

Abstract
Glioma-initiating cells (GIC), which reside within the perivascular microenvironment to maintain self-renewal capacity, are responsible for glioblastoma initiation, progression, and recurrence. However, the molecular mechanisms controlling crosstalk between GICs and endothelial cells are poorly understood. Here, we report that, in both GICs and endothelial cells, platelet-derived growth factor (PDGF)–driven activation of nitric oxide (NO) synthase increases NO-dependent inhibitor of differentiation 4 (ID4) expression, which in turn promotes JAGGED1–NOTCH activity through suppression of miR129 that specifically represses JAGGED1 suppression. This signaling axis promotes tumor progression along with increased GIC self-renewal and growth of tumor vasculature in the xenograft tumors, which is dramatically suppressed by NOTCH inhibitor. ID4 levels correlate positively with NOS2 (NO synthase-2), HES1, and HEY1 and negatively with miR129 in primary GICs. Thus, targeting the PDGF–NOS–ID4–miR129 axis and NOTCH activity in the perivascular microenvironment might serve as an efficacious therapeutic modality for glioblastoma.
Introduction
Glioblastoma multiforme (GBM) is the most frequent and aggressive of brain malignancies, with a median survival of only 14.6 months after diagnosis, despite modern surgical and medical therapies (1). Recently, a subpopulation (glioma-initiating cells, GIC) with augmented tumor-initiating potential and stem cell behavior has been identified in glioblastomas (2, 3), suggesting that therapeutic approaches targeting GICs would have enhanced antitumor efficacy (4).
Adult neural stem cells (NSC) are located around capillaries in the subventricular zone (SVZ) and subgranular zone (SGZ). Interactions between NSCs and the vasculature in the SVZ and SGZ maintain NSC properties (5, 6). Similar to NSCs, GICs are enriched in the perivascular microenvironment and interact with the vasculature to maintain self-renewal and proliferative capacities (7). Recent studies have suggested that nitric oxide (NO) secreted from endothelial cells enhances the self-renewal capacity of GICs through the activation of JAGGED1–NOTCH signaling in the platelet-derived growth factor (PDGF)–induced murine glioma model (8). PDGF signaling is altered in various tumors, including glioblastoma, and promotes self-renewal and tumorigenesis in GICs (9–11). In addition, PDGF signaling is involved in endothelial cell functions, such as migration, proliferation, and tube formation (12). However, the molecular mechanisms controlling GICs and the vasculature remain poorly understood.
Interestingly, PDGF autocrine signaling promotes the proliferation of astrocytes and neural progenitors (13). Inhibitor of differentiation 4 (ID4) promotes tumorigenesis in PDGF-induced oligodendroglioma (14), induces dedifferentiation of Ink4a/Arf−/− mouse astrocytes, and generates GICs through the activation of cyclin E and NOTCH signaling (15). NOTCH signaling plays a crucial role in inducing angiogenesis in the tumor microenvironment (16, 17) and in maintaining stem cell traits in GICs (8, 18–20). However, the specific roles of ID4 in the perivascular microenvironments have not been fully established. In this study, we hypothesized that ID4 functions as a key regulator in connecting PDGF signaling and NO activity in GICs and tumor endothelium.
Materials and Methods
Cell culture and conditions
Human glioma cell lines (A172, A1207, and LN229) were purchased from the ATCC and maintained in high-glucose Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Hyclone), 1% penicillin and streptomycin (Life Technologies), and 2 mmol/L L-glutamine (Life Technologies). Human umbilical vein endothelial cells (HUVEC) and human retina endothelial cells (HREC) were grown in endothelial cell growth medium (Lonza). Several glioma-initiating cell lines [X01, X02, X03 (ref. 21), GSC1T, GSC2, Aju14 (ref. 22), AC17, AC20, 84NS, 528NS, 13NS, 30NS, 83NS, 1123NS (refs. 23, 24), 559, 559T, and 448 (ref. 25)] were established from human brain tumors. All GICs tested were cultured using neurobasal medium (Invitrogen) supplemented with modified N2, B27, EGF (20 ng/mL; R&D Systems), and bFGF (basic fibroblast growth factor; 20 ng/mL; R&D Systems) in suspension or in an adherent culture with laminin-coated flasks as described previously (26). For neurosphere formation assays, cells were seeded at a density of 2 cells per mm2 in 12-well plates and then grown in suspension with NBE supplemented with N2, B27, EGF (20 ng/mL), bFGF (20 ng/mL), PDGFB (50 ng/mL; R&D Systems), TGFβ (20 nmol/L; Sigma), leukemia inhibitory factor (LIF; 20 ng/mL; Chemicon), interleukin 6 (IL6 and 10 ng/mL; R&D Systems), TNFα (10 ng/mL; R&D Systems), and lipopolysaccharide (LPS; 100 ng/mL; Sigma) as described previously (22). Cytokines were replaced every 3 days, and the neurosphere numbers were determined after 14 days. Cells were treated with LNAME (L-NG-nitroarginine methyl ester; 200 μmol/L; Sigma; ref. 8), ODQ ([1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one; 2 μmol/L; Sigma; ref. 27), GSNO (100 and 200 μmol/L; Sigma; ref. 8), DAPT (100 and 1,000 nmol/L, Sigma; ref.18).
Immunofluorescence assay
Mice harboring tumors were perfused with PBS and 4% paraformaldehyde. Mouse tumor tissues embedded in paraffin were sliced into 5-μm-thick sections and stained with primary antibodies against NESTIN (MAB1259; 1:200; R&D Systems), CD15 (BD559045; 1:200; BD Biosciences), HES1 (AB5702; 1:200; Chemicon), cleaved caspase-3 (9664; 1:100; Cell Signaling Technology), Ki67 (NCL-Ki67p; 1:500; Novocastra), and vWF (M0616; 1:100; DaKo) for 12 hours at 4°C. Frozen brain tumor tissue slides (12–16 μm) were incubated with the following antibodies: ID4 (ab49261; 1:200; Abcam), JAGGED1 (2155; 1:200; Cell Signaling Technology), NOS2 (NO synthase-2; ABN26; 1:200; Millipore), PDGFB (SC-7878; 1:300; Santa Cruz Biotechnology), PDGFRα (SC-3381; 500; Santa Cruz Biotechnology), NICD (2421; 1:100; Cell Signaling Technology), CD31 (3528; 1:200; Cell Signaling Technology) for 12 hours at 4°C. Nuclei were then stained with 4′,6-diamidino-2-phenylindole (DAPI; 1 μg/mL) for 5 minutes. Fluorescence images were obtained using a confocal laser scanning microscope (LSM5 Pascal, Carl Zeiss).
FACS assays
Cells fixed with 4% paraformaldehyde were incubated with antibodies against NESTIN (MAB1259; 1:100; R&D Systems), SOX2 (AF2018; 1:200; R&D systems), CD133 (AC133; 1:10; MACS), TUJ1 (MMS-435p; 1:1,000; Covance), GFAP (691102; 1:100; MP Biomedicals Immuno), and OLIG2 (AB9610; 1:100; Millipore) for 12 hours at 4°C. Propidium iodide (2 μg/mL; Sigma-Aldrich) was added 5 minutes before FACS analysis (Beckman Coulter).
In vitro angiogenesis assay
In vitro tube formation of HUVECs and HRECs was evaluated using an in vitro angiogenesis assay kit (Chemicon). A172-CON and A172-ID4 cells were seeded at 5 × 105 cells per 10-cm plate. After 1 day, the conditioned media were harvested and filtered through a 0.2-μm filter (Sartorius Stedim Biotech). Endothelial cells (1 × 104 cells/well) were cultured on Matrigel in basal media with PDGF, EGM (Lonza), or conditioned media collected from A172-CON and A172-ID4 cells at 37°C for 12 hours. The cultures were then photographed (×40 magnification). Three random view fields per well were examined, and the tubes were counted.
In vivo tumorigenicity assay
To establish xenograft models, A1207-CON/ID4 (2 × 106) or GSC1T (5 × 104) cells were subcutaneously injected into nude mice (BALB/c nu/nu mice) along with HUVEC-CON/ID4 cells (2 × 106 or 2 × 105 with A1207 cells and 5 × 103 with GSC1T cells). Twenty-one days after tumor implantation, 40% ethanol or 5 mg/kg of MK0752 (in 40% ethanol/60% saline, 50 μL, 100 μg total) was injected directly into the tumor mass using a 1-mL disposable syringe once daily for 7 days, and tumor progression was monitored by measuring tumor size and weight. All mouse experiments were approved by the animal care committee at the College of Life Science and Biotechnology, Korea University (Seoul, Republic of Korea) and were performed in accordance with government and institutional guidelines and regulations.
Statistical analysis
Data were analyzed by the two-tailed Student t test; *, P < 0.05 and **, P < 0.01 were considered statistically significant. Data are presented as the mean ± SD. A Pearson product–moment correlation coefficient (r) was used to analyze the linear correlation between ID4 and NOS2, NOS1, NOS3, miR129, HES1, and HEY1 levels.
RNA and protein analyses, plasmids and gene transfection, luciferase reporter gene assay, and nitrite level determination by 4,5-diaminofluorescein
Detailed experimental procedures are provided in the Supplementary Information.
Results
PDGF regulates tumorsphere-forming ability of primary GICs
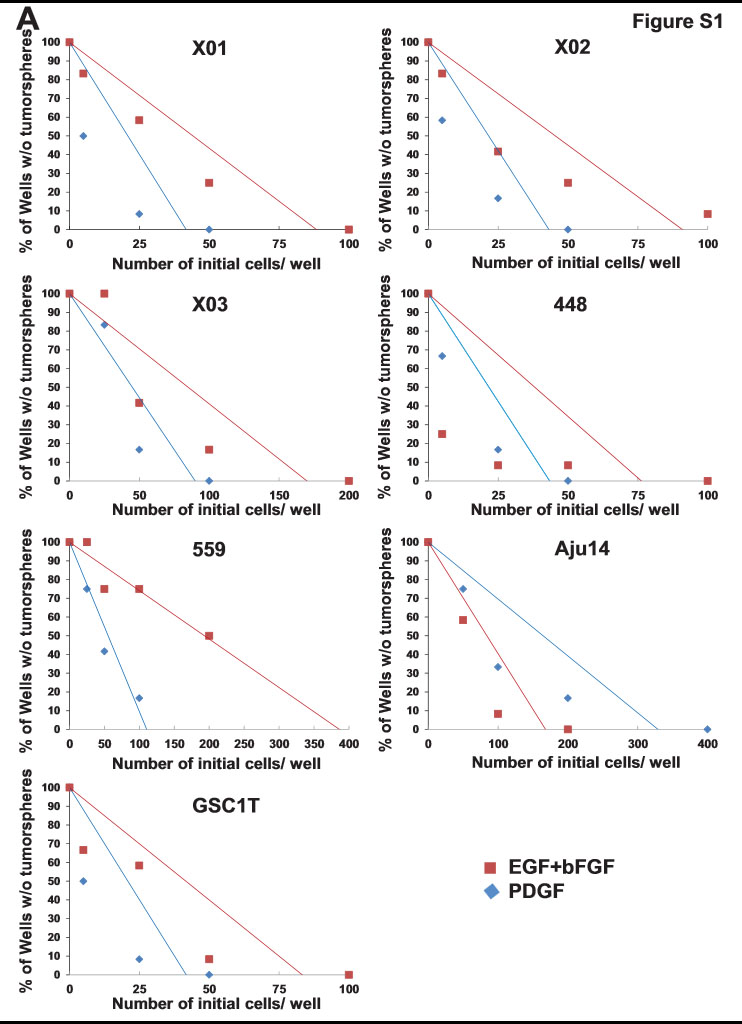
To explore which growth factors and cytokines enable patient-derived GICs to sustain stemness traits, we examined the tumorsphere-forming ability of GICs grown in serum-free medium supplemented with various growth factors and cytokines for 14 days. Notably, similar to EGF and bFGF, growth factors known to maintain stemness in GICs, PDGF promoted tumorsphere formation in all GICs tested (Fig. 1A and Supplementary Fig. S1A) with increased expression of stem cell markers (NESTIN, SOX2, CD133, and CD15; Supplementary Fig. S1B). Ectopic expression of PDGFB has been shown to induce ID4 in cultured astrocytes (13), and ID4 induces dedifferentiation of Ink4a/Arf−/− mouse astrocytes and gives rise to GICs (15). Thus, we investigated ID4 expression levels in GICs grown with PDGF by qRT-PCR. ID4 mRNA levels were induced in various GICs treated with PDGF (Supplementary Fig. S1C), suggesting that PDGF might regulate GIC stemness by inducing ID4.
Figure 1.
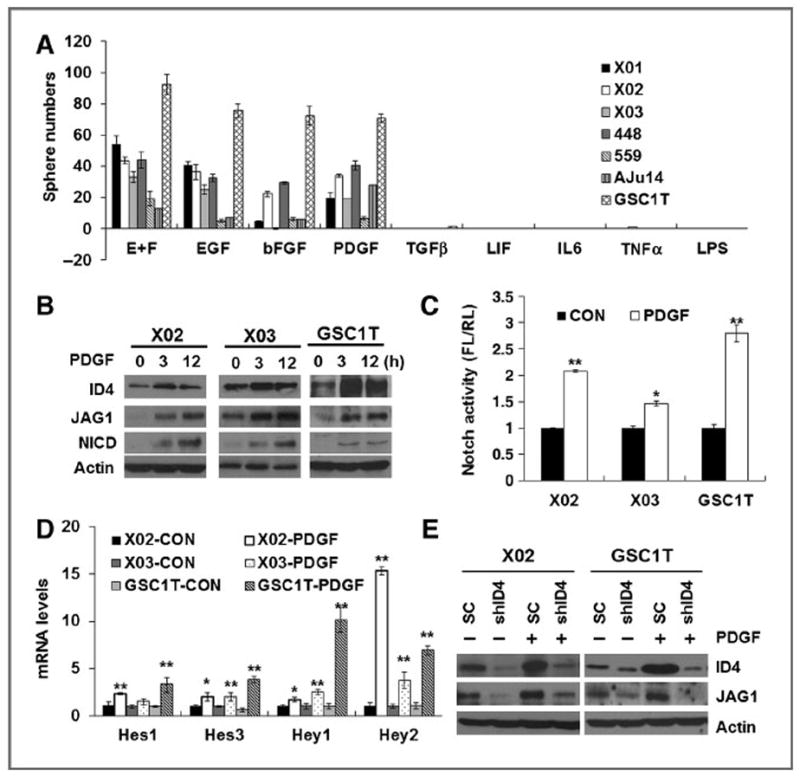
PDGF induces ID4 expression and JAGGED1–NOTCH1 signaling. A, tumorsphere numbers of primary GICs grown for 14 days upon treatment of EGF, bFGF, PDGF, TGFβ, LIF, IL6, TNFα, and LPS. B, Western blot analysis of ID4, JAGGED1, and NICD expression in GICs upon PDGF treatment. C, NOTCH luciferase activity increased in GICs (X02, X03, and GSC1T) 3 hours after PDGF treatment. D, qRT-PCR of genes downstream of NOTCH in GICs following PDGF treatment. E, Western blot analysis of ID4 and JAGGED1 expression in X02-shID4 and GSC1T-shID4 cells upon PDGF treatment (50 ng/mL); *, P < 0.05 (n = 3); **, P < 0.01 (n = 3); data, mean ± SD.
PDGF induces ID4 expression and JAGGED1–NOTCH signaling
Our previous studies have demonstrated that ID4 gives rise to GICs from Ink4a/Arf−/− mouse astrocytes by inducing cyclin E and JAGGED1–NOTCH signaling (15). Thus, we performed Western blot analysis to examine ID4 and JAGGED1 protein expression in GICs treated with PDGF. ID4 and JAGGED1 protein levels increased significantly at 3 and 12 hours after PDGF treatment in X02, X03, and GSC1T GICs (Fig. 1B). Along with inducing JAGGED1, NOTCH receptor activity was also markedly elevated by PDGF treatment, as determined by NOTCH-dependent luciferase activity and NOTCH downstream target gene (HES1, HES3, HEY1, and HEY2) expression (Fig. 1C and D). Furthermore, inhibition of NOTCH activity by DAPT (a γ-secretase inhibitor) sharply reduced the tumor-sphere-forming ability in X01, X02, X03, and GSC1T cells (Supplementary Fig. S1D). To test whether PDGF directly regulates GIC stemness through ID4, we established ID4-depleted GICs (X02-shID4 and GSC1T-shID4) using ID4-specific short hairpin RNA interference (shRNAi; Supplementary Fig. S1E). Tumorsphere numbers of X02-shID4 and GSC1T-shID4 cells were markedly decreased in the presence of PDGF compared with their control counterparts (Supplementary Fig. S1F). We also found that PDGF failed to induce JAGGED1 protein in ID4-depleted GICs (Fig. 1E). These results suggest that PDGF regulates GIC traits through ID4-mediated activation of JAGGED1–NOTCH signaling activity.
PDGF regulates ID4 by activating NOS–NO signaling
Recent studies revealed that NO secreted from endothelial cells promotes stemness in PDGF-induced gliomas, but not in EGFR-amplified gliomas (8), and that NOS2 plays a pivotal role in GIC proliferation and tumor growth (28). Thus, we examined whether PDGF upregulates ID4 and JAGGED1–NOTCH signaling by activating NOS–NO signaling in GICs. We first measured nitrite (NO2−) and NOS levels in X02, X03, and GSC1T GICs 3 hours after PDGF treatment, and found that PDGF induced NO synthesis in GICs (Fig. 2A). Upon PDGF treatment, NOS2 and NOS3 mRNA levels increased in all five GICs tested (Supplementary Fig. S2A), and NOS2 protein elevated in all three GICs tested (Supplementary Fig. S2A, inset). Depletion of NOS2 and NOS3 expression in GICs by siRNAs deceased ID4 expression levels (Supplementary Fig. S2B). Depletion of NOS2 and ID4 in GICs led to decrease in cells expressing stem cell markers (CD133, SOX2, and Nestin) in the stem cell culture conditions, whereas markedly increased in cells expressing differentiated astrocyte marker (GFAP), but not neuron and oligodendrocyte markers, in the differentiation culture conditions (DMEM supplemented with 5% FBS; Supplementary Fig. S2C). To further examine whether NO directly enhances ID4 expression, we treated S-nitrosoglutathione (GSNO; a NO donor) in all four GICs. GSNO induced ID4 expression in all X02, X03, GSC1T, and 448 GICs tested (Fig. 2B and Supplementary Fig. S2D). Next, we treated PDGF-pretreated GICs with LNAME (a pan-NOS inhibitor) and 1H-ODQ [a soluble guanylate cyclase (sGC) inhibitor]. NOS and sGC inhibitors significantly suppressed PDGF-induced ID4 mRNA and protein expression in X02, X03, and GSC1T GICs (Fig. 2C and D). These findings suggest that PDGF activates ID4 through the NOS–NO–sGC signaling pathway.
Figure 2.
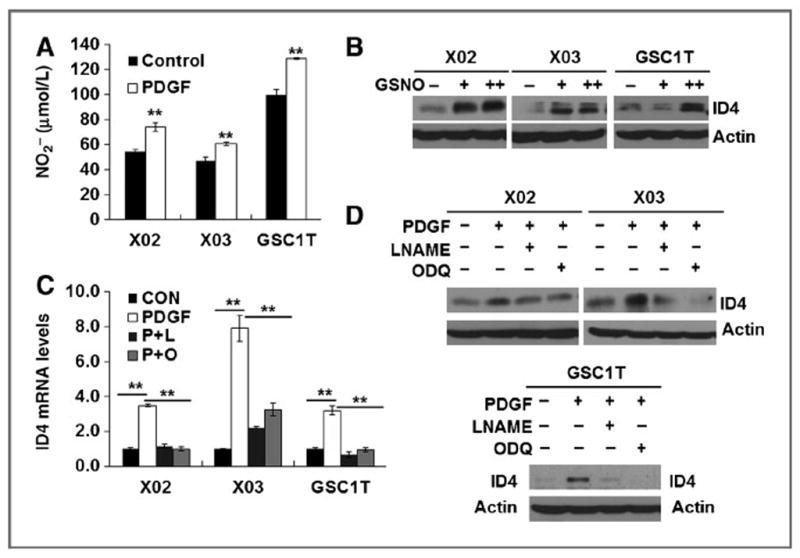
PDGF induces ID4 via NOS-NO signaling. A, nitrite (NO2−) levels in X02, X03, and GSC1T GICs 12 hours after PDGF (50 ng/mL) treatment. B, ID4 expression after treatment with GSNO (100 and 200 μmol/L) in GICs (X02, X03, and GSC1T). C and D, NOS inhibitor (LNAME; 200 μmol/L) and sGC inhibitor (ODQ; 1 μmol/L) markedly suppressed PDGF-mediated induction of ID4 mRNA (C) and protein (D) expression in GICs; **, P < 0.01 (n = 3); data, mean ± SD.
ID4 activates JAGGED1 by suppressing miR129
Previously, we found that ID4 regulates NOTCH transcription activity, cell proliferation, and tumorsphere formation in human glioma cells A172 and LN229, which express the lower and higher endogenous ID4 levels, respectively (15). In the present study, we overexpressed ID4 in the A172 glioma cell line for a gain of ID4 function studies, whereas we depleted ID4 expression in two GICs (X02 and GSC1T) and LN229 glioma cells by an shRNAi method for a loss of ID4 function studies. ID4 increased JAGGED1 protein levels without any remarkable change in the JAGGED1 mRNA level in ID4-overepxressing A172 cells (A172-ID4), indicating that ID4 modulates JAGGED1 at the posttranscriptional level (Supplementary Fig. S3SA). ID4 significantly activated JAGGED1 3′ untranslated region (UTR)–luciferase activity, but not JAGGED1 promoter–luciferase activity (Supplementary Fig. S3SB). Because microRNAs (miRNA) mediate translation repression by binding imperfectly matched sequences in the 3′UTR of target mRNA and ID4 enhanced SOX2 protein expression by suppressing miR9* (22); thus, we speculated that ID4-repressible miRs may regulate translational repression of JAGGED1. Using in silico prediction programs, such as Pictar (29), TargetScan (30), rnaHybrid (31), and miRanda (32), we selected several candidate JAGGED1-targeting miRs, including miR26, miR129, and miR449, that contain E-box sites on their promoter, which are well-known cis-acting elements repressed by the ID family. To determine which miRs are negatively regulated by ID4, we compared their expression in A172-ID4 and LN229-shID4 cells. Only miR129 was significantly suppressed by ID4 (Supplementary Fig. S3C). The miR129 promoter–luciferase activity analysis also showed that ID4 represses miR129 expression in a dose-dependent manner (Fig. 3A). PDGF treatment suppressed miR129 levels in X02 and GSC1T GICs as well as LN229 glioma cells (Fig. 3B). Expression levels of miR129 significantly increased in ID4-depleted cells and did not decrease in these cells upon PDGF treatment (Fig. 3B), indicating that PDGF decreases miR129 expression through ID4 induction. There are two putative miR129-binding sites in the JAGGED1 3′UTR, which are completely conserved across several species (Supplementary Fig. S3D). To determine whether JAGGED1 is specifically regulated by miR129, we generated three JAGGED1 3′UTR constructs with mutations in the putative miR129-binding sites (190 MUT, 1330 MUT, and 190+1330 double MUT; Supplementary Fig. S3E). As compared with JAGGED1 3′UTR–WT luciferase activity, only JAGGED1 3′UTR–190+1330 double MUT–luciferase activity was not regulated by miR129 (Fig. 3C), indicating that miR129 suppresses JAGGED1 by paring with two binding sites. In addition, transfection of XO2, GSC1T, and LN229 cells with mature miR129 decreased JAGGED1 protein levels (Fig. 3D). Transfection of 2′-O-methyl antisense RNA oligonucleotides (33, 34) against miR129 (2′-O-methyl-miR129) in A172 glioma cells increased JAGGED1 protein levels in a dose-dependent manner (Supplementary Fig. S3F). Reconstitution of miR129 in A172-ID4 cells decreased JAGGED1 protein levels, and PDGF failed to induce JAGGED1 expression in miR129-transfected A172 cells (Supplementary Fig. S3G). Thus, PDGF-induced ID4 increases JAGGED1 by repressing miR129, which would otherwise suppress JAGGED1 protein expression by targeting two binding sites in its 3′UTR.
Figure 3.
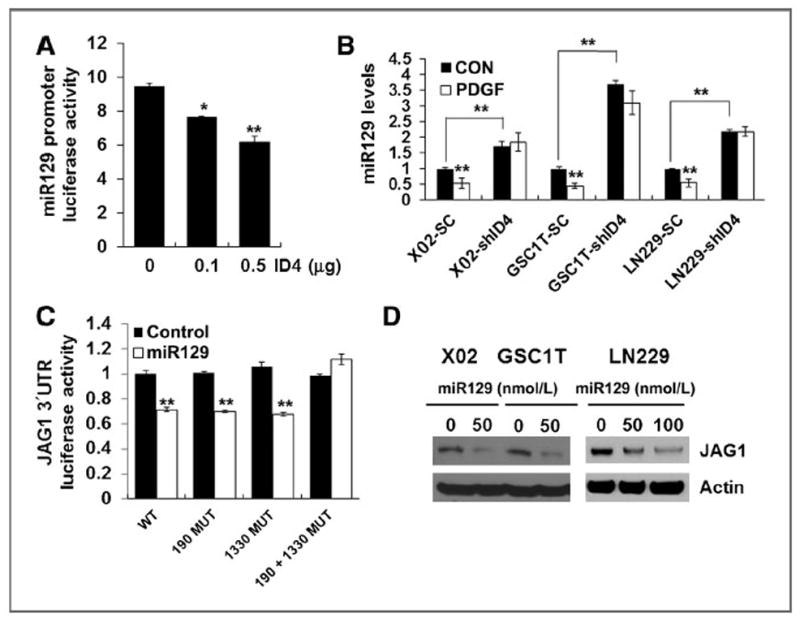
ID4 induces JAGGED1 protein by suppression of miR129. A, ID4 transfection led to reduced miR129 promoter–luciferase activity in a dose-dependent manner in 293T cells. B, PDGF suppressed miR129 expression. PDGF failed to reduce miR129 expression increased by depletion of ID4 in GICs and glioma cells. C, luciferase activity of wild-type or mutant (190 MUT and 1330 MUT) JAGGED1 3′UTR reporter genes in 293T cells transfected with miR129 or empty vector. D, Western blot analysis of JAGGED1 expression upon mature miR129 transfection into GICs and glioma cells; *, P < 0.05 (n = 3); **, P < 0.01 (n = 3); data, mean ± SD.
The PDGF–NOS–ID4 signaling axis also regulates JAGGED1–NOTCH signaling by suppressing miR129 in endothelial cells
Because perivasculature is known as a niche for GICs (7), we examined whether ID4 is expressed in cells located around the perivascular microenvironment of human GBM specimens. As expected, most of ID4+ cells localized around vWF+ endothelial cells (Fig. 4A). Of interest, ID4 was colocalized with vWF (Fig. 4A), indicating that vWF+ endothelial cells also express ID4. These results raise a question whether the PDGF–NOS–ID4–miR129 axis also occurs in endothelial cells. Previous studies have demonstrated that JAGGED1–NOTCH (35, 36) and NO signaling (37, 38) play essential roles in angiogenesis of endothelial cells. Upon PDGF treatment, ID4 mRNA, not JAGGED1 mRNA, was increased, whereas both ID4 and JAG-GED1 proteins were increased in HRECs and HUVECs, indicating that similar to GICs, PDGF also regulates JAGGED1 expression at the posttranscriptional level in endothelial cells (Fig. 4B and Supplementary Fig. S4A). PDGF suppressed miR129 expression (Fig. 4C and Supplementary Fig. S4B) and increased NOTCH transcriptional activity in HUVECs (Fig. 4D and Supplementary Fig. S4C). Overexpression of ID4 and JAGGED1 in HUVECs also increased NOTCH transcriptional activity and NOTCH downstream target genes (HES1, HES3, HEY1, and HEY2; Supplementary Fig. S4C–S4E). ID4 overexpression in HUVECs or PDGF treatment in HUVECs and HRECs increased nitrite production (Fig. 4E and Supplementary Fig. S4F). We further investigated whether NOS–NO signaling is also involved in PDGF-driven induction of ID4 expression and NOTCH signaling in the endothelial cells. NOS2 levels were upregulated in PDGF-treated endothelial cells (Fig. 4F and Supplementary Fig. S4H). In the endothelial cells, GSNO induced ID4 expression; NOS and sGC inhibitors suppressed PDGF-induced ID4 protein expression (Fig. 4F and Supplementary Fig. S4H). Finally, in vitro the tube-forming ability of endothelial cells was significantly increased by PDGF treatment, ID4 or JAGGED1 overexpression, or growing in the conditioned medium derived from ID4-overexpressing glioma cells (Fig. 4G and Supplementary Fig. S4I). Taken together, our results indicate that the PDGF–NOS–ID4–miR129 axis and JAGGED1–NOTCH activity play crucial roles in not only GICs but also endothelial cells.
Figure 4.

ID4-driven suppression of miR129 regulates the PDGF–NOS–ID4–miR129 axis and regulates JAGGED1–NOTCH signaling in endothelial cells. A, immunofluorescence analysis revealed that distribution of ID4 (green) and vWF (red) is closely associated in the perivasculature of human GBM specimens. DAPI was used for nuclear staining. B, PDGF induced ID4 (mRNA and protein) and JAGGED1 (protein) in HRECs. C, PDGF repressed miR129 levels in HRECs. D, NOTCH luciferase activity was significantly elevated in HRECs treated with PDGF. E, PDGF increased NO levels in HRECs. F, in HRECs, PDGF increased NOS2 protein; GSNO induced ID4 expression; LNAME and ODQ suppressed PDGF-mediated induction of ID4. G, PDGF alone increased the in vitro tube-forming ability of HRECs at higher levels than shown by EGM culture conditions (supplemented with cocktail of angiogenic factors). The conditioned medium prepared from A172-ID4 cells increased the in vitro tube-forming ability of HRECs; *, P < 0.05 (n =3); **, P < 0.01 (n =3); data, mean ± SD.
ID4-driven activation of JAGGED1–NOTCH signaling is a key between glioma and endothelial cells during tumorigenesis
To understand the significance of ID4-driven activation of JAGGED1–NOTCH signaling in both GICs and endothelial cells of the perivascular microenvironment in tumorigenesis, we first examined in vitro NOTCH activity in A1207–NOTCH–CSL luciferase reporter cells that are cocultured with HUVEC-CON and HUVEC-ID4 cells or HUVEC–NOTCH–CSL luciferase reporter cells that are cocultured with A1207–CON and A1207–ID4 cells. NOTCH activity in glioma cells and HUVECs was significantly elevated as cocultured with HUVEC-ID4 cells and A1207–ID4 cells, respectively (Supplementary Fig. S5A). Next, we assessed the effect of ID4 activation in glioma and endothelial cells on in vivo tumorigenesis by coinjecting A1207–CON or A1207–ID4 cells with HUVEC-CON or HUVEC-ID4 cells, as performed previously (20). HUVEC-ID4 cells accelerated A1207–CON and A1207–ID4 tumor growth compared with HUVEC-CON. The most significant increase in tumor growth was observed in the coinjection of A1207–ID4 and HUVEC-ID4 cells (Supplementary Fig. S5B), indicating that ID4 activation in glioma and endothelial cells promotes tumor growth in vivo. Next, we examined the in vitro tube-forming ability of HUVEC-CON and HUVEC-ID4 cells by treatment of NOTCH (DAPT) and VEGF (bevasizumab) inhibitor, and found that NOTCH inhibition suppressed in vitro tube formation of HUVEC-ID4 cells more dramatically than with VEGF inhibition (Supplementary Fig. S5C). These results suggest that NOTCH inhibition is more effective way to suppress the ID4-driven angiogenesis.
To directly address the effect of NOTCH inhibition during ID4-driven tumor progression and angiogenesis, we subcutaneously coinjected GSC1T and HUVEC-ID4 cells, intratumorally treated with MK0752 (a NOTCH inhibitor; ref. 39) for 7 days, and then examined tumor growth. MK0752 significantly reduced tumor growth at levels similar to those observed from coinjection of GSC1T and HUVEC-CON cells (Fig. 5A). In addition, MK0752 also dramatically suppressed tumor growth from the coinjection of A1207–ID4 and HUVEC-ID4 cells (Supplementary Fig. S5D). Immunofluorescence analysis revealed that glioma stem-like (NESTIN+; Fig. 5B), proliferating (Ki67+; Fig. 5C), and NOTCH-active (HES1+; Fig. 5D) cells were increased in tumors derived from the coinjection of GSC1T and HUVEC-ID4 cells. However, MK0752 treatment caused marked increase of apoptotic cells (cleaved caspase-3+; Fig. 5B) with decreases of NESTIN+, Ki67+, and HES1+ cells in tumors derived from the coinjection of GSC1T and HUVEC-ID4 cells (Fig. 5B–D). The similar results were also observed in tumors derived from the coinjection of A1207–ID4 and HUVEC-ID4 cells by MK0752 treatment (Supplementary Fig. S5E for merged images). Taken together, these results show that the ID4-driven activation of JAGGED1–NOTCH signaling in GICs and endothelial cells plays a crucial role in tumor progression.
Figure 5.
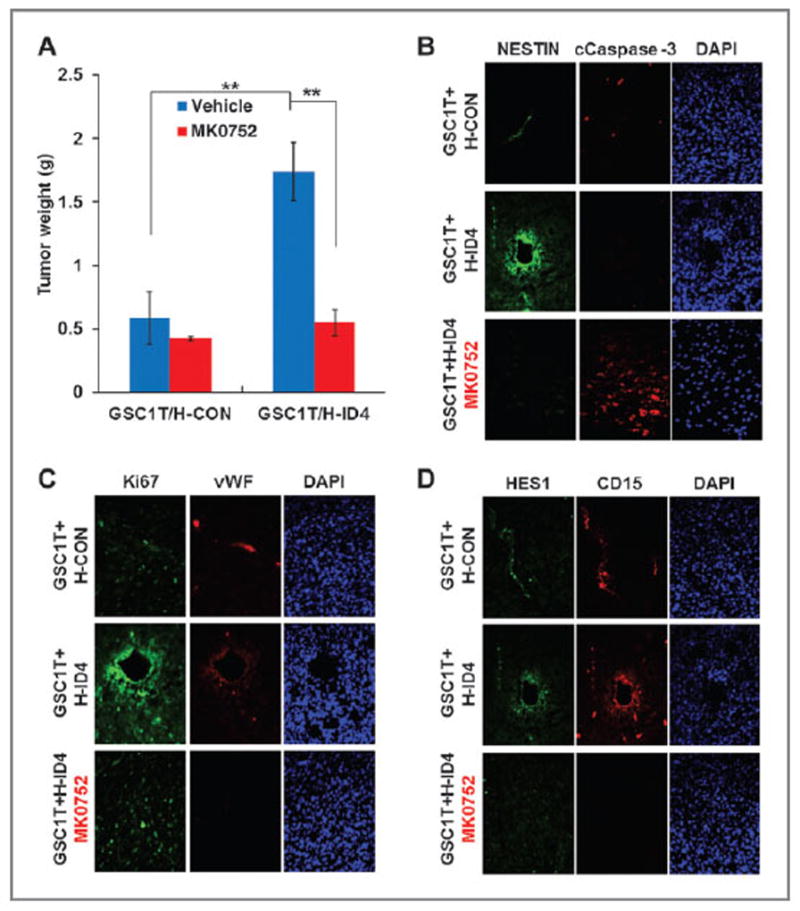
Activation of NOTCH signaling by ID4 in GICs and endothelial cells plays a crucial role in tumor progression. A, NOTCH inhibitor (MK0752, 5mg/kg) significantly suppressed tumorigenesis of GSC1T GICs coinjected with HUVEC-ID4 cells. B–D, immunofluorescence was used to observe cells positive for NESTIN and cleaved caspase-3 (B); vWF and Ki67 (C); and HES1 and CD15 (D) in tumors derived from GSC1T coinjected with either HUVEC-CON or HUVEC-ID4, followed by MK0752 treatment or not. DAPI was used for nuclear staining; **, P < 0.01 (n = 6); data, mean ± SD.
PDGF–NOS–ID4–miR129 axis-relevant gene signatures in primary GICs
To evaluate whether expression of genes involved in the PDGF–NOS–ID4–miR129 axis and NOTCH signaling is specific for GICs, Western blot and qRT-PCR analyses were conducted with 17 GICs cultured under stem cell culture conditions or differentiation culture conditions (23, 40, 41). All ID4, NOS2, PDGFB, PDGFRα, JAGGED1, and NICD protein levels decreased in seven of the 17 GICs grown in differentiation culture conditions (Fig. 6A). ID4 expression showed a significant positive correlation with NOS2, but not NOS1 and NOS3, in GICs (Fig. 6B and Supplementary Figs. S6A and S4B). An inverse correlation was observed between ID4 and miR129 expression (Fig. 6C), whereas positive correlations were observed between ID4 and NOTCH downstream target genes (HES1 and HEY1; Fig. 6D and E).
Figure 6.

Expression of genes involved in the PDGF–NOS–ID4–miR129 axis and JAGGED1–NOTCH signaling in human patient–derived GICs. A, comparison of ID4, JAGGED1, NOS2, PDGFB, PDGFRα, and NICD protein levels between 17 primary GICs grown under stem cell (N; NBE with EGF and bFGF) and differentiation (F; 5% FBS) culture conditions. The closed circle indicates GICs displaying decrease in protein expression tested by GIC differentiation. B–E, ID4 mRNA levels in 34 primary GICs grown in NBE (open, n = 17) and 5% FBS cultures (closed, n = 17) correlated with NOS2 (B), HES1 (D), and HEY1 (E) mRNA levels. C, ID4 inversely correlated with miR129 levels. A Pearson product–moment correlation coefficient (r) was used to analyze the linear correlation between two variables.
PDGF–NOS–ID4–miR129 axis-relevant gene expression in human GBM tumor tissues and in PDGFB-driven glioma mice
Next, we determined whether cells located around the perivasculatures of human GBM express ID4, PDGFB, PDGFRα, NOS2, JAGGED1, and NICD. Human GBM cells expressing ID4, PDGFB, PDGFRα, NOS2, JAGGED1, and NICD were abundant around CD31+ endothelial cells. Some of CD31+ endothelial cells also expressed ID4, PDGFB, PDGFRα, NOS2, JAGGED1, and NICD (Supplementary Fig. S7). Oncogenic role of PDGF in gliomagenesis has been well demonstrated in RCAS–hPDGFB/Nestin-tva:Cdkn2a−/− mice, in which PDGF is induced in Nestin+ neural progenitor cells and gives rise to mouse GBM (42). Thus, we examined whether our PDGF–NOS–ID4–miR129 axis-relevant genes are upregulated in PDGFB-driven mouse GBM. Immunofluorescence analysis showed ID4, PDGFB, PDGFRα, NOS2, JAGGED1, and NICD were highly expressed in this mouse GBM (Supplementary Fig. S8).
Taken together, our results indicate that the PDGF–NOS–ID4–miR129 axis and JAGGED1–NOTCH signaling might specifically occur in GICs and endothelial cells, suggesting that this signaling axis serves as a promising therapeutic target to suppress both GICs and tumor endothelial cells in the peri-vascular microenvironment.
Discussion
In the present study, we demonstrate that the PDGF–NOS–ID4–miR129 regulatory axis activates JAGGED1–NOTCH signaling in GICs and endothelial cells. Furthermore, activation of JAGGED1–NOTCH signal in GICs and endothelial cells promotes tumor progression along with increased GIC proliferation and angiogenesis. Our findings also provide an anticancer therapeutic rationale targeting NOTCH signaling that is required for the maintenance of the tumor perivascular micro-environments composed of GICs and endothelial cells.
Similar to EGF and bFGF (40), PDGF alone is sufficient to maintain the undifferentiated status of GICs by promoting nonadherent neurosphere growth and inducing stem cell markers, such as NESTIN, SOX2, CD133, and CD15, in several GICs derived from patients with GBM. In agreement with our findings, PDGF signaling has been shown to stimulate neuro-sphere formation and proliferation and suppress differentiation of PDGFRA-expressing B cells (also known as NSCs) in the adult murine SVZ (11, 43). Furthermore, mouse models with PDGF overexpression give rise to brain tumors, and its overexpression occurs with equal frequency in both low- and high-grade gliomas, indicating that PDGF signaling may be implicated in tumor initiation (44). However, precise mechanisms underlying PDGF-driven tumorigenesis are not well understood. In the present study, we demonstrate that PDGF signaling plays a crucial role in ID4-mediated regulation of GICs and endothelial cells by promoting the PDGF–NOS–ID4 signaling axis. This effect might lead to signal transduction in GICs and endothelial cells by maintaining cancer stemness and angiogenesis, respectively. Two previous studies have shown that GIC-specific endogenous NOS2 modulates GIC proliferation and tumor growth in perivascular microenvironment (28) and that transient activation of the NO/cGMP pathway is sufficient to impart a more stem cell–like phenotype to glioma cells (8). However, our findings provide mechanistic insight for understanding how the perivascular microenvironment is maintained during tumorigenesis.
Although a few studies have revealed that suppression of miR129 promotes tumor progression in gastric, endometrial, and liver cancer (45–47), there are no mechanistic explanations demonstrating its oncogenic function. We found that ID4-mediated suppression of miR129, a JAGGED1-targeting miR, induces constitutive activation of JAGGED1–NOTCH signaling in GICs and endothelial cells. In silico miRNA prediction software programs show that miR129 could also regulate other stemness regulatory genes, such as SOX2 and TCF4. Thus, miR129 plays more diverse roles in controlling normal stem cells and cancer stem cells.
Similar to previous studies that the PDGF and NOS signaling pathways are potential therapeutic targets in GICs (8, 29, 48), targeting NOTCH signaling dramatically reduced tumor growth by suppressing GICs and angiogenesis. We used a subcutaneous xenograft system to examine the effects of Notch signaling inhibitor in both GICs and endothelial cells. However, this system could not completely represent the brain tumor microenvironment. Giannini and colleagues have compared the histopathologic and genetic features of patient GBMs, subcutaneous, and intracranial xenograft tumors and demonstrated how subcutaneous and intracranial xenograft models display several similarities and differences in reflecting human brain tumors (49). In their study, two types of xenograft tumors displayed similar mitotic activity and gene alterations, and these features could reflect their patient tumor characteristics. However, there were several discrepancies observed between subcutaneous and intracranial xenograft tumors: (i) a relative scarcity of histopathologic features of GBM patient tumors, such as presence of mild microvascular proliferation and necrosis, in intracranial xenograft tumors, and (ii) the limited ability of subcutaneous xenograft tumors in evaluating mouse survival and infiltrating properties. Having the strengths and weaknesses of each model in mind, an orthotopic xenograft model will also be applied in the future studies to validate the effect of ID4–Notch–signaling inhibition on tumor progression.
Our results showed that genes relevant to the PDGF–NOS–ID4 signaling axis and JAGGED1–NOTCH pathway are specifically expressed in primary GICs and that ID4 expression levels correlate positively with NOS2, HES1, and HEY1 and negatively with miR129 in GICs. Thus, these results suggest that combinatorial therapy using a NOTCH inhibitor for targeting GICs and endothelial cells of the perivascular microenvironment and conventional chemo- or radiotherapy for targeting non-GIC glioma cells may provide a novel therapeutic approach to more effectively eliminate tumors like GBM.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors thank all members of the Cell Growth Regulation Laboratory for helpful discussion and technical assistance, and Dr. Eric Holland for providing PDGFB-driven glioma in the RCAS-hPDGFB/Nestin-tva:Cdkn2a−/− mice.
Grant Support
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MEST; 2011-0017544), the American Cancer Society (MRSG-08-108-01), and NIH/NCI (1R21CA135013-01A1).
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authors’ Contributions
Conception and design: H.-M. Jeon, H. Kim
Development of methodology: H.-M. Jeon, S.-H. Kim
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): H.-M. Jeon, S.-H. Kim, J.B. Park, S.H. Kim, K. Joshi, I. Nakano
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): H.-M. Jeon, S.-H. Kim, X. Jin
Writing, review, and/or revision of the manuscript: H.-M. Jeon, S.-H. Kim, H. Kim
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): H.-M. Jeon, S.-H. Kim, S.H. Kim, K. Joshi, I. Nakano, H. Kim
Study supervision: H. Kim
References
- 1.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 2.Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A. 2003;100:15178–83. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–8. [PubMed] [Google Scholar]
- 4.Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nat Rev Cancer. 2006;6:425–36. doi: 10.1038/nrc1889. [DOI] [PubMed] [Google Scholar]
- 5.Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramova N, et al. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science. 2004;304:1338–40. doi: 10.1126/science.1095505. [DOI] [PubMed] [Google Scholar]
- 6.Tavazoie M, Van der Veken L, Silva-Vargas V, Louissaint M, Colonna L, Zaidi B, et al. A specialized vascular niche for adult neural stem cells. Cell Stem Cell. 2008;3:279–88. doi: 10.1016/j.stem.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 8.Charles N, Ozawa T, Squatrito M, Bleau AM, Brennan CW, Hambardzumyan D, et al. Perivascular nitric oxide activates NOTCH signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell. 2010;6:141–52. doi: 10.1016/j.stem.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calzolari F, Malatesta P. Recent insights into PDGF-induced glioma-genesis. Brain Pathol. 2010;20:527–38. doi: 10.1111/j.1750-3639.2009.00335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang Y, Boije M, Westermark B, Uhrbom L. PDGF-B can sustain self-renewal and tumorigenicity of experimental glioma-derived cancer-initiating cells by preventing oligodendrocyte differentiation. Neoplasia. 2011;13:492–503. doi: 10.1593/neo.11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim Y, Kim E, Wu Q, Guryanova O, Hitomi M, Lathia JD, et al. Platelet-derived growth factor receptors differentially inform intertumoral and intratumoral heterogeneity. Genes Dev. 2012;26:1247–62. doi: 10.1101/gad.193565.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hellström M, Kalén M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047–55. doi: 10.1242/dev.126.14.3047. [DOI] [PubMed] [Google Scholar]
- 13.Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 2001;15:1913–25. doi: 10.1101/gad.903001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Appolloni I, Calzolari F, Barilari M, Terrile M, Daga A, Malatesta P. Antagonistic modulation of gliomagenesis by Pax6 and Olig2 in PDGF-induced oligodendroglioma. Int J Cancer. 2012;131:E1078–87. doi: 10.1002/ijc.27606. [DOI] [PubMed] [Google Scholar]
- 15.Jeon HM, Jin X, Lee JS, Oh SY, Sohn YW, Park HJ, et al. Inhibitor of differentiation 4 drives brain tumor-initiating cell genesis through cyclin E and NOTCH signaling. Genes Dev. 2008;22:2028–33. doi: 10.1101/gad.1668708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeng Q, Li S, Chepeha DB, Giordano TJ, Li J, Zhang H, et al. Crosstalk between tumor and endothelial cells promotes tumor angiogenesis by MAPK activation of NOTCH signaling. Cancer Cell. 2005;8:13–23. doi: 10.1016/j.ccr.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 17.Gridley T. NOTCH signaling in vascular development and physiology. Development. 2007;134:2709–18. doi: 10.1242/dev.004184. [DOI] [PubMed] [Google Scholar]
- 18.Jin X, Kim SH, Jeon HM, Beck S, Sohn YW, Yin J, et al. Interferon regulatory factor 7 regulates glioma stem cells via interleukin-6 and NOTCH signalling. Brain. 2012;135:1055–69. doi: 10.1093/brain/aws028. [DOI] [PubMed] [Google Scholar]
- 19.Androutsellis-Theotokis A, Leker RR, Soldner F, Hoeppner DJ, Ravin R, Poser SW, et al. NOTCH signalling regulates stem cell numbers in vitro and in vivo. Nature. 2006;442:823–6. doi: 10.1038/nature04940. [DOI] [PubMed] [Google Scholar]
- 20.Zhu TS, Costello MA, Talsma CE, Flack CG, Crowley JG, Hamm LL, et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011;71:6061–72. doi: 10.1158/0008-5472.CAN-10-4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD, et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene. 2009;28:3949–59. doi: 10.1038/onc.2009.252. [DOI] [PubMed] [Google Scholar]
- 22.Jeon HM, Sohn YW, Oh SY, Kim SH, Beck S, Kim S, et al. ID4 imparts chemoresistance and cancer stemness to glioma cells by derepressing miR9*-mediated suppression of SOX2. Cancer Res. 2011;71:3410–21. doi: 10.1158/0008-5472.CAN-10-3340. [DOI] [PubMed] [Google Scholar]
- 23.Miyazaki T, Pan Y, Joshi K, Purohit D, Hu B, Demir H, et al. Telomestatin impairs glioma stem cell survival and growth through the disruption of telomeric G-quadruplex and inhibition of the protooncogene, c-Myb. Clin Cancer Res. 2012;18:1268–80. doi: 10.1158/1078-0432.CCR-11-1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jijiwa M, Demir H, Gupta S, Leung C, Joshi K, Orozco N, et al. CD44v6 regulates growth of brain tumor stem cells partially through the AKT-mediated pathway. PLoS ONE. 2011;6:e24217. doi: 10.1371/journal.pone.0024217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joo KM, Kim SY, Jin X, Song SY, Kong DS, Lee JI, et al. Clinical and biological implications of CD133-positive and CD133-negative cells in glioblastomas. Lab Invest. 2008;88:808–15. doi: 10.1038/labinvest.2008.57. [DOI] [PubMed] [Google Scholar]
- 26.Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R, et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell. 2009;4:568–80. doi: 10.1016/j.stem.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 27.Moro MA, Russel RJ, Cellek S, Lizasoain I, Su Y, Darley-Usmar VM, et al. cGMP mediates the vascular and platelet actions of nitric oxide: confirmation using an inhibitor of the soluble guanylyl cyclase. Proc Natl Acad Sci U S A. 1996;93:1480–5. doi: 10.1073/pnas.93.4.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eyler CE, Wu Q, Yan K, MacSwords JM, Chandler-Militello D, Misuraca KL, et al. Glioma stem cell proliferation and tumor growth are promoted by nitric oxide synthase-2. Cell. 2011;146:53–66. doi: 10.1016/j.cell.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 30.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–98. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 31.Krüger J, Rehmsmeier M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006;34:W451–4. doi: 10.1093/nar/gkl243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Betel D, Wilson M, Gabow A, Marks DS, Sander C. The microRNA org resource: targets and expression. Nucleic Acids Res. 2008;36:D149–53. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meister G, Landthaler M, Dorsett Y, Tuschl T. Sequence-specific inhibition of microRNAs and siRNA-induced RNA silencing. RNA. 2004;10:544–50. doi: 10.1261/rna.5235104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.György H, Simard MJ, Mello CC, Zamore PD. Sequence-specific inhibition of small RNA function. PLoS Biol. 2004;2:e98. doi: 10.1371/journal.pbio.0020098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krebs LT, Xue Y, Norton CR, Shutter JR, Maguire M, Sundberg JP, et al. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000;14:1343–52. [PMC free article] [PubMed] [Google Scholar]
- 36.Benedito R, Roca C, Sorensen I, Adams S, Gossler A, Fruttiger M, et al. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell. 2009;137:1124–35. doi: 10.1016/j.cell.2009.03.025. [DOI] [PubMed] [Google Scholar]
- 37.Kawasaki K, Smith RS, Jr, Hsieh CM, Sun J, Chao J, Liao JK. Activation of the phosphatidylinositol 3-kinase/protein kinase Akt pathway mediates nitric oxide-induced endothelial cell migration and angiogenesis. Mol Cell Biol. 2003;23:5726–37. doi: 10.1128/MCB.23.16.5726-5737.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun CO, et al. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc Natl Acad Sci U S A. 2001;98:2604–9. doi: 10.1073/pnas.041359198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fouladi M, Stewart CF, Olson J, Wagner LM, Onar-Thomas A, Kocak M, et al. Phase I trial of MK-0752 in children with refractory CNS malignancies: a pediatric brain tumor consortium study. J Clin Oncol. 2011;29:3529–34. doi: 10.1200/JCO.2011.35.7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee J, Kotliarovam S, Kotliarov Y, Li A, Su Q, Donin NM, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 41.Nakano I, Joshi K, Visnyei K, Hu B, Watanabe M, Lam D, et al. Siomycin A targets brain tumor stem cells partially through a MELK-mediated pathway. Neuro Oncol. 2011;13:622–34. doi: 10.1093/neuonc/nor023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hambardzumyan D, Amankulor NM, Helmy KY, Becher OJ, Holland EC. Modeling adult gliomas using RCAS/t-va technology. Transl Oncol. 2009;2:89–95. doi: 10.1593/tlo.09100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jackson EL, Garcia-Verdugo JM, Gil-Perotin S, Roy M, Quinones-Hinojosa A, VandenBerg S, et al. PDGFR alpha-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron. 2006;51:187–99. doi: 10.1016/j.neuron.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 44.Varela M, Ranuncolo SM, Morand A, Lastiri J, De Kier Joffé EB, Puricelli LI, et al. EGF-R and PDGF-R, but not bcl-2, overexpression predict overall survival in patients with low-grade astrocytomas. J Surg Oncol. 2004;86:34–40. doi: 10.1002/jso.20036. [DOI] [PubMed] [Google Scholar]
- 45.Huang YW, Liu JC, Deatherage DE, Luo J, Mutch DG, Goodfellow PJ, et al. Epigenetic repression of microRNA-129–2 leads to overexpression of SOX4 oncogene in endometrial cancer. Cancer Res. 2009;69:9038–46. doi: 10.1158/0008-5472.CAN-09-1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Y, Hei Y, Shu Q, Dong J, Gao Y, Fu H, et al. VCP/p97, down-regulated by microRNA-129–5p, could regulate the progression of hepatocellular carcinoma. PLoS ONE. 2012;7:e35800. doi: 10.1371/journal.pone.0035800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu J, Qian J, Li C, Kwok L, Cheng F, Liu P, et al. miR129 regulates cell proliferation by downregulating Cdk6 expression. Cell Cycle. 2010;9:1809–18. doi: 10.4161/cc.9.9.11535. [DOI] [PubMed] [Google Scholar]
- 48.Desjardins A, Quinn JA, Vredenburgh JJ, Sathornsumetee S, Friedman AH, Herndon JE, et al. Phase II study of imatinib mesylate and hydroxyurea for recurrent grade III malignant gliomas. J Neurooncol. 2008;83:53–60. doi: 10.1007/s11060-006-9302-2. [DOI] [PubMed] [Google Scholar]
- 49.Giannini C, Sarkaria JN, Saito A, Uhm JH, Galanis E, Carlson BL, et al. Patient tumor EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro Oncol. 2005;7:164–76. doi: 10.1215/S1152851704000821. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.