

Abstract
A key element to delineate the biology of individual tumors is the regulation of apoptosis. In this work, we functionally characterize two breast cancer associated genes, the proteasome 26S subunit ATPase 3 interacting protein (PSMC3IP) and the epithelial-stromal interaction 1 (EPSTI1), to explore their potential apoptotic role in breast cancer. We first explore the existence of direct physical interactions with annotated BC-apoptotic genes. Based on the generated interaction network, we examine several apoptotic markers to determine the effect of PSMC3IP and EPSTI1 gene expression modulation in two different human breast cancer cell lines to suggest potential molecular mechanisms to unveil their role in the disease. Our results show that PSMC3IP and EPSTI1 are able to modulate the extrinsic apoptotic pathway in estrogen receptor positive and triple negative breast cancer cell lines, highlighting them as potential therapeutic targets.
Introduction
Due to its complexity, breast cancer (BC) is often considered a broad set of diseases including multiple, distinct biological subtypes with diverse natural histories that present a varied spectrum of clinical, pathological and molecular features with different prognostic and therapeutic implications [1]. The poor prognostic outcome of breast cancer is largely due to its resistance to current cancer therapies, where the balance between cell proliferation and apoptosis plays a critical role and it is crucial in determining the overall growth or regression of the tumor in response to treatments [2]. Hence, identifying proteins involved in apoptosis resistance developed by tumorigenic cells has an essential importance in order to find new therapeutic approaches. Two major apoptosis pathways have been described: the mitochondria mediated (intrinsic) and the death receptor mediated (extrinsic), which is initiated by the binding of ligands such as TNF-α or TNF-related apoptosis inducing ligand (TRAIL) to death receptors [3]. Once the receptors are activated, they oligomerize and form complexes that recruit and activate the initiator caspase-8. Active caspase-8 subsequently cleaves effector caspases like caspase-3 and caspase-7, resulting in activation or inactivation (as well as translocation), of several substrates such as the poly ADP ribose polymerase (PARP), with the consequent induction of cell death [4, 5].
In the last decade, network biology approaches have contributed to identify novel causative and susceptibility oncogenes, as well as secondary effectors that could not be highlighted by conventional analysis based on differential expression [6] Therefore, this novel approach can provide a deeper understanding of the molecular mechanisms underlying complex pathological processes, offering new biomarkers that may help to improve breast cancer diagnosis.
PSMC3IP gene is located on chromosome 17q21, proximal to BRCA1 [7] and previously linked to breast cancer predisposition [8]. It has been characterized as a nuclear receptor participating in estrogen, androgen, glucocorticoid and progesterone receptor–mediated gene regulation [9, 10]. PSMC3IP is upregulated in breast cancer [7, 11] and in addition, inactivating mutations [12] have also been shown to regulate DNA recombination in DNA repair [13], potentially contributing to an increased risk in familial breast and ovarian cancers.
On the other hand, EPSTI1, an interferon (IFN) response gene [14], has been identified as a stromal fibroblast-induced gene in breast cancer, being highly upregulated in invasive breast carcinomas as compared with normal breast [15, 16]. More recently, it has been shown that expression levels of EPSTI1 associate with tumor initiation and migration, stem cell–like properties, epithelial-mesenchymal transition (EMT) [17] and breast cancer invasion and metastasis [18], with the highest expression observed in basal-like subtype breast cancer cells exhibiting a poor prognosis [17].
Although the relationship of PSMC3IP and EPSTI1 with BC is well established, the underlying molecular mechanisms are still unknown. In the present study, we describe novel interactions between PSMC3IP and EPSTI1 with well-established BC genes which are also related to apoptosis and cell proliferation processes. We explore the anti-apoptotic role of PSMC3IP and EPSTI1 and their contribution in breast cancer development. We have carried out a functional characterization associated to cell apoptosis by means of caspase-8 and-3 activation, PARP cleavage and DNA integrity, based on gene overexpression and silencing in two different human breast cancer derived cell lines under both basal and apoptotic-induced conditions.
Materials and Methods
Subcloning of human cDNAs into Y2H plasmids
Human ORF clones were cloned into pENTR⁄D-TOPO vector (pENTR Directional TOPO cloning kit; Life Technologies) and sequence verified. AKT1, BCAR3 and EPSTI1 clones derived from the human ORFeome v1.1 [19]; CASP8 and PSMC3IP from Life Technologies Ultimate ORF Clones [20]. All ORFs were individually transferred into yeast two-hybrid (Y2H) destination vectors by Gateway recombinational cloning (ProQuest Two-Hybrid System, Life Technologies). BC-apoptosis genes were cloned into pDEST32 to generate bait plasmids and the BC associated genes (EPSTI1 and PSMC3IP) were cloned into pDEST22 to obtain prey plasmids.
Y2H matrix screens
Bait and prey plasmids were pair-wise co-transformed into MaV203 yeast strain in 96-well arrays and plated onto selective SD2 (lacking Leu and Trp amino acids) agar media and incubated for 48 hours at 30°C to detect colony growth. Co-transformant arrays were then replica plated onto different selective media for interaction screening. To assay the activation of the HIS3 reporter gene, SD3 (lacking Leu, Trp, His) agar plates were supplemented with 12 to 100 mM of 3-aminotriazole (3AT, Sigma-Aldrich), being 50 mM 3AT the optimal concentration for positive HIS3 activation colonies. Similarly, we assayed the activation of the URA3 reporter gene by plating onto SD3 (lacking Leu, Trp, Uracil). Double reporter HIS3/URA3 activation was evaluated by SD4 (lacking Leu, Trp, His, Uracil) agar plates supplemented with 20 mM of 3AT and LacZ reporter gene was tested by the β-galactosidase assay on a nylon membrane placed onto a SD2 agar plate.
In order to minimize the number of false positives (at the cost of penalizing potential false negatives as well), we subsequently scored the positive interactions based on their ability to activate at least two reporter genes or being repeatedly observed in biological replica screens, which defined our high-confidence (HC) interaction set.
Cell culture
MDA-MB-231 and MCF-7 cells were kindly provided by Dr. Violeta Serra (VHIO, Barcelona, Spain). MDA-MB-231 were cultured in Dulbecco’s Modified Eagle Medium (DMEM)/F12 and MCF-7 were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (GIBCO Life Technologies), both supplemented with 10% Fetal Bovine Serum (FBS) and 1% Penicillin-Streptomycin (10,000 U/mL) purchased from GIBCO Life Technologies.
Plasmids and siRNAs
Human ORF cDNA clones were acquired from Thermo Fisher Scientific. siRNAs targeting PSMC3IP and LUCIFERASE (non-targeting control siRNA) gene expression were purchased from Life Technologies. XIAP derived siRNA II was from Cell Signaling Technology and EPSTI1 derived siRNA was designed as described before [17].
Cell transfection and treatment
For expression of Myc-tagged fusion proteins, cDNA clones were subcloned into pDEST-Myc-tagged vector using the Gateway cloning system (Life Technologies). Cell transfection using X-tremeGENE 9 (Roche) was performed according to the manufacturer’s instructions. After 24 hours of cell transfection, cells were treated for another 24 hours with recombinant human TRAIL/Apo2 ligand (PeproTech) at final concentration of 80ng/ml in MDA-MB-231 and 100ng/ml in MCF-7.
For RNA interference-mediated gene silencing, cells were seeded and exposed to 50 nM of either gene-specific siRNA or non-targeting control siRNA (siLUC), using Lipofectamine RNAiMAX transfection reagent (Life Technologies) for 48 h. For EPSTI1 silencing, IFN-α (Chemicon, Millipore) was added to a final concentration of 1000 U/ml 8 hours before harvesting the cells. TRAIL treatment was conducted in the same conditions as in overexpression assays (see above).
Caspase-8 and caspase-3 activity assays
The activities of caspase 3 and 8 were measured using APOPCYTO Caspase-3 Colorimetric Assay Kit (Medical and Biological Laboratories) and Caspase-8 Colorimetric Assay Kit (BioVision), respectively. Briefly, total cell protein was extracted using ice-cold cell lysis buffer. Then, 100–200 μg of total protein was diluted in 50 μl of lysis buffer and 50 μl of 2× reaction buffer containing 10 mM DTT. 5 μl of caspase 3 or 8 substrate were added into each well of a 96-well microplate. After incubation at 37°C for 3 hours, the absorbance was measured at 405 nm.
Immunoblot analysis
Protein concentration was determined using the Bio-Rad DC protein assay (Bio-Rad Laboratories). 20 µg of total protein were electrophoresed in 8% SDS-PAGE gel and transferred to Immobilon-P Membrane, PVDF (Millipore). Membranes were incubated overnight at 4°C with primary antibodies. Rabbit polyclonal anti-PSMC3IP, Sigma-Aldrich (HPA044439) at dilution (1:1000), rabbit polyclonal anti-EPSTI1, Sigma Aldrich (SAB2100696) at dilution (1:2000), both from Sigma Aldrich, rabbit polyclonal anti-PARP, Cell Signaling Technology (#9542) at dilution (1:1000) and mouse monoclonal anti-β-actin, Abcam (ab20272), at dilution (1:20000) followed by incubation with the appropriated HRP-conjugated secondary antibody plus enhanced chemiluminescence substrate (GE Healthcare). Protein band amounts were roughly quantified by densitometry using ImageJ (http://rsb.info.nih.gov/ij/), following standard procedures. Loading control protein β-actin was used as a reference to compare relative protein amounts.
Propidium iodide staining and flow cytometry analysis
Apoptotic cells were quantified by flow cytometry as previously described [21]. Briefly, cells were washed with PBS, fixed in cold 70% ethanol, and then stained with propidium iodide (Sigma Aldrich) while being treated with RNase (Sigma-Aldrich). Quantitative analysis of sub-G0/G1 cells was carried out in a CouLter XL cytometer using FlowJo software (Tree Star).
TUNEL assay
Apoptosis was assayed in cell culture using a commercially available kit (In Situ Cell Death Detection kit, fluorescein; Roche, Nutley, NJ) designed to detect terminal deoxynucleotidyl transferase (TdT)–mediated nick end labeling (TUNEL). Cells were fixed in 4% paraformaldehyde and permeabilized in 0.1% Triton X-100 in 0.1% sodium citrate. After washing in PBS, cells were incubated in TUNEL reaction mixture at 37°C for 60 minutes, washed, and mounted. Nuclei were counterstained with Hoechst 33342 Trihydrochloride, trihidrate (Invitrogen).
Cell viability assay
Cell viability was determined by adding a final concentration of 1.1 mg/mL MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Sigma Aldrich) into the cell culture. Cells were incubated for 2 hours at 37°C, then the medium was removed, MTT was dissolved in DMSO (Sigma Aldrich) and its absorbance was determined at 570nm.
Correlation in gene expression profiles
We used the microarray data from [22], a compendium of gene expression profiles from 73 normal tissue and cell types, and the protein data from the Human Protein Atlas [23]. We applied a mixture model in order to obtain correlation coefficients that are robust under the presence of noise. We fit the model using the Expectation-Maximization (EM) algorithm [24]. We defined two genes as co-expressed if their EM correlation coefficient was greater than 0.5 and the probability of noise less than 0.5.
Statistical analysis
Results are expressed as the mean ± standard deviation (SD) of three independent experiments. Statistically significant differences were determined by one-way ANOVA followed by Tukey-Kramer post-test to identify pair wise differences. Differences were considered significant at P < 0.05*. Statistical analyses were carried out using GraphPad Prism (GraphPad Software Inc V4.03, San Diego, CA, USA) software.
Results and Discussion
PSMC3IP and EPSTI1 interact with key apoptotic proteins
PSMC3IP and EPSTI1 present relevant transcriptomics/genomics aberrations in breast cancer [7, 11, 15–18], although the molecular mechanisms are still unknown. Since cell proliferation and apoptosis are key for the development of breast tumorigenesis, we sought to explore the eventual implication of these genes in the regulation of these processes. To this end, we exploited the high interconnectivity observed among disease genes [25] to reveal novel direct relationships between well-established BC genes and PSMC3IP and EPSTI1, which could provide a molecular rationale for the implication of these genes in the disease.
An important factor when performing interaction discovery strategies is the selection of the core genes. We extensively mined the literature and the OMIM database [26] to select the most relevant BC genes that are involved in apoptosis or cell proliferation, ending up with 9 BC-apoptosis genes to be included in our interaction screen: AKT1, BAG4, BCAR3, CASP8, CDKN2A, CKN2C, CHEK2, IGF1R and PARP1 (See S1 Table).
We then performed pairwise yeast two-hybrid (Y2H) assays to identify novel interactions between BC-apoptosis genes and PSMC3IP and EPSTI1. Among the 7 high-confidence interactions we identified (see Methods and S1 Table), those of PSMC3IP and EPSTI1 with central proteins in the apoptosis extrinsic pathway, i.e. caspase 8, appeared as the most promising ones. Interestingly, PSMC3IP and EPSTI1 both show a strong co-expression profile with CASP8 in breast normal and cancer tissues, also reflected in higher protein levels (see Methods and S1 Table for further details). In addition, PSMC3IP and EPSTI1 also interact with breast cancer anti-estrogen resistance 3 (BCAR3), which is known to regulate proliferation and induce anti-estrogen resistance in ZR75–1 and MCF-7 breast cancer cells [27]. Finally, we also found EPSTI1 to interact with AKT1 (unpublished data), an antiapoptotic protein that in response to specific stimuli phosphorylates and inactivates certain components of the apoptotic machinery, such as the Bcl2 antagonist of cell death (BAD) and caspase-9 [28, 29]. In breast cancer, the PI3K-Akt pathway is a critical downstream effector of growth factor receptors such as HER2/ErbB2, insulin-like growth factor receptor (IGFR) and epidermal growth factor receptor (EGFR) [30–33]. Taken together, these findings suggest that PSMC3IP and EPSTI1 could play a role in the regulation of the apoptotic response.
Modulation of PSMC3IP and EPSTI1 expression in breast cancer cells
We first investigated whether PSMC3IP and EPSTI1 endogenous protein levels are indeed up-regulated in carcinoma cells such as MCF-7 and MDA-MB-231 (Fig. 1), which represent common breast cancer subtypes differently graded upon hormone dependency and aggressiveness. MCF-7 is a weakly invasive luminal cell line, representative of estrogen receptor (ER)-positive tumors [34]. On the other hand, MDA-MB-231 is a highly invasive basal cell line, and it is often used as model for ER-negative tumors [35]. PSMC3IP is highly overexpressed in both MDA-MB-231 and MCF-7, respectively, compared to the non-tumorigenic breast cancer cell line (MCF-10A) (Fig. 1A). Conversely, EPSTI1 shows a more moderate increase, particularly in MCF-7 cells (1.3-fold, Fig. 1B), as previously observed [17], providing evidence about the heteroclonal nature of MCF-7 sublines [36].
Figure 1. Expression of PSMC3IP and EPSTI1 in normal and breast cancer cell lines.

We inspected the endogenous expression of PSMC3IP (A) and EPSTI1 (B) in two types of breast cancer cell lines, MDA-MB-231 and MCF-7, as compared to a normal breast epithelial cell line, MCF-10A. Estimated protein levels based on densitometry (right) of the immunoblots (left) show a PSMC3IP 19- and 15-fold expression in MDA-MB-231 and MCF-7 cells, while EPSTI1 only shows 1.9- and 1.3-fold in each cell line, respectively. Protein levels were normalized based on the loading control protein β-actin. (*P <0.05, **P<0.01, ***P <0.001 vs MCF-10A cells).
Furthermore, to enhance the phenotypic response that PSMC3IP and EPSTI1 might have on caspase-8, we induced the extrinsic apoptotic pathway using TRAIL, which typically generated about 25 and 35% cell viability decrease in non-transfected MDA-MB-231 and MCF-7 cells, respectively (Fig. 2), in agreement with the data reported in the literature [37–39].
Figure 2. TRAIL-induced apoptosis in breast cancer cells.
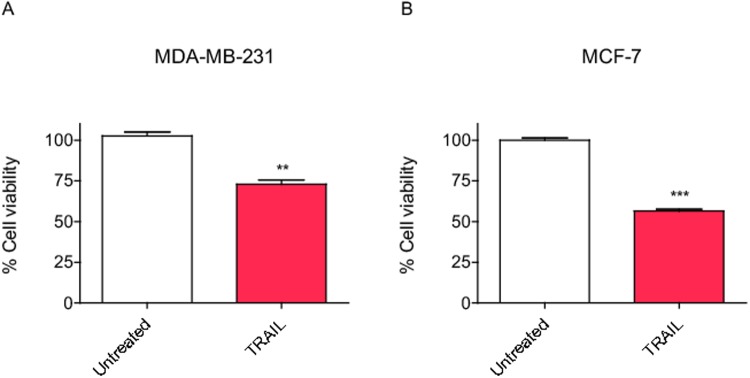
(A) MDA-MB-231 cells treated with the apoptosis inducing ligand TRAIL at 80ng/mL for 24h show a moderate decrease in cell viability while (B) MCF-7 cells treated with TRAIL at 100ng/mL.for 24h show a more pronounced decrease in viability. Each bar represents the mean ±SD of three experiments performed in duplicate (*P <0.05, **P <0.01, ***P <0.001 vs untreated cells).
We then modulated PSMC3IP and EPSTI1 differential expression by either cDNA or siRNA knockout cell transfection. We included the X-linked inhibitor of apoptosis protein (XIAP) as an anti-apoptotic reference gene, since it is a well-characterized inhibitor of caspase-3, caspase-7 and caspase-9 [40, 41] (Fig. 3). Compared to cells transfected with empty vectors, we observed a highly overexpression of PSMC3IP in both cell lines (MDA-MB-231, 6.5-fold; MCF-7, 13-fold) (Fig. 3A–3B), albeit we only achieved a moderate EPSTI1 overexpression (2.1-fold and 2.6-fold, respectively (Fig. 3A–3B). On the other hand, by siRNA transfection, we were able to reduce PSMC3IP levels by 70% in MDA-MB-231 and 50% in MCF-7 cells compared to cells transfected with control siRNA (siLUC) (Fig. 3C–3D). To maximize and visualize the effect of EPSTI1 depletion, we induced its endogenous expression with IFN-α prior to gene silencing in both cell lines (Fig. 3C–3D), as previously reported [17].
Figure 3. Modulation of PSMC3IP and EPSTI1 expression in breast cancer cells.

(A-B) Genes were overexpressed as Myc-tagged fusion proteins in different cell lines and protein relative levels were analysed based on MYC-tag empty transfection vector (Vector). (C-D) Endogenous gene expression was silenced using specific siRNA and depletion levels were analysed based on siRNA against luciferase expression (siLUC) as a negative control. Prior to depletion experiments, EPSTI1 expression was induced by treating cells with IFN-α at 1000 U/ml for 8h. XIAP was used as a reference anti-apoptotic protein in all experiments. Each bar represents the mean ±SD of three experiments performed in duplicate (*P <0.05, **P <0.01, ***P <0.001 vs MYC-tag vector in overexpression assays and vs siLUCIFERASE in silencing).
We subsequently performed a variety of functional assays to measure the apoptotic activity of the two candidate genes in BC cells under basal or apoptotic induced conditions.
PSMC3IP and EPSTI1 expression regulates caspase-8 activity
Since PSMC3IP and EPSTI1 both interact with caspase-8, we first sought to examine the influence of these interactions on caspase-8 activity, which is upstream of the apoptotic cascade. We only observe significant caspase-8 activity decrease upon PSMC3IP overexpression in MDA-MB-231 cells (1.6-fold P<0.05) (Fig. 4A). But interestingly, both individual candidates do decrease caspase-8 activity in MCF-7 cells (PSMC3IP, 1.2-fold P<0.05; EPSTI1, 1.5-fold P<0.001) (Fig. 4B). In agreement with the overexpression results, caspase-8 activity increases after PSMC3IP or EPSTI1 gene silencing in both cell lines (Fig. 4C–D), although preeminently in MCF-7 TRAIL-treated cells upon PSMC3IP depletion (1.3-fold, P<0.01) and under basal conditions upon EPSTI1 depletion (1.3-fold, P<0.05) (Fig. 4D). As expected, XIAP overexpression or silencing does not affect caspase-8 activity, since the inhibitory effect of XIAP is on downstream caspases like caspase-3 or caspase-7.
Figure 4. Caspase-8 activity modulation.

Caspase-8 activity was quantified by measuring the chromophore levels released from caspase-8 cleaved substrates. Overexpression of PSMC3IP or EPSTI1 in TRAIL-treated MDA-MB-231 (A) and MCF-7 cells (B) decrease caspase-8 activity based on the MYC-tag empty transfection vector (Vector) as control. Caspase-8 activity was also measured after gene silencing in MDA-MB-231 (C) and MCF-7 (D) cells, under basal or TRAIL-treated conditions. Genes were silenced using specific siRNAs targeting XIAP, PSMC3IP or EPSTI1 and siRNA against luciferase expression (siLUC) was used as a negative control. EPSTI1-depleted cells were previously treated with IFN-α at 1000 U/ml for 8h. MDA-MB-231 and MCF-7 cells were treated with TRAIL for 24h at 80 or 100ng/mL, respectively. XIAP was used as an anti-apoptotic reference in all experiments. Each bar represents the mean ±SD of three experiments performed in duplicate (*P <0.05, **P <0.01, ***P <0.001 vs MYC-tag vector in overexpression assays and vs siLUCIFERASE in silencing).
Collectively, these results indicate that PSMC3IP and EPSTI1 do modulate caspase-8 activity, suggesting their involvement in the extrinsic apoptotic pathway in breast cancer cells.
PSMC3IP and EPSTI1 expression modulates caspase-3 activity and PARP cleavage
It is well known that the activation of initiator caspases, like caspase-8, leads to the activation of the executioner caspases, such as caspase-3 in MDA-MB-231 cells [42]. Therefore, we investigated whether PSMC3IP or EPSTI1 expression affects caspase-3 activity under basal or apoptotic conditions in MDA-MB-231 cells. We observed that overexpression of either gene does not alter caspase-3 activity levels (Fig. 5A). Yet, EPSTI1 silencing results in an increased caspase-3 activity in both basal conditions (2.6-fold, P<0.001) and upon TRAIL treatment (2.4-fold, P<0.001), giving similar results as the silencing of the anti-apoptotic gene XIAP. On other hand, PSMC3IP silencing is only able to increase caspase-3 activity under TRAIL treatment (1.5-fold, P<0.05) (Fig. 5B). These results indicate that indeed EPSTI1 and PSMC3IP modulate caspase-3 activity in MDA-MB.231 cells, albeit at varying degrees of apoptotic stimulation.
Figure 5. Caspase-3 activity modulation and analysis of cleaved PARP protein levels.

(A) Caspase-3 activity was measured by colorimetric quantification of fluorescent products released from caspase-3 cleaved substrates.in TRAIL-treated MDA-MB-231 cells overexpressing XIAP, PSMC3IP or EPSTI1. None of the overexpressed genes was able to significantly decrease the activity relative to MYC-tag empty transfection vector (Vector) as control. (B) Caspase-3 activity was also measured in MDA-MB-231 cells under basal or TRAIL-treated conditions after gene silencing using specific siRNA targeting XIAP, PSMC3IP or EPSTI1. (C) Immunoblot analysis of cleaved PARP protein levels in gene-overexpressing MDA-MB-231 cells under TRAIL conditions, using MYC-tag transfection vector (Vector) as a negative control, reveals an attenuation of downstream apoptotic cascades. (D) This effect is much more pronounced in TRAIL-treated MCF-7 cells. (E) Analysis of cleaved PARP protein levels after gene silencing in MDA-MB-231 cells. siLUC was used as a negative control. (F) The same analysis in MCF-7 cells shows a more pronounced effect, even under basal conditions. EPSTI1-depleted cells were previously treated with IFN-α at 1000 U/ml for 8h. In apoptosis-induced conditions, MDA-MB-231 and MCF-7 cells were treated with TRAIL for 24h at 80 or 100ng/mL, respectively. XIAP was used as an anti-apoptotic reference in all experiments. Each bar represents the mean ±SD of three experiments performed in duplicate (*P <0.05, **P <0.01, ***P <0.001 vs MYC-tag vector in overexpression assays and vs siLUCIFERASE in silencing).
Caspase-7 and caspase-3 coordinate the last phase of apoptosis by cleaving protein substrates such as PARP [43, 44], which has an essential role in repairing single-strand breaks (SSBs). PARP is inactivated by caspase cleavage, causing SSB repair inhibition that can result in lethal DNA damage [45]. Interestingly, overexpressing PSMC3IP or EPSTI1 show a significant decrease in PARP cleavage in MDA-MB-231 and MCF-7 cells (1.3-fold, P<0.01; 1.8-fold, P<0.01) (1.2-fold, P<0.05; 2.9-fold, P<0.001) (Fig. 5C–D). Furthermore, in agreement with our caspase-3 activity results, there is an increase of cleaved PARP levels after PSMC3IP or EPSTI1 gene depletion in MDA-MB-231 cells upon TRAIL induction (PSMC3IP, 3.2-fold, P<0.001; EPSTI1, 1.5-fold, P<0.05) (Fig. 5E). This effect is more pronounced in MCF-7 cells, where PARP cleavage is increased even in basal conditions (EPSTI1, 4.2-fold, P<0.001; PSMC3IP, 3.6-fold, P<0.05) (Fig. 5F). Taken together, although preclinical studies have shown that ER-negative breast cancer cell lines are more sensitive to PARP inhibitors compared to luminal cells [46], our results indicate that PARP cleavage is similarly affected by the expression of putative extrinsic regulators such as PSMC3IP and EPSTI1 in both ER-negative (i.e. MDA-MB-231) and in luminal breast cancer cell line (i.e. MCF-7). In addition, since EPSTI1 also interacts with AKT1, it could play an alternative role in modulating apoptosis through PI3K pathway, in line with preclinical data that demonstrate synergistic activity when PARP inhibitors are combined with PI3K inhibitors [47].
Increased DNA fragmentation and reduced cell viability are associated with PSMC3IP and EPSTI1 down-regulation
DNA fragmentation, resulting from apoptotic signalling cascades, is a hallmark of late-stage apoptosis [48]. Hence, we wanted to examine whether EPSTI1 and PSMC3IP are able to alter the final apoptotic response beyond the modification of caspase activity. To this end, we quantified the number of apoptotic cells by flow cytometry (i.e. measurement of the sub-G0/G1 peak in the fluorescence histograms) and we further examined DNA fragmentation by TUNEL assays (Fig. 6).
Figure 6. Detection of DNA fragmentation.

(A) The number of apoptotic MDA-MB-231 cells was quantified by flow cytometry using propidium iodide DNA staining after gene depletion under basal or TRAIL-treated conditions (i.e. measurement of the sub-G0/G1 peak in the fluorescence DNA histograms, right panels). (B) Apoptosis was also evaluated in MDA-MB-231 by the inspection of DNA fragmentation by TUNEL (fluorescein-12-dUTP labeled fragmented DNA) staining (right panels). Cell nuclei were stained with Hoechst (blue fluorescence) to estimate the number of total cells. (C) A similar analysis was carried out in MCF-7 cells. The measurement of the sub-G0/G1 peaks (right panels) indicates a significantly higher number of apoptotic gene-depleted cells even under basal conditions (left). (D) TUNEL positive nuclei displaying green fluorescence are observed under basal conditions (right), although a higher number is clearly observed under TRAIL-induced conditions (left). siLUC was used as a negative control, XIAP was used as an anti-apoptotic reference in all experiments. EPSTI1-depleted cells were previously treated with IFN-α at 1000 U/ml for 8h. In apoptosis-induced conditions, cells were treated with TRAIL for 24h, at 80 or 100ng/mL respectively. Each bar represents the mean ±SD of three experiments performed in duplicate (*P <0.05, **P <0.01, ***P <0.001 vs siLUC).
In MDA-MB-231 cells, we detect an increased number of apoptotic cells upon PSMC3IP silencing under TRAIL conditions (1.6-fold, P<0.01), while EPSTI1 depletion has already a similar effect under basal conditions (1.7-fold, P<0.05) (Fig. 6A). Yet, TUNEL-based fluorescent microscopy images do not show conclusive results (Fig. 6B). Interestingly, MCF-7 cells show a clearer phenotype, where PSMC3IP depletion is able to increase the number of apoptotic cells in basal conditions (4.3-fold, P<0.01) (Fig. 6C) and EPSTI1 silencing is now able to induce a high increase of apoptotic cells under both conditions (17.7-fold, P<0.001; 3.5-fold, P<0.001) (Fig. 6C). These findings are in accordance with apoptotic positive cells observed in TUNEL assays (Fig. 6D).
Lastly, we sought to determine breast cancer cells viability after PSMC3IP or EPSTI1 expression modulation. As observed in Fig. 7, up-regulation of either gene is not able to recapitulate the viability of neither TRAIL-treated MDA-MB-231 (Fig. 7A) nor MCF-7 cells (Fig. 7B). Conversely, EPSTI1 down-regulation does decrease the viability of MDA-MB-231 cells treated with TRAIL (1.9-fold, P<0.001) (Fig. 7C). Intriguingly, PSMC3IP or EPSTI1 silencing only induces significant MCF-7 cell viability decrease in basal conditions (1.3-fold, P<0.05 and 1.5-fold, P<0.01 respectively) (Fig. 7D).
Figure 7. Cell viability and recovery.

Cell viability was determined by MTT absorbance assays (A) Histograms showing the viability of PSMC3IP or EPSTI1-overexpressing MDA-MB-231 cells and (B) MCF-7 cells under TRAIL-induced conditions. Based on empty vector (MYC-tag) as a negative control, we do not observe a significant recovery of apoptosis-induced cells after gene overexpression. (C) Viability measurement of gene-depleted MDA-MB-231 cells reveals that EPSTI1 depletion reduces about 50% of viability as compared to siLUC negative control. (D) Intriguingly, in MCF-7 cells, both PSMC3IP and EPSTI1 depletion lead to a decreased viability under basal but not under TRAIL-treated conditions. XIAP was used as an anti-apoptotic reference in all experiments. EPSTI1-depleted cells were previously treated with IFN-α at 1000 U/ml for 8h. In apoptosis-induced conditions, cells were treated with TRAIL for 24h, at 80 or 100ng/mL respectively. Each bar represents the mean ±SD of three experiments performed in duplicate (*P <0.05, **P <0.01, ***P <0.001 vs siLUC).
Concluding Remarks
As shown in the summary Tables 1 and 2, collectively, our findings reveal that PSMC3IP and EPSTI1 have a strong anti-apoptotic role in breast cancer cells, particularly in estrogen receptor positive and triple negative, by means of physical interaction with the apoptosis initiator caspase-8 (Fig. 8). Interestingly, PSCM3IP or EPSTI1 depletion in breast cancer cells show increased DNA fragmentation and reduced cell viability even in the absence of apoptotic stimuli, indicating that they might also modulate the apoptotic pathway trough alternative mechanisms, such as by BCAR3 or AKT1 interaction, particularly in case of EPSTI1. Although further studies are required to gain deeper insight into the molecular mechanisms underlying the anti-apoptotic role of PSMC3IP and EPSTI1 in breast cancer, our findings highlight them beforehand as very interesting therapeutic targets, preeminently for their ability to apoptosis sensitization.
Table 1. Overview of the results obtained from apoptosis-related functional assays upon modulation of PSMC3IP expression in breast cancer cells.
| PSMC3IP | BASAL | TRAIL | ||||
|---|---|---|---|---|---|---|
| MDA-MB-231 silencing | MCF-7 silencing | MDA-MB-231 overexpression | MDA-MB-231 silencing | MCF-7 overexpression | MCF-7 silencing | |
| caspase 8 activity | ns | ns | - | ns | - | + + |
| caspase 3 activity | ns | na | ns | + | na | na |
| cleaved PARP | ns | + | -- | + + + | -- | + |
| apoptotic cells | ns | + + | nd | + + | nd | ns |
| cell viability | ns | - | ns | ns | ns | ns |
Table 2. Overview of the results obtained from apoptosis-related functional assays upon modulation of EPSTI1 expression in breast cancer cells.
| EPSTI1 | BASAL | TRAIL | ||||
|---|---|---|---|---|---|---|
| MDA-MB-231 silencing | MCF-7 silencing | MDA-MB-231 overexpression | MDA-MB-231 silencing | MCF-7 overexpression | MCF-7 silencing | |
| caspase 8 activity | ns | + | ns | ns | --- | ns |
| caspase 3 activity | + + + | na | ns | + + + | na | na |
| cleaved PARP | ns | + + + | - | + | --- | + + |
| apoptotic cells | + | + + + | nd | ns | nd | + + + |
| cell viability | ns | -- | ns | --- | ns | ns |
Statistically significant increase or decrease is highlighted +P <0.05, ++P <0.01, +++P <0.001 and - P <0.05, - - P <0.01, - - - P <0.001 respectively.
nd = no data available, na = not applicable, ns = no significant differences.
Figure 8. Mechanistic model of PSMC3IP and EPSTI1 as putative apoptotic factors.

The extrinsic apoptosis pathway is initiated by the ligand binding to the death receptor, forming the DISC complex and leading to the activation of the caspase signaling cascade. The overexpression of EGFR and HER2 or activation of components of their downstream signaling pathways (i.e. PI3K pathway) induce an anti-apoptotic signaling through PI3K-Akt pathway in endocrine resistant breast cancer cells. Based on our findings, we suggest that PSMC3IP and EPSTI1 may regulate the apoptotic pathway via the physical interaction mainly with the apoptosis initiator CASP8, but also with AKT1 and BCAR3. Pro-apoptotic proteins are displayed in red and anti-apoptotic in green. Candidate proteins are displayed in purple.
Supporting Information
List of selected BC-apoptosis genes and detected interactions with PSMC3IP and EPSTI1 observed by matrix Y2H screens. Candidate gene expression in healthy and carcinoma breast tissues and their co-expression with core BC-apoptotic genes.
(XLSX)
Acknowledgments
We thank M. Duran-Frigola and A. R. Nebreda (IRB Barcelona) for fruitful discussions and support. This work was partially supported by the Spanish Ministerio de Ciencia e Innovación (BIO2010–22073). EC-B is a recipient of an FPI fellowship.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was partially supported by the Spanish Ministerio de Ciencia e Innovación (BIO2010-22073). EC-B is a recipient of an FPI fellowship.
References
- 1. Yang KM, Jung Y, Lee JM, Kim W, Cho JK, et al. (2013) Loss of TBK1 induces epithelial-mesenchymal transition in the breast cancer cells by ERalpha downregulation. Cancer Res 73: 6679–6689. 10.1158/0008-5472.CAN-13-0891 [DOI] [PubMed] [Google Scholar]
- 2. Wong RS (2011) Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res 30: 87 10.1186/1756-9966-30-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ashkenazi A, Holland P, Eckhardt SG (2008) Ligand-based targeting of apoptosis in cancer: the potential of recombinant human apoptosis ligand 2/Tumor necrosis factor-related apoptosis-inducing ligand (rhApo2L/TRAIL). J Clin Oncol 26: 3621–3630. 10.1200/JCO.2007.15.7198 [DOI] [PubMed] [Google Scholar]
- 4. Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, et al. (1998) Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9: 267–276. 10.1016/S1074-7613(00)80609-3 [DOI] [PubMed] [Google Scholar]
- 5. Juo P, Kuo CJ, Yuan J, Blenis J (1998) Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade. Curr Biol 8: 1001–1008. 10.1016/S0960-9822(07)00420-4 [DOI] [PubMed] [Google Scholar]
- 6. Zanzoni A, Soler-Lopez M, Aloy P (2009) A network medicine approach to human disease. FEBS Lett 583: 1759–1765. 10.1016/j.febslet.2009.03.001 [DOI] [PubMed] [Google Scholar]
- 7. Rommens JM, Durocher F, McArthur J, Tonin P, LeBlanc JF, et al. (1995) Generation of a transcription map at the HSD17B locus centromeric to BRCA1 at 17q21. Genomics 28: 530–542. 10.1006/geno.1995.1185 [DOI] [PubMed] [Google Scholar]
- 8. Hall JM, Lee MK, Newman B, Morrow JE, Anderson LA, et al. (1990) Linkage of early-onset familial breast cancer to chromosome 17q21. Science 250: 1684–1689. 10.1126/science.2270482 [DOI] [PubMed] [Google Scholar]
- 9. Ko L, Cardona GR, Henrion-Caude A, Chin WW (2002) Identification and characterization of a tissue-specific coactivator, GT198, that interacts with the DNA-binding domains of nuclear receptors. Mol Cell Biol 22: 357–369. 10.1128/MCB.22.1.357-369.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Satoh T, Ishizuka T, Tomaru T, Yoshino S, Nakajima Y, et al. (2009) Tat-binding protein-1 (TBP-1), an ATPase of 19S regulatory particles of the 26S proteasome, enhances androgen receptor function in cooperation with TBP-1-interacting protein/Hop2. Endocrinology 150: 3283–3290. 10.1210/en.2008-1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sircoulomb F, Bekhouche I, Finetti P, Adelaide J, Ben Hamida A, et al. (2010) Genome profiling of ERBB2-amplified breast cancers. BMC Cancer 10: 539 10.1186/1471-2407-10-539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peng M, Bakker JL, Dicioccio RA, Gille JJ, Zhao H, et al. (2013) Inactivating Mutations in GT198 in Familial and Early-Onset Breast and Ovarian Cancers. Genes Cancer 4: 15–25. 10.1177/1947601913486344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Petukhova GV, Romanienko PJ, Camerini-Otero RD (2003) The Hop2 protein has a direct role in promoting interhomolog interactions during mouse meiosis. Dev Cell 5: 927–936. 10.1016/S1534-5807(03)00369-1 [DOI] [PubMed] [Google Scholar]
- 14. Buess M, Nuyten DS, Hastie T, Nielsen T, Pesich R, et al. (2007) Characterization of heterotypic interaction effects in vitro to deconvolute global gene expression profiles in cancer. Genome Biol 8: R191 10.1186/gb-2007-8-9-r191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nielsen HL, Ronnov-Jessen L, Villadsen R, Petersen OW (2002) Identification of EPSTI1, a novel gene induced by epithelial-stromal interaction in human breast cancer. Genomics 79: 703–710. 10.1006/geno.2002.6755 [DOI] [PubMed] [Google Scholar]
- 16. Gudjonsson T, Ronnov-Jessen L, Villadsen R, Bissell MJ, Petersen OW (2003) To create the correct microenvironment: three-dimensional heterotypic collagen assays for human breast epithelial morphogenesis and neoplasia. Methods 30: 247–255. 10.1016/S1046-2023(03)00031-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. de Neergaard M, Kim J, Villadsen R, Fridriksdottir AJ, Rank F, et al. (2010) Epithelial-stromal interaction 1 (EPSTI1) substitutes for peritumoral fibroblasts in the tumor microenvironment. Am J Pathol 176: 1229–1240. 10.2353/ajpath.2010.090648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li T, Lu H, Shen C, Lahiri SK, Wason MS, et al. (2013) Identification of epithelial stromal interaction 1 as a novel effector downstream of Kruppel-like factor 8 in breast cancer invasion and metastasis. Oncogene. [DOI] [PMC free article] [PubMed]
- 19. Lamesch P, Li N, Milstein S, Fan C, Hao T, et al. (2007) hORFeome v3.1: a resource of human open reading frames representing over 10,000 human genes. Genomics 89: 307–315. 10.1016/j.ygeno.2006.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liang F, Matrubutham U, Parvizi B, Yen J, Duan D, et al. (2004) ORFDB: an information resource linking scientific content to a high-quality Open Reading Frame (ORF) collection. Nucleic Acids Res 32: D595–599. 10.1093/nar/gkh118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gong J, Traganos F, Darzynkiewicz Z (1994) A selective procedure for DNA extraction from apoptotic cells applicable for gel electrophoresis and flow cytometry. Anal Biochem 218: 314–319. 10.1006/abio.1994.1184 [DOI] [PubMed] [Google Scholar]
- 22. Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, et al. (2004) A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A 101: 6062–6067. 10.1073/pnas.0400782101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, et al. (2010) Towards a knowledge-based Human Protein Atlas. Nat Biotechnol 28: 1248–1250. 10.1038/nbt1210-1248 [DOI] [PubMed] [Google Scholar]
- 24. Dempster A, Laird N, Rubin D (1977) Maximum Likelihood from Incomplete Data via the EM Algorithm. Journal of the Royal Statistical Society Series B (Methodological) 39: 1–38. [Google Scholar]
- 25. Oti M, Brunner HG (2007) The modular nature of genetic diseases. Clin Genet 71: 1–11. 10.1111/j.1399-0004.2006.00708.x [DOI] [PubMed] [Google Scholar]
- 26. McKusick VA (2007) Mendelian Inheritance in Man and its online version, OMIM. Am J Hum Genet 80: 588–604. 10.1086/514346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van Agthoven T, van Agthoven TL, Dekker A, van der Spek PJ, Vreede L, et al. (1998) Identification of BCAR3 by a random search for genes involved in antiestrogen resistance of human breast cancer cells. EMBO J 17: 2799–2808. 10.1093/emboj/17.10.2799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Datta SR, Dudek H, Tao X, Masters S, Fu H, et al. (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91: 231–241. 10.1016/S0092-8674(00)80405-5 [DOI] [PubMed] [Google Scholar]
- 29. Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, et al. (1998) Regulation of cell death protease caspase-9 by phosphorylation. Science 282: 1318–1321. 10.1126/science.282.5392.1318 [DOI] [PubMed] [Google Scholar]
- 30. Lobenhofer EK, Huper G, Iglehart JD, Marks JR (2000) Inhibition of mitogen-activated protein kinase and phosphatidylinositol 3-kinase activity in MCF-7 cells prevents estrogen-induced mitogenesis. Cell Growth Differ 11: 99–110. [PubMed] [Google Scholar]
- 31. Tsai EM, Wang SC, Lee JN, Hung MC (2001) Akt activation by estrogen in estrogen receptor-negative breast cancer cells. Cancer Res 61: 8390–8392. [PubMed] [Google Scholar]
- 32. Lenferink AE, Busse D, Flanagan WM, Yakes FM, Arteaga CL (2001) ErbB2/neu kinase modulates cellular p27(Kip1) and cyclin D1 through multiple signaling pathways. Cancer Res 61: 6583–6591. [PubMed] [Google Scholar]
- 33. Yang HY, Shao R, Hung MC, Lee MH (2001) p27 Kip1 inhibits HER2/neu-mediated cell growth and tumorigenesis. Oncogene 20: 3695–3702. 10.1038/sj.onc.1204472 [DOI] [PubMed] [Google Scholar]
- 34. Thomas RJ, Guise TA, Yin JJ, Elliott J, Horwood NJ, et al. (1999) Breast cancer cells interact with osteoblasts to support osteoclast formation. Endocrinology 140: 4451–4458. 10.1210/endo.140.10.7037 [DOI] [PubMed] [Google Scholar]
- 35. Zhang Q, Fan H, Shen J, Hoffman RM, Xing HR (2010) Human breast cancer cell lines co-express neuronal, epithelial, and melanocytic differentiation markers in vitro and in vivo. PLoS One 5: e9712 10.1371/journal.pone.0009712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nugoli M, Chuchana P, Vendrell J, Orsetti B, Ursule L, et al. (2003) Genetic variability in MCF-7 sublines: evidence of rapid genomic and RNA expression profile modifications. BMC Cancer 3: 13 10.1186/1471-2407-3-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu J, Zhou JY, Tainsky MA, Wu GS (2007) Evidence that tumor necrosis factor-related apoptosis-inducing ligand induction by 5-Aza-2’-deoxycytidine sensitizes human breast cancer cells to adriamycin. Cancer Res 67: 1203–1211. 10.1158/0008-5472.CAN-06-2310 [DOI] [PubMed] [Google Scholar]
- 38. Xu J, Zhou JY, Wei WZ, Wu GS (2010) Activation of the Akt survival pathway contributes to TRAIL resistance in cancer cells. PLoS One 5: e10226 10.1371/journal.pone.0010226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rahman M, Davis SR, Pumphrey JG, Bao J, Nau MM, et al. (2009) TRAIL induces apoptosis in triple-negative breast cancer cells with a mesenchymal phenotype. Breast Cancer Res Treat 113: 217–230. 10.1007/s10549-008-9924-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Deveraux QL, Takahashi R, Salvesen GS, Reed JC (1997) X-linked IAP is a direct inhibitor of cell-death proteases. Nature 388: 300–304. 10.1038/40901 [DOI] [PubMed] [Google Scholar]
- 41. Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, et al. (1998) IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J 17: 2215–2223. 10.1093/emboj/17.8.2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Janicke RU, Sprengart ML, Wati MR, Porter AG (1998) Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem 273: 9357–9360. 10.1074/jbc.273.16.9357 [DOI] [PubMed] [Google Scholar]
- 43. Germain M, Affar EB, D’Amours D, Dixit VM, Salvesen GS, et al. (1999) Cleavage of automodified poly(ADP-ribose) polymerase during apoptosis. Evidence for involvement of caspase-7. J Biol Chem 274: 28379–28384. 10.1074/jbc.274.40.28379 [DOI] [PubMed] [Google Scholar]
- 44. Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, et al. (1995) Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 81: 801–809. 10.1016/0092-8674(95)90541-3 [DOI] [PubMed] [Google Scholar]
- 45. Lord CJ, Garrett MD, Ashworth A (2006) Targeting the double-strand DNA break repair pathway as a therapeutic strategy. Clin Cancer Res 12: 4463–4468. 10.1158/1078-0432.CCR-06-1269 [DOI] [PubMed] [Google Scholar]
- 46. Hastak K, Alli E, Ford JM (2010) Synergistic chemosensitivity of triple-negative breast cancer cell lines to poly(ADP-Ribose) polymerase inhibition, gemcitabine, and cisplatin. Cancer Res 70: 7970–7980. 10.1158/0008-5472.CAN-09-4521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ibrahim YH, Garcia-Garcia C, Serra V, He L, Torres-Lockhart K, et al. (2012) PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov 2: 1036–1047. 10.1158/2159-8290.CD-11-0348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Collins JA, Schandi CA, Young KK, Vesely J, Willingham MC (1997) Major DNA fragmentation is a late event in apoptosis. J Histochem Cytochem 45: 923–934. 10.1177/002215549704500702 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of selected BC-apoptosis genes and detected interactions with PSMC3IP and EPSTI1 observed by matrix Y2H screens. Candidate gene expression in healthy and carcinoma breast tissues and their co-expression with core BC-apoptotic genes.
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.