

Abstract
Aims
Long QT syndrome 1 (LQT1) mutations in KCNQ1 that decrease cardiac IKs (slowly activating delayed rectifier K+ current) underlie ventricular arrhythmias and sudden death. LQT1 mutations may suppress IKs by preventing KCNQ1 assembly, disrupting surface trafficking, or inhibiting gating. We investigated mechanisms underlying how three LQT1 mutations in KCNQ1 C-terminus assembly domain (R555H/G589D/L619M) decrease IKs in heterologous cells and cardiomyocytes.
Methods and results
In Chinese hamster ovary (CHO) cells, mutant KCNQ1 + KCNE1 channels either produced no currents (G589D/L619M) or displayed markedly reduced IKs with a right-shifted voltage-dependence of activation (R555H). When co-expressed with wild-type (wt) KCNQ1, the mutant KCNQ1s displayed varying intrinsic dominant-negative capacities that were affected by auxiliary KCNE1. All three mutant KCNQ1s assembled with wt KCNQ1 as determined by fluorescence resonance energy transfer (FRET). We developed an optical quantum dot labelling assay to measure channel surface density. G589D/R555H displayed substantial reductions in surface density, which were either partially (G589D) or fully (R555H) rescued by wt KCNQ1. Unexpectedly, L619M showed no trafficking defect. In adult rat cardiomyocytes, adenovirus-expressed homotetrameric G589D/L619M + KCNE1 channels yielded no currents, whereas R555H + KCNE1 produced diminished IKs with a right-shifted voltage-dependence of activation, mimicking observations in CHO cells. In contrast to heterologous cells, homotetrameric R555H channels showed no trafficking defect in cardiomyocytes.
Conclusion
Distinct LQT1 mutations in KCNQ1 assembly domain decrease IKs using unique combinations of biophysical and trafficking mechanisms. Functional deficits in IKs observed in heterologous cells are mostly, but not completely, recapitulated in adult rat cardiomyocytes. A ‘methodological chain’ combining approaches in heterologous cells and cardiomyocytes provides mechanistic insights that may help advance personalized therapy for LQT1 mutations.
Keywords: Long QT syndrome, KCNQ1, Channel trafficking, Cardiac myocyte, Ion channel
1. Introduction
In the human heart, the slowly activating delayed rectifier K+ current (IKs) plays an important role in action potential repolarization.1,2 The IKs channel complex contains four pore-forming KCNQ1 (or Kv7.1) subunits assembled with auxiliary KCNE1 peptides.3–7 Channels reconstituted with KCNQ1 alone in heterologous cells display a rapidly activating outward K+ current.3,4 Co-expressing KCNE1 increases current density three- to four-fold, right-shifts voltage-dependence of activation, and dramatically slows kinetics of channel activation to produce the characteristic IKs waveform.3,4 KCNE1 increases KCNQ1 single-channel conductance8,9 and induces a 100-fold increase in KCNQ1 affinity for phosphatidylinositol 4,5-bisphosphate (PIP2),10 which is necessary for effective channel gating.11,12 KCNE1 slows the movement of KCNQ1 voltage sensors,13,14 and may also affect coupling between voltage sensor movement and channel opening.15
Long QT syndrome (LQTS), a condition characterized by a prolonged QT interval on the ECG, underlies lethal cardiac arrhythmias. Mutations in several different ion channel subunits and scaffold proteins have been linked to LQTS.16,17 Congenital loss-of-function mutations in KCNQ1 (LQT1) that reduce cardiac IKs are the most common form of LQTS, accounting for >50% of genotyped patients.18,19 Reduced IKs delays repolarization and prolongs the cardiac action potential to cause LQT1, a serious condition that predisposes some affected individuals to exertion-triggered ventricular arrhythmias and sudden cardiac death.17,20,21 Over 250 LQT1-causing mutations have been mapped throughout the entire KCNQ1 coding sequence.19,22 LQT1 mutations could lead to suppressed IKs via several distinct mechanisms: (i) preventing channel assembly; (ii) impaired channel trafficking to the cell surface; and (iii) defective biophysical properties, including reduced open probability, diminished single-channel conductance, or a rightward shift in voltage-dependence of activation. Because most LQT1 cases arise in an autosomal dominant fashion with affected individuals having one wild-type (wt) and one mutant gene, it is also essential to determine the propensity of mutant KCNQ1 subunits to act as dominant negatives. Overall, a comprehensive approach evaluating channel biophysics, surface density, and dominant-negative propensities is necessary to define mechanisms distinct LQT1 mutations use to decrease IKs. This is important because the particular mechanisms individual LQT1 mutations utilize to suppress IKs could inform on appropriate therapeutic options.23–26
The functional impact of several LQT1 mutations has been studied by different groups using heterologous expression.27–31 We sought to build on these previous studies in two critical ways. First, we took a holistic approach towards identifying underlying mechanisms of three distinct LQT1 mutations by examining their impact on ionic current, voltage-dependence of activation, channel assembly, surface trafficking, and dominant-negative propensities in the absence and presence of KCNE1. Secondly, we determined how faithfully the functional impact and underlying mechanisms of LQT1 mutations identified using heterologous expression are translated in actual adult cardiomyocytes. An enigmatic feature of LQT1 is inconsistent correlation between severity of biophysical defects determined in vitro and risk of arrhythmias in patients.23,32 A potential explanation is the existence of cardiomyocyte-specific modifying factors that either exacerbate or diminish the functional impact of particular LQT1 mutations in the heart. Adult ventricular cardiomyocytes have a unique cytoarchitecture, intracellular milieu, and complement of proteins that are different from those of widely used heterologous expression systems. Hence, understanding how specific LQT1 mutations suppress IKs in cardiomyocytes, and how these contrast with heterologous cells, is important for deepened insights into factors governing disease penetrance and risk of arrhythmias in humans.
Here, we investigated how three LQT1 mutations (R555H, G589D, and L619M) located in KCNQ1 C-terminus decrease IKs. We found that the three mutations use distinct combinations of biophysical and trafficking mechanisms to suppress IKs, and differ in their capacity to act as dominant negatives in a KCNE1-dependent manner. The functional deficits in IKs observed in heterologous cells were, for the most part, recapitulated in adult rat cardiomyocytes. However, for homotetrameric R555H channels, a trafficking defect observed in heterologous cells was not evident in cardiomyocytes.
2. Methods
2.1. Generation of plasmid constructs
Human KCNQ1 cDNA was cloned into the bicistronic plasmid pShuttle CMV ires-EGFP (enhanced green fluorescent protein) to generate KCNQ1-ires-GFP. Human KCNE1 cDNA was subcloned into pcDNA3.1 using NheI and ApaI restriction sites. Overlap extension PCR was used to fuse enhanced cyan and yellow fluorescent proteins (ECFP and EYFP) in frame to the C-terminus of KCNE1 and KCNQ1, respectively. A 13-residue bungarotoxin-binding site (BBS; TGGCGGTACTACGAGAGCAGCCTGGAGCCCTACCCCGAC)33–35 was introduced between residues 148–149 in the extracellular S1–S2 loop of KCNQ1 using the QuikChange Lightning Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA) according to the manufacturer's instructions. Missense mutations in KCNQ1 were introduced using the QuikChange Lightning Site-Directed Mutagenesis Kit. All constructs were verified by sequencing.
2.2. Generation of adenoviruses
Adenoviral vectors were generated using the pAdEasy system (Stratagene) as previously described.36 Plasmid shuttle vectors (pShuttle CMV) containing cDNA for wt and mutant KCNQ1 or KCNE1 subunits were linearized with PmeI and electroporated into BJ5183-AD-1 electrocompetent cells pre-transformed with the pAdEasy-1 viral plasmid (Stratagene). PacI restriction digestion was used to identify transformants with successful recombination. Positive recombinants were amplified using XL-10-Gold bacteria, and the recombinant adenoviral plasmid DNA linearized with PacI digestion. Human embryonic kidney 293 (HEK293) cells cultured in 60 mm diameter dishes at 70–80% confluency were transfected with linearized adenoviral DNA and monitored for cytopathic effects and adenoviral plaques 7–10 days after transfection. Cells were harvested and subjected to three freeze–thaw cycles and centrifuged (3452 g) to remove cellular debris. The supernatant (2 mL) was used to infect a 10 cm dish of 90% confluent HEK293 cells. Viral expansion and purification were carried out as previously described.37 Briefly, ten 15 cm tissue culture dishes containing 90% confluent HEK293 cells were infected with viral supernatant (1 mL). After 48 h, cells were harvested, combined, pelleted by centrifugation, and resuspended in 8 mL of buffer containing (in mM) Tris–HCl 20, CaCl2 1, and MgCl2 1 (pH 8.0). Cells were lysed using four freeze–thaw cycles and cellular debris pelleted by centrifugation. Virus-laden supernatant was purified on a caesium chloride (CsCl) discontinuous gradient by layering three densities of CsCl (1.25, 1.33, and 1.45 g/mL). After centrifugation (50 000 rpm; SW41Ti Rotor, Beckman-Coulter Optima L-100K ultracentrifuge; 1 h, 4°C), a band of virus at the interface between the 1.33 and 1.45 g/mL layers was removed and dialyzed against PBS (12 h, 4°C).
2.3. Cell culture and transfection
Low-passage-number Chinese hamster ovary (CHO) cells were cultured at 37°C in Ham F12 supplemented with 10% foetal bovine serum (FBS), 100 µg/mL of normocin, and 3.8 mM NaHCO3. CHO cells were transiently transfected with desired constructs in 6 cm tissue culture dishes—KCNQ1-YFP (yellow fluorescent protein; 5 µg) and KCNE1-CFP (5 µg) using FUGENE or X-tremeGENE HP according to the manufacturers' instructions (Roche). Low-passage-number HEK 293 cells were maintained in DMEM supplemented with 10% FBS and 100 μg/mL of penicillin–streptomycin at 37°C. HEK 293 cells were transiently transfected with the appropriate constructs together with 1 μg T-antigen cDNA using calcium phosphate precipitation. Primary ventricular cardiac myocytes were isolated and cultured as previously described.36,37 Briefly, adult male Sprague–Dawley rats were deeply anaesthetized with isoflurane in accordance with the guidelines of the Columbia University Animal Care and Use Committee. Hearts were excised, and ventricular myocytes were isolated by enzymatic digestion using a Langendorff perfusion apparatus and cultured on laminin-coated coverslips. Myocytes were infected with adenovirus 3–5 h after initial plating.
2.4. Electrophysiology
Whole-cell membrane currents were recorded at room temperature in CHO cells or cardiomyocytes using an EPC-10 patch-clamp amplifier (HEKA Electronics) controlled by the PatchMaster software (HEKA). A coverslip with adherent CHO cells or myocytes was placed on the glass bottom of a recording chamber (0.7–1 mL in volume) mounted on the stage of an inverted Nikon Eclipse Ti-U microscope. Micropipettes were fashioned from 1.5 mm thin-walled glass and fire-polished. Internal solution contained (in mM): 133 KCl, 0.4 GTP, 10 EGTA, 1 MgSO4, 5 K2ATP (added on day of experimentation), 0.5 CaCl2, and 10 HEPES (pH 7.2). External solution contained (in mM): 147 NaCl, 4 KCl, 2 CaCl2, and 10 HEPES (pH 7.4). Pipette resistance was typically 1.5−2 MΩ when filled with internal solution. I–V curves were generated from a family of step depolarizations (−40 to +100 mV in 10 mV steps from a holding potential of −50 mV). Currents were sampled at 20 kHz and filtered at 5 or 10 kHz. Traces were acquired at a repetition interval of 10 s.
2.5. Detection of cell surface KCNQ1 channels with quantum dots
Surface BBS-tagged wt or mutant KCNQ1 subunits expressed in HEK239 cells and cardiomyocytes were labelled with quantum dot as previously described.33,35,36 Fluorescence signals were assayed by flow cytometry (HEK293 cells) using a BD LSR II Cell Analyzer (BD Biosciences) or confocal microscopy (HEK293 cells and cardiomyocytes). Briefly, HEK 293 cells expressing BBS-tagged wt or mutant KCNQ1 subunits were washed twice with PBS containing Ca2+ and Mg2+, and sequentially incubated with 1 μM biotinylated α-bungarotoxin (BTX) in DMEM/3% BSA at room temperature for 1 h followed by 5 nM streptavidin-conjugated quantum dot (QD655, Invitrogen) at 4°C for 1 h in the dark. Surface-labelled HEK293 cells were harvested with trypsin, washed with PBS, and assayed by flow cytometry. Fluorescence signals from flow cytometry were analysed using the FloJo Software. For each group of experiments, we used isochronal untransfected and single colour controls to manually set threshold and gain settings for each fluorophore to ensure signals remained in the linear range.
2.6. Confocal imaging
Confocal images were obtained with a Leica SP2 confocal microscope using a ×63 oil immersion objective (NA 1.4). Laser lines and detector settings were: CFP: 405 nm diode laser, 465–502 nm detection; YFP: 514 nm argon, 520–560 nm detection; and QD655: 405 nm diode laser, 620–700 nm detection. Laser power was set at 30%, and detector gain at 550–700 V to reduce noise and prevent saturation of image pixels. Pinhole size was set at 1 Airy unit.
2.7. Detection of wt and mutant KCNQ1 subunit assembly by FRET
We used a three-cube fluorescence resonance energy transfer (FRET) method adapted from Chen et al.38,39 to determine whether CFP-tagged mutant KCNQ1s co-assemble with wt KCNQ1-YFP. As a negative control, wt KCNQ1-YFP was transfected with the auxiliary voltage-dependent Ca2+ channel subunit, CaVβ3-CFP. HEK293 cells cultured on fibronectin-coated MaTek dishes were transfected with XFP-tagged proteins. Forty-eight hours post-transfection, dishes were washed with PBS and mounted on a Nikon Eclipse Ti-U, inverted microscope. Cells were imaged using a ×40 oil objective (NA 1.3). Experiments were performed using an EasyRatioPro Fluorescence Imaging System (Photon Technology International). Three separate signals were acquired for each donor–acceptor pair condition: the donor channel (DD) which excites and detects donor emission, the acceptor channel (AA) which excites and detects acceptor emission, and the FRET channel (DA) which excites the donor, but detects acceptor emission. Excitation wavelengths of 440 nm (CFP and FRET cubes) and 500 nm (YFP cube) were applied using a random access monochromator with a 75 W Xenon Arc lamp housing (PTI DeltaRam X, Photon Technology International). Filter cubes used were (dichroic, emission): DD (455DCLP, D480/30M); AA (525DRLP, 530EFLP); and DA (455DRLP, 535DF25). Individual cells were segmented by drawing region of interest (ROI) areas. ROIs with no cells were used to assess background levels which were subtracted from experimental signals. Cross-talk parameters were determined by imaging cells expressing either donor (CFP) or acceptor (YFP) fluorescent proteins alone. Constants that describe the ratio of sensitized acceptor emission to donor fluorescence quenching (G) and the fluorescence intensity ratio of a 1 : 1 donor-to-acceptor concentration (k) were determined using donor–acceptor fusion proteins and used to calculate the FRET efficiency as described.38,39
2.8. Data and statistical analyses
Electrophysiological data were analysed offline using built-in functions in Fitmaster (HEKA), Microsoft Excel, and Origin software. Current amplitudes (pA) were divided by the cell size (pF) and expressed as current densities (pA/pF). Data are reported as means ± SEM. Statistical differences between means were determined using one-way ANOVA with Bonferroni post hoc analyses or two-tailed unpaired t-test for comparisons between two groups. A value of P < 0.05 was considered significant.
3. Results
3.1. Impact of distinct A-domain LQT1 mutations on KCNQ1 alone currents
Human KCNQ1 C-terminus is 322 amino acids long (residues 354–676) and contains four helices with distinct functions: helices A (residues 370–389) and B (residues 506–532) are important for calmodulin (CaM) binding and regulation;29,31 helices C (residues 548–562) and D (residues 587–620) together comprise the A-domain critical for channel assembly and trafficking—helix C forms dimers40 and interacts with KCNE1 distal C-terminus,41 whereas helix D forms a coiled-coil structure essential for channel tetramerization.40,42,43 We focused our studies on three LQT1 mutations (R555H, G589D, and L619M) spanning helices C and D in the A-domain (Figure 1A).19,27,44 G589D has been previously studied by various groups under different conditions that have produced discrepant results, with some studies finding this mutation gives rise to non-functional channels while others do not.28–31,40 To our knowledge, R555H and L619M mutations have not been studied, although several groups have examined functional properties of R555C,27,45 which provides a nice frame of reference for our studies on R555H.
Figure 1.

Electrophysiological properties of wt and mutant KCNQ1 in the absence of KCNE1. (A) Schematic showing the modular structure of KCNQ1 C-terminus and relative locations of LQT1 mutations evaluated in this study. The structure of the coiled-coil tetramerization domain (helix D) is reproduced from PDB ID: 3BJ4. (B) Representative traces from CHO cells either untransfected (left panel) or expressing wt KCNQ1-YFP (middle). (Right panel) Population Ipeak–V curves for untransfected (open circle) or wt KCNQ1-expressing (filled square, n = 47) cells. (C) Exemplar currents from CHO cells expressing R555H-YFP alone (left) or with wt KCNQ1-YFP (middle). (Right panel) Population Ipeak–V curves for R555H expressed alone (open diamond, n = 12) or with wt KCNQ1 (filled diamond, n = 5). Ipeak–V curve for wt KCNQ1-YFP is reproduced (red line) to facilitate visual comparison. (D and E) Exemplar currents and Ipeak–V relationships for indicated KCNQ1 mutants, same format as C. Data points are means ± SEM, n = 6–10. Scale bars, 10 pA/pF, 1 s. Displayed exemplar currents were obtained from test pulses to −40, −10, +10, +40, +70, and +100 mV for all experimental conditions.
We first examined how these LQT1 mutations affected whole-cell currents in channels reconstituted in CHO cells with wt or mutant KCNQ1 subunits alone (no KCNE1). To facilitate visual identification of transfected cells and monitor subcellular localization of channels, we fused YFP to the C-terminus of wt and mutant KCNQ1 subunits. Fusing YFP to KCNQ1 C-terminus did not grossly affect channel properties (see Supplementary material online, Figure S1), in agreement with previous reports.46 CHO cells expressing wt KCNQ1 alone displayed voltage-dependent, rapidly activating outward whole-cell currents that were absent in non-transfected cells (Figure 1B). Compared with wt KCNQ1, homotetrameric R555H displayed significantly depressed steady-state currents at all voltages (Figure 1C), whereas homotetrameric G589D or L619M channels yielded essentially no currents above background (Figure 1D and E; see Supplementary material online, Table S1).
We next determined the capacity of the different LQT1 mutant KCNQ1 subunits to act as dominant negatives when co-expressed in a 1 : 1 molar ratio with wt KCNQ1. Currents through R555H and L619M were partially rescued by wt KCNQ1, while full recovery was obtained with G589D (Figure 1C–E; see Supplementary material online, Table S1). Hence, in this context, R555H and L619M, but not G589D, were moderately dominant negative. All three mutant proteins interacted with wt KCNQ1 as determined by FRET (see Supplementary material online, Figure S2), indicating that the mutations did not grossly disrupt channel assembly.
3.2. Impact of KCNE1 on biophysical properties of A-domain LQT1 mutants
Co-expressing wt KCNQ1-YFP (5 µg) with KCNE1 (5 µg) yielded dramatically larger currents with markedly slow activation kinetics compared with KCNQ1 alone channels (Figure 2A–C; see Supplementary material online, Table S1). Homotetrameric R555H-YFP + KCNE1 channels displayed significantly depressed current amplitude compared with wt KCNQ1-YFP + KCNE1 (Figure 2D–F; see Supplementary material online, Table S2). Normalizing activation curves to the peak current obtained at +100 mV indicated that homotetrameric R555H + KCNE1 channels displayed a rightward shift in the voltage-dependence of activation compared with wt KCNQ1 + KCNE1 (see Supplementary material online, Figure S3). Homotetrameric G589D or L619M subunits co-expressed with KCNE1 yielded virtually no currents (Figure 2G–L).
Figure 2.

Electrophysiological properties of wt and mutant KCNQ1 in the presence of KCNE1. (A) Exemplar currents from CHO cells expressing KCNQ1 alone (left) or KCNQ1-YFP + KCNE1 (right). (B) Population Ipeak–V curves for KCNQ1-YFP + KCNE1 channels (filled square, n = 11). Data for KCNQ1 alone channels are reproduced from Figure 1B (grey trace). (C) Voltage-dependence of time constants for activation (τrise) of wt KCNQ1 alone (open circle, n = 47) and wt KCNQ1 + KCNE1 (filled square, n = 11) currents. (D) Exemplar currents from CHO cells expressing (left) R555H + KCNE1 (homotetrameric) or (right) R555H + wt KCNQ1 + KCNE1 (heterotetrameric). (E) Population Ipeak–V curves for homotetrameric (open diamond, n = 18) and heterotetrameric (filled diamond, n = 7) R555H channels. Data for control wt KCNQ1 + KCNE1 channels are reproduced (red trace) for comparison. (F) Voltage-dependence of τrise for homotetrameric (open diamond, n = 18) and heterotetrameric (filled diamond, n = 7) R555H channels. Data for wt KCNQ1 alone (cyan trace) and wt KCNQ1 + KCNE1 (red trace) channels are shown for comparison. (G–L) Exemplar currents, Ipeak–V relationships, and τrise–V curves for homotetrameric and heterotetrameric channels containing the indicated KCNQ1 mutants, same format as D–F. Data points are mean ± SEM, n = 5–21. Scale bars, 20 pA/pF, 1 s. Displayed exemplar currents were obtained from test pulses to −40, −10, +10, +40, +70, and +100 mV for all experimental conditions.
The dominant-negative propensities of the mutant KCNQ1s in the presence of KCNE1 were of particular interest given that most LQT1 patients are heterozygous for the disease mutation. To examine this, we measured biophysical properties of heterotetrameric channels containing mutant KCNQ1 (2.5 µg) + wt KCNQ1 (2.5 µg) + KCNE1 (5 µg). R555H + wt KCNQ1 + KCNE1 channels displayed similar biophysical properties to R555H + KCNE1 channels with respect to the depressed current amplitude, slowed activation kinetics, and right-shifted voltage-dependence of activation compared with wt KCNQ1 + KCNE1 (Figure 2D–F; see Supplementary material online, Figure S3 and Table S1). G589D + wt KCNQ1 + KCNE1 yielded currents that reached ∼30% amplitude of wt KCNQ1 + KCNE1 channels (Figure 2G and H), and displayed similar activation kinetics (Figure 2I; see Supplementary material online, Table S2) and voltage-dependence of activation (see Supplementary material online, Figure S3). In contrast, L619M + KCNE1 channels were completely rescued by wt KCNQ1 (Figure 2J–L). Overall, these results indicate that, in the presence of KCNE1, R555H and G589D impose strong and moderate dominant-negative effects, respectively, on IKs amplitude. In contrast, L619M does not exert a dominant-negative effect on IKs. Similar data were obtained when the ratios of transfected plasmids were changed for homotetrameric (6 µg KCNQ1 and 3 µg KCNE1) and heterotetrameric (3 µg mutant KCNQ1, 3 µg wt KCNQ1, and 3 µg KCNE1) channels, demonstrating the robustness of the results (see Supplementary material online, Figure S4).
3.3. Impact of A-domain LQT1 mutations on surface density of KCNQ1 channels
Beyond biophysical properties, complete understanding of mechanisms by which LQT1 mutations reduce IKs requires knowledge of their impact on channel surface density. We developed an optical approach that permits quantitative assessment of KCNQ1 relative surface density in live cells and eliminates potentially confounding factors, such as differences in transfection efficiency, protein stability, and expression levels. To implement the approach, we introduced a 13-residue high-affinity BBS33,34,47 into the KCNQ1 extracellular S1–S2 loop (Figure 3A). This modification did not grossly change channel properties as BBS-tagged KCNQ1 yielded rapidly activating currents when expressed alone, and was modulated by KCNE1 in a manner similar to untagged channels (see Supplementary material online, Figure S5). Moreover, mutant BBS-tagged KCNQ1 + KCNE1 channels mimicked the deficits in current amplitude observed with their untagged counterparts (see Supplementary material online, Figure S6).
Figure 3.

Optical detection of cell surface BBS-tagged KCNQ1 with quantum dot. (A, left) Schematic showing quantum dot (QD655) labelling of cell surface BBS-tagged KCNQ1-YFP. (Middle) Confocal images of non-permeabilized cells expressing BBS-KCNQ1-YFP labelled with QD655. Panels show channels for YFP (total expression), CFP, and QD655 (surface channels). Scale bar, 20 µm. (B) Flow cytometry dot plot showing QD655 vs. YFP fluorescence intensity signals for cells expressing BBS-KCNQ1-YFP. About 50 000 cells were counted. Vertical and horizontal lines represent threshold values set based on isochronal untransfected cells. Each dot represents a single cell. Dots have been arbitrarily coloured to facilitate visualization of distinct populations. Green dots represent BBS-KCNQ1-YFP-expressing cells with little QD655 signal, indicating low channel surface density. Red dots denote BBS-KCNQ1-YFP-positive cells with robust channel trafficking to the surface. Black dots in bottom left quadrant are untransfected cells. (C) Normalized mean QD655 signals from YFP-positive cells provide an index of relative surface density. (D and E) Data for cells expressing BBS-KCNQ1-YFP + KCNE1-CFP, same format as A–C. (F and G) Negative control. Data for cells expressing untagged KCNQ1-YFP, same format as A–C. *P < 0.05 compared with BBS-KCNQ1-YFP, unpaired Student's t-test (scale bars, 20 μm). (H) Computed relative surface density for BBS-tagged wt and LQT1 mutant KCNQ1-YFP subunits in the absence and presence of untagged wt KCNQ1.
To selectively label surface channels, we exposed non-permeabilized cells expressing BBS-KCNQ1 sequentially to biotinylated bungarotoxin (BTX-biotin) and streptavidin-conjugated quantum dot (QD655), respectively.33,35 Confocal images of cells expressing BBS-KCNQ1-YFP alone displayed robust QD655 fluorescence staining around the periphery (Figure 3A). Flow cytometry permitted rapid, high throughput quantification of fluorescence signals from tens of thousands of QD655-labelled cells, but with single-cell resolution. A dot plot representation of flow cytometry data for cells expressing BBS-KCNQ1-YFP shows robust QD655 staining in a large population of YFP-positive cells (Figure 3B). We computed the mean QD655 fluorescence of YFP-positive cells as an index of channel surface density (Figure 3C). QD655 signals are normalized to YFP fluorescence, which directly corresponds to KCNQ1 expression levels. Relative to cells expressing BBS-KCNQ1-YFP alone, co-expressing KCNE1-CFP caused a 20% decrease in BBS-KCNQ1-YFP surface density (Figure 3C and E). This result is broadly consistent with the idea that KCNE1 subunits may regulate KCNQ1 surface density by acting as endocytic chaperones.48 Negative control cells expressing KCNQ1-YFP lacked QD655 fluorescence (Figure 3F and G), indicating negligible non-specific staining.
With this method in hand, we evaluated the impact of the distinct LQT1 mutations on KCNQ1 surface density. Compared with wt KCNQ1, homotetrameric R555H and G589D showed a relative surface density of 59 ± 3 and 31 ± 13%, respectively, indicating these mutations caused significant trafficking defects (Figure 3H). Surprisingly, homotetrameric L619M channels exhibited no trafficking deficits and had a similar cell surface density as wt KCNQ1 (Figure 3H). We examined the extent to which trafficking deficits were dominant negative by co-expressing BBS-tagged mutant KCNQ1-YFP with wt KCNQ1-CFP. Controls for these experiments were provided by cells expressing wt BBS-KCNQ1-YFP + wt KCNQ1-CFP. Wt KCNQ1 completely rescued channel surface density for R555H (99 ± 5%), whereas G589D was only partially recovered (58 ± 2%), indicating this mutation had a dominant-negative trafficking effect on wt KCNQ1 (Figure 3H). In mirror experiments, we evaluated the impact of mutant KCNQ1-YFP subunits on BBS-tagged wt KCNQ1-CFP. This approach also showed a prominent dominant-negative trafficking effect for G589D, but not for R55H or L619M (see Supplementary material online, Figure S7).
3.4. Relative contributions of trafficking and gating deficits in A-domain LQT1 mutations
We plotted mean Ipeak,+30 mV as a function of relative surface density to gain insights into relative contribution of biophysical and trafficking defects to loss of IKs in the physiological voltage range for the distinct KCNQ1 mutations (Figure 4). Control wt KCNQ1 + KCNE1 channels had a relative surface density = 1 and Ipeak,+30 mV = 106 ± 18 pA/pF. A line from the origin to this point tracks the expected relationship between Ipeak,+30 mV and relative surface density if mutations affected only channel trafficking with no impact on biophysical properties. This representation of the data explicitly shows that, in the physiological voltage range, homotetrameric mutant KCNQ1 + KCNE1 channels display a dramatic loss of IKs that is underlain by unique combinations of biophysical and trafficking defects (Figure 4, open symbols). Homotetrameric R555H (open diamond) and G589D (open inverted triangles) co-expressed with KCNE1 display loss of IKs due to both biophysical and trafficking abnormalities, whereas L619M + KCNE1 channels (left-pointing open triangle) lack current purely due to biophysical mechanisms. The underlying mechanisms were altered in heterotetrameric channels co-expressing wt KCNQ1 with mutant KCNQ1 + KCNE1subunits (Figure 4, solid symbols). Heterotetrameric channels containing R555H showed deeply inhibited IKs, despite all the channels being present at the cell surface. Hence, this mutation exerts a purely dominant-negative biophysical effect on wt KCNQ1 + KCNE1 channels. In contrast, deficits in channel trafficking prominently contribute to the moderate dominant-negative effects of G589D channels on IKs (Figure 4).
Figure 4.
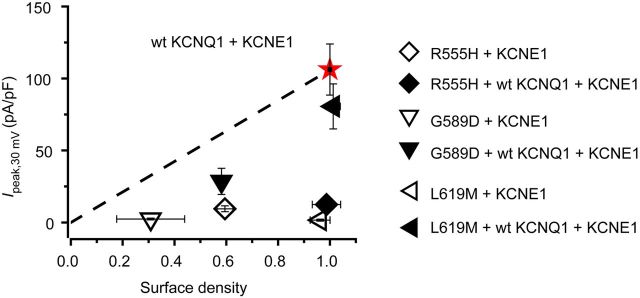
Relative contributions of biophysical and trafficking mechanisms to IKs suppression for distinct LQT1 mutations. Plot of peak IKs recorded at +30 mV (Ipeak, 30mV) vs. relative surface density for wt KCNQ1 + KCNE1 (star) and indicated mutant KCNQ1 + KCNE1 ± wt KCNQ1. Dotted line from origin to wt KCNQ1 data point tracks expected relationship between Ipeak,+30 mV and relative surface density if mutations affected only channel trafficking with no biophysical effects. Deviations from this line report on biophysical mechanisms contributing to observed changes in IKs.
3.5. Impact of A-domain LQT1 mutations in cardiomyocytes
We chose to evaluate functional properties of wt and mutant KCNQ1 subunits in adult rat ventricular cardiomyocytes because these cells lack appreciable endogenous IKs, which would otherwise contaminate measurements of exogenously expressed channels and complicate data interpretation.1 We used adenoviral vectors to express wt and mutant KCNQ1 and KCNE subunits in acutely cultured adult rat ventricular cardiomyocytes. Because these cardiomyocytes contain several different K+ channel types,1 we used the KCNQ1 inhibitor, chromanol 293B, as a tool to isolate IKs. In cardiomyocytes expressing wt KCNQ1 + KCNE1, membrane depolarization evoked large outward currents characterized by rapid and slowly activating components, and slowly deactivating tail currents (Figure 5A, trace a). Chromanol 293B blocked a significant proportion of the outward and tail currents (Figure 5A, trace b). The difference traces (a–b) revealed a large outward current with the hallmark slow activation kinetics, tail currents, and voltage-dependence of IKs (Figure 5A and B). As a negative control, cardiomyocytes expressing KCNE1 alone displayed voltage-dependent rapidly activating outward currents that steadily declined to a plateau and were insensitive to 30 µM chromanol 293B, leading to essentially zero difference currents across all examined voltages (Figure 5C and D). The waveforms of endogenous currents likely reflect a mixture of the diverse repolarizing K+ currents known to occur in rodent cardiomyocytes.1 Cardiomyocytes expressing homotetrameric R555H + KCNE1 channels generated IKs with smaller amplitude, slower activation kinetics, and right-shifted V0.5 of activation compared with wt KCNQ1 + KCNE1 (Figure 5E and F). In contrast, homotetrameric G589D or L619M + KCNE1 yielded channels that on average produced virtually no IKs (Figure 5G–J). These functional outcomes are in close agreement with the observations in CHO cells.
Figure 5.
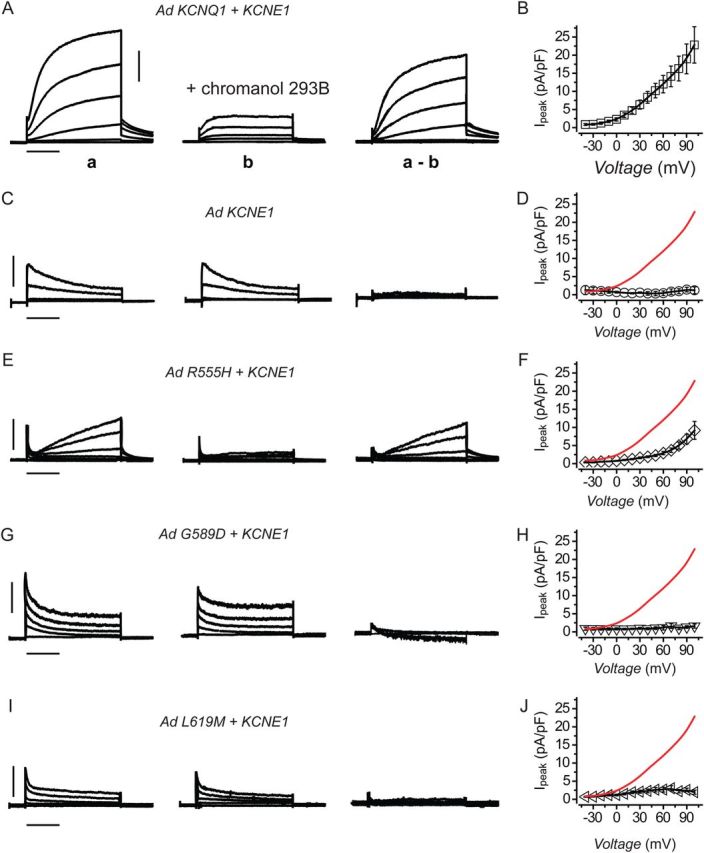
Reconstitution of wt and mutant IKs in adult rat ventricular cardiomyocytes. (A) Exemplar current traces measured in the absence (traces a) and presence (traces b) of 30 µM chromanol 293B in a cardiomyocyte expressing wt KCNQ1 + KCNE1. The difference traces (a–b) represent the chromanol 293B-sensitive currents, i.e. IKs. (B) Population I–V curves for chromanol 293B-sensitive currents in adult rat cardiomyocytes expressing wt KCNQ1 + KCNE1, n = 26. (C and D) Data for cardiomyocytes expressing KCNE1 alone indicate the lack of IKs (n = 4), same format as A and B. (E–J) Data obtained from cardiomyocytes expressing R555H + KCNE1 (n = 21), G589D + KCNE1 (n = 10), and L619M + KCNE1 (n = 16), respectively, same format as A and B. Data for wt KCNQ1 + KCNE1 channels are reproduced (red trace) in E–J for visual comparison. Data points are mean ± SEM. Data were generated from three different rat cardiomyocyte preparations. Scale bars, 5 pA/pF, 1 s.
To determine whether the trafficking effects of the LQT1 mutations observed in CHO cells were similarly recapitulated in cardiomyocytes, we turned to the QD655 labelling assay. Non-infected control cardiomyocytes showed little QD655 signal (Figure 6A and C), indicating low background fluorescence. Cardiomyocytes expressing BBS-KCNQ1-YFP displayed robust QD655 fluorescence staining of the sarcolemma (Figure 6). In contrast to observations in CHO cells, homotetrameric BBS-R555H-YFP targeted to the cell surface even better (32% increase) than wt KCNQ1 (Figure 6). Similarly, L619M channel cell surface density was the same as wt KCNQ1. In contrast, G589D channels displayed a 58% lower sarcolemmal QD655 signal compared with wt KCNQ1, indicating this mutation impaired trafficking in cardiomyocytes (Figure 6). A scatter plot of QD655 vs. YFP fluorescence signals indicated that the results were not due to differences in KCNQ1 expression among the different conditions. Curiously, this representation of the data also showed that, within an experimental condition, KCNQ1 surface density remained relatively constant even as protein expression levels varied up to five-fold (Figure 6C). This finding suggests tight homeostatic mechanisms in cardiomyocytes that regulate KCNQ1 channel surface density over a wide range of protein expression levels. Alternatively, the results could reflect a saturation of the forward trafficking pathway for these channels in cardiomyocytes.
Figure 6.
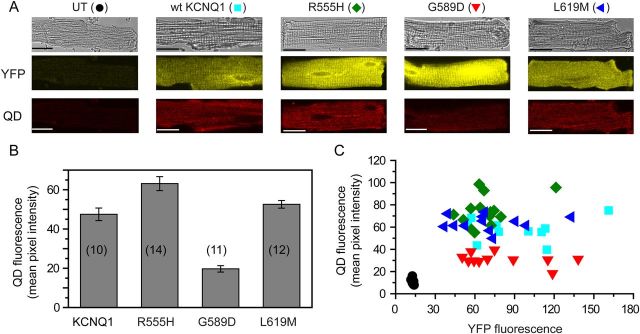
Detection of surface KCNQ1 channels with quantum dot in cardiomyocytes. (A) Confocal images of non-permeabilized adult rat ventricular cardiomycytes showing bright-field, YFP, and QD655 fluorescence images. Panels show uninfected control myocytes and cardiomyocytes expressing wt and mutant BBS-tagged KCNQ1 subunits. Scale bars, 20 µm. (B) QD655 fluorescence mean pixel intensity for the different conditions. (C) Scatter plots of QD655 (surface channels) vs. YFP fluorescence (total expression) for the different BBS-tagged wt (filled square) and mutant KCNQ1-YFP subunits (R555H, filled diamond; G589D, inverted triangle; L619M, left-pointing filled triangle). Data were generated from two different rat cardiomyocyte preparations.
4. Discussion
We investigated mechanisms by which three LQT1 mutations in KCNQ1 C-terminus lead to suppressed IKs in heterologous cells and cardiomyocytes. Taking a comprehensive approach, we found that though all three mutations are in the KCNQ1 A-domain, they suppressed IKs using unique combinations of biophysical and trafficking mechanisms. The distinct mutant KCNQ1 subunits displayed differing dominant-negative propensities that were affected by KCNE1. Finally, we show that most, but not all of the functional observations and underlying mechanisms observed in heterologous cells are recapitulated in cardiomyocytes.
Several LQT1 mutations map to KCNQ1 A-domain.19 The mechanism of some of these can be easily rationalized because they disrupt the helix D coiled-coil and thus prevent channel assembly.42 However, several A-domain mutations including the three studied here do not conform to this simple explanation. We show here that R555H, G589D, and L619M are all assembly-competent as indicated by FRET and their dominant-negative effects when expressed with wt KCNQ1 + KCNE1. While the R555H mutation has not been previously investigated, the functional properties of the related R555C have been studied by several groups.10,27,45 Homotetrameric R555C + KCNE1 channels display a reduced IKs amplitude, a more than +40 mV rightward shift in V0.5 of activation,27,45 and exhibit a strong dominant-negative phenotype in heterotetrameric channels.27 Our results with R555H in CHO cells are largely consistent with these prior observations. We further discovered that homotetrameric R555H subunits display a significant trafficking defect that is completely rescued by wt KCNQ1. This finding provides the new insights that R555H exerts a dominant-negative effect of on IKs through a purely biophysical mechanism. Importantly, the functional deficits of homotetrameric R555H IKs currents are recapitulated in cardiomyocytes. Previous studies indicate that PIP2 is necessary for effective gating of KCNQ1 + KCNE1 channels,10,12,45 and that R555C decreases the affinity of KCNQ1 for PIP2.10,45 We speculate that a decreased affinity for PIP2 may underlie the rightward shift in V0.5 for activation and slowed activation kinetics of R555H channels.
There is conflicting data on the viability of channels containing G589D.30,40,42,44 Kanki et al.42 observed that homomeric G589D + KCNE1 channels expressed in CHO cells yielded no currents, and did not traffic to the cell surface. Similarly, Hirsch and colleagues40 found that channels reconstituted in CHO cells with G589D alone (without KCNE1) produced no currents. In COS cells, G589D + KCNE1 channels were observed to produce small currents with right-shifted V0.5 of activation in a small fraction of transfected cells.44 In contrast, Marx et al.30 showed that heterotetrameric channels containing G589D + wt KCNQ1 + KCNE1 displayed robust IKs, albeit with a reduced amplitude compared with wt channels. Our comprehensive approach reconciles these divergent viewpoints by demonstrating that G589D produces measurable currents only when co-assembled with wt KCNQ1 in heterotetrameric channels. The data further provides the new insights that the dominant-negative effect of G589D in heterotetrameric channels can be at least partially explained by a prominent trafficking defect. The non-viability of homotetrameric G589D channels and their defective trafficking is recapitulated in cardiomyocytes. G589D has been shown to disrupt PKA regulation of IKs due to decreased binding of the A-kinase anchoring protein, yotiao.30 Our findings suggest an additional KCNQ1 trafficking defect in individuals with G589D mutation.
Homotetrameric L619M + KCNE1 channels displayed little current in CHO cells and cardiomyocytes, and this effect was attributable to solely biophysical mechanisms. This observation was unexpected given the position of the L619 at the distal end of helix D. The multi-tiered, modular structure of KCNQ1 C-terminus encourages a view where distinct functions are assigned to separate domains.40 From that perspective, mutations in helix D might be expected to disrupt channel assembly or trafficking. The surprisingly strong biophysical consequences of L619M suggest an unappreciated link between helix D and channel gating that is beyond the role of this region in channel assembly.
Our results indicate that individuals homozygous for any of the three KCNQ1 C-terminus mutations we studied would exhibit a profound loss of IKs. Nevertheless, such homozygous LQT1 mutations are quite rare.44 Our results predict that heterozygous individuals with any of the three mutations would lose cardiac IKs repolarization capacity with the following rank order of severity: R555H > G589D > L619M. The results further suggest that different therapeutic strategies may be more efficacious for distinct mutations. For L619M and R555H, small molecules that enhance channel opening and shift the V0.5 of activation to the normal range would be predicted to correct the loss-of-function phenotype. In contrast, for G589D, our data suggest small molecule chaperones that boost channel surface expression would most appropriately normalize basal cardiac IKs. It is worth mentioning that, for LQT1 patients, lethal ventricular arrhythmias are typically triggered by exertion when adrenaline (via PKA activation) stimulates the heartbeat. Hence, it will be important to investigate the impact of these mutations on PKA regulation of IKs. In this regard, G589D has been shown to disrupt PKA regulation of IKs, due to decreased binding of the A kinase anchoring protein, yotiao, to channels containing this mutation.30
In conclusion, this work advances a ‘methodological chain’ that can be widely applied to studies of disease-causing mutations of cardiovascular ion channels. The comprehensive approach, together with direct comparisons of results between heterologous cells and cardiomyocytes, can yield mechanistic insights that help advance personalized therapy for distinct arrhythmogenic mutations.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by the National Institutes of Health (RC1-HL099759 to W.R.K. and H.M.C., and RO1-GM070650 to W.R.K.); an American Heart Association Scientist Development Grant (13SDG16850065 to A.S.A.); and an American Heart Association Established Investigator Award (0940002N to H.M.C.).
Acknowledgements
We thank Timothy Kernan, Ming Chen, and Dr. Kun Fang for technical assistance. We also thank an anonymous reviewer for coining the term ‘methodological chain’.
Conflict of interest: none declared.
References
- 1.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–1253. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 2.Charpentier F, Merot J, Loussouarn G, Baro I. Delayed rectifier K(+) currents and cardiac repolarization. J Mol Cell Cardiol. 2010;48:37–44. doi: 10.1016/j.yjmcc.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 3.Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 4.Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 5.Takumi T, Ohkubo H, Nakanishi S. Cloning of a membrane protein that induces a slow voltage-gated potassium current. Science. 1988;242:1042–1045. doi: 10.1126/science.3194754. [DOI] [PubMed] [Google Scholar]
- 6.Morin TJ, Kobertz WR. Counting membrane-embedded KCNE beta-subunits in functioning K+ channel complexes. Proc Natl Acad Sci USA. 2008;105:1478–1482. doi: 10.1073/pnas.0710366105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Plant LD, Xiong D, Dai H, Goldstein SA. Individual IKs channels at the surface of mammalian cells contain two KCNE1 accessory subunits. Proc Natl Acad Sci USA. 2014;111:E1438–E1446. doi: 10.1073/pnas.1323548111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sesti F, Goldstein SA. Single-channel characteristics of wild-type IKs channels and channels formed with two minK mutants that cause long QT syndrome. J Gen Physiol. 1998;112:651–663. doi: 10.1085/jgp.112.6.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang Y, Sigworth FJ. Single-channel properties of IKs potassium channels. J Gen Physiol. 1998;112:665–678. doi: 10.1085/jgp.112.6.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y, Zaydman MA, Wu D, Shi J, Guan M, Virgin-Downey B, Cui J. KCNE1 enhances phosphatidylinositol 4,5-bisphosphate (PIP2) sensitivity of IKs to modulate channel activity. Proc Natl Acad Sci USA. 2011;108:9095–9100. doi: 10.1073/pnas.1100872108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loussouarn G, Park KH, Bellocq C, Baro I, Charpentier F, Escande D. Phosphatidylinositol-4,5-bisphosphate, PIP2, controls KCNQ1/KCNE1 voltage-gated potassium channels: a functional homology between voltage-gated and inward rectifier K+ channels. EMBO J. 2003;22:5412–5421. doi: 10.1093/emboj/cdg526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang H, Craciun LC, Mirshahi T, Rohacs T, Lopes CM, Jin T, Logothetis DE. PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron. 2003;37:963–975. doi: 10.1016/s0896-6273(03)00125-9. [DOI] [PubMed] [Google Scholar]
- 13.Osteen JD, Gonzalez C, Sampson KJ, Iyer V, Rebolledo S, Larsson HP, Kass RS. KCNE1 alters the voltage sensor movements necessary to open the KCNQ1 channel gate. Proc Natl Acad Sci USA. 2010;107:22710–22715. doi: 10.1073/pnas.1016300108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruscic KJ, Miceli F, Villalba-Galea CA, Dai H, Mishina Y, Bezanilla F, Goldstein SA. IKs channels open slowly because KCNE1 accessory subunits slow the movement of S4 voltage sensors in KCNQ1 pore-forming subunits. Proc Natl Acad Sci USA. 2013;110:E559–E566. doi: 10.1073/pnas.1222616110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun X, Zaydman MA, Cui J. Regulation of voltage-activated K(+) channel gating by transmembrane beta subunits. Front Pharmacol. 2012;3:63. doi: 10.3389/fphar.2012.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morita H, Wu J, Zipes DP. The QT syndromes: long and short. Lancet. 2008;372:750–763. doi: 10.1016/S0140-6736(08)61307-0. [DOI] [PubMed] [Google Scholar]
- 17.Moss AJ, Kass RS. Long QT syndrome: from channels to cardiac arrhythmias. J Clin Invest. 2005;115:2018–2024. doi: 10.1172/JCI25537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldenberg I, Moss AJ. Long QT syndrome. J Am Coll Cardiol. 2008;51:2291–2300. doi: 10.1016/j.jacc.2008.02.068. [DOI] [PubMed] [Google Scholar]
- 19.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 20.Ackerman MJ. The long QT syndrome: ion channel diseases of the heart. Mayo Clin Proc. 1998;73:250–269. doi: 10.4065/73.3.250. [DOI] [PubMed] [Google Scholar]
- 21.Choi G, Kopplin LJ, Tester DJ, Will ML, Haglund CM, Ackerman MJ. Spectrum and frequency of cardiac channel defects in swimming-triggered arrhythmia syndromes. Circulation. 2004;110:2119–2124. doi: 10.1161/01.CIR.0000144471.98080.CA. [DOI] [PubMed] [Google Scholar]
- 22.Peroz D, Rodriguez N, Choveau F, Baro I, Merot J, Loussouarn G. Kv7.1 (KCNQ1) properties and channelopathies. J Physiol. 2008;586:1785–1789. doi: 10.1113/jphysiol.2007.148254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moss AJ, Goldenberg I. Importance of knowing the genotype and the specific mutation when managing patients with long QT syndrome. Circ Arrhythm Electrophysiol. 2008;1:213–226. doi: 10.1161/CIRCEP.108.796599. discussion 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimizu W. The long QT syndrome: therapeutic implications of a genetic diagnosis. Cardiovasc Res. 2005;67:347–356. doi: 10.1016/j.cardiores.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 25.Priori SG, Barhanin J, Hauer RN, Haverkamp W, Jongsma HJ, Kleber AG, McKenna WJ, Roden DM, Rudy Y, Schwartz K, Schwartz PJ, Towbin JA, Wilde AM. Genetic and molecular basis of cardiac arrhythmias: impact on clinical management part III. Circulation. 1999;99:674–681. doi: 10.1161/01.cir.99.5.674. [DOI] [PubMed] [Google Scholar]
- 26.Priori SG, Barhanin J, Hauer RN, Haverkamp W, Jongsma HJ, Kleber AG, McKenna WJ, Roden DM, Rudy Y, Schwartz K, Schwartz PJ, Towbin JA, Wilde AM. Genetic and molecular basis of cardiac arrhythmias: impact on clinical management parts I and II. Circulation. 1999;99:518–528. doi: 10.1161/01.cir.99.4.518. [DOI] [PubMed] [Google Scholar]
- 27.Chouabe C, Neyroud N, Guicheney P, Lazdunski M, Romey G, Barhanin J. Properties of KvLQT1 K+ channel mutations in Romano-Ward and Jervell and Lange-Nielsen inherited cardiac arrhythmias. EMBO J. 1997;16:5472–5479. doi: 10.1093/emboj/16.17.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chouabe C, Neyroud N, Richard P, Denjoy I, Hainque B, Romey G, Drici MD, Guicheney P, Barhanin J. Novel mutations in KvLQT1 that affect Iks activation through interactions with Isk. Cardiovasc Res. 2000;45:971–980. doi: 10.1016/s0008-6363(99)00411-3. [DOI] [PubMed] [Google Scholar]
- 29.Ghosh S, Nunziato DA, Pitt GS. KCNQ1 assembly and function is blocked by long-QT syndrome mutations that disrupt interaction with calmodulin. Circ Res. 2006;98:1048–1054. doi: 10.1161/01.RES.0000218863.44140.f2. [DOI] [PubMed] [Google Scholar]
- 30.Marx SO, Kurokawa J, Reiken S, Motoike H, D'Armiento J, Marks AR, Kass RS. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science. 2002;295:496–499. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- 31.Shamgar L, Ma L, Schmitt N, Haitin Y, Peretz A, Wiener R, Hirsch J, Pongs O, Attali B. Calmodulin is essential for cardiac IKS channel gating and assembly: impaired function in long-QT mutations. Circ Res. 2006;98:1055–1063. doi: 10.1161/01.RES.0000218979.40770.69. [DOI] [PubMed] [Google Scholar]
- 32.Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long-QT syndrome: clinical impact. Circulation. 1999;99:529–533. doi: 10.1161/01.cir.99.4.529. [DOI] [PubMed] [Google Scholar]
- 33.Yang T, Xu X, Kernan T, Wu V, Colecraft HM. Rem, a member of the RGK GTPases, inhibits recombinant CaV1.2 channels using multiple mechanisms that require distinct conformations of the GTPase. J Physiol. 2010;588:1665–1681. doi: 10.1113/jphysiol.2010.187203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sekine-Aizawa Y, Huganir RL. Imaging of receptor trafficking by using alpha-bungarotoxin-binding-site-tagged receptors. Proc Natl Acad Sci USA. 2004;101:17114–17119. doi: 10.1073/pnas.0407563101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fang K, Colecraft HM. Mechanism of auxiliary beta-subunit-mediated membrane targeting of L-type (Ca(V)1.2) channels. J Physiol. 2011;589:4437–4455. doi: 10.1113/jphysiol.2011.214247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subramanyam P, Chang DD, Fang K, Xie W, Marks AR, Colecraft HM. Manipulating L-type calcium channels in cardiomyocytes using split-intein protein transsplicing. Proc Natl Acad Sci USA. 2013;110:15461–15466. doi: 10.1073/pnas.1308161110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Colecraft HM, Alseikhan B, Takahashi SX, Chaudhuri D, Mittman S, Yegnasubramanian V, Alvania RS, Johns DC, Marban E, Yue DT. Novel functional properties of Ca(2+) channel beta subunits revealed by their expression in adult rat heart cells. J Physiol. 2002;541:435–452. doi: 10.1113/jphysiol.2002.018515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen H, Puhl HL, III, Koushik SV, Vogel SS, Ikeda SR. Measurement of FRET efficiency and ratio of donor to acceptor concentration in living cells. Biophys J. 2006;91:L39–L41. doi: 10.1529/biophysj.106.088773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen H, Puhl HL, III, Ikeda SR. Estimating protein-protein interaction affinity in living cells using quantitative Forster resonance energy transfer measurements. J Biomed Opt. 2007;12:054011. doi: 10.1117/1.2799171. [DOI] [PubMed] [Google Scholar]
- 40.Wiener R, Haitin Y, Shamgar L, Fernandez-Alonso MC, Martos A, Chomsky-Hecht O, Rivas G, Attali B, Hirsch JA. The KCNQ1 (Kv7.1) COOH terminus, a multitiered scaffold for subunit assembly and protein interaction. J Biol Chem. 2008;283:5815–5830. doi: 10.1074/jbc.M707541200. [DOI] [PubMed] [Google Scholar]
- 41.Haitin Y, Wiener R, Shaham D, Peretz A, Cohen EB, Shamgar L, Pongs O, Hirsch JA, Attali B. Intracellular domains interactions and gated motions of I(KS) potassium channel subunits. EMBO J. 2009;28:1994–2005. doi: 10.1038/emboj.2009.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanki H, Kupershmidt S, Yang T, Wells S, Roden DM. A structural requirement for processing the cardiac K+ channel KCNQ1. J Biol Chem. 2004;279:33976–33983. doi: 10.1074/jbc.M404539200. [DOI] [PubMed] [Google Scholar]
- 43.Howard RJ, Clark KA, Holton JM, Minor DL., Jr Structural insight into KCNQ (Kv7) channel assembly and channelopathy. Neuron. 2007;53:663–675. doi: 10.1016/j.neuron.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Piippo K, Swan H, Pasternack M, Chapman H, Paavonen K, Viitasalo M, Toivonen L, Kontula K. A founder mutation of the potassium channel KCNQ1 in long QT syndrome: implications for estimation of disease prevalence and molecular diagnostics. J Am Coll Cardiol. 2001;37:562–568. doi: 10.1016/s0735-1097(00)01124-4. [DOI] [PubMed] [Google Scholar]
- 45.Park KH, Piron J, Dahimene S, Merot J, Baro I, Escande D, Loussouarn G. Impaired KCNQ1-KCNE1 and phosphatidylinositol-4,5-bisphosphate interaction underlies the long QT syndrome. Circ Res. 2005;96:730–739. doi: 10.1161/01.RES.0000161451.04649.a8. [DOI] [PubMed] [Google Scholar]
- 46.Nakajo K, Ulbrich MH, Kubo Y, Isacoff EY. Stoichiometry of the KCNQ1—KCNE1 ion channel complex. Proc Natl Acad Sci USA. 2010;107:18862–18867. doi: 10.1073/pnas.1010354107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilkins ME, Li X, Smart TG. Tracking cell surface GABAB receptors using an alpha-bungarotoxin tag. J Biol Chem. 2008;283:34745–34752. doi: 10.1074/jbc.M803197200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu X, Kanda VA, Choi E, Panaghie G, Roepke TK, Gaeta SA, Christini DJ, Lerner DJ, Abbott GW. MinK-dependent internalization of the IKs potassium channel. Cardiovasc Res. 2009;82:430–438. doi: 10.1093/cvr/cvp047. [DOI] [PMC free article] [PubMed] [Google Scholar]