

Abstract
Introduction
Chemotherapy-related endothelial damage contributes to the early development of cardiovascular morbidity in testicular cancer patients. We aimed to identify relevant mechanisms of and search for candidate biomarkers for this endothelial damage.
Methods
Human micro-vascular endothelial cells (HMEC-1) were exposed to bleomycin or cisplatin with untreated samples as control. 18k cDNA microarrays were used. Gene expression differences were analysed at single gene level and in gene sets clustered in biological pathways and validated by qRT-PCR. Protein levels of a candidate biomarker were measured in testicular cancer patient plasma before, during and after bleomycin-etoposide-cisplatin chemotherapy, and related to endothelial damage biomarkers (von Willebrand Factor (vWF), high-sensitivity C-Reactive Protein (hsCRP)).
Results
Microarray data identified several genes with highly differential expression; e.g. Growth Differentiation Factor 15 (GDF-15), Activating Transcription Factor 3 (ATF3) and Amphiregulin (AREG). Pathway analysis revealed strong associations with ‘p53’ and ‘Diabetes Mellitus’ gene sets. Based on known function, we measured GDF-15 protein levels in 41 testicular patients during clinical follow-up. Pre-chemotherapy GDF-15 levels equalled controls. Throughout chemotherapy GDF-15, vWF and hsCRP levels increased, and were correlated at different time-points.
Conclusion
An unbiased approach in a preclinical model revealed genes related to chemotherapy-induced endothelial damage, like GDF-15. The increases in plasma GDF-15 levels in testicular cancer patients during chemotherapy and its association with vWF and hsCRP suggest that GDF-15 is a potentially useful biomarker related to endothelial damage.
Introduction
The introduction of platinum-based chemotherapy in the late seventies has resulted in high cure rates in patients with metastatic testicular cancer. However, this chemotherapy causes side-effects, resulting in morbidity in successfully treated survivors [1]. Relevant issues are the increased risks for second malignancies and cardiovascular disease (CVD) [2–5]. CVD can arise during or shortly after treatment [6, 7], as well as years to decades later [3–5, 8–11].
One of the mechanisms involved in the development of this treatment-related CVD is direct endothelial damage. In addition to induction of endothelial cell death [12–14], both bleomycine [14–16] and cisplatin [14, 17–19] indirectly influence endothelial cell function, e.g. through interference with inflammatory and fibrinolytic factors. Ultimately, this chemotherapy-induced cellular activation can progress to endothelial dysfunction, accelerated atherosclerosis and overt CVD.
Early recognition and possibly prevention of these treatment-related complications are critical to maintain an optimal health condition of testicular cancer survivors. The development of CVD is a gradual process, and early interventions or intensified screening may slow down or stop the progression towards overt clinical morbidity. Biomarkers for treatment-related endothelial damage can identify those patients at increased risk for CVD.
In this study we back-translated the clinical finding that bleomycin and cisplatin induce endothelial damage. We used an unbiased approach by analysing cDNA microarray results to identify novel genes associated with chemotherapy-related endothelial damage. Gene expression profiles were generated from the human microvascular endothelial cell line (HMEC-1) before and after treatment with bleomycin and cisplatin at different time points and concentrations. Quantitative Real Time PCR (qRT-PCR) was performed to confirm genes with significant expression differences in several experimental settings. Next, based on known function of these genes in the literature, we selected one of these candidate genes for further validation as proof of principle at the protein level in a testicular cancer patient cohort. With this translational approach we aimed to identify mechanisms of and potential biomarkers for chemotherapy-related endothelial damage.
Materials and Methods
Cell line model
HMEC-1 is an immortalised human dermal micro-vascular endothelial cell line that retains its morphologic and functional endothelial cell characteristics during several passages [20]. Cells were grown as a monolayer in MCDB-131 medium (Invitrogen, Merelbeke, Belgium) supplemented with 10% foetal calf serum (Bodinco, Alkmaar, the Netherlands), 10 mM L-glutamine (Invitrogen, Merelbeke, Belgium), 1 μg/mL hydrocortisone (Sigma-Aldrich, Amsterdam, the Netherlands) and 10 ng/mL human epidermal growth factor (R&D Systems, Abingdon, UK), and were cultured at 37°C in a humidified atmosphere containing 5% CO2. Experiments were performed between passages 15–30.
In an “acute”-exposure setting, HMEC-1 were left untreated as controls, or were treated with 0.3 (IC50 (concentration inhibiting cell survival by 50%)) or 1.5 μg/mL (IC90) bleomycin and 2.6 (IC50) or 12.9 μM (IC90) cisplatin for 6, 24, and 48 hours (S1A Fig.). The IC50 values for both bleomycin and cisplatin fall within the plasma physiological concentrations of these drugs in patients during active treatment [14, 21–22]. In addition, in a “chronic”-exposure setting, lower doses were administered (IC10; bleomycin 0.06 μg/mL or cisplatin 0.52 μM) two times a week; cells were collected for analysis at day 30 (S1B Fig.). Administration of cisplatin had to be withheld at the 7th administration because of considerable cell death, but was continued at full dose thereafter. Bleomycin could be administered without interruption.
cDNA microarray experiments
Total RNA was isolated from HMEC-1 by a RNeasy kit (Qiagen, Venlo, the Netherlands) and pooled for each time-point and drug from 2 independent experiments. After purification (Qiaquick PCR purification kit, Qiagen, Venlo, the Netherlands), amplified RNA (cRNA) samples were transformed to cDNA with reverse transcriptase, independently labelled with Cy3 (green) and Cy5 (red), and randomly hybridised to the custom-made 18K cDNA microarrays. Fluorescent images of the microarray slides were obtained with the Affymetrix GMS428 scanner (Affymetrix, Santa Clara, CA) for both fluorophores, signal intensities for each spot were quantified by dedicated IMAGENE 5.6 software (Biodiscovery, Marina del Rey, CA).
Quantile normalisation was applied to log2 transformed Cy3 and Cy5 intensities. Operon v2.0 (Human Genome Oligo Set V2) probe identifiers were converted to official HUGO gene symbols. Expression values of multiple probes targeting a single gene were averaged, resulting in a total of 15,950 unique genes. Subsequently, expression data obtained from multiple hybridisations (n = 4) of the same HMEC-1 specimen were averaged.
The data have been deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO Series Accession number GSE62523 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE62523).
Class comparison
In the “acute” exposure setting, differentially expressed genes between HMEC-1 untreated samples and samples exposed to the different drug dosages (i.e. IC50 and IC90) were tested with the non-parametric Cuzick test for linear trend, resulting in a Z score and a P value. This test was done for cells collected after 6 (t = 6), 24 (t = 24) and 48 (t = 48) hours. For all three time-points the Z score resulting from the Cuzick tests for linear trend was summed (i.e. ΣZ = Zt = 6 + Zt = 24 + Zt = 48); thereby selecting on genes with the constraint that changes in expression in a consistent direction with increasing concentrations over time. Genes were ranked according to their ΣZ score.
In the “chronic” exposure setting, a T-test was performed on gene expression levels obtained from samples exposed to the drugs (IC10 cisplatin or IC10 bleomycin) versus the untreated control samples that were collected after 30 days incubation. Results of these genes were ranked according to P-value.
Gene Set Enrichment Analysis
Gene Set Enrichment Analysis (GSEA) [23] was executed with GSEA 2.0 software package (Broad Institute, Cambridge, MA). Expression data of all 15,950 genes were compared against functional gene sets to determine whether any of these sets were enriched in HMEC-1 treated with bleomycin or cisplatin in the “acute” as well as the “chronic” exposure setting. The comparison was performed using 169 gene sets from Kyoto Encyclopedia of Genes and Genomes database (KEGG; http://www.genome.jp/kegg/). Statistical significance of enrichment was determined using an empirical gene-based permutation test using 1000 permutations. A false discovery rate (FDR) was calculated for each functional gene set, which represent the estimated probability that a given enrichment score represents a false positive finding. We report gene sets with a FDR ≤ 0.10 and P ≤ 0.025.
Quantitative Real Time PCR
Differential expression of three genes was validated by qRT-PCR. For this purpose, RNA samples included in the cDNA microarray analysis were used. In addition, two independent experiments were performed in which HMEC-1 was exposed to cisplatin and bleomycin according to the “acute”-exposure setting. RNA samples were isolated after 6, 24 and 48 hours exposure to the drugs (S1A Fig.). All RNA samples were DNase treated to eliminate genomic DNA-contamination, and subsequently, RNA was reverse transcribed into cDNA. qRT-PCR was performed using Applied Biosystems TaqMan assays, according to the manufacturers protocol. Master Mix, primers and TaqMan probes were purchased from Applied Biosystems (Nieuwerkerk a/d IJssel, the Netherlands). Three genes, with highly differential expression in three out of four experimental settings, were considered plausible candidates for qRT-PCR validation. The genes and their respective Taqman gene expression assay numbers were Growth Differentiation Factor 15 (GDF-15; Hs00171132_m1), Activating Transcription Factor 3 (ATF3; Hs00231069_m1), Amphiregulin (AREG; Hs00155832_m1); in addition expression of the housekeeping gene Glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Hs02758991_g1) was determined. All experiments were performed in triplicate using ABI PRISM 7900 HT Sequence Detection System, with the following cycling conditions: 2 min at 50°C, 10 min at 95°C, followed by 40 cycles of 15 sec at 95°C and 1 min at 60°C. Relative quantity of target genes was calculated by dividing the mean cycle threshold (CT) for the gene of interest by the mean CT-value for the housekeeping gene GAPDH. Relative expression-differences were calculated by comparing expression to the baseline time-point (t = 6, S1A Fig.). Two-sided t-test was used to compare differences in expression. P-values < 0.05 were considered to indicate a significant difference.
GDF-15 protein levels in testicular cancer patient plasma during and after bleomycin- and cisplatin-based chemotherapy
To clinically validate the findings from the cell line model, we used a cohort of 41 testicular cancer patients who participated in a prospective study on early chemotherapy-related cardiovascular changes during bleomycin- and cisplatin-based regimens. Patients eligible for the study had metastatic testicular cancer, were 18–50 years old and were receiving first line cisplatin-based chemotherapy at the University Medical Centre Groningen, the Netherlands. Exclusion criteria were previous chemotherapy or radiotherapy, presence of CVD, use of erythropoietin and glomerular filtration rate < 60 mL/min. The local ethics committee approved the study, and written informed consent was obtained from all participants. Depending on their International Germ Cell Cancer Collaborative Group (IGCCCG) prognosis group, patients received either three or four BEP courses lasting 3 weeks each (bleomycin—30 USP, days 2, 8 and 15; etoposide—100 mg/m2, days 1–5) and cisplatin—20 mg/m2, days 1–5). During the first 6 days patients were hydrated with 4 L NaCl 0.9%/day and received daily anti-emetic therapy (dexamethason, ondansetron). Blood samples were drawn at day 1, 8 and 15 of the first chemotherapy course, day 1 and 8 of the second and third course, (c1d1 (= baseline), c1d8, c1d15, c2d1, etc.), one month after completion and one year after start of chemotherapy. EDTA plasma was serially collected and stored in -20°C until analysis. Reference data were obtained from healthy male siblings of adult childhood cancer survivors, who had participated as control subjects in a cross-sectional study on late cardiovascular sequelae of treatment for childhood cancer [24]. Out of these healthy male siblings, a control group with a comparable median age as the testicular cancer patients was selected. Measurements in the controls were performed as described above.
Plasma GDF-15 protein levels were determined by sandwich enzyme-linked immunosorbent assay (ELISA) with a commercially available kit (R&D Systems, Abingdon, UK). Furthermore, these GDF-15 protein levels were related to plasma markers for endothelial damage (von Willebrand Factor (vWF), measured as described earlier) [25] and systemic inflammation (high-sensitivity C-Reactive Protein (hsCRP), as described earlier) [26]. For analysis of changes in these markers, non-normally distributed data are represented as median (range). For comparisons between groups the non-parametric Mann-Whitney U test was applied; the Wilcoxon’s signed rank test was used for paired changes. Two-sided P-values ≤ 0.05 were considered to indicate significance, SPSS software package version 22 (SPSS Inc., Chicago, IL) was used.
Results
cDNA microarray
Class comparison. The top 50 of most differentially expressed genes in the “acute” and “chronic” exposure setting for bleomycin and cisplatin are summarised in Table 1. Fig. 1 shows a Venn diagram of the overlapping genes in the top 50’s of the four different exposure settings. From this analysis three genes, e.g. GDF-15, ATF3 and AREG, were found in the top 50 of three out of four exposure settings. Because of this overlap in the different exposure settings we considered these three genes plausible candidates, and selected these for validation by qRT-PCR.
Table 1. Top 50 genes with largest difference in expression in the different exposition settings in HMEC-1.
| BLEOMYCIN - ACUTE | BLEOMYCIN - CHRONIC | CISPLATIN - ACUTE | CISPLATIN - CHRONIC | ||||
|---|---|---|---|---|---|---|---|
| Gene | ΣZ | Gene | P-value | Gene | ΣZ | Gene | P-value |
| ATF3 * ‡ | ↑ 9.574 | CYP1B1 | ↑ <0.0001 | CORT | ↑ 9.577 | MX1† | ↓ <0.0001 |
| KIAA1370 | ↑ 9.574 | KLF12 | ↑ <0.0001 | TREM2 | ↑ 9.222 | GPR87 | ↓ <0.0001 |
| RRAD | ↑ 9.552 | PRSS35 ‡ | ↑ <0.001 | ATF3 * § | ↑ 9.039 | IFI27 | ↓ <0.0001 |
| C12orf5 | ↑ 9.476 | GLS2 | ↓ <0.001 | AVPI1 | ↑ 8.757 | JMJD4 | ↑ <0.001 |
| GDF-15 ‡ | ↑ 9.454 | ENPP4 | ↑ <0.001 | AK1* | ↑ 8.658 | FAM155A | ↓ <0.001 |
| GPR87 | ↑ 9.454 | ZMAT1 | ↑ <0.001 | VASN | ↑ 8.653 | OAS3 | ↓ <0.001 |
| MDM2 | ↑ 9.356 | TMEM184C | ↑ <0.001 | SESN2 * | ↑ 8.633 | MX2 | ↓ <0.001 |
| SESN2 * | ↑ 9.258 | ADAM12† | ↑ <0.001 | LRDD | ↑ 8.633 | HLA-F | ↓ <0.001 |
| COL7A1 | ↑ 9.16 | RAB11FIP1 | ↑ <0.001 | AREG§ | ↑ 8.633 | AREG † § | ↓ <0.001 |
| ATG16L2 | ↑ 9.16 | PTGER2 | ↑ <0.001 | PLCD1 | ↑ 8.586 | LOC158376 | ↑ <0.001 |
| AK1* | ↑ 9.16 | SERPINB2 | ↑ <0.001 | ADM | ↑ 8.549 | ALKBH8 | ↑ <0.001 |
| FAS | ↑ 8.964 | SCFD2 | ↑ <0.01 | LRRTM2 | ↑ 8.524 | ABCC4 | ↑ <0.001 |
| LIF | ↑ 8.964 | KRTAP4-8 | ↑ <0.01 | DEDD2 | ↑ 8.457 | HIST2H2BE | ↓ <0.001 |
| VWCE | ↑ 8.866 | C10orf136 | ↑ <0.01 | RALGDS | ↑ 8.37 | RFT1 | ↑ <0.001 |
| TP53INP1 | ↑ 8.817 | HES1† | ↓ <0.01 | DNAJB2 | ↑ 8.364 | LRRC38 | ↑ <0.001 |
| VDR | ↑ 8.811 | COL1A2 | ↑ <0.01 | MSX1 | ↑ 8.328 | SNAI1† | ↓ <0.001 |
| C4orf18 | ↑ 8.768 | HIST1H2BJ | ↑ <0.01 | MST150 | ↑ 8.322 | C19orf42 | ↑ <0.01 |
| FERMT1 | ↑ 8.734 | RBPJL | ↑ <0.01 | NR4A3 | ↑ 8.157 | LOC151171 | ↓ <0.01 |
| FUCA1 | ↑ 8.691 | CRTAC1 | ↓ <0.01 | FDXR | ↑ 8.143 | SIAE | ↓ <0.01 |
| MCC | ↑ 8.691 | CCDC148† | ↓ <0.01 | ITPKA | ↑ 8.143 | IQGAP2 | ↓ <0.01 |
| NELF | ↑ 8.691 | ROR1 | ↑ <0.01 | KREMEN2§ | ↑ 8.143 | POLA1 | ↑ <0.01 |
| BTG2 | ↑ 8.691 | EDN1 | ↓ <0.01 | SLC31A2 | ↑ 8.126 | PDK4 | ↓ <0.01 |
| TGFBR1 | ↑ 8.691 | TGFB2 ‡ | ↑ <0.01 | CEACAM1 | ↑ 8.107 | ATF3 † § | ↓ <0.01 |
| TMEM131 | ↑ 8.572 | PRKCZ | ↑ <0.01 | IRF5 | ↑ 8.101 | TNFSF10 | ↓ <0.01 |
| CDH10 | ↑ 8.572 | MARVELD2 | ↑ <0.01 | FOXL2 | ↑ 8.101 | DHX37 | ↑ <0.01 |
| FZD2 | ↓ -7.775 | ATF3 † ‡ | ↓ <0.01 | VCAM1 * | ↓ -8.432 | TOMM40L | ↑ <0.01 |
| C3orf36 | ↓ -7.809 | CD82 | ↓ <0.01 | C7orf10 | ↓ -8.437 | IFIT3 | ↓ <0.01 |
| TRIB2 * | ↓ -7.817 | XTP3TPA | ↑ <0.01 | C3orf26 | ↓ -8.438 | IFI44L | ↓ <0.01 |
| SMA5 | ↓ -7.835 | SPTAN1 | ↓ <0.01 | MYRIP | ↓ -8.535 | TNC† | ↑ <0.01 |
| NDRG4 | ↓ -7.869 | KIAA1655 | ↑ <0.01 | ROR1 | ↓ -8.56 | KREMEN2 § | ↓ <0.01 |
| EBPL | ↓ -7.873 | DDB2 | ↓ <0.01 | QKI | ↓ -8.597 | HNMT | ↓ <0.01 |
| TGFB2 ‡ | ↓ -7.912 | SNAI1† | ↓ <0.01 | TRIB2 * | ↓ -8.628 | BRD9 | ↑ <0.01 |
| MYCN | ↓ -7.971 | STC2 | ↑ <0.01 | CDKAL1 | ↓ -8.647 | BEX2 | ↓ <0.01 |
| SEMA3A * | ↓ -7.971 | TNC† | ↑ <0.01 | TFPI | ↓ -8.658 | MGC33894 | ↓ <0.01 |
| GIMAP2 | ↓ -7.992 | LRRIQ1 | ↓ <0.01 | LTBP1 | ↓ -8.664 | DUSP1† | ↓ <0.01 |
| ZFP36 | ↓ -8.001 | ASMT | ↑ <0.01 | PLK1 | ↓ -8.686 | C7orf54 | ↓ <0.01 |
| UBE2G2 | ↓ -8.044 | GATA6 | ↑ <0.01 | NAV1 | ↓ -8.714 | ID4 | ↑ <0.01 |
| RNASE1 | ↓ -8.069 | GDF-15 † ‡ | ↓ <0.01 | PAPPA | ↓ -8.726 | CCDC148† | ↓ <0.01 |
| MXD3 | ↓ -8.21 | PCDHGA3 | ↓ <0.01 | PIF1* | ↓ -8.765 | MDM2 | ↓ <0.01 |
| HIST1H2AM | ↓ -8.218 | DDX19A | ↑ <0.01 | DHRSX | ↓ -8.891 | PRAP1 | ↓ <0.01 |
| C14orf94 | ↓ -8.222 | RARB | ↑ <0.01 | GMDS | ↓ -8.928 | FOS | ↓ <0.01 |
| PIF1* | ↓ -8.261 | CCND2 | ↓ <0.01 | LAMA4 | ↓ -9.02 | HES1† | ↓ <0.01 |
| OASL | ↓ -8.265 | BCS1L | ↑ <0.01 | DKFZP0335 | ↓ -9.02 | ASPA | ↓ <0.01 |
| PRSS35 ‡ | ↓ -8.44 | BIRC7 | ↑ <0.01 | CD9 | ↓ -9.051 | GDF-15 † | ↓ <0.01 |
| CLEC14A * | ↓ -8.482 | MX1† | ↓ <0.01 | FAT4 | ↓ -9.124 | COL9A3 | ↓ <0.01 |
| HOXD8 | ↓ -8.679 | RERG | ↑ <0.01 | DOCK1 | ↓ -9.222 | LGALS9 | ↓ <0.01 |
| HIST1H1D | ↓ -8.713 | ADAMTS12 | ↑ <0.01 | SMAD7 | ↓ -9.32 | NR4A1 | ↓ <0.01 |
| CLEC3B | ↓ -8.866 | DUSP1† | ↓ <0.01 | FHOD3 | ↓ -9.381 | ADAM12† | ↑ <0.01 |
| VCAM1 * | ↓ -9.16 | AREG† | ↓ <0.01 | CLEC14A * | ↓ -9.418 | RNASE7 | ↓ <0.01 |
| C9orf3 | ↓ -9.552 | OSBPL1A | ↑ <0.01 | SEMA3A * | ↓ -9.418 | HHATL | ↓ <0.01 |
* Overlapping genes in the “acute” exposure setting for both drugs,
† Overlapping genes in the “chronic” exposure setting for both drugs.
‡ Overlapping genes in the “acute” and “chronic” exposure setting for bleomycin.
§ Overlapping genes in the “acute” and “chronic” exposure setting for cisplatin.
Figure 1. Overlapping genes in top 50 of most differentially expressed genes in HMEC-1 exposed to bleomycin and cisplatin.

cDNA microarray—GSEA. Pathways enriched at a FDR ≤ 0.10 and P ≤ 0.025 in the GSEA are summarised in Table 2. In the “acute”-exposure setting to bleomycin, six pathways were enriched (all up-regulated), while no pathways were enriched in the “chronic” setting with the set criteria for FDR. Cisplatin exposure resulted in 12 enriched pathways in the “acute”-exposure setting (up-regulated n = 3, down-regulated n = 9) while six pathways were enriched in the “chronic”-exposure setting (all down-regulated). The ‘p53’ and the ‘Type I Diabetes Mellitus’ gene sets were enriched in three out of four exposure settings; genes included in this gene set are summarised in S1 Table.
Table 2. Gene Set Enrichment Analysis on gene expression profiles from HMEC-1 following “acute” and “chronic” exposure to bleomycin and cisplatin, using pathway definitions from KEGG.
| BLEOMYCIN - ACUTE | CISPLATIN - ACUTE | CISPLATIN - CHRONIC | ||||
|---|---|---|---|---|---|---|
| FDR | P-value | FDR | P-value | FDR | P-value | |
| Cellular Processes; Cell Communication | ||||||
| Adherens junction | ↓ 0.07 | 0.003 | ||||
| Focal adhesion | ↓ 0.04 | <0.0001 | ||||
| Cellular Processes; Cell Growth and Death | ||||||
| p53 signalling pathway | ↑ 0.001 | <0.0001 | ↑ 0.02 | <0.0001 | ↓ 0.1 | <0.0001 |
| Environmental Information Processing; Signal Transduction | ||||||
| TGF-beta signalling pathway | ↓ 0.05 | <0.0001 | ||||
| Genetic Information Processing; Folding, Sorting and Degradation | ||||||
| Ubiquitin mediated proteolysis | ↓ 0.04 | 0.001 | ||||
| Human Diseases | ||||||
| Endometrial cancer | ↓ 0.04 | <0.0001 | ||||
| Cholera infection | ↑ 0.10 | 0.005 | ||||
| Type I Diabetes Mellitus | ↑ 0.07 | <0.0001 | ↑ 0.05 | <0.0001 | ↓ 0.01 | <0.0001 |
| Neurodegerative disease | ↓ 0.09 | 0.008 | ||||
| Prion diseases | ↓ 0.08 | 0.01 | ||||
| Metabolism; Carbohydrate Metabolism | ||||||
| Butanoate metabolism | ↑ 0.02 | <0.0001 | ||||
| Glyoxylate and dicarboxylate metabolism | ↓ 0.09 | 0.02 | ||||
| Reductive carboxylate cycle | ↓ 0.04 | 0.002 | ||||
| Glycosylphosphatidylinositol(GPI)-anchor biosynthesis | ↑ 0.03 | 0.002 | ||||
| N-Glycan biosynthesis | ↓ 0.08 | <0.0001 | ||||
| Linoleic acid metabolism | ↑ 0.08 | 0.005 | ↑ 0.09 | 0.01 | ||
| Polyunsatyrated fatty acid biosynthesis | ↓ 0.04 | 0.002 | ||||
| Organismal Systems; Immune System | ||||||
| Antigen processing and presentation | ↓ 0.0 | <0.0001 | ||||
| Toll-like receptor signalling pathway | ↓ 0.08 | <0.0001 | ||||
No pathways were enriched according to these criteria after “chronic” exposure to bleomycin.
qRT-PCR
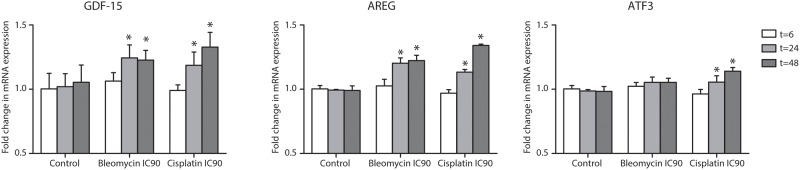
To validate changes in expression of GDF-15, ATF3 and AREG qRT-PCR was performed. In the “acute”-exposure setting, mRNA-expression of all three genes increased in time after exposure to bleomycin and cisplatin, in concordance with the microarray data. After 48 hours exposure to both drugs, mRNA expression of all three genes was significantly higher compared to untreated control cells. No change in mRNA expression of these three genes occurred in untreated control cells in time (Fig. 2).
Figure 2. Relative differences (mean ± standard deviation) in gene expression of GDF-15, ATF3, AREG in HMEC-1 after “acute” exposure to bleomycin and cisplatin measured by qRT-PCR.
Plasma GDF-15 protein levels in testicular cancer patients treated with BEP-chemotherapy
Based on data from the literature we selected GDF-15 for further validation on the protein level in plasma of testicular cancer patient during and after treatment, and related GDF-15 levels to known plasma endothelial damage biomarkers (vWF, hsCRP) [25, 26]. Baseline characteristics of the patients are summarised in Table 3. Although short from significance (p = 0.06), baseline (i.e. before start of chemotherapy) GDF-15 protein levels in testicular cancer patients were not different from healthy age-matched males (Fig. 3A). Patients in the IGCCCG good prognosis group had slightly lower baseline GDF-15 protein levels than patients in intermediate or poor prognosis groups (good prognosis: median 362.9 pg/mL (range 197.6–1059.5; n = 33), intermediate/poor prognosis: median 689.1 pg/mL (range 186.8–1935.0; n = 8); P = 0.04). Median baseline GDF-15 level was not related to tumor stage (stage 2 disease: median 373.0 pg/mL (range 197.6–1935.0; n = 30), stage 3&4 disease: median 486.4 pg/mL (range 186.8–1875.9; n = 11); P = 0.40). Pre-chemotherapy GDF-15 protein levels were not related to age (rs = 0.08; P = 0.61).
Table 3. Characteristics of 41 patients with disseminated testicular cancer and treated with cisplatin containing combination chemotherapy.
| Median (range) | Number (%) | |
|---|---|---|
| Number of patients | 41 | |
| Age at start of treatment, years | 31 (18–46) | |
| Diagnosis | ||
| Non-seminoma | 35 (85.4) | |
| Seminoma | 6 (14.6) | |
| IGCCCG prognosis group | ||
| Good | 33 (80.5) | |
| Intermediate | 7 (17.1) | |
| Poor | 1 (2.4) | |
| Tumor Stage | ||
| II | 30 (73.2) | |
| III | 4 (9.8) | |
| IV | 7 (17.1) | |
| Treatment regimen | ||
| 3 cycles BEP | 29 (70.7) | |
| 4 cycles BEP | 11 (26.8) | |
| 4 cycles EP | 1 (2.4) |
Abbreviations: International Germ Cell Cancer Collaborative Group (IGCCCG); bleomycin etoposide cisplatin chemotherapy (BEP).
Figure 3. GDF-15.

A. Plasma GDF-15 protein levels in patients with metastatic testicular cancer (n = 41) prior to start of bleomycin- and cisplatin-based chemotherapy, compared to healthy age-matched males (n = 10); B. Plasma GDF-15 protein levels before, during and after completion of bleomcyin- and cisplatin-based chemotherapy for testicular cancer. The sample at c1d1 is drawn before initiation of chemotherapy. (*) p < 0,05 compared to baseline value or indicated time-point.
During BEP-chemotherapy, GDF-15 protein levels increased compared to baseline, with significantly higher levels 1 months and 1 year post-chemotherapy (Fig. 3B, Table 4). Compared to pre-chemotherapy, plasma levels of vWF and hsCRP changed significantly during treatment (Table 4). After completion of chemotherapy, vWF-levels remained persistently elevated, whereas hsCRP returned to pre-chemotherapy values. At baseline, levels of GDF-15 were related to levels of vWF (rs = 0.35; P = 0.03) and hsCRP (rs = 0.39; P = 0.014). During chemotherapy, levels of GDF-15 correlated with hsCRP at c1d8 (rs = 0.44; P = 0.01) and with vWF at c3d8 (rs = 0.40; P = 0.017). At the follow-up visit one month after completion of chemotherapy, levels of GDF-15 and vWF were strongly correlated (rs = 0.56; P = 0.001), whereas this relation was not found for GDF-15 and hsCRP (rs = 0.18; P = 0.28). One year after start of chemotherapy, no relation between GDF-15 levels and vWF or hsCRP (rs = 0.28; P = 0.12; rs = 0.10; P = 0.57) was found.
Table 4. Plasma levels of GDF-15 (pg/mL), vWF (%) and hsCRP (mg/L) in testicular cancer patients (n = 41) before, during and after completion of bleomycin- and cisplatin-based chemotherapy.
| GDF-15 (pg/mL) | vWF (%) | hsCRP (mg/L) | ||||
|---|---|---|---|---|---|---|
| Median | Range | Median | Range | Median | Range | |
| Course1 | ||||||
| Day 1 (= baseline) | 383.1 | 186.8–1935.0 | 100 | 42–297 | 2.0 | 0.2–87.1 |
| Day 8 | 3473.7 * | 1344.5–9028.3 | 164* | 56–319 | 0.6* | 0.2–4.9 |
| Day 15 | 1587.2 * | 655.2–4014.9 | 145* | 57–394 | 4.2† | 1.1–101.0 |
| Course 2 | ||||||
| Day 1 | 1145.5 * | 593.6–3074.5 | 143* | 34–360 | 5.1 | 1.1–39.8 |
| Day 8 | 4898.0 * | 2183.0–10794.9 | 197* | 66–464 | 0.6* | 0.2–21.1 |
| Course 3 | ||||||
| Day 1 | 2067.7 * | 639.0–4918.0 | 194* | 66–440 | 3.7 | 0.3–39.9 |
| Day 8 | 5542.8 * | 702.3–18958.9 | 197* | 81–419 | 0.6 | 0.2–25.7 |
| One month after completion of chemotherapy | ||||||
| 1009.6 * | 409.7–4737.1 | 135* | 56–249 | 2.2 | 0.4–29.1 | |
| One year after start of chemotherapy | ||||||
| 395.2 * ‡ | 246.9–913.2 | 115* ‡ | 49–218 | 1.5§ | 0.2–14.3 | |
* P < 0.01 compared to baseline, Wilcoxon signed rank test
† P < 0.05 compared to baseline, Wilcoxon signed rank test
‡ P < 0.01 compared to one month after completion of chemotherapy, Wilcoxon signed rank test
§ P < 0.05 compared to one month after completion of chemotherapy, Wilcoxon signed rank test
Discussion
In this study we used an unbiased translational approach with cDNA microarray as a tool to find novel mechanisms related to and select candidate biomarkers involved in chemotherapy-induced endothelial damage. With this in vitro strategy, we found several single genes with significant changes in expression upon exposure to bleomycin and cisplatin. Three genes with strong expression differences in three out of four experimental settings, GDF-15, ATF3 and AREG, were validated by qRT-PCR. In addition, GSEA revealed clusters of genes involved in several pathways, including ‘p53’ and ‘Type I Diabetes Mellitus’ gene sets, which were affected in this model. Furthermore, we showed that BEP-chemotherapy (Bleomycin; Etoposide; Cisplatin) did indeed affect plasma GDF-15 protein levels in testicular cancer patients and that these levels related to known endothelial damage biomarkers such as vWF and hsCRP.
In the single gene analysis several genes were significantly differentially expressed in the different experimental settings in the HMEC-1 in vitro model. The genes GDF-15, ATF3 and AREG were in the top 50’s of most differentially expressed genes in three or four exposure settings.
The cytokine GDF-15 (also known as Macrophage Inhibitory Cytokine 1 (MIC-1) or NSAID activated gene (NAG-1)) is a member of the Transforming Growth Factor β (TGFβ) family. GDF15 is induced by all sorts of stimuli including cytokines, chemo- and/or radiotherapy and injury in all sorts of different cells and tissues [27–30]. Release of GDF-15 can induce anti-inflammatory, anti-apoptotic and anti-proliferative effects, thereby exerting vasculoprotective mechanisms. Therefore, increases in GDF-15 plasma levels in our patient cohort may well result from increased production by endothelial cells and/or macrophages, to compensate for chemotherapy-induced damage. In a study evaluating gene expression changes in prostate cancer samples pre- and post docetaxel/mitoxantrone treatment, GDF-15 was one of highest up-regulated genes post-treatment [31], illustrating that cytostatics can influence GDF-15 expression in both malignant and non-malignant cells.
Elevated levels of GDF-15 have been associated with increased risk of diseases hypothesised to result from chronic inflammation [29, 32] and numerous studies showed that GDF-15 is a valuable biomarker for cardiovascular disease [29, 30, 32–34]. GDF-15 is thought to play a relevant role in cardiovascular damage responses [35], possibly through a pro-angiogenic response as was demonstrated in a pre-clinical model with HUVEC [36]. GDF-15 is also described as biomarker/predictor of albuminuria, known to reflect established micro-vascular damage [37]. In addition, several studies implicate GDF-15 in aspects of metabolic disorders e.g. insulin resistance and obesity [30, 38, 39]. Recently, higher levels of circulating MIC-1/GDF-15 were also associated with an increased risk of colorectal cancer supporting a role of chronic inflammation in the development of colorectal cancer [40].
ATF3 is a downstream member of the MAP-kinase signalling pathway that encodes for a nuclear factor that stimulates transcription upon cellular stress. Fast up-regulation of ATF3 is a central stress response in different endothelial cell models, induced by various noxious stimuli [41–46]. Interference with ATF3-levels protected cells from apoptosis induction, e.g. induced by TNFα in HUVEC [47], by cisplatin in a human glioblastoma cell line [42], or related to doxorubicin in cardiomyocytes [48]. Interestingly, immunohistochemically measured ATF3-expression is increased in atherosclerotic areas of human iliac arteries [43]. Few studies addressed AREG, a member of the epidermal growth factor receptors that plays an important role in cellular proliferation and survival. Breast cancer cells exposed to cisplatin secreted the AREG-protein over extended periods of time, i.e. up to 72 hours after exposition [49].
Based on the available data we decided to study plasma protein levels of GDF-15 in testicular cancer patients before, during and after treatment with BEP-chemotherapy, and relate changes to levels of known endothelial damage biomarkers [25, 26]. The increases in GDF-15 throughout treatment as well as post-chemotherapy may partly relate to cancer-related mechanisms, as the GDF-15 protein level wasslightly higher in patients with more advanced disease stage as measured by IGCCCG score, which may relate to release of GDF-15 from apoptotic tumour cells. However, its role in endothelial damage is supported by the fact that GDF-15 levels correlated with proteins known to reflect chemotherapy-related endothelial damage in testicular cancer patients, vWF and hsCRP [25, 26]. Therefore, it is conceivable that the observed rise in plasma GDF15 is involved in chemotherapy-related endothelial damage, however it is probably not due to endothelial injury alone. As this protein is mechanistically related to chemotherapy-related endothelial damage it may be a potentially sensitive biomarker for detecting this damage. Further extended analysis of GDF15 levels in combination with phenotyping of cardiovascular risk factors in a larger cohort of patients is warranted. Although in this study no relation was found between pretreatment levels of GDF15 and age, it is known that this is the case in the normal population and should be taken into account when analyzing GDF15 levels in time. Moreover, the fact that the TGFβ-pathway is involved in this damage may be a clue to interventions that decrease the amount of endothelial damage related to treatment with bleomycin and cisplatin.
GSEA showed significantly enriched of several pathways, including ‘p53’ and ‘Type I Diabetes Mellitus’ in “acute” exposure to bleomycin and cisplatin, and “chronic” exposure to cisplatin. The finding that p53-related genes were affected by bleomycin as well as cisplatin illustrates the validity of our approach, as both drugs exert their key therapeutic effect by cellular apoptosis induction. The enriched ‘Type I Diabetes Mellitus’ gene set includes several genes involved in inflammatory processes, e.g. Human Leucocyte Antigen-molecules, interleukins and TNF, indicating that bleomycin and cisplatin induce an inflammatory response in endothelial cells. This finding is completely in line with studies in testicular cancer patients treated with cisplatin-based regimens, which showed higher rates of systemic inflammation and endothelial dysfunction [25, 26, 50]. As circulating platinum remains detectable in the circulation years to decades after cisplatin treatment [51–52], long-term testicular cancer survivors may well have ongoing vascular damage and chronic low-grade endothelial inflammation. When chronic inflammatory responses prove to be an important pathogenic factor for the development of chemotherapy-induced endothelial damage, intervention with anti-inflammatory drugs is a rationale approach to alleviate these effects.
In conclusion, we utilised cDNA microarray to detect in an unbiased way genes and pathways associated with chemotherapy-related endothelial damage, to find biomarkers for and mechanisms involved in this damage. In HMEC-1 exposed to bleomycin and cisplatin, several genes were strongly differentially expressed, e.g. GDF-15, ATF3 and AREG. In GSEA, clusters of genes involved in cell death and inflammation were affected. The observed changes in plasma GDF-15 protein levels in testicular cancer patients induced by cisplatin- and bleomycin-containing chemotherapy indicate that this informative pre-clinical approach can be translated to a clinical setting, and that GDF-15 may be a potential biomarker of interest that is mechanistically involved in chemotherapy-related healthy tissue damage such as endothelial damage. Further in vitro and in vivo exploration is warranted. This facilitates the rationale towards selection of targets for intervention, with early surrogate biomarkers for chemotherapy-related endothelial damage.
Supporting Information
A. “acute” exposure setting: immortalised HMEC-1 were exposed to bleomycin (0.3 μg/ml (IC50), 1.5 μg/mL (IC90)) or cisplatin (2.6 μM (IC50), 12.9 μM (IC90)) for 6, 24 and 48 hours; B. “chronic” exposure setting: over the course of 30 days HMEC-1 was exposed to 0.06 μg/mL bleomycin (IC10) or 0.52 μM cisplatin (IC10) twice weekly). In both experiments untreated samples served as controls. (*) RNA-isolation and cDNA microarray experiments; (†) RNA isolation and qRT-PCR.
(TIF)
(DOC)
Acknowledgments
We thank Kevin Bouma, Gert Jan Meersma, Haukeline Volders and Nynke Zwart for their technical assistance and for the collection of patient samples.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
Support was provided by RUG 2009-4365 from the Dutch Cancer Society. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Feldman DR, Bosl GJ, Sheinfeld J, Motzer RJ (2008) Medical treatment of advanced testicular cancer. JAMA 299: 672–684. 10.1001/jama.299.6.672 [DOI] [PubMed] [Google Scholar]
- 2. van den Belt-Dusebout AW, de Wit R, Gietema JA, Horenblas S, Louwman MW, et al. (2007) Treatment-specific risks of second malignancies and cardiovascular disease in 5-year survivors of testicular cancer. J Clin Oncol 25: 4370–4378. 10.1200/JCO.2006.10.5296 [DOI] [PubMed] [Google Scholar]
- 3. Huddart RA, Norman A, Shahidi M, Horwich A, Coward D, et al. (2003) Cardiovascular disease as a long-term complication of treatment for testicular cancer. J Clin Oncol 21: 1513–1523. 10.1200/JCO.2003.04.173 [DOI] [PubMed] [Google Scholar]
- 4. Meinardi MT, Gietema JA, van der Graaf WT, van Veldhuisen DJ, Runne MA, et al. (2000) Cardiovascular morbidity in long-term survivors of metastatic testicular cancer. J Clin Oncol 18: 1725–1732. [DOI] [PubMed] [Google Scholar]
- 5. Strumberg D, Brügge S, Korn MW, Koeppen S, Ranft J, et al. (2002) Evaluation of long-term toxicity in patients after cisplatin-based chemotherapy for non-seminomatous testicular cancer. Ann Oncol 13: 229–236. 10.1093/annonc/mdf058 [DOI] [PubMed] [Google Scholar]
- 6. Weijl NI, Rutten MF, Zwinderman AH, Keizer HJ, Nooy MA, et al. (2000) Thromboembolic events during chemotherapy for germ cell cancer: a cohort study and review of the literature. J Clin Oncol 18: 2169–2178. [DOI] [PubMed] [Google Scholar]
- 7. Stefenelli T, Kuzmits R, Ulrich W, Glogar D (1988) Acute vascular toxicity after combination chemotherapy with cisplatin, vinblastine, and bleomycin for testicular cancer. Eur Heart J 9: 552–556. [DOI] [PubMed] [Google Scholar]
- 8. Nuver J, Smit AJ, Sleijfer DT, van Gessel AI, van Roon AM, et al. (2005) Left ventricular and cardiac autonomic function in survivors of testicular cancer. Eur J Clin Invest 35: 99–103. 10.1111/j.1365-2362.2005.01460.x [DOI] [PubMed] [Google Scholar]
- 9. van den Belt-Dusebout AW, Nuver J, de Wit R, Gietema JA, ten Bokkel Huinink WW, et al. (2006) Long-term risk of cardiovascular disease in 5-year survivors of testicular cancer. J Clin Oncol 24: 467–475. 10.1200/JCO.2005.02.7193 [DOI] [PubMed] [Google Scholar]
- 10. Fosså SD, Aass N, Harvei S, Tretli S (2004) Increased mortality rates in young and middle-aged patients with malignant germ cell tumours. Br J Cancer 90: 607–612. 10.1038/sj.bjc.6601558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Haugnes HS, Wethal T, Aass N, Dahl O, Klepp O, et al. (2010) Cardiovascular risk factors and morbidity in long-term survivors of testicular cancer: A 20-year follow-up study. J Clin Oncol 28: 4649–4657. 10.1200/JCO.2010.29.9362 [DOI] [PubMed] [Google Scholar]
- 12. Lechner D, Kollars M, Gleiss A, Kyrle PA, Weltermann A (2007) Chemotherapy-induced thrombin generation via procoagulant endothelial microparticles is independent of tissue factor activity. J Thromb Haemost 5: 2445–2452. 10.1111/j.1538-7836.2007.02788.x [DOI] [PubMed] [Google Scholar]
- 13. L’Azou B, Fernandez P, Bareille R, Beneteau M, Bourget C, et al. (2005) In vitro endothelial cell susceptibility to xenobiotics: comparison of three cell types. Cell Biol Toxicol 21: 127–137. 10.1007/s10565-005-0172-8 [DOI] [PubMed] [Google Scholar]
- 14. Nuver J, De Haas EC, Van Zweeden M, Gietema JA, Meijer C (2010) Vascular damage in testicular cancer patients: A study on endothelial activation by bleomycin and cisplatin in vitro. Oncol Rep 23: 247–253. [PubMed] [Google Scholar]
- 15. Ishii H, Takada K (2002) Bleomycin induces E-selectin expression in cultured umbilical vein endothelial cells by increasing its mRNA levels through activation of NF-kappaB/Rel. Toxicol Appl Pharmacol 184: 88–97. 10.1016/S0041-008X(02)99499-8 [DOI] [PubMed] [Google Scholar]
- 16. Dirix LY, Libura M, Libura J, Vermeulen PB, De Bruijn EA, et al. (1997) In vitro toxicity studies with mitomycins and bleomycin on endothelial cells. Anticancer Drugs 8: 859–868. 10.1097/00001813-199710000-00007 [DOI] [PubMed] [Google Scholar]
- 17. Yu M, Han J, Cui P, Dai M, Li H, et al. (2008) Cisplatin up-regulates ICAM-1 expression in endothelial cell via a NF-kappaB dependent pathway. Cancer Sci 99: 391–397. 10.1111/j.1349-7006.2008.00696.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shi Y, Inoue S, Shinozaki R, Fukue K, Kougo T (1998) Release of cytokines from human umbilical vein endothelial cells treated with platinum compounds in vitro. Jpn J Cancer Res 89: 757–767. 10.1111/j.1349-7006.1998.tb03281.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Montiel M, Urso L, de la Blanca EP, Marsigliante S, Jiménez E (2009) Cisplatin reduces endothelial cell migration via regulation of type 2-matrix metalloproteinase activity. Cell Physiol Biochem 23: 441–448. 10.1159/000218191 [DOI] [PubMed] [Google Scholar]
- 20. Ades EW, Candal FJ, Swerlick RA, George VG, Summers S, et al. (1992) HMEC-1: establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol 99: 683–690. 10.1111/1523-1747.ep12613748 [DOI] [PubMed] [Google Scholar]
- 21. Alberts DS, Chen HSG, Liu R, Himmelstein KJ, Mayersohn M, et al. (1978) Bleomycin pharmacokinetics in man. Cancer Chemother Pharmacol 1: 177–181. 10.1007/BF00253118 [DOI] [PubMed] [Google Scholar]
- 22. Urien S, Brain E, Bugat R, Pivot X, Lochon I, et al. (2005) Pharmacokinetics of platinum after oral or intravenous cisplatin: a phase 1 study in 32 adult patients. Cancer Chemother Pharmacol 55: 55–60. 10.1007/s00280-004-0852-8 [DOI] [PubMed] [Google Scholar]
- 23. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102: 15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brouwer CAJ, Postma A, Hooimeijer HLH, Smit AJ, Vonk JM, et al. (2013) Endothelial Damage in Long-Term Survivors of Childhood Cancer. J Clin Oncol 31: 3906–3913. 10.1200/JCO.2012.46.6086 [DOI] [PubMed] [Google Scholar]
- 25. Nuver J, Smit AJ, van der Meer J, van den Berg MP, van der Graaf WT, et al. (2005) Acute chemotherapy-induced cardiovascular changes in patients with testicular cancer. J Clin Oncol 23: 9130–9137. 10.1200/JCO.2005.01.4092 [DOI] [PubMed] [Google Scholar]
- 26. Nuver J, Smit AJ, Sleijfer DT, van Gessel AI, van Roon AM, et al. (2004) Microalbuminuria, decreased fibrinolysis, and inflammation as early signs of atherosclerosis in long-term survivors of disseminated testicular cancer. Eur J Cancer 40: 701–706. 10.1016/j.ejca.2003.12.012 [DOI] [PubMed] [Google Scholar]
- 27. Ago T, Sadoshima J (2006) GDF15, a cardioprotective TGF-β superfamily protein. Circ Res 98: 294–297. 10.1161/01.RES.0000207919.83894.9d [DOI] [PubMed] [Google Scholar]
- 28. Wiklund FE, Bennet AM, Magnusson PKE, Eriksson UK, Lindmark F, et al. Macrophage inhibitory cytokine-1 (MIC-1/GDF15): a new marker of all-cause mortality. Aging Cell 2010; 9: 1057–64. 10.1111/j.1474-9726.2010.00629.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Breit SN, Johnen H, Cook AD, Tsai VWW, Mohammed MG, et al. (2011) The TGF-β superfamily cytokine, MIC-1/GDF15: A pleotrophic cytokine with roles in inflammation, cancer and metabolism. Growth Factors 29: 187–195. 10.3109/08977194.2011.607137 [DOI] [PubMed] [Google Scholar]
- 30. Unsicker K, Spittau B, Krieglstein K (2013) The multiple facets of the TGF-b family cytokine growth/ differentiation factor-15/macrophage inhibitory cytokine-1. Cytokine Growth Factor Rev 24: 373–384. 10.1016/j.cytogfr.2013.05.003 [DOI] [PubMed] [Google Scholar]
- 31. Huang CY, Beer TM, Higano CS, True LD, Vessella R, et al. (2007) Molecular alterations in prostate carcinomas that associate with in vivo exposure to chemotherapy: identification of a cytoprotective mechanism involving growth differentiation factor 15. Clin Cancer Res 13: 5825–5833. 10.1158/1078-0432.CCR-07-1037 [DOI] [PubMed] [Google Scholar]
- 32. Brown DA, Breit SN, Buring J, Fairlie WD, Bauskin AR, et al. (2002) Concentration in plasma of macrophage inhibitory cytokine-1 and risk of cardiovascular events in women: a nested case-control study. Lancet 359: 2159–2163. 10.1016/S0140-6736(02)09093-1 [DOI] [PubMed] [Google Scholar]
- 33. Eggers KM, Kempf T, Lagerqvist B, Lindahl B, Olofsson S, et al. (2012) Relations of growth-differentiation factor-15 to biomarkers reflecting vascular pathologies in a population-based sample of elderly subjects. Scand J Clin Lab Invest 72: 45–51. 10.3109/00365513.2011.626072 [DOI] [PubMed] [Google Scholar]
- 34. Lok DJ, Klip YT, Lok SI, Bruggink-André de la Porte PW, Badings E, et al. (2013) Incremental Prognostic Power of Novel Biomarkers (Growth-Differentiation Factor-15, High-Sensitivity C-Reactive Protein, Galectin-3, and High-Sensitivity Troponin-T) in Patients With Advanced Chronic Heart Failure—. Am J Cardiol 112: 831–837. 10.1016/j.amjcard.2013.05.013 [DOI] [PubMed] [Google Scholar]
- 35. Kempf T, Eden M, Strelau J, Naguib M, Willenbockel C, et al. (2006) The transforming growth factor-β superfamily member growth-differentiation factor-15 protects the heart from ischemia/reperfusion injury. Circ Res 98: 351–360. 10.1161/01.RES.0000202805.73038.48 [DOI] [PubMed] [Google Scholar]
- 36. Song H, Yin D, Liu Z (2012) GDF-15 promotes angiogenesis through modulating p53/HIF-1α signaling pathway in hypoxic human umbilical vein endothelial cells. Mol Biol Rep 39: 4017–4022. 10.1007/s11033-011-1182-7 [DOI] [PubMed] [Google Scholar]
- 37. Hellemons ME, Mazagova M, Gansevoort RT, Henning RH, de Zeeuw D, et al. (2012) Growth-Differentiation Factor 15 Predicts Worsening of Albuminuria in PatientsWith Type 2 Diabetes. Diabetes Care 35: 2340–2346. 10.2337/dc12-0180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kempf T, Guba-Quint A, Torgerson J, Magnone MC, Haefliger C, et al. (2012) Growth differentiation factor 15 predicts future insulin resistance and impaired glucose control in obese nondiabetic individuals: results from the XENDOS trial. Eur J of Endocrinology 167: 671–678. 10.1530/EJE-12-0466 [DOI] [PubMed] [Google Scholar]
- 39. Herder M, Carstensen DM (2013) Ouwens. Anti-inflammatory cytokines and risk of type 2 diabetes. C. Obesity and Metabolism 15 (Suppl. 3): 39–50. [DOI] [PubMed] [Google Scholar]
- 40. Mehta RS, Song M, Bezwada N, Wu K, Garcia-Albeniz X, et al. (2014) A prospective study of macrophage inhibitory cytokine-1 (MIC-1/GDF15) and risk of colorectal cancer. J Natl Cancer Inst (in press). 10.1093/jnci/dju016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fu M, Zhu X, Zhang J, Liang J, Lin Y, et al. (2003) Egr-1 target genes in human endothelial cells identified by microarray analysis. Gene 315: 33–41. 10.1016/S0378-1119(03)00730-3 [DOI] [PubMed] [Google Scholar]
- 42. Hamdi M, Popeijus HE, Carlotti F, Janssen JM, van der Burgt C, et al. (2008) ATF3 and Fra1 have opposite functions in JNK- and ERK-dependent DNA damage responses. DNA Repair (Amst) 7: 487–496. 10.1016/j.dnarep.2007.12.004 [DOI] [PubMed] [Google Scholar]
- 43. Nawa T, Nawa MT, Adachi MT, Uchimura I, Shimokawa R, et al. (2002) Expression of transcriptional repressor ATF3/LRF1 in human atherosclerosis: colocalization and possible involvement in cell death of vascular endothelial cells. Atherosclerosis 161: 281–291. 10.1016/S0021-9150(01)00639-6 [DOI] [PubMed] [Google Scholar]
- 44. Kool J, Hamdi M, Cornelissen-Steijger P, van der Eb AJ, Terleth C, et al. (2005) Induction of ATF3 by ionizing radiation is mediated via a signaling pathway that includes ATM, Nibrin1, stress-induced MAPkinases and ATF-2. Oncogene 22: 4235–4242. 10.1038/sj.onc.1206611 [DOI] [PubMed] [Google Scholar]
- 45. Chen SC, Liu YC, Shyu KG, Wang DL (2008) Acute hypoxia to endothelial cells induces activating transcription factor 3 (ATF3) expression that is mediated via nitric oxide. Atherosclerosis 201: 281–288. 10.1016/j.atherosclerosis.2008.02.014 [DOI] [PubMed] [Google Scholar]
- 46. Cai Y, Zhang C, Nawa T, Aso T, Tanaka M, et al. (2000) Homocysteine-responsive ATF3 gene expression in human vascular endothelial cells: activation of c-Jun NH(2)-terminal kinase and promoter response element. Blood 96: 2140–2148. [PubMed] [Google Scholar]
- 47. Kawauchi J, Zhang C, Nobori K, Hashimoto Y, Adachi MT, et al. (2002) Transcriptional repressor activating transcription factor 3 protects human umbilical vein endothelial cells from tumor necrosis factor-alpha-induced apoptosis through down-regulation of p53 transcription. J Biol Chem 277: 39025–39034. 10.1074/jbc.M202974200 [DOI] [PubMed] [Google Scholar]
- 48. Nobori K, Ito H, Tamamori-Adachi M, Adachi S, Ono Y, et al. (2002) ATF3 inhibits doxorubicin-induced apoptosis in cardiac myocytes: a novel cardioprotective role of ATF3. J Mol Cell Cardiol 34: 1387–1397. 10.1006/jmcc.2002.2091 [DOI] [PubMed] [Google Scholar]
- 49. Eckstein N, Servan K, Girard L, Cai D, von Jonquieres G, et al. (2008) Epidermal growth factor receptor pathway analysis identifies amphiregulin as a key factor for cisplatin resistance of human breast cancer cells. J Biol Chem 283: 739–750. 10.1074/jbc.M706287200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vaughn DJ, Palmer SC, Carver JR, Jacobs LA, Mohler ER (2008) Cardiovascular risk in long-term survivors of testicular cancer. Cancer 112: 1949–1953. 10.1002/cncr.23389 [DOI] [PubMed] [Google Scholar]
- 51. Gietema JA, Meinardi MT, Messerschmidt J, Gelevert T, Alt F, et al. (2000) Circulating plasma platinum more than 10 years after cisplatin treatment for testicular cancer. Lancet 355: 1075–1076. 10.1016/S0140-6736(00)02044-4 [DOI] [PubMed] [Google Scholar]
- 52. Sprauten M, Darrah TH, Peterson DR, Campbell ME, Hannigan RE, et al. (2012) Impact of long-term serum platinum concentrations on neuro- and ototoxicity in cisplatin-treated survivors of testicular cancer. J Clin Oncol 30: 300–307. 10.1200/JCO.2011.37.4025 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. “acute” exposure setting: immortalised HMEC-1 were exposed to bleomycin (0.3 μg/ml (IC50), 1.5 μg/mL (IC90)) or cisplatin (2.6 μM (IC50), 12.9 μM (IC90)) for 6, 24 and 48 hours; B. “chronic” exposure setting: over the course of 30 days HMEC-1 was exposed to 0.06 μg/mL bleomycin (IC10) or 0.52 μM cisplatin (IC10) twice weekly). In both experiments untreated samples served as controls. (*) RNA-isolation and cDNA microarray experiments; (†) RNA isolation and qRT-PCR.
(TIF)
(DOC)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.