

Abstract
A mutation update on the nebulin gene (NEB) is necessary because of recent developments in analysis methodology, the identification of increasing numbers and novel types of variants, and a widening in the spectrum of clinical and histological phenotypes associated with this gigantic, 183 exons containing gene. Recessive pathogenic variants in NEB are the major cause of nemaline myopathy (NM), one of the most common congenital myopathies. Moreover, pathogenic NEB variants have been identified in core-rod myopathy and in distal myopathies. In this update, we present the disease-causing variants in NEB in 159 families, 143 families with NM, and 16 families with NM-related myopathies. Eighty-eight families are presented here for the first time. We summarize 86 previously published and 126 unpublished variants identified in NEB. Furthermore, we have analyzed the NEB variants deposited in the Exome Variant Server (http://evs.gs.washington.edu/EVS/), identifying that pathogenic variants are a minor fraction of all coding variants (~7%). This indicates that nebulin tolerates substantial changes in its amino acid sequence, providing an explanation as to why variants in such a large gene result in relatively rare disorders. Lastly, we discuss the difficulties of drawing reliable genotype–phenotype correlations in NEB-associated disease.
Keywords: nebulin, NEB, nemaline (rod) myopathy, actin–myosin, sarcomere
Introduction
The nebulin gene (NEB), with its 183 exons, encodes one of the biggest proteins in vertebrates (600–900 kDa). This protein is one of the key components of the skeletal muscle sarcomere, where it determines the minimum lengths of the actin filaments and regulates actin–myosin interactions and the calcium sensitivity of force generation [Bang et al., 2006; Witt et al., 2006; Chandra et al., 2009]. Nebulin is a highly repetitive protein with the potential to bind more than 200 actin monomers. The major part of nebulin consists of 30–35 amino acids-long simple repeats, which are arranged into super repeats, each super repeat consisting of seven simple repeats. An actin-binding motif, SDXXYK, is present once in each simple repeat, and a putative tropomyosin-binding motif, WLKGIGW, is present once in each super repeat [Labeit & Kolmerer, 1995]. The expression of nebulin is greatest in skeletal muscle, but the protein has also been detected at low abundance in heart and brain [Kazmierski et al., 2003; Joo et al., 2004; Laitila et al., 2012].
All pathogenic variants hitherto known in NEB have been recessive, mostly compound heterozygous, and the most common cause of autosomal-recessive nemaline myopathy (NM; MIM #256030) [Pelin et al., 1999; Pelin et al., 2002; Anderson et al., 2004; Lehtokari et al., 2006; Lawlor et al., 2011; Ochala et al., 2011; Kapoor et al., 2013; Malfatti et al., 2014]. Other, less common entities include early-onset distal myopathy without nemaline bodies [Wallgren-Pettersson et al., 2007], a distal form of NM [Lehtokari et al., 2011], core-rod myopathy with generalized muscle weakness [Romero et al., 2009], and a childhood-onset distal myopathy with rods and cores [Scoto et al., 2013].
In addition to NEB (MIM #161650), variants in eight different genes are currently known to cause NM; skeletal muscle α-actin (ACTA1; MIMs #102610, #161800), the most frequent cause of sporadic NM, α-slow tropomyosin (TPM3; MIMs #191030, #609284), β-tropomyosin (TPM2; MIMs #190990, #609285), troponin T1 (TNNT1; MIMs #191041, #605355), cofilin-2 (CFL2; MIMs #601443, #610687), Kelch repeat and BTB/POZ domains-containing protein 13 (KBTBD13; MIMs #613727, #609273), Kelch-like family member 40 (KLHL40; MIMs #615340, #615384, also known as Kelch repeat and BTB/POZ domains containing protein 5, KBTBD5), and Kelch-like family member 41 (KLHL41; MIMs #607701, #615731, also known as Kelch repeat and BTB/POZ domains containing protein 10, KBTBD10) [Laing et al., 1995; Nowak et al., 1999; Pelin et al., 1999; Johnston et al., 2000; Donner et al., 2002; Agrawal et al., 2007; Sambuughin et al., 2010; Ravenscroft et al., 2013; Gupta et al., 2013, respectively].
The Nebulin Gene and the Reference Sequences
The gene-encoding nebulin covers 249 kb in the chromosomal region 2q23 and has 183 exons. Translation initiates in exon 3 and continues to the last exon. A repetitive region in the middle of the nebulin gene complicates analytical testing. The triplicated region (TRI) of 24 exons is arranged as three sets of eight nearly perfectly repetitive exons (82–89, 90–97, and 98–105). Among each set of repetitive exons, there are too few unique nucleotides to differentiate one copy from the other two using most analysis methods, for example, differentiating exon 82 from 90 or 98. By convention, therefore, a novel variant identified in “exon” 82–90–98 is described as if it were located in exon 82, and not 90 or 98. The triplicated region is also thought to undergo alternative splicing, but firm experimental evidence of this is still lacking.
Alternatively spliced exons include exons 63–66, 143–144, and 167–177. Exons 63–66 seem to be expressed together, being either present or absent from the transcript. The expression of the exon pair 143 and 144 is mutually exclusive, whereas exons 167–177 are independently spliced [Donner et al., 2004; Pelin and Wallgren-Pettersson, 2008; Laitila et al., 2012]. It has been hypothesized that such a large variety of different nebulin isoforms is needed because of the diverse requirements on muscle tissue at different stages of development, in various muscle fiber types and in different muscles [Kazmierski et al., 2003; McElhinny et al., 2003; Labeit et al., 2011; Laitila et al., 2012].
We have updated the location of all NEB exons in the genomic reference sequence NG_009382.1, and deposited two NEB mRNA reference sequences, NM_001164507 (transcript variant 1) and NM_001164508 (transcript variant 2) into GenBank at NCBI (http://www.ncbi.nlm.nih.gov/). The transcript variant 1 sequence includes the alternative exon 143, and the transcript variant 2 sequence includes the alternative exon 144. To simplify the description of variants, although data suggest exons 143 and 144 are never expressed in the same transcript, we use NM_001271208.1 as a reference covering 26,307 bp (coding region 25,683 bp) including both exons 143 and 144.
Nebulin Variants and the LOVDdatabase
To date, we have ascertained 212 (126 novel and 86 published) different pathogenic variants in NEB in 159 families in which both pathogenic variants have been identified using a variety of different analysis methods: single-strand conformation polymorphism, denaturing high-performance liquid chromatography, Sanger sequencing of genomic or cDNA, next-generation sequencing (NGS) of NEB alone or of whole exomes, our self-designed microarray for detecting copy-number variants (NM-CGH), and targeted next-NGS of a panel of known neuromuscular genes, including all known NM genes. The identified variants are dispersed all along NEB.
In the majority (83%) of families, the patients are compound heterozygous for two different variants. Twelve variants have only been seen in homozygous form and nine variants in both heterozygous and homozygous forms.
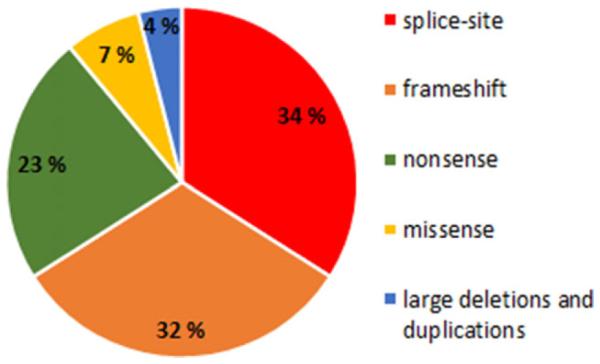
The most common types of variants are splice-site mutations (34%), followed by frameshift mutations (32%) caused by small (<20 bp) deletions or insertions, nonsense mutations (23%), and missense mutations (7%). Large deletions and duplications (>1 kb) are rare (4%) (Fig. 1).
Figure 1.
Different types of variants in NEB. The distribution of 212 different NEB variants identified in this study cohort of 159 families.
In our study, only those missense variants that are predicted to affect nebulin–actin or nebulin–tropomyosin interactions have been interpreted as pathogenic. It is likely that some missense sequence variants outside these regions are also disease causing, but it is very difficult to distinguish these variants from rare polymorphisms that occur in NM patients by chance. Therefore, we have adopted a conservative approach categorizing missense changes to minimize the chance of misinterpreting rare benign variants as being pathogenic. Both nonsense and frameshift variants are predicted to lead to truncation and/or degradation of the isoform where the mutated exon is expressed.
In the current paper, all identified variants, both previously published and novel, have been mapped according to the coding sequence of NCBI GenBank cDNA reference sequence NM_001271208.1. All variants have been deposited in the LOVD-powered NEB variant database (http://NEB.LOVD.nl).
The Spectrum of Myopathies Caused by Variants in NEB
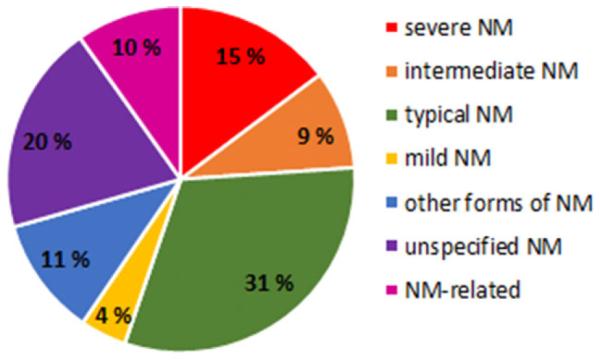
Most commonly, variants in NEB have been associated with NM. Out of 159 families included in this article, 143 (90%) have NM, whereas only 16 families with variants in NEB have other disorders (Fig. 2); early-onset distal myopathy without nemaline bodies (four families), a distal form of NM (three families), core-rod myopathy with generalized muscle weakness (three families), and a childhood-onset distal myopathy with rods and cores (three families). In addition, we report three families with fetal akinesia/lethal multiple pterygium syndrome.
Figure 2.
Clinical entities caused by variants in NEB. The different forms of NM and their incidence in this study cohort of 159 families. Unspecified NM, clinical data were not available. NM related, families with distal myopathy without nemaline rods, distal forms of NM, core myopathy, distal form of core-rod myopathy, and fetal akinesia/lethal multiple pterygium syndrome.
Nemaline Myopathy
NM was described by two separate groups in 1963 as a novel disease entity [Conen et al., 1963; Shy et al., 1963]. The most common clinical presentation is early-onset muscle weakness predominantly affecting proximal limb muscles. The hallmark feature on muscle pathology is the accumulation of Z-disk and thin filament proteins into aggregates named nemaline bodies or rods, usually accompanied by disorganization of the muscle Z-disks [Magee and Shy, 1956; Conen et al., 1963; Shy et al., 1963; Wallgren-Pettersson et al., 1995]. However, NM varies in clinical severity. Six clinical subgroups are used to classify the disorder: the severe, intermediate, and typical congenital forms of NM, a mild childhood/juvenile-onset form; adult-onset NM; and other (atypical) forms of NM (Table 1) [Wallgren-Pettersson and Laing, 2000]. Clinical classification of the disorder in individual patients may be difficult early in life, as progression during childhood is one of the features distinguishing between categories. Also, patients sharing the same variants may have different disease severities, even within the same family [Lehtokari et al., 2006].
Table 1.
Clinical (Severity) Classification of NM and Other Disorders Caused by NEB Mutations (Modified from Wallgren-Pettersson et al., 2000)
| Form of congenital myopathy | Clinical inclusion (and exclusion) criteria |
|---|---|
| Severe NM | Onset at or before birth: no spontaneous movements; no spontaneous respiration, or with severe contractures or fractures at birth |
| Intermediate NM | Infantile onset: patient breathing and moving at birth, but unable to maintain respiratory independence, or to sit and walk independently; use of wheelchair before the age of 11 years; contractures developing in early childhood |
| Typical NM | Onset in infancy: typical distribution of muscle weakness (weakness most pronounced in facial, bulbar, and respiratory muscles, neck flexors, and limb-girdle muscles; initially proximal, later also distal limb involvement); motor milestones delayed but reached; course slowly progressive or nonprogressive |
| Exclusion criteria: contractures/fractures at birth; lack of spontaneous movements and/or respiration at birth; failure to sit/walk independently; use of wheelchair before 11 years |
|
| Mild childhood or juvenile-onset NM | Childhood or juvenile onset |
| Adult onset NM | Adult onset |
| Other forms of NM | Unusual associated features such as cardiomyopathy, ophthalmoplegia, CNS involvement, or unusual distribution of muscle weakness |
| Exclusion criteria: distal myopathy caused by nebulin mutations | |
| Core-rod myopathy | |
| Distal myopathy | Mainly distal involvement at presentation |
| Core-rod myopathy | Definite cores and rods on biopsy |
The variants identified in the 143 NM families are summarized in Supp. Table S1A–F. Patients from 24 families had severe congenital NM, patients from 15 families had the intermediate congenital form, 51 patients had the typical congenital form (individual sibs from four families had either the typical or the intermediate forms of NM), and seven had mild childhood-onset NM. None had adult-onset NM, whereas 18 were classified into the group of “other forms,” and 32 had an unspecified form of NM, that is, clinical data were lacking (Fig. 2). In 50 NM families, both pathogenic variants had been published previously, whereas in 12 families, only one of two compound heterozygous variants had been published. Eighty-one nebulin-related NM families with both pathogenic variants identified are presented here for the first time.
Distal Nebulin Myopathy Without Nemaline Rods
Variants in NEB were long believed only to cause NM. However, in 2007, we discovered two different homozygous NEB missense variants, causing a novel disease entity in four Finnish families with a clinically mild distal myopathy [Wallgren-Pettersson et al., 2007]. The patients presented with only distal weakness, in a pattern similar to that of Laing myopathy [Lamont et al., 2014], and no nemaline bodies were apparent at light microscopy. In one of the patients, no rods were found in any of the four biopsies taken over the years of diagnostic workup (at ages of 14, 16, 27, and 29 years). Both variants were present in homozygous form in two Finnish families each, in five and two individuals, respectively (Table 2) [Wallgren-Pettersson et al., 2007]. When present in a compound heterozygous state with more disruptive variants, the same variants are known to cause NM. One of the missense variants, p.Ser6366Ile, resides in NEB exon 122 and causes increased nebulin–actin affinity, whereas the second missense variant, p.Thr7382Pro, in NEB exon 151 reduces the affinity of nebulin for tropomyosin [Marttila et al., 2014]. The differences in muscle involvement in NM and distal nebulin myopathy, as well as the milder phenotype are intriguing. A possible explanation could be that the missense variants in NEB, being less disruptive, would cause milder, distal-only myopathy without nemaline rods.
Table 2.
Families with NM-Related Myopathies (n = 16)
| Family ID | Exon/intron; variant in cDNA; in protein sequence | Additional details |
|---|---|---|
| Families with distal myopathy without nemaline rods (mild) (n = 4) | ||
| Dist1 (mild) | ex151; c.22249A>C; p.Thr7417ProE-FAM I | FIN2 |
| Dist2 (mild) | ex151; c.22249A>C; p.Thr7417ProE-FAM II | FIN2 |
| Dist3 (mild) | ex122; c.19097G>T; p.Ser6366IleE-FAM III | FIN1 |
| Dist4 (mild) | ex122; c.19097G>T; p.Ser6366IleE-FAM IV | FIN1 |
| Distal nemaline myopathy (n = 3) | ||
| F156 (mild) | ex122; c.19097G>T; p.Ser6366IleD-Fam33 | FIN1 |
| int53;c.7228–1G>T;p.? | ||
| F227 (mild) | ex163; c.23525_23526del; p.Arg7842fsJ-Fam1 | |
| ex156;c.22993T>C;p.Leu7597ProJ-Fam1 | ||
| F343 (mild) | ex18; c.1622dup; p.Asp542*J-Fam2 | |
| int23; c.2211+5G>A; p.?J-Fam2 | ||
| Core-rod myopathy with generalized muscle weakness (n = 3) | ||
| F220 (severe) | ex140; c.21006_21007dup; p.Lys7003fsG | ALT3 |
| ex171; c.24294_24297dup; p.Glu8100fsG | ||
| F378 (severe) | int122; c.19102-19102_10del; p.? | Rods in addition to the cores |
| ex1l9;c.18676C>T;p.Gln6226* | ||
| F400 (mild) | int25; c.2415+1G>A; p.? | Normal strength in lower limbs. pseudodominant; father affected, mother carrier. rods in addition to the cores |
| Distal form of core-rod myopathy (n = 3) | ||
| CR2 (typical) | del ex55; c.7431+1916_7536+372del; p.Arg2478_Asp2512delN | Ashk. 2.5 kb del |
| ex172; c.24327_24375dup; p.Val8126fsN | ALT3 | |
| F282 (typical) | int106; c.16909-2A>G; p.?R | ALT3 |
| ex171; c.24294_24297dup; p.Glu8098fsR | ||
| F410 (typical) | ex129;c.19944G>A;p.?R | 133 kb dup |
| dup 5’–ex81; chr2.hg19:g.(152,465,598_152,465,794)_(152,599, 010_152,608,650)dupR |
||
| Families with fetal akinesia/lethal multiple pterygium syndrome (n = 3) | ||
| D12–060 cons | ex29; c.2920C>T; p.Arg974* | |
| F412 | ex120; c.18786 C>G; p.Tyr6262* | |
| ex121; C.18981 OG; p.Tyr6327* | ||
| F384 | del ex55; c.7431+1916_7536+372del, p.Arg2478_Asp2512delL-P4 | Ashk. 2.5 kb del |
| int66; c.9619–2A>G; p.?L-P4 |
Severity is in parentheses. Reference sequence: c.NM_001271208.1 (contains all of the 183 exons including the triplicated region).
cons, consanguineous; HOZ, homozygous mutation, del ex55, Ashk., Ashkenazi Jewish founder mutation; Finnish founder mutations: FIN1, a missense mutation Ser > Ile in exon 122; FIN2, a missense mutation Thr > Pro in exon 151; FIN3, a frameshift mutation in exon 122; ALT1, mutation in the alternatively spliced exons 63–66 (always expressed together); ALT2, mutation in the alternatively spliced exons 143 or 144; ALT3, mutation in the alternatively spliced exons 167–177 (expressed independently), TRI, mutation in the triplicate region.
Published mutations in Table 2: A, Pelin et al. (1999); B, Pelin et al. (2002); C, Anderson et al. (2004); D, Lehtokari et al. (2006); E, Wallgren-Pettersson et al. (2007); F, Lehtokari and Greenleaf et al. (2009); G, Romero and Lehtokari et al. (2009); H, Lawlor et al. (2011); I, Ochala et al. (2011); J, Lehtokari et al. (2011); K, Kapoor et al. (2013); L, Yonath et al. (2012); M, Böhm et al. (2013); N, Scoto et al. (2013); O, Gajda et al. (2013); P, Kiiski et al. (2013); Q, Malfatti et al. (2014); R, Malfatti et al. (in preparation).
Distal NM
A few years after the identification of the above group of patients, we identified NEB variants in three non-Finnish patients in two unrelated families, presenting in childhood with distal weakness and showing nemaline bodies in their first, diagnostic biopsy (F227 and F343; Table 2) [Lehtokari et al., 2011]. Here, we report one more patient with a distal form of NM. This Finnish patient (F156; Table 2) is compound heterozygous for an exon 122 missense variant (p.Ser6366Ile) and a splice-site variant in intron 53 (c.7228-1G>T). We can thus confirm that NM and distal myopathies caused by NEB variants form a clinical and histological continuum, and that NM should be considered as a differential diagnosis in patients presenting with an early-onset, predominantly distal myopathy.
Core-Rod Myopathy
We previously published NEB variants underlying a severe form of core-rod myopathy in one patient (F220; Table 2) [Romero et al., 2009]. Here, we report two additional families, F400 and F378, with clinically mild and severe forms, respectively, in which the patients have core structures in their biopsies in addition to nemaline rods. Both the father and the three affected children of the consanguineous family F400 are affected and homozygous for a recessive variant in the donor splice site of exon 25 (c.2415+1G>A; Table 2), resulting in a pseudodominant pattern of inheritance. In this family, the distribution of muscle weakness was unusual in that the patients had normal strength in the leg muscles. So far, to our knowledge, no other patients with the histological picture of core-rod myopathy and upper limb-only proximal muscle weakness have been found to have variants in NEB.
Distal Core-Rod Myopathy
Recently, Scoto et al. (2013) reported a childhood-onset distal myopathy with rods and cores, caused by compound heterozygous NEB variants (the Ashkenazi founder deletion of exon 55 and a frameshift mutation in the alternatively spliced exon 172, family CR2, Table 2). In the current paper, we report compound heterozygous NEB variants identified in two French families in which the patients had a similar distal phenotype (F282, F410; Table 2). Interestingly, the patients in family F282 share a frameshift variant in exon 171 with the core-rod myopathy patient of F220 and F127 with severe NM. The patients of family F282 had a distal distribution of limb muscle weakness, combined in one of them with neck flexor and respiratory muscle weakness. The histological pattern was a combination of definite rods and cores. One of the patients, like the one presented in the paper by Scoto et al. (2013), has required nocturnal noninvasive ventilatory support. Both patients are ambulant, at the ages of 11 and 15 years, respectively (Malfatti et al., in preparation).
Fetal Akinesia
We have identified NEB variants with fetal akinesia/lethal multiple pterygium syndrome (D12–060 and F412), and fetal akinesia, respectively (F384) (Table 2). The muscle biopsy performed on the fetuses from F412 showed almost complete replacement of muscle with adipose tissue.
Genotype–Phenotype Correlations in Patients with NEB Variants
Although the number of NEB variants identified is now substantial, it is not adequate for statistical analyses. We were not able to obtain statistically significant genotype–phenotype correlations in terms of the type of variant in relation to the clinical classification of the patient’s disease. It is to be noted that the same variant can be associated with different forms of NM, and even with different myopathies.
We examined the variants, their locations, and the phenotypes they cause from different angles. Variants in alternatively spliced exons might be hypothesized to help us understand the function of the different exons and isoforms in different muscles or muscle fibers. However, the variants are most commonly heterozygous, and combined with a variant in an exon expressed in all isoforms. Also, the variants in alternatively spliced exons are still rather small in number, making it impossible at this time to draw conclusions about the functions of different exons based on the variants identified in patients. Variants in exons 63–66 and 143–144 may, in the future, shed light on the function of these exons.
One approach toward finding genotype–phenotype correlations was to compare the 48 families with pathogenic variants in the alternatively spliced exons; 35 families had one variant, and 13 families had both variants residing in the alternatively spliced exons 63–66, 143–144, or 167–177 (ALT1, ALT2, and ALT3, respectively, in Tables 2–3; Supp. Table S1). Of these, patients in two families had severe presentations, patients in three families had intermediate congenital NM, 11 had typical congenital NM, and 15 had an unspecified form of NM (Supp. Table S1). Interestingly, we noticed that many of these patients (14 families) presented exceptional clinical and/or histological features. Furthermore, three showed unusual histological features; one had core-rod (F220) and two distal core-rod myopathy (CR2 and F410; Table 2).
Table 3.
NEB Variants Shared by Two or More Families
| Shared variant | Number of families |
|---|---|
| del ex55 (Ashkenazi founder mutation) | 13 |
| ex122; c.19097G>T; p.Ser6366Ile (Finnish mut 1) | 10 |
| int32; c.3255+1G>A; p.? (hotspot) | 8 |
| int32; c.3255+1G>T; p.? (hotspot) | 6 |
| ex151; c.22144A>C; p.Thr7382Pro (Finnish mut 2) | 5 |
| ex119; c.18676C>T; p.Gln6226* | 4 |
| int4;c.78+1G>A;p.? | 3 |
| intl3;c.1l52+1G>A;p.? | 3 |
| int36;c.3987+1G>A; p.? | 3 |
| int43; c.5343+5G>A; p.? | 3 |
| ex58; c.8031_8041del; p.Lys2677Asnfs*7 | 3 |
| ex171 ALT; c.24189_24192dup; p.Glu8063Serfs*5 | 3 |
| ex173 ALT; c.24302_24305dup; p.Leu8102Phefs*18 | 3 |
| ex175 ALT; c.24527_24528del; p.Pro8176Argfs*4 | 3 |
| ex18; c.1623del; p.Pro541Profs*16 | 2 |
| int25;c.2415+1G>A; p.? | 2 |
| int29;c.2943+1G>A; p.? | 2 |
| ex38; c.4337G>T; p.Gly1446Val | 2 |
| int36; c.3987+1_3987+2inv; p.? | 2 |
| ex58; c.7964A>G; p.Tyr2655Cys | 2 |
| int66; c.9619–2A>G; p.? | 2 |
| ex122; c.19048_19057del; p.Thr6350Profs*4 (Finnish mut 3) | 2 |
| ex129; c.19944G>A; p.Ser6648 | 2 |
| ex138; c.20845dup;p.Thr6949Asnfs*16 | 2 |
| ex158; C.228310T; p.Arg7611* | 2 |
| ex163;c.23420_23421del;p.Arg7807Serfs*16 | 2 |
| ex170 ALT; c.24113C>A; p.Ser8038* | 2 |
| ex175 ALT; c.24549_24550del; p.Arg8183Serfs*8 | 2 |
| ex175 ALT; c.24579G>A; p.? | 2 |
| ex177 ALT; c.24735_24736del; p.Ala8246* | 2 |
| del ex43—45 | 2 |
| del ex77 | 2 |
Two families with patients assigned to the group of “other forms of NM” had variants in the triplicate region (TRI) of NEB. One Finnish patient (F316) belonging to the group of “other forms of NM” was compound heterozygous for variants in nonalternatively spliced exons (exons 68 and 122), and another (non-Finnish) patient (F309) was compound heterozygous for a nonsense variant in exon 78 and a very unusual large duplication (>103 kb altogether), with one breakpoint upstream of NEB and the other breakpoint in intron 51 (Supp. Table S1E).
In addition to the Ashkenazi Jewish founder mutation (exon 55 deletion), we have identified seven other large deletions or duplications (1–133 kb) in nine families. The clinical severity varied in these patients from severe congenital NM (F181) through typical congenital NM (F45 and F318) to mild clinical phenotypes (F309). One patient had core-rod myopathy (F410). Two families, F211 with typical, and F333 with mild NM, shared a deletion of exons 43–45, and two families shared a deletion of exon 77, but had unspecified forms of NM (PG-F19 and PG-F20).
Variants in the Alternatively Splice Exons 63–66
Patients in seven families had variants in the alternatively spliced exons 63–66. Patients from families F270, F284, and F386 were classified as having “other forms” of NM (Supp. Table S1E). They all had pronounced weakness of axial muscles, with relative sparing of limb muscles, as opposed to the usual distribution of axial, proximal limb muscle, and later distal involvement. Patients from family F4 had the typical congenital (Supp. Table S1C) and Bos227-1, the severe congenital form of NM (Supp. Table S1A). No tissue samples were available from a fetus from family F384, where the pregnancy was terminated because of fetal akinesia, (Table 2). One patient (PG-F5), with a splice-site variant in intron 66, had an unspecified form of NM (Supp. Table S1F).
Exons 63–66 are always expressed together, that is, all four exons are either present in the transcript producing the isoform, or are absent from it, and they encode one super-repeat highly homologous to super-repeat 11 [Donner et al., 2004]. Biopsies from eight different skeletal muscles analyzed by reverse-transcription PCR in the study of Laitila et al. (2012) expressed both types of transcripts, that is, those including and those excluding exons 63–66. Furthermore, no significant differences in expression levels of exons 63–66 were observed in an expression microarray analysis of 21 different types of leg muscles [Laitila et al., 2012]. These results indicate that both transcripts expressing exons 63–66 and transcripts lacking exons 63–66 are needed for normal muscle function in many different types of muscles. All patients with truncating variants in exons 63–66 are expected to preferentially express nebulin isoforms lacking these exons.
Variants in Exons 143–144
Altogether, three families had variants in exons 143 or 144, which are alternatively spliced. An adult sibling pair with a heterozygous nonsense mutation of the alternatively spliced exon 143 had ophthalmoplegia, which is very unusual in NM (F187; Supp. Table S1E). In this family, and in the family with a homozygous nonsense mutation of the alternative exon 144 (F390; Supp. Table S1E), one of the sibs had undergone anesthesia and had had difficulty waking up from it, although the situation did not progress to overt malignant hyperthermia. Again, this is an unusual phenomenon in NM [Cunliffe and Burrows, 1985; Stackhouse et al., 1994; Shenkman et al., 2000]. Other unusual features in F390 with a truncating mutation in exon 144 were that the biopsy showed dystrophic features, and that the neck flexors, which are almost always weak in NM, had normal muscle strength. Additionally, the patient of family F114, with an unspecified form of NM, had a heterozygous frameshift mutation in exon 144 (Supp. Table S1F). We have no clinical or histological data on this family.
It seems that exons 143 and 144 are never expressed together in the same isoform, but both types of transcripts are usually present at different expression levels in the same muscle [Donner et al., 2006; Laitila et al., 2012]. The patients with truncating mutations in exon 144 are expected only to express nebulin exon 143 isoforms, whereas patients with the truncating mutation in exon 143 are expected only to express exon 144 isoforms. The protein domains encoded by exons 143 and 144 differ in charge and hydrophobicity, and they appear to have different and important regulatory functions in developing muscles [Donner et al., 2004; Donner et al., 2006].
Variants in Exons 167–177
Patients from 38 families had variants in the alternatively spliced exons 167–177: patients from one family (F407) had severe, three families had intermediate (G22593, G36659, and P8), and 10 families had typical NM (SYD-F1, SYD-F2, SYD-F3, F328, AG-F1, F318, F340, F349, G19106, and F238).
Nine families with variants in the alternatively spliced exons 167–177 had been assigned to the category “other forms” of NM: patients from three families had fasciculations (F127, F206, and F382), and three a selective pattern of muscle weakness different from that of the typical form of NM (F189, F389, and F324). Two other families (F79 and F81) with different homozygous frameshift variants in the same exon (171) had NM with rigid neck and/or spine. One patient (F31) had a very severe, unusual form of NM and was born with hypospadias and micropenis (Supp. Table S1E).
Patients from one family had severe core-rod myopathy (F220) and two a distal form of core-rod myopathy (F282 and CR2). Families F220 and F282 share a frameshift mutation in exon 171 (p.Glu8098fs) (Table 2). The same variation was also identified in the cousin of the patient in F282 who had a severe, unusual form of NM with fasciculations (F127; Supp. Table S1E).
No clinical details are available of patients in 14 of the families with variants in exons 167–177 (Supp. Table S1F).
Exons 167–177 are spliced independently of each other, and all encode 31 amino acid long simple repeats of the Z-disk region of nebulin [Donner et al., 2004]. The beginning of this region binds desmin at the edge of the Z-disk, and the simple repeats extending into the Z-disk are thought to specify Z-disk width in different fiber types [Bang et al., 2002; Tonino et al., 2010]. The variants are expected to reduce nebulin isoform diversity, and may affect nebulin–desmin interactions as well as Z-disk assembly.
Of the 67 different frameshift mutations identified in NEB to date, 29 reside in alternatively spliced exons, and 27 of them in exons 167–177. All variants identified in exons 167–177 are truncating (frameshift or nonsense). Notably, only one patient (F88, severe NM; Supp. Table S1A) had a homozygous frameshift variant in an exon that is not alternatively spliced (c.25200del in exon 180), suggesting that homozygous frameshift variants are not tolerated in ubiquitously expressed exons, unless at the very 3tend of the gene.
Shared, Recurrent, and Founder Mutations in NEB
There is only one relatively common founder mutation known in NEB, with world-wide occurrence, that is, the 2.5-kb deletion of the entire exon 55 (p.Arg2478_Asp2512del) identified in the Ashkenazi Jewish population [Anderson et al., 2004]. The carrier frequency of this variant in the Ashkenazi Jewish population is 1:108–168 [Anderson et al., 2004; Lehtokari et al., 2009; Scott et al., 2010]. In our cohort, this variant was identified in 13 families (in homozygous form in four and in heterozygous form in nine families).
Three different variants identified in Finnish families may be regarded as founder mutations in the Finnish population (p.Ser6366Ile in ex122, p.Thr7382Pro in ex151, and p.Thr6350Profs*4 in ex122). The missense variants were both present in homozygous form in two distal nebulin myopathy families each. The exon 122 missense variant was identified in heterozygous form in eight NM families, including one with distal NM, and in homozygous form in two families with distal nebulin myopathy. The exon 151 missense variant was found in heterozygous form in three NM families and in homozygous form in two families with distal nebulin myopathy. The carrier frequencies of these two variants in Finland were estimated based on the Sequencing Initiative Suomi (SISU) project data (http://www.sisuproject.fi/). According to SISU, the carrier frequency of the p.Ser6366Ile variant is 1:653, and of the p.Thr738Pro 1:1,108 in the Finnish population. The third Finnish founder mutation, a frameshift variant, was identified in two NM families. One of the Finnish families, F1 shows compound heterozygosity for the p.Thr7382Pro and the p.Thr6350Profs*4 variants (Supp. Table S1C) [Lehtokari et al., 2006; Wallgren-Pettersson et al., 2007].
In addition to the four founder mutations, 28 different variants were shared between two or more families, and 11 different variants between three or more families. Our haplotype results show that the shared variants in 10 families occur on different haplotypes, indicating that these variants have arisen de novo more than once in humans, whereas in 15 families the haplotype is shared (Table 3).
Only one location in NEB can be considered to be a true mutational hotspot; nucleotide c.3255+1 (donor splice site of intron 32) was mutated in 15 unrelated families, and c.3255+2 in two unrelated families. Recurrent mutations were also detected in the donor splice site of intron 36. Three families showed c.3987+1G>A, and two families c.3987+1_3987+2inv (Table 3). There is no obvious explanation, for example, any repetitive sequence, of why these nucleotides seem to be mutated recurrently.
A nonsense variant in exon 119 (p.Gln6226*) was identified in four families. In two families, the patients are cousins (F127 with severe other form of NM; Supp. Table S1E; and F378 with severe NM with cores; Table 2). In our data set, there are currently only two families sharing the same two compound heterozygous variants. Families F349 and G19106 share a nonsense variant in exon 119 (p.Gln6226*) and also a splice-site variant c.24684G>A in exon 175 (Supp. Table S1C). Another pair of families sharing the same variants is F17 and F267 (c.3987+1G>A and c.20845dup; Supp. Table S1B and F). F17 was sent from United States and F267 is from The Netherlands. These families share the haplotype (markers D2S2277, D2S2275, and D2S2299 used). No haplotype data were available for the families F349 (from Switzerland) and G19106 (from France).
NEB Variants in Healthy Individuals and Disease Incidence Estimates
The Exome Variant Server (http://evs.gs.washington.edu/EVS/), a collection of exome-sequencing data of 6,503 DNA samples from unrelated individuals of African-American and European-American origin, contains 1,295 variants in NEB (ESP6500 data release, February, 2014). To the best of our knowledge, the EVS cohorts do not include individuals with neuromuscular disorders. Of the 1,295 variants, 522 represent missense changes, 488 variants are within introns (excluding splice sites), 262 are synonymous changes in the coding region, 10 frameshift mutations, six in-frame deletions or insertions, three intronic splice-site mutations, and two nonsense mutations. The in-frame deletions and insertions affect one to five amino acids each, and we consider them as being nonpathogenic due to their high-occurrence rate in the study population, with several homozygotes identified. Some of the indels may also represent sequencing or annotation errors. Of the synonymous changes, two are part of 5′ splice signals and may affect splicing. Of the 522 missense variants, 31 reside in conserved actin-binding sites of nebulin and could be considered to affect actin binding, whereas seven are in putative tropomyosin-binding sites and may affect nebulin–tropomyosin interaction. Of the approximately 6,000 individuals whose samples were successfully exome sequenced, 69 were heterozygous carriers of pathogenic NEB variants, which equal a carrier frequency of approximately 1/87, and a disease incidence of approximately 1 in 30,000 for recessively inherited myopathies caused by NEB variants. The incidence of NM in one population has previously been estimated as 0.02 per 1,000 live births (1 in 50,000) [Wallgren-Pettersson, 1990].
Clinical and Diagnostic Relevance
Variants in NEB may cause NM with a wide range of clinical severities as well as other related entities. If the other known NM genes have been ruled out as the cause of the disease in a patient with NM, the next step in the diagnostic procedure would be to analyze the patient for NEB variants. Most often NEB variants are associated with the typical form of recessive NM, in which the clinical picture consists of myopathic facies, nasal voice or even dysarthria, pronounced neck flexor, axial muscle, and limb girdle weakness in comparison with the less affected, but weak proximal limb muscles. Distal muscle involvement, including foot drop, is often noted later. Respiratory involvement is often out of proportion to the weakness of the other muscles [Wallgren-Pettersson, 1989; Lehtokari et al., 2006].
In some cases, histological findings may help direct mutation detection. Nebulin positivity is the rule, but if the muscle biopsy shows uneven staining for nebulin, a nebulin variant appears more likely than an ACTA1 variant [Pelin et al., 1999; Gurgel-Giannetti et al., 2001; Sewry et al., 2001]. If there are intranuclear nemaline bodies, an excess of thin filaments, retained expression of cardiac-actin in muscle, or zebra bodies, an ACTA1 variant is likely [Nowak et al., 1999; Nowak et al., 2007].
Molecular verification of the diagnosis is a prerequisite for determining the mode of inheritance, for accurate genetic counseling and, where desired, for enabling prenatal diagnosis in future pregnancies. The carrier frequency of the exon 55 deletion in the Ashkenazi Jewish population may be as high as 1:110, so after exclusion of the smaller genes as the causative gene in the family, this variant would be a first choice in further molecular diagnostic analysis of patients in many parts of the world. In Finnish patients, the Finnish founder mutations can be sought for first. Attention should also be paid to the donor splice sites of intron 32 and intron 36, which are frequently mutated.
It has become evident that even considering the now large number of patients with clarified genotypes, it is extremely difficult to discern any detailed genotype–phenotype correlations in NEB-caused NM. This renders variant types unreliable as a basis for determining the prognosis in individual patients. Missense variants, however, do not seem to be commonly pathogenic, and in order to underlie NM, it is likely that a missense variant needs to be combined with a more disruptive variant.
Relevant Animal Models
Four nebulin mouse models have been published; two Neb knock-out models with very severe phenotypes [Bang et al., 2006; Witt et al., 2006], one homozygous for the deletion of exon 55 (Ashkenazi founder mutation) also with a very severe phenotype [Ottenheijm et al., 2013], and a recent one with a deletion of the SH3-domain of nebulin, with no structural or histological skeletal muscle abnormalities, but slight impact on muscle function [Yamamoto et al., 2013]. Furthermore, a zebrafish model homozygous for a splice-site mutation in neb also shows a severe phenotype [Telfer et al., 2012].
Conclusions and Future Prospects
With NGS techniques becoming more common in the diagnostic field, it is important to bear in mind that no technique alone reveals all variants in the gigantic NEB gene. There are regions of great homology that cause difficulties in sequencing, such as the NEB triplicate region consisting of exons 82–89, 90–97, and 98–105. Copy-number variants spanning the entire NEB triplicate region block have, however, been identified with our NM-CGH microarray [Kiiski et al., 2013]. Small variants in this region can be unraveled through whole-genome sequencing and subsequent Sanger sequencing. Studies of the triplicate region are ongoing to shed light on the relevance of this region for normal and altered gene function.
Missense changes and small in-frame deletions and insertions in NEB should be verified using functional analyses in order to determine the consequence of the change in the protein. Variants affecting highly conserved regions of nebulin, such as those altering actin-binding sites, may be regarded as pathogenic.
The clinical and histological spectrum of entities caused by variants in NEB is a continuum, ranging in severity from the severe form with perinatal onset to the mild forms. The distribution of weakness can vary from generalized muscle weakness, more pronounced in proximal limb muscles, to distal-only involvement, although neck flexor weakness appears to be rather consistent. Histological patterns range from a severe, but almost invariably nondystrophic disturbance of the myofibrillar pattern to an almost normal picture on hematoxylin-eosin staining, with or without nemaline bodies, sometimes combined with cores.
Genotype–phenotype correlations remain mostly vague. Although variants in NEB have been unambiguously shown to be a common cause of recessive NM, these variants may not be the only factors determining the severity of the disease. Modifying genetic factors are currently being revealed by the use of new techniques such as whole-exome and whole-genome sequencing. In addition, other determinants including good clinical care, nutrition and other factors influence the well-being of the patients.
Ethical Approval
The study has been approved by the Ethics Committee of the Children’s Hospital, University of Helsinki, Helsinki, Finland.
Supplementary Material
Acknowledgments
We thank the following clinicians/colleagues for providing data and/or DNA samples on one or two patients each: Drs. C. Angelini, N. Barisic, N. Belart, J. Bohm, L. Brueton, K. Bushby, C. Dias, A. Echaniz-Laguna, A. Forjaz de Sampaio, M. Garcia-Barcina, C. Gardiner, H. Gervais-Bernard, C. Graziano, J. Greenberg, S. Iannaccone, L. Jarre, P.-Y. Jeannet, P.S. Jouk, F. Juul-Hansen, T. Homfray, E. Honey, I. Hughes, R. Khajuria, C. Kubisch, S. Kapoor, T. Löppönen, L. Mehta, J. Monteiro, V. Lacroze, H. Lochmüller, S. Pajusalu, S. Penttilä, B. Porfirio, H. Roper, S. Rudnik-Schöneborn, N. Safina, U. Schaeffer, T. Stojkovic, K. Swoboda, H. Topaloglu, C. Vial, M. de Visser, D. Wenger, J. Wilmshurst, H. Yonath, K. Yau, and K. Õunap. Dr. Luisa Mota-Vieira excluded consanguinity in family F378, and coordinated the mutation detection efforts relating to this family.
We also thank Vandana Gupta, Emily Todd, Gina Ravenscroft, and Bjarne Udd for identifying mutations in one family each. We are grateful to Gunilla Donner and Lotta Eklundh for meticulous secretarial assistance in handling the clinical database.
Contract grant sponsors: Association Française contre les Myopathies; Sigrid Jusélius Foundation; Academy of Finland; Finska Läkaresällskapet; Medicinska understödsföreningen Liv och Hälsa r.f; National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIH grant R01 AR044345); Muscular Dystrophy Association (USA; MDA201302); NHMRC of Australia (APP1035828, APP1026933); Foundation for Building Strength for Nemaline Myopathy; Australian National Health and Medical Research Council (NH&MRC) Fellowship (APP1002147); NH&MRC EU Collaborative Grant APP1055295.
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- Agrawal PB, Greenleaf RS, Tomczak KK, Lehtokari VL, Wallgren-Pettersson C, Wallefeld W, Laing NG, Darras BT, Maciver SK, Dormitzer PR, Beggs AH. Nemaline myopathy with minicores caused by mutation of the CFL2 gene encoding the skeletal muscle actin-binding protein, cofilin-2. Am J Hum Genet. 2007;80:162–167. doi: 10.1086/510402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SL, Ekstein J, Donnelly MC, Keefe EM, Toto NR, LeVoci LA, Rubin BY. Nemaline myopathy in the Ashkenazi Jewish population is caused by a deletion in the nebulin gene. Hum Genet. 2004;115:185–190. doi: 10.1007/s00439-004-1140-8. [DOI] [PubMed] [Google Scholar]
- Bang ML, Gregorio C, Labeit S. Molecular dissection of the interaction of desmin with the C-terminal region of nebulin. J Struct Biol. 2002;137:119–127. doi: 10.1006/jsbi.2002.4457. [DOI] [PubMed] [Google Scholar]
- Bang ML, Li X, Littlefield R, Bremner S, Thor A, Knowlton KU, Lieber RL, Chen J. Nebulin-deficient mice exhibit shorter thin filament lengths and reduced contractile function in skeletal muscle. J Cell Biol. 2006;173:905–916. doi: 10.1083/jcb.200603119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohm J, Vasli N, Malfatti E, Le Gras S, Feger C, Jost B, Monnier N, Brocard J, Karasoy H, Gerard M, Walter MC, Reilich P, et al. An integrated diagnosis strategy for congenital myopathies. PLoS One. 2013;8:e67527. doi: 10.1371/journal.pone.0067527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra M, Mamidi R, Ford S, Hidalgo C, Witt C, Ottenheijm C, Labeit S, Granzier H. Nebulin alters cross-bridge cycling kinetics and increases thin filament activation: a novel mechanism for increasing tension and reducing tension cost. J Biol Chem. 2009;284:30889–30896. doi: 10.1074/jbc.M109.049718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conen PE, Murphy EG, Donohue WL. Light and electron microscopic studies of "myogranules" in a child with hypotonia and muscle weakness. Can Med Assoc J. 1963;89:983–986. [PMC free article] [PubMed] [Google Scholar]
- Cunliffe M, Burrows FA. Anaesthetic implications of nemaline rod myopathy. Can Anaesth Soc J. 1985;32:543–547. doi: 10.1007/BF03010806. [DOI] [PubMed] [Google Scholar]
- Donner K, Nowak KJ, Aro M, Pelin K, Wallgren-Pettersson C. Developmental and muscle-type-specific expression of mouse nebulin exons 127 and 128. Genomics. 2006;88:489–495. doi: 10.1016/j.ygeno.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Donner K, Ollikainen M, Ridanpaa M, Christen HJ, Goebel HH, de Visser M, Pelin K, Wallgren-Pettersson C. Mutations in the beta-tropomyosin (TPM2) gene—a rare cause of nemaline myopathy. Neuromuscul Disord. 2002;12:151–158. doi: 10.1016/s0960-8966(01)00252-8. [DOI] [PubMed] [Google Scholar]
- Donner K, Sandbacka M, Lehtokari VL, Wallgren-Pettersson C, Pelin K. Complete genomic structure of the human nebulin gene and identification of alternatively spliced transcripts. Eur J Hum Genet. 2004;12:744–751. doi: 10.1038/sj.ejhg.5201242. [DOI] [PubMed] [Google Scholar]
- Gajda A, Horvath E, Hortobagyi T, Gergev G, Szabo H, Farkas K, Nagy N, Szell M, Sztriha L. Nemaline myopathy type 2 (NEM2): two novel mutations in the nebulin (NEB) gene. J Child Neurol. 2013 doi: 10.1177/0883073813494476. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Gupta VA, Ravenscroft G, Shaheen R, Todd EJ, Swanson LC, Shiina M, Ogata K, Hsu C, Clarke NF, Darras BT, Farrar MA, Hashem A, et al. Identification of KLHL41 mutations implicates BTB-Kelch-mediated ubiquitination as an alternate pathway to myofibrillar disruption in nemaline myopathy. Am J Hum Genet. 2013;93:1108–1117. doi: 10.1016/j.ajhg.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurgel-Giannetti J, Reed U, Bang ML, Pelin K, Donner K, Marie SK, Carvalho M, Fireman MA, Zanoteli E, Oliveira AS, Zatz M, Wallgren-Pettersson C, et al. Nebulin expression in patients with nemaline myopathy. Neuromuscul Disord. 2001;11:154–162. doi: 10.1016/s0960-8966(00)00177-2. [DOI] [PubMed] [Google Scholar]
- Johnston JJ, Kelley RI, Crawford TO, Morton DH, Agarwala R, Koch T, Schaffer AA, Francomano CA, Biesecker LG. A novel nemaline myopathy in the Amish caused by a mutation in troponin T1. Am J Hum Genet. 2000;67:814–821. doi: 10.1086/303089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo YM, Lee MA, Lee YM, Kim MS, Kim SY, Jeon EH, Choi JK, Kim WH, Lee HC, Min BI, Kang HS, Kim CR. Identification of chicken nebulin isoforms of the 31-residue motifs and non-muscle nebulin. Biochem Biophys Res Commun. 2004;325:1286–1291. doi: 10.1016/j.bbrc.2004.10.153. [DOI] [PubMed] [Google Scholar]
- Kapoor S, Singh A, Lehtokari VL, Wallgren-Pettersson C, Batra VV. Mutations in the nebulin gene in a child with nemaline (rod) myopathy. Indian J Pediatr. 2013;80:691–693. doi: 10.1007/s12098-012-0867-9. [DOI] [PubMed] [Google Scholar]
- Kazmierski ST, Antin PB, Witt CC, Huebner N, McElhinny AS, Labeit S, Gregorio CC. The complete mouse nebulin gene sequence and the identification of cardiac nebulin. J Mol Biol. 2003;328:835–846. doi: 10.1016/s0022-2836(03)00348-6. [DOI] [PubMed] [Google Scholar]
- Kiiski K, Laari L, Lehtokari VL, Lunkka-Hytonen M, Angelini C, Petty R, Hackman P, Wallgren-Pettersson C, Pelin K. Targeted array comparative genomic hybridization—a new diagnostic tool for the detection of large copy number variations in nemaline myopathy-causing genes. Neuromuscul Disord. 2013;23:56–65. doi: 10.1016/j.nmd.2012.07.007. [DOI] [PubMed] [Google Scholar]
- Labeit S, Kolmerer B. The complete primary structure of human nebulin and its correlation to muscle structure. J Mol Biol. 1995;248:308–315. doi: 10.1016/s0022-2836(95)80052-2. [DOI] [PubMed] [Google Scholar]
- Labeit S, Ottenheijm CA, Granzier H. Nebulin, a major player in muscle health and disease. FASEB J. 2011;25:822–829. doi: 10.1096/fj.10-157412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing NG, Wilton SD, Akkari PA, Dorosz S, Boundy K, Kneebone C, Blumbergs P, White S, Watkins H, Love DR. A mutation in the alpha tropomyosin gene TPM3 associated with autosomal dominant nemaline myopathy NEM1. Nat Genet. 1995;10:249. doi: 10.1038/ng0695-249. [DOI] [PubMed] [Google Scholar]
- Laitila J, Hanif M, Paetau A, Hujanen S, Keto J, Somervuo P, Huovinen S, Udd B, Wallgren-Pettersson C, Auvinen P, Hackman P, Pelin K. Expression of multiple nebulin isoforms in human skeletal muscle and brain. Muscle Nerve. 2012;46:730–737. doi: 10.1002/mus.23380. [DOI] [PubMed] [Google Scholar]
- Lamont PJ, Wallefeld W, Hilton-Jones D, Udd B, Argov Z, Barboi AC, Bonneman C, Boycott KM, Bushby K, Connolly AM, Davies N, Beggs AH, et al. Novel mutations widen the phenotypic spectrum of slow skeletal/beta-cardiac myosin (MYH7) distal myopathy. Hum Mutat. 2014;37:868–879. doi: 10.1002/humu.22553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor MW, Ottenheijm CA, Lehtokari VL, Cho K, Pelin K, Wallgren-Pettersson C, Granzier H, Beggs AH. Novel mutations in NEB cause abnormal nebulin expression and markedly impaired muscle force generation in severe nemaline myopathy. Skelet Muscle. 2011;1:23. doi: 10.1186/2044-5040-1-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtokari VL, Greenleaf RS, DeChene ET, Kellinsalmi M, Pelin K, Laing NG, Beggs AH, Wallgren-Pettersson C. The exon 55 deletion in the nebulin gene—one single founder mutation with world-wide occurrence. Neuromuscul Disord. 2009;19:179–181. doi: 10.1016/j.nmd.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtokari VL, Pelin K, Herczegfalvi A, Karcagi V, Pouget J, Franques J, Pellissier JF, Figarella-Branger D, von der Hagen M, Huebner A, Schoser B, Lochmuller H, et al. Nemaline myopathy caused by mutations in the nebulin gene may present as a distal myopathy. Neuromuscul Disord. 2011;21:556–562. doi: 10.1016/j.nmd.2011.05.012. [DOI] [PubMed] [Google Scholar]
- Lehtokari VL, Pelin K, Sandbacka M, Ranta S, Donner K, Muntoni F, Sewry C, Angelini C, Bushby K, Van den Bergh P, Iannaccone S, Laing NG, et al. Identification of 45 novel mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Hum Mutat. 2006;27:946–956. doi: 10.1002/humu.20370. [DOI] [PubMed] [Google Scholar]
- Magee KR, Shy GM. A new congenital non-progressive myopathy. Brain. 1956;79:610–621. doi: 10.1093/brain/79.4.610. [DOI] [PubMed] [Google Scholar]
- Malfatti E, Lehtokari VL, Bohm J, De Winter JM, Schaffer U, Estournet B, Quijano-Roy S, Monges S, Lubieniecki F, Bellance R, Viou MT, Madelaine A, et al. Muscle histopathology in nebulin-related nemaline myopathy: ultrastrastructural findings correlated to disease severity and genotype. Acta Neuropathol Commun. 2014;2:44. doi: 10.1186/2051-5960-2-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marttila M, Hanif M, Lemola E, Nowak KJ, Laitila J, Grönholm M, Wallgren-Pettersson C, Pelin K. Nebulin interactions with actin and tropomyosin are altered by disease-causing mutations. Skelet Muscle. 2014;4:15. doi: 10.1186/2044-5040-4-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElhinny AS, Kazmierski ST, Labeit S, Gregorio CC. Nebulin: the nebulous, multifunctional giant of striated muscle. Trends Cardiovasc Med. 2003;13:195–201. doi: 10.1016/s1050-1738(03)00076-8. [DOI] [PubMed] [Google Scholar]
- Nowak KJ, Sewry CA, Navarro C, Squier W, Reina C, Ricoy JR, Jayawant SS, Childs AM, Dobbie JA, Appleton RE, Mountford RC, Walker KR, et al. Nemaline myopathy caused by absence of alpha-skeletal muscle actin. Ann Neurol. 2007;61:175–184. doi: 10.1002/ana.21035. [DOI] [PubMed] [Google Scholar]
- Nowak KJ, Wattanasirichaigoon D, Goebel HH, Wilce M, Pelin K, Donner K, Jacob RL, Hubner C, Oexle K, Anderson JR, Verity CM, North KN, et al. Mutations in the skeletal muscle alpha-actin gene in patients with actin myopathy and nemaline myopathy. Nat Genet. 1999;23:208–212. doi: 10.1038/13837. [DOI] [PubMed] [Google Scholar]
- Ochala J, Lehtokari VL, Iwamoto H, Li M, Feng HZ, Jin JP, Yagi N, Wallgren-Pettersson C, Penisson-Besnier I, Larsson L. Disrupted myosin cross-bridge cycling kinetics triggers muscle weakness in nebulin-related myopathy. FASEB J. 2011;25:1903–1913. doi: 10.1096/fj.10-176727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottenheijm CA, Buck D, de Winter JM, Ferrara C, Piroddi N, Tesi C, Jasper JR, Malik FI, Meng H, Stienen GJ, Beggs AH, Labeit S, et al. Deleting exon 55 from the nebulin gene induces severe muscle weakness in a mouse model for nemaline myopathy. Brain. 2013;136:1718–1731. doi: 10.1093/brain/awt113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelin K, Donner K, Holmberg M, Jungbluth H, Muntoni F, Wallgren-Pettersson C. Nebulin mutations in autosomal recessive nemaline myopathy: an update. Neuromuscul Disord. 2002;12:680–686. doi: 10.1016/s0960-8966(02)00066-4. [DOI] [PubMed] [Google Scholar]
- Pelin K, Hilpela P, Donner K, Sewry C, Akkari PA, Wilton SD, Wattanasirichaigoon D, Bang ML, Centner T, Hanefeld F, Odent S, Fardeau M, et al. Mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Proc Natl Acad Sci USA. 1999;96:2305–2310. doi: 10.1073/pnas.96.5.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelin K, Wallgren-Pettersson C. Nebulin—a giant chameleon. Adv Exp Med Biol. 2008;642:28–39. [PubMed] [Google Scholar]
- Ravenscroft G, Miyatake S, Lehtokari VL, Todd EJ, Vornanen P, Yau KS, Hayashi YK, Miyake N, Tsurusaki Y, Doi H, Saitsu H, Osaka H, et al. Mutations in KLHL40 are a frequent cause of severe autosomal-recessive nemaline myopathy. Am J Hum Genet. 2013;93:6–18. doi: 10.1016/j.ajhg.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero NB, Lehtokari VL, Quijano-Roy S, Monnier N, Claeys KG, Carlier RY, Pellegrini N, Orlikowski D, Barois A, Laing NG, Lunardi J, Fardeau M, et al. Core-rod myopathy caused by mutations in the nebulin gene. Neurology. 2009;73:1159–1161. doi: 10.1212/WNL.0b013e3181bacf45. [DOI] [PubMed] [Google Scholar]
- Sambuughin N, Yau KS, Olive M, Duff RM, Bayarsaikhan M, Lu S, Gonzalez-Mera L, Sivadorai P, Nowak KJ, Ravenscroft G, Mastaglia FL, North KN, et al. Dominant mutations in KBTBD13, a member of the BTB/Kelch family, cause nemaline myopathy with cores. Am J Hum Genet. 2010;87:842–847. doi: 10.1016/j.ajhg.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoto M, Cullup T, Cirak S, Yau S, Manzur AY, Feng L, Jacques TS, Anderson G, Abbs S, Sewry C, Jungbluth H, Muntoni F. Nebulin (NEB) mutations in a childhood onset distal myopathy with rods and cores uncovered by next generation sequencing. Eur J Hum Genet. 2013;21:1249–1252. doi: 10.1038/ejhg.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott SA, Edelmann L, Liu L, Luo M, Desnick RJ, Kornreich R. Experience with carrier screening and prenatal diagnosis for 16 Ashkenazi Jewish genetic diseases. Hum Mutat. 2010;31:1240–1250. doi: 10.1002/humu.21327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewry CA, Brown SC, Pelin K, Jungbluth H, Wallgren-Pettersson C, Labeit S, Manzur A, Muntoni F. Abnormalities in the expression of nebulin in chromosome-2 linked nemaline myopathy. Neuromuscul Disord. 2001;11:146–153. doi: 10.1016/s0960-8966(00)00172-3. [DOI] [PubMed] [Google Scholar]
- Shenkman Z, Sheffer O, Erez I, Litmanovitc I, Jedeikin R. Spinal anesthesia for gastrostomy in an infant with nemaline myopathy. Anesth Analg. 2000;91:858–859. doi: 10.1097/00000539-200010000-00016. [DOI] [PubMed] [Google Scholar]
- Shy GM, Engel WK, Somers JE, Wanko T. Nemaline myopathy. A new congenital myopathy. Brain. 1963;86:793–810. doi: 10.1093/brain/86.4.793. [DOI] [PubMed] [Google Scholar]
- Stackhouse R, Chelmow D, Dattel BJ. Anesthetic complications in a pregnant patient with nemaline myopathy. Anesth Analg. 1994;79:1195–1197. doi: 10.1213/00000539-199412000-00028. [DOI] [PubMed] [Google Scholar]
- Telfer WR, Nelson DD, Waugh T, Brooks SV, Dowling JJ. Neb: a zebrafish model of nemaline myopathy due to nebulin mutation. Dis Model Mech. 2012;5:389–396. doi: 10.1242/dmm.008631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonino P, Pappas CT, Hudson BD, Labeit S, Gregorio CC, Granzier H. Reduced myofibrillar connectivity and increased Z-disk width in nebulin-deficient skeletal muscle. J Cell Sci. 2010;123:384–391. doi: 10.1242/jcs.042234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallgren-Pettersson C. Congenital nemaline myopathy. A clinical follow-up of twelve patients. J Neurol Sci. 1989;89:1–14. doi: 10.1016/0022-510x(89)90002-6. [DOI] [PubMed] [Google Scholar]
- Wallgren-Pettersson C. Congenital nemaline myopathy: a longitudinal study. Societas scientiarum Fennica; Helsinki, Finland: 1990. [Google Scholar]
- Wallgren-Pettersson C, Jasani B, Newman GR, Morris GE, Jones S, Singhrao S, Clarke A, Virtanen I, Holmberg C, Rapola J. Alpha-actinin in nemaline bodies in congenital nemaline myopathy: immunological confirmation by light and electron microscopy. Neuromuscul Disord. 1995;5:93–104. doi: 10.1016/0960-8966(94)00035-8. [DOI] [PubMed] [Google Scholar]
- Wallgren-Pettersson C, Laing NG. Report of the 70th ENMC International Work-shop: nemaline myopathy, 11–13 June 1999, Naarden, The Netherlands. Neuromuscul Disord. 2000;10:299–306. doi: 10.1016/s0960-8966(99)00129-7. [DOI] [PubMed] [Google Scholar]
- Wallgren-Pettersson C, Lehtokari VL, Kalimo H, Paetau A, Nuutinen E, Hackman P, Sewry C, Pelin K, Udd B. Distal myopathy caused by homozygous missense mutations in the nebulin gene. Brain. 2007;130:1465–1476. doi: 10.1093/brain/awm094. [DOI] [PubMed] [Google Scholar]
- Witt CC, Burkart C, Labeit D, McNabb M, Wu Y, Granzier H, Labeit S. Nebulin regulates thin filament length, contractility, and Z-disk structure in vivo. EMBO J. 2006;25:3843–3855. doi: 10.1038/sj.emboj.7601242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto DL, Vitiello C, Zhang J, Gokhin DS, Castaldi A, Coulis G, Piaser F, Filomena MC, Eggenhuizen PJ, Kunderfranco P, Camerini S, Takano K, et al. The nebulin SH3 domain is dispensable for normal skeletal muscle structure but is required for effective active load bearing in mouse. J Cell Sci. 2013;126:5477–5489. doi: 10.1242/jcs.137026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonath H, Reznik-Wolf H, Berkenstadt M, Eisenberg-Barzilai S, Lehtokari VL, Wallgren-Pettersson C, Mehta L, Achiron R, Gilboa Y, Polak-Charcon S, Winder T, Frydman M, et al. Carrier state for the nebulin exon 55 deletion and abnormal prenatal ultrasound findings as potential signs of nemaline myopathy. Prenat Diagn. 2012;32:70–74. doi: 10.1002/pd.2905. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.