensional structure and survival of newly formed blood vessels after focal cerebral ischemia. Neuroreport. 2003;14:1171–1176. doi: 10.1097/01.wnr.0000075304.76650.29. [DOI] [PubMed] [Google Scholar]
- Kucinski T, Koch C, Eckert B, Becker V, Kromer H, Heesen C, Grzyska U, Freitag HJ, Rother J, Zeumer H. Collateral circulation is an independent radiological predictor of outcome after thrombolysis in acute ischaemic stroke. Neuroradiology. 2003;45:11–18. doi: 10.1007/s00234-002-0881-0. [DOI] [PubMed] [Google Scholar]
- Kuwabara Y, Ichiya Y, Sasaki M, Yoshida T, Masuda K. Time dependency of the acetazolamide effect on cerebral hemodynamics in patients with chronic occlusive cerebral arteries. Early steal phenomenon demonstrated by [15O]H2O positron emission tomography. Stroke. 1995;26:1825–1829. doi: 10.1161/01.str.26.10.1825. [DOI] [PubMed] [Google Scholar]
- la Sala A, Pontecorvo L, Agresta A, Rosano G, Stabile E. Regulation of collateral blood vessel development by the innate and adaptive immune system. Trends Mol Med. 2012;18:494–501. doi: 10.1016/j.molmed.2012.06.007. [DOI] [PubMed] [Google Scholar]
- Lanahan AA, Hermans K, Claes F, Kerley-Hamilton JS, Zhuang ZW, Giordano FJ, Carmeliet P, Simons M. VEGF receptor 2 endocytic trafficking regulates arterial morphogenesis. Dev Cell. 2010;18:713–724. doi: 10.1016/j.devcel.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannutti BJ, Gately ST, Quevedo ME, Soff GA, Paller AS. Human angiostatin inhibits murine hemangioendothelioma tumor growth in vivo. Cancer Res. 1997;57:5277–5280. [PubMed] [Google Scholar]
- Larrivee B, Freitas C, Suchting S, Brunet I, Eichmann A. Guidance of vascular development: lessons from the nervous system. Circ Res. 2009;104:428–441. doi: 10.1161/CIRCRESAHA.108.188144. [DOI] [PubMed] [Google Scholar]
- Leblanc GG, Golanov E, Awad IA, Young WL. Biology of vascular malformations of the brain. Stroke. 2009;40:e694–702. doi: 10.1161/STROKEAHA.109.563692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrin F, Deckers M, Bertolino P, Ten Dijke P. TGF-beta receptor function in the endothelium. Cardiovasc Res. 2005;65:599–608. doi: 10.1016/j.cardiores.2004.10.036. [DOI] [PubMed] [Google Scholar]
- Lebrin F, Srun S, Raymond K, Martin S, van den Brink S, Freitas C, Breant C, Mathivet T, Larrivee B, Thomas JL, Arthur HM, Westermann CJ, Disch F, Mager JJ, Snijder RJ, Eichmann A, Mummery CL. Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia. Nat Med. 2010;16:420–428. doi: 10.1038/nm.2131. [DOI] [PubMed] [Google Scholar]
- Lee CW, Stabile E, Kinnaird T, Shou M, Devaney JM, Epstein SE, Burnett MS. Temporal patterns of gene expression after acute hindlimb ischemia in mice: insights into the genomic program for collateral vessel development. J Am Coll Cardiol. 2004;43:474–482. doi: 10.1016/j.jacc.2003.09.033. [DOI] [PubMed] [Google Scholar]
- Lee KY, Latour LL, Luby M, Hsia AW, Merino JG, Warach S. Distal hyperintense vessels on FLAIR: an MRI marker for collateral circulation in acute stroke? Neurology. 2009;72:1134–1139. doi: 10.1212/01.wnl.0000345360.80382.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, Ferrara N, Nagy A, Roos KP, Iruela-Arispe ML. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130:691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardo ED, Hinck L, Masu M, Keino-Masu K, Ackerman SL, Tessier-Lavigne M. Vertebrate homologues of C. elegans UNC-5 are candidate netrin receptors. Nature. 1997;386:833–838. doi: 10.1038/386833a0. [DOI] [PubMed] [Google Scholar]
- Li F, Lan Y, Wang Y, Wang J, Yang G, Meng F, Han H, Meng A, Yang X. Endothelial Smad4 maintains cerebrovascular integrity by activating N-cadherin through cooperation with Notch. Dev Cell. 2011;20:291–302. doi: 10.1016/j.devcel.2011.01.011. [DOI] [PubMed] [Google Scholar]
- Liebeskind DS. Collateral circulation. Stroke. 2003;34:2279–2284. doi: 10.1161/01.STR.0000086465.41263.06. [DOI] [PubMed] [Google Scholar]
- Liebeskind DS. Collaterals in acute stroke: beyond the clot. Neuroimaging Clin N Am. 2005a;15:553–573. x. doi: 10.1016/j.nic.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Liebeskind DS. Neuroprotection from the collateral perspective. IDrugs. 2005b;8:222–228. [PubMed] [Google Scholar]
- Liebeskind DS. Of mice and men: essential considerations in the translation of collateral therapeutics. Stroke. 2008;39:e187–188. doi: 10.1161/STROKEAHA.108.522888. author reply e189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebeskind DS. Imaging the future of stroke: I. Ischemia. Ann Neurol. 2009;66:574–590. doi: 10.1002/ana.21787. [DOI] [PubMed] [Google Scholar]
- Liebeskind DS. Reperfusion for acute ischemic stroke: arterial revascularization and collateral therapeutics. Curr Opin Neurol. 2010;23:36–45. doi: 10.1097/WCO.0b013e328334da32. [DOI] [PubMed] [Google Scholar]
- Liebeskind DS. Collateral perfusion: time for novel paradigms in cerebral ischemia. Int J Stroke. 2012;7:309–310. doi: 10.1111/j.1747-4949.2012.00818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebeskind DS. Trials of endovascular therapies or collaterals? Int J Stroke. 2013;8:258–259. doi: 10.1111/ijs.12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebeskind DS, Cotsonis GA, Saver JL, Lynn MJ, Cloft HJ, Chimowitz MI Warfarin-Aspirin Symptomatic Intracranial Disease I. Collateral circulation in symptomatic intracranial atherosclerosis. J Cereb Blood Flow Metab. 2011a;31:1293–1301. doi: 10.1038/jcbfm.2010.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebeskind DS, Cotsonis GA, Saver JL, Lynn MJ, Turan TN, Cloft HJ, Chimowitz MI Warfarin-Aspirin Symptomatic Intracranial Disease I. Collaterals dramatically alter stroke risk in intracranial atherosclerosis. Ann Neurol. 2011b;69:963–974. doi: 10.1002/ana.22354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima FO, Furie KL, Silva GS, Lev MH, Camargo EC, Singhal AB, Harris GJ, Halpern EF, Koroshetz WJ, Smith WS, Yoo AJ, Nogueira RG. The pattern of leptomeningeal collaterals on CT angiography is a strong predictor of long-term functional outcome in stroke patients with large vessel intracranial occlusion. Stroke. 2010;41:2316–2322. doi: 10.1161/STROKEAHA.110.592303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Chang C, Cheung WM, Lin MH, Chen JJ, Hsu CY, Chen JH, Lin TN. Dynamic changes in vascular permeability, cerebral blood volume, vascular density, and size after transient focal cerebral ischemia in rats: evaluation with contrast-enhanced magnetic resonance imaging. J Cereb Blood Flow Metab. 2008;28:1491–1501. doi: 10.1038/jcbfm.2008.42. [DOI] [PubMed] [Google Scholar]
- Lin TN, Sun SW, Cheung WM, Li F, Chang C. Dynamic changes in cerebral blood flow and angiogenesis after transient focal cerebral ischemia in rats. Evaluation with serial magnetic resonance imaging. Stroke. 2002;33:2985–2991. doi: 10.1161/01.str.0000037675.97888.9d. [DOI] [PubMed] [Google Scholar]
- Lin W, Xiong L, Han J, Leung TW, Soo YO, Chen X, Wong KS. External counterpulsation augments blood pressure and cerebral flow velocities in ischemic stroke patients with cerebral intracranial large artery occlusive disease. Stroke. 2012;43:3007–3011. doi: 10.1161/STROKEAHA.112.659144. [DOI] [PubMed] [Google Scholar]
- Liu Y, Belayev L, Zhao W, Busto R, Belayev A, Ginsberg MD. Neuroprotective effect of treatment with human albumin in permanent focal cerebral ischemia: histopathology and cortical perfusion studies. Eur J Pharmacol. 2001;428:193–201. doi: 10.1016/s0014-2999(01)01255-9. [DOI] [PubMed] [Google Scholar]
- Lo EH. Experimental models, neurovascular mechanisms and translational issues in stroke research. Br J Pharmacol. 2008;153(Suppl 1):S396–405. doi: 10.1038/sj.bjp.0707626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- Lu H, Wang Y, He X, Yuan F, Lin X, Xie B, Tang G, Huang J, Tang Y, Jin K, Chen S, Yang GY. Netrin-1 hyperexpression in mouse brain promotes angiogenesis and long-term neurological recovery after transient focal ischemia. Stroke. 2012;43:838–843. doi: 10.1161/STROKEAHA.111.635235. [DOI] [PubMed] [Google Scholar]
- Maas MB, Lev MH, Ay H, Singhal AB, Greer DM, Smith WS, Harris GJ, Halpern E, Kemmling A, Koroshetz WJ, Furie KL. Collateral vessels on CT angiography predict outcome in acute ischemic stroke. Stroke. 2009;40:3001–3005. doi: 10.1161/STROKEAHA.109.552513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchal G, Serrati C, Rioux P, Petit-Taboue MC, Viader F, de la Sayette V, Le Doze F, Lochon P, Derlon JM, Orgogozo JM, et al. PET imaging of cerebral perfusion and oxygen consumption in acute ischaemic stroke: relation to outcome. Lancet. 1993;341:925–927. doi: 10.1016/0140-6736(93)91214-7. [DOI] [PubMed] [Google Scholar]
- Marti HJ, Bernaudin M, Bellail A, Schoch H, Euler M, Petit E, Risau W. Hypoxia-induced vascular endothelial growth factor expression precedes neovascularization after cerebral ischemia. Am J Pathol. 2000;156:965–976. doi: 10.1016/S0002-9440(10)64964-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVerry F, Liebeskind DS, Muir KW. Systematic review of methods for assessing leptomeningeal collateral flow. AJNR Am J Neuroradiol. 2012;33:576–582. doi: 10.3174/ajnr.A2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisner JK, Price RJ. Spatial and temporal coordination of bone marrow-derived cell activity during arteriogenesis: regulation of the endogenous response and therapeutic implications. Microcirculation. 2010;17:583–599. doi: 10.1111/j.1549-8719.2010.00051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon BK, Smith EE, Coutts SB, Welsh DG, Faber JE, Goyal M, Hill MD, Demchuk AM, Damani Z, Cho KH, Chang HW, Hong JH, Sohn SI. Leptomeningeal collaterals are associated with modifiable metabolic risk factors. Ann Neurol. 2013 doi: 10.1002/ana.23906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SW, Tessier-Lavigne M, Kennedy TE. Netrins and their receptors. Adv Exp Med Biol. 2007;621:17–31. doi: 10.1007/978-0-387-76715-4_2. [DOI] [PubMed] [Google Scholar]
- Morioka M, Hamada J, Kawano T, Todaka T, Yano S, Kai Y, Ushio Y. Angiographic dilatation and branch extension of the anterior choroidal and posterior communicating arteries are predictors of hemorrhage in adult moyamoya patients. Stroke. 2003;34:90–95. doi: 10.1161/01.str.0000047120.67507.0d. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA. Brain protection: maybe yes, maybe no. Stroke. 2010;41:S85–86. doi: 10.1161/STROKEAHA.110.598458. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M, Schimrigk K. Vasomotor reactivity and pattern of collateral blood flow in severe occlusive carotid artery disease. Stroke. 1996;27:296–299. doi: 10.1161/01.str.27.2.296. [DOI] [PubMed] [Google Scholar]
- Murakami M, Nguyen LT, Zhuang ZW, Moodie KL, Carmeliet P, Stan RV, Simons M. The FGF system has a key role in regulating vascular integrity. J Clin Invest. 2008;118:3355–3366. doi: 10.1172/JCI35298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy JA, Dvorak AM, Dvorak HF. VEGF-A and the induction of pathological angiogenesis. Annu Rev Pathol. 2007;2:251–275. doi: 10.1146/annurev.pathol.2.010506.134925. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Suga S, Kawase T, Toda M. Intracarotid injection of granulocyte-macrophage colony-stimulating factor induces neuroprotection in a rat transient middle cerebral artery occlusion model. Brain Res. 2006;1089:179–185. doi: 10.1016/j.brainres.2006.03.059. [DOI] [PubMed] [Google Scholar]
- Nanba R, Tada M, Kuroda S, Houkin K, Iwasaki Y. Sequence analysis and bioinformatics analysis of chromosome 17q25 in familial moyamoya disease. Childs Nerv Syst. 2005;21:62–68. doi: 10.1007/s00381-004-1005-4. [DOI] [PubMed] [Google Scholar]
- Navaratna D, Guo S, Arai K, Lo EH. Mechanisms and targets for angiogenic therapy after stroke. Cell Adh Migr. 2009;3:216–223. doi: 10.4161/cam.3.2.8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmagadda A, Park HP, Prado R, Ginsberg MD. Albumin therapy improves local vascular dynamics in a rat model of primary microvascular thrombosis: a two-photon laser-scanning microscopy study. Stroke. 2008;39:198–204. doi: 10.1161/STROKEAHA.107.495598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura N, Schaffer CB, Friedman B, Lyden PD, Kleinfeld D. Penetrating arterioles are a bottleneck in the perfusion of neocortex. Proc Natl Acad Sci U S A. 2007;104:365–370. doi: 10.1073/pnas.0609551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama M, Hoshino A, Tsai L, Henley JR, Goshima Y, Tessier-Lavigne M, Poo MM, Hong K. Cyclic AMP/GMP-dependent modulation of Ca2+ channels sets the polarity of nerve growth-cone turning. Nature. 2003;423:990–995. doi: 10.1038/nature01751. [DOI] [PubMed] [Google Scholar]
- O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- Omura-Matsuoka E, Yagita Y, Sasaki T, Terasaki Y, Oyama N, Sugiyama Y, Todo K, Sakoda S, Kitagawa K. Hypertension impairs leptomeningeal collateral growth after common carotid artery occlusion: restoration by antihypertensive treatment. J Neurosci Res. 2011;89:108–116. doi: 10.1002/jnr.22522. [DOI] [PubMed] [Google Scholar]
- Ovbiagele B, Saver JL, Starkman S, Kim D, Ali LK, Jahan R, Duckwiler GR, Vinuela F, Pineda S, Liebeskind DS. Statin enhancement of collateralization in acute stroke. Neurology. 2007;68:2129–2131. doi: 10.1212/01.wnl.0000264931.34941.f0. [DOI] [PubMed] [Google Scholar]
- Ozgur HT, Kent Walsh T, Masaryk A, Seeger JF, Williams W, Krupinski E, Melgar M, Labadie E. Correlation of cerebrovascular reserve as measured by acetazolamide-challenged SPECT with angiographic flow patterns and intra- or extracranial arterial stenosis. AJNR Am J Neuroradiol. 2001;22:928–936. [PMC free article] [PubMed] [Google Scholar]
- Palmer TD, Willhoite AR, Gage FH. Vascular niche for adult hippocampal neurogenesis. J Comp Neurol. 2000;425:479–494. doi: 10.1002/1096-9861(20001002)425:4<479::aid-cne2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Pardali E, Goumans MJ, ten Dijke P. Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol. 2010;20:556–567. doi: 10.1016/j.tcb.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Parthasarathy AB, Tom WJ, Gopal A, Zhang X, Dunn AK. Robust flow measurement with multi-exposure speckle imaging. Opt Express. 2008;16:1975–1989. doi: 10.1364/oe.16.001975. [DOI] [PubMed] [Google Scholar]
- Penumbra Pivotal Stroke Trial I. The penumbra pivotal stroke trial: safety and effectiveness of a new generation of mechanical devices for clot removal in intracranial large vessel occlusive disease. Stroke. 2009;40:2761–2768. doi: 10.1161/STROKEAHA.108.544957. [DOI] [PubMed] [Google Scholar]
- Peppiatt CM, Howarth C, Mobbs P, Attwell D. Bidirectional control of CNS capillary diameter by pericytes. Nature. 2006;443:700–704. doi: 10.1038/nature05193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petcu EB, Smith RA, Miroiu RI, Opris MM. Angiogenesis in old-aged subjects after ischemic stroke: a cautionary note for investigators. J Angiogenes Res. 2010;2:26. doi: 10.1186/2040-2384-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phng LK, Gerhardt H. Angiogenesis: a team effort coordinated by notch. Dev Cell. 2009;16:196–208. doi: 10.1016/j.devcel.2009.01.015. [DOI] [PubMed] [Google Scholar]
- Pipp F, Boehm S, Cai WJ, Adili F, Ziegler B, Karanovic G, Ritter R, Balzer J, Scheler C, Schaper W, Schmitz-Rixen T. Elevated fluid shear stress enhances postocclusive collateral artery growth and gene expression in the pig hind limb. Arterioscler Thromb Vasc Biol. 2004;24:1664–1668. doi: 10.1161/01.ATV.0000138028.14390.e4. [DOI] [PubMed] [Google Scholar]
- Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873–887. doi: 10.1016/j.cell.2011.08.039. [DOI] [PubMed] [Google Scholar]
- Quaegebeur A, Lange C, Carmeliet P. The neurovascular link in health and disease: molecular mechanisms and therapeutic implications. Neuron. 2011;71:406–424. doi: 10.1016/j.neuron.2011.07.013. [DOI] [PubMed] [Google Scholar]
- Quaegebeur A, Segura I, Carmeliet P. Pericytes: blood-brain barrier safeguards against neurodegeneration? Neuron. 2010;68:321–323. doi: 10.1016/j.neuron.2010.10.024. [DOI] [PubMed] [Google Scholar]
- Redlitz A, Daum G, Sage EH. Angiostatin diminishes activation of the mitogen-activated protein kinases ERK-1 and ERK-2 in human dermal microvascular endothelial cells. J Vasc Res. 1999;36:28–34. doi: 10.1159/000025623. [DOI] [PubMed] [Google Scholar]
- Reinhart WH, Nagy C. Albumin affects erythrocyte aggregation and sedimentation. Eur J Clin Invest. 1995;25:523–528. doi: 10.1111/j.1365-2362.1995.tb01739.x. [DOI] [PubMed] [Google Scholar]
- Ribo M, Flores A, Rubiera M, Pagola J, Sargento-Freitas J, Rodriguez-Luna D, Coscojuela P, Maisterra O, Pineiro S, Romero FJ, Alvarez-Sabin J, Molina CA. Extending the time window for endovascular procedures according to collateral pial circulation. Stroke. 2011;42:3465–3469. doi: 10.1161/STROKEAHA.111.623827. [DOI] [PubMed] [Google Scholar]
- Ricard N, Bidart M, Mallet C, Lesca G, Giraud S, Prudent R, Feige JJ, Bailly S. Functional analysis of the BMP9 response of ALK1 mutants from HHT2 patients: a diagnostic tool for novel ACVRL1 mutations. Blood. 2010;116:1604–1612. doi: 10.1182/blood-2010-03-276881. [DOI] [PubMed] [Google Scholar]
- Robin AM, Zhang ZG, Wang L, Zhang RL, Katakowski M, Zhang L, Wang Y, Zhang C, Chopp M. Stromal cell-derived factor 1alpha mediates neural progenitor cell motility after focal cerebral ischemia. J Cereb Blood Flow Metab. 2006;26:125–134. doi: 10.1038/sj.jcbfm.9600172. [DOI] [PubMed] [Google Scholar]
- Rordorf G, Cramer SC, Efird JT, Schwamm LH, Buonanno F, Koroshetz WJ. Pharmacological elevation of blood pressure in acute stroke. Clinical effects and safety. Stroke. 1997;28:2133–2138. doi: 10.1161/01.str.28.11.2133. [DOI] [PubMed] [Google Scholar]
- Rordorf G, Koroshetz WJ, Ezzeddine MA, Segal AZ, Buonanno FS. A pilot study of drug-induced hypertension for treatment of acute stroke. Neurology. 2001;56:1210–1213. doi: 10.1212/wnl.56.9.1210. [DOI] [PubMed] [Google Scholar]
- Rosell A, Lo EH. Multiphasic roles for matrix metalloproteinases after stroke. Curr Opin Pharmacol. 2008;8:82–89. doi: 10.1016/j.coph.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Saharinen P, Eklund L, Miettinen J, Wirkkala R, Anisimov A, Winderlich M, Nottebaum A, Vestweber D, Deutsch U, Koh GY, Olsen BR, Alitalo K. Angiopoietins assemble distinct Tie2 signalling complexes in endothelial cell-cell and cell-matrix contacts. Nat Cell Biol. 2008;10:527–537. doi: 10.1038/ncb1715. [DOI] [PubMed] [Google Scholar]
- Santisakultarm TP, Schaffer CB. Optically quantified cerebral blood flow. J Cereb Blood Flow Metab. 2011;31:1337–1338. doi: 10.1038/jcbfm.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalzo F, Liebeskind D, Hu X. Reducing false intracranial pressure alarms using morphological waveform features. IEEE Trans Biomed Eng. 2013;60:235–239. doi: 10.1109/TBME.2012.2210042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schabitz WR, Kruger C, Pitzer C, Weber D, Laage R, Gassler N, Aronowski J, Mier W, Kirsch F, Dittgen T, Bach A, Sommer C, Schneider A. A neuroprotective function for the hematopoietic protein granulocyte-macrophage colony stimulating factor (GM-CSF) J Cereb Blood Flow Metab. 2008;28:29–43. doi: 10.1038/sj.jcbfm.9600496. [DOI] [PubMed] [Google Scholar]
- Schaffer CB, Friedman B, Nishimura N, Schroeder LF, Tsai PS, Ebner FF, Lyden PD, Kleinfeld D. Two-photon imaging of cortical surface microvessels reveals a robust redistribution in blood flow after vascular occlusion. PLoS Biol. 2006;4:e22. doi: 10.1371/journal.pbio.0040022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaper W. Collateral circulation: past and present. Basic Res Cardiol. 2009;104:5–21. doi: 10.1007/s00395-008-0760-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaper W, Buschmann I. Arteriogenesis, the good and bad of it. Cardiovasc Res. 1999;43:835–837. doi: 10.1016/s0008-6363(99)00191-1. [DOI] [PubMed] [Google Scholar]
- Schatteman GC, Hanlon HD, Jiao C, Dodds SG, Christy BA. Blood-derived angioblasts accelerate blood-flow restoration in diabetic mice. J Clin Invest. 2000;106:571–578. doi: 10.1172/JCI9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellinger PD, Shuaib A, Kohrmann M, Liebeskind DS, Jovin T, Hammer MD, Sen S, Huang DY, Solander S, Gupta R, Leker RR, Saver JL for the STI. Reduced Mortality and Severe Disability Rates in the SENTIS Trial. AJNR Am J Neuroradiol. 2013 doi: 10.3174/ajnr.A3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiekofer S, Galasso G, Sato K, Kraus BJ, Walsh K. Impaired revascularization in a mouse model of type 2 diabetes is associated with dysregulation of a complex angiogenic-regulatory network. Arterioscler Thromb Vasc Biol. 2005;25:1603–1609. doi: 10.1161/01.ATV.0000171994.89106.ca. [DOI] [PubMed] [Google Scholar]
- Schierling W, Troidl K, Apfelbeck H, Troidl C, Kasprzak PM, Schaper W, Schmitz-Rixen T. Cerebral arteriogenesis is enhanced by pharmacological as well as fluid-shear-stress activation of the Trpv4 calcium channel. Eur J Vasc Endovasc Surg. 2011;41:589–596. doi: 10.1016/j.ejvs.2010.11.034. [DOI] [PubMed] [Google Scholar]
- Schierling W, Troidl K, Mueller C, Troidl C, Wustrack H, Bachmann G, Kasprzak PM, Schaper W, Schmitz-Rixen T. Increased intravascular flow rate triggers cerebral arteriogenesis. J Cereb Blood Flow Metab. 2009a;29:726–737. doi: 10.1038/jcbfm.2008.165. [DOI] [PubMed] [Google Scholar]
- Schierling W, Troidl K, Troidl C, Schmitz-Rixen T, Schaper W, Eitenmuller IK. The role of angiogenic growth factors in arteriogenesis. J Vasc Res. 2009b;46:365–374. doi: 10.1159/000189797. [DOI] [PubMed] [Google Scholar]
- Schirmer SH, van Royen N, Moerland PD, Fledderus JO, Henriques JP, van der Schaaf RJ, Vis MM, Baan J, Jr, Koch KT, Horrevoets AJ, Hoefer IE, Piek JJ. Local cytokine concentrations and oxygen pressure are related to maturation of the collateral circulation in humans. J Am Coll Cardiol. 2009;53:2141–2147. doi: 10.1016/j.jacc.2009.02.049. [DOI] [PubMed] [Google Scholar]
- Schneider UC, Schilling L, Schroeck H, Nebe CT, Vajkoczy P, Woitzik J. Granulocyte-macrophage colony-stimulating factor-induced vessel growth restores cerebral blood supply after bilateral carotid artery occlusion. Stroke. 2007;38:1320–1328. doi: 10.1161/01.STR.0000259707.43496.71. [DOI] [PubMed] [Google Scholar]
- Scholz D, Ito W, Fleming I, Deindl E, Sauer A, Wiesnet M, Busse R, Schaper J, Schaper W. Ultrastructure and molecular histology of rabbit hind-limb collateral artery growth (arteriogenesis) Virchows Arch. 2000;436:257–270. doi: 10.1007/s004280050039. [DOI] [PubMed] [Google Scholar]
- Schwarz S, Georgiadis D, Aschoff A, Schwab S. Effects of induced hypertension on intracranial pressure and flow velocities of the middle cerebral arteries in patients with large hemispheric stroke. Stroke. 2002;33:998–1004. doi: 10.1161/01.str.0000014584.17714.2e. [DOI] [PubMed] [Google Scholar]
- Scott RM, Smith ER. Moyamoya disease and moyamoya syndrome. N Engl J Med. 2009;360:1226–1237. doi: 10.1056/NEJMra0804622. [DOI] [PubMed] [Google Scholar]
- Scott RM, Smith JL, Robertson RL, Madsen JR, Soriano SG, Rockoff MA. Long-term outcome in children with moyamoya syndrome after cranial revascularization by pial synangiosis. J Neurosurg. 2004;100:142–149. doi: 10.3171/ped.2004.100.2.0142. [DOI] [PubMed] [Google Scholar]
- Shen F, Fan Y, Su H, Zhu Y, Chen Y, Liu W, Young WL, Yang GY. Adeno-associated viral vector-mediated hypoxia-regulated VEGF gene transfer promotes angiogenesis following focal cerebral ischemia in mice. Gene Ther. 2008;15:30–39. doi: 10.1038/sj.gt.3303048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin HK, Nishimura M, Jones PB, Ay H, Boas DA, Moskowitz MA, Ayata C. Mild induced hypertension improves blood flow and oxygen metabolism in transient focal cerebral ischemia. Stroke. 2008;39:1548–1555. doi: 10.1161/STROKEAHA.107.499483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shovlin CL. Hereditary haemorrhagic telangiectasia: pathophysiology, diagnosis and treatment. Blood Rev. 2010;24:203–219. doi: 10.1016/j.blre.2010.07.001. [DOI] [PubMed] [Google Scholar]
- Shuaib A, Bornstein NM, Diener HC, Dillon W, Fisher M, Hammer MD, Molina CA, Rutledge JN, Saver JL, Schellinger PD, Shownkeen H, Investigators ST. Partial aortic occlusion for cerebral perfusion augmentation: safety and efficacy of NeuroFlo in Acute Ischemic Stroke trial. Stroke. 2011a;42:1680–1690. doi: 10.1161/STROKEAHA.110.609933. [DOI] [PubMed] [Google Scholar]
- Shuaib A, Butcher K, Mohammad AA, Saqqur M, Liebeskind DS. Collateral blood vessels in acute ischaemic stroke: a potential therapeutic target. Lancet Neurol. 2011b;10:909–921. doi: 10.1016/S1474-4422(11)70195-8. [DOI] [PubMed] [Google Scholar]
- Sieczkiewicz GJ, Herman IM. TGF-beta 1 signaling controls retinal pericyte contractile protein expression. Microvasc Res. 2003;66:190–196. doi: 10.1016/s0026-2862(03)00055-4. [DOI] [PubMed] [Google Scholar]
- Smith ER, Scott RM. Surgical management of moyamoya syndrome. Skull Base. 2005;15:15–26. doi: 10.1055/s-2005-868160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smrcka M, Ogilvy CS, Crow RJ, Maynard KI, Kawamata T, Ames A., 3rd Induced hypertension improves regional blood flow and protects against infarction during focal ischemia: time course of changes in blood flow measured by laser Doppler imaging. Neurosurgery. 1998;42:617–624. doi: 10.1097/00006123-199803000-00032. discussion 624–615. [DOI] [PubMed] [Google Scholar]
- Srinivasan VJ, Atochin DN, Radhakrishnan H, Jiang JY, Ruvinskaya S, Wu W, Barry S, Cable AE, Ayata C, Huang PL, Boas DA. Optical coherence tomography for the quantitative study of cerebrovascular physiology. J Cereb Blood Flow Metab. 2011;31:1339–1345. doi: 10.1038/jcbfm.2011.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockmann C, Doedens A, Weidemann A, Zhang N, Takeda N, Greenberg JI, Cheresh DA, Johnson RS. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature. 2008;456:814–818. doi: 10.1038/nature07445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storkebaum E, Quaegebeur A, Vikkula M, Carmeliet P. Cerebrovascular disorders: molecular insights and therapeutic opportunities. Nat Neurosci. 2011;14:1390–1397. doi: 10.1038/nn.2947. [DOI] [PubMed] [Google Scholar]
- Stratman AN, Malotte KM, Mahan RD, Davis MJ, Davis GE. Pericyte recruitment during vasculogenic tube assembly stimulates endothelial basement membrane matrix formation. Blood. 2009;114:5091–5101. doi: 10.1182/blood-2009-05-222364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchting S, Bicknell R, Eichmann A. Neuronal clues to vascular guidance. Exp Cell Res. 2006;312:668–675. doi: 10.1016/j.yexcr.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Sugiyama Y, Yagita Y, Oyama N, Terasaki Y, Omura-Matsuoka E, Sasaki T, Kitagawa K. Granulocyte colony-stimulating factor enhances arteriogenesis and ameliorates cerebral damage in a mouse model of ischemic stroke. Stroke. 2011;42:770–775. doi: 10.1161/STROKEAHA.110.597799. [DOI] [PubMed] [Google Scholar]
- Suzuki J, Takaku A. Cerebrovascular “moyamoya” disease. Disease showing abnormal net-like vessels in base of brain. Arch Neurol. 1969;20:288–299. doi: 10.1001/archneur.1969.00480090076012. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Gotoh F, Gotoh J, Koto A. Evidence for in vivo cerebrovascular neurogenic vasodilatation in the rat. Clin Auton Res. 1991;1:23–26. doi: 10.1007/BF01826054. [DOI] [PubMed] [Google Scholar]
- Taguchi A, Soma T, Tanaka H, Kanda T, Nishimura H, Yoshikawa H, Tsukamoto Y, Iso H, Fujimori Y, Stern DM, Naritomi H, Matsuyama T. Administration of CD34+ cells after stroke enhances neurogenesis via angiogenesis in a mouse model. J Clin Invest. 2004;114:330–338. doi: 10.1172/JCI20622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda Y, Costa S, Delamarre E, Roncal C, Leite de Oliveira R, Squadrito ML, Finisguerra V, Deschoemaeker S, Bruyere F, Wenes M, Hamm A, Serneels J, Magat J, Bhattacharyya T, Anisimov A, Jordan BF, Alitalo K, Maxwell P, Gallez B, Zhuang ZW, Saito Y, Simons M, De Palma M, Mazzone M. Macrophage skewing by Phd2 haplodeficiency prevents ischaemia by inducing arteriogenesis. Nature. 2011;479:122–126. doi: 10.1038/nature10507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita S, Zheng LP, Brogi E, Kearney M, Pu LQ, Bunting S, Ferrara N, Symes JF, Isner JM. Therapeutic angiogenesis. A single intraarterial bolus of vascular endothelial growth factor augments revascularization in a rabbit ischemic hind limb model. J Clin Invest. 1994;93:662–670. doi: 10.1172/JCI117018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, Shimizu K. Hypoplasia of the bilateral internal carotid arteries. Brain Nerve. 1957;9:37–43. [Google Scholar]
- Tanghetti B, Capra R, Giunta F, Marini G, Orlandini A. Moyamoya syndrome in only one of two identical twins. Case report. J Neurosurg. 1983;59:1092–1094. doi: 10.3171/jns.1983.59.6.1092. [DOI] [PubMed] [Google Scholar]
- Tchaikovski V, Olieslagers S, Bohmer FD, Waltenberger J. Diabetes mellitus activates signal transduction pathways resulting in vascular endothelial growth factor resistance of human monocytes. Circulation. 2009;120:150–159. doi: 10.1161/CIRCULATIONAHA.108.817528. [DOI] [PubMed] [Google Scholar]
- Todo K, Kitagawa K, Sasaki T, Omura-Matsuoka E, Terasaki Y, Oyama N, Yagita Y, Hori M. Granulocyte-macrophage colony-stimulating factor enhances leptomeningeal collateral growth induced by common carotid artery occlusion. Stroke. 2008;39:1875–1882. doi: 10.1161/STROKEAHA.107.503433. [DOI] [PubMed] [Google Scholar]
- Tomita M, Suzuki N, Hamel E, Busija D, Lauritzen M. Regulation of cerebral microcirculation--update. Keio J Med. 2000;49:26–34. doi: 10.2302/kjm.49.26. [DOI] [PubMed] [Google Scholar]
- Troidl C, Jung G, Troidl K, Hoffmann J, Mollmann H, Nef H, Schaper W, Hamm CW, Schmitz-Rixen T. The temporal and spatial distribution of macrophage subpopulations during arteriogenesis. Curr Vasc Pharmacol. 2013;11:5–12. [PubMed] [Google Scholar]
- Troidl C, Nef H, Voss S, Schilp A, Kostin S, Troidl K, Szardien S, Rolf A, Schmitz-Rixen T, Schaper W, Hamm CW, Elsasser A, Mollmann H. Calcium-dependent signalling is essential during collateral growth in the pig hind limb-ischemia model. J Mol Cell Cardiol. 2010a;49:142–151. doi: 10.1016/j.yjmcc.2010.03.021. [DOI] [PubMed] [Google Scholar]
- Troidl C, Troidl K, Schierling W, Cai WJ, Nef H, Mollmann H, Kostin S, Schimanski S, Hammer L, Elsasser A, Schmitz-Rixen T, Schaper W. Trpv4 induces collateral vessel growth during regeneration of the arterial circulation. J Cell Mol Med. 2009a;13:2613–2621. doi: 10.1111/j.1582-4934.2008.00579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troidl K, Ruding I, Cai WJ, Mucke Y, Grossekettler L, Piotrowska I, Apfelbeck H, Schierling W, Volger OL, Horrevoets AJ, Grote K, Schmitz-Rixen T, Schaper W, Troidl C. Actin-binding rho activating protein (Abra) is essential for fluid shear stress-induced arteriogenesis. Arterioscler Thromb Vasc Biol. 2009b;29:2093–2101. doi: 10.1161/ATVBAHA.109.195305. [DOI] [PubMed] [Google Scholar]
- Troidl K, Schaper W. Arteriogenesis versus angiogenesis in peripheral artery disease. Diabetes Metab Res Rev. 2012;28(Suppl 1):27–29. doi: 10.1002/dmrr.2232. [DOI] [PubMed] [Google Scholar]
- Troidl K, Tribulova S, Cai WJ, Ruding I, Apfelbeck H, Schierling W, Troidl C, Schmitz-Rixen T, Schaper W. Effects of endogenous nitric oxide and of DETA NONOate in arteriogenesis. J Cardiovasc Pharmacol. 2010b;55:153–160. doi: 10.1097/FJC.0b013e3181c9556f. [DOI] [PubMed] [Google Scholar]
- van Laar PJ, Hendrikse J, Klijn CJ, Kappelle LJ, van Osch MJ, van der Grond J. Symptomatic carotid artery occlusion: flow territories of major brain-feeding arteries. Radiology. 2007;242:526–534. doi: 10.1148/radiol.2422060179. [DOI] [PubMed] [Google Scholar]
- van Oostrom MC, van Oostrom O, Quax PH, Verhaar MC, Hoefer IE. Insights into mechanisms behind arteriogenesis: what does the future hold? J Leukoc Biol. 2008;84:1379–1391. doi: 10.1189/jlb.0508281. [DOI] [PubMed] [Google Scholar]
- Vander Eecken HM. Morphological significance of leptomeningeal anastomoses confined to the territory of cerebral arteries. Acta Neurol Psychiatr Belg. 1954;54:525–532. [PubMed] [Google Scholar]
- Vinukonda G, Dummula K, Malik S, Hu F, Thompson CI, Csiszar A, Ungvari Z, Ballabh P. Effect of prenatal glucocorticoids on cerebral vasculature of the developing brain. Stroke. 2010;41:1766–1773. doi: 10.1161/STROKEAHA.110.588400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walshe TE, Saint-Geniez M, Maharaj AS, Sekiyama E, Maldonado AE, D’Amore PA. TGF-beta is required for vascular barrier function, endothelial survival and homeostasis of the adult microvasculature. PLoS One. 2009;4:e5149. doi: 10.1371/journal.pone.0005149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltenberger J, Lange J, Kranz A. Vascular endothelial growth factor-A-induced chemotaxis of monocytes is attenuated in patients with diabetes mellitus: A potential predictor for the individual capacity to develop collaterals. Circulation. 2000;102:185–190. doi: 10.1161/01.cir.102.2.185. [DOI] [PubMed] [Google Scholar]
- Wang RK, An L. Doppler optical micro-angiography for volumetric imaging of vascular perfusion in vivo. Opt Express. 2009;17:8926–8940. doi: 10.1364/oe.17.008926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Huang J, Li Y, Yang GY. Roles of chemokine CXCL12 and its receptors in ischemic stroke. Curr Drug Targets. 2012;13:166–172. doi: 10.2174/138945012799201603. [DOI] [PubMed] [Google Scholar]
- Wayner DD, Burton GW, Ingold KU, Locke S. Quantitative measurement of the total, peroxyl radical-trapping antioxidant capability of human blood plasma by controlled peroxidation. The important contribution made by plasma proteins. FEBS Lett. 1985;187:33–37. doi: 10.1016/0014-5793(85)81208-4. [DOI] [PubMed] [Google Scholar]
- Weber M, Baker MB, Moore JP, Searles CD. MiR-21 is induced in endothelial cells by shear stress and modulates apoptosis and eNOS activity. Biochem Biophys Res Commun. 2010;393:643–648. doi: 10.1016/j.bbrc.2010.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesley RC, Jr, Morgan DB. Effect of continuous intra-aortic balloon inflation in canine open chest cardiopulmonary resuscitation. Crit Care Med. 1990;18:630–633. doi: 10.1097/00003246-199006000-00011. [DOI] [PubMed] [Google Scholar]
- Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci. 2011a;14:1398–1405. doi: 10.1038/nn.2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler EA, Bell RD, Zlokovic BV. Lack of Smad or Notch leads to a fatal game of brain pericyte hopscotch. Dev Cell. 2011b;20:279–280. doi: 10.1016/j.devcel.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Wityk RJ. Blood pressure augmentation in acute ischemic stroke. J Neurol Sci. 2007;261:63–73. doi: 10.1016/j.jns.2007.04.033. [DOI] [PubMed] [Google Scholar]
- Yamada I, Himeno Y, Suzuki S, Matsushima Y. Posterior circulation in moyamoya disease: angiographic study. Radiology. 1995;197:239–246. doi: 10.1148/radiology.197.1.7568830. [DOI] [PubMed] [Google Scholar]
- Yarnitsky D, Lorian A, Shalev A, Zhang ZD, Takahashi M, Agbaje-Williams M, Macdonald RL. Reversal of cerebral vasospasm by sphenopalatine ganglion stimulation in a dog model of subarachnoid hemorrhage. Surg Neurol. 2005;64:5–11. doi: 10.1016/j.surneu.2004.09.029. discussion 11. [DOI] [PubMed] [Google Scholar]
- Yin KJ, Hamblin M, Chen YE. Angiogenesis-Regulating microRNAs and Ischemic Stroke. Curr Vasc Pharmacol. 2013 doi: 10.2174/15701611113119990016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan F, Wang Y, Guan Y, Ren Y, Lu H, Xiao T, Xie H, Vosler PS, Chen J, Yang GY. Real-time imaging of mouse lenticulostriate artery following brain ischemia. Front Biosci (Elite Ed) 2013;5:517–524. doi: 10.2741/e633. [DOI] [PubMed] [Google Scholar]
- Zbinden R, Vogel R, Meier B, Seiler C. Coronary collateral flow and peripheral blood monocyte concentration in patients treated with granulocyte-macrophage colony stimulating factor. Heart. 2004;90:945–946. doi: 10.1136/hrt.2003.018259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao BQ, Wang S, Kim HY, Storrie H, Rosen BR, Mooney DJ, Wang X, Lo EH. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med. 2006;12:441–445. doi: 10.1038/nm1387. [DOI] [PubMed] [Google Scholar]