

Abstract
Aims
Cardiac β-adrenergic receptors (β-AR) are key regulators of cardiac haemodynamics and size. The scaffolding protein A-kinase anchoring protein 79/150 (AKAP5) is a key regulator of myocardial signalling by β-ARs. We examined the function of AKAP5 in regulating cardiac haemodynamics and size, and the role of β-ARs and Ca2+-regulated intracellular signalling pathways in this phenomenon.
Methods and results
We used echocardiographic, histological, genetic, and biochemical methods to examine the effect of ablation of AKAP5 on cardiac haemodynamics, size, and signalling in mice. AKAP5−/− mice exhibited enhanced signs of cardiac dilatation and dysfunction that progressed with age. Infusions of isoprenaline worsened cardiac haemodynamics in wild-type (WT) mice only, but increased the ratio of heart-to-body weight equally in WT and in AKAP5−/− mice. Mechanistically, loss of AKAP5 was associated with enhanced activity of cardiac calmodulin kinase II (CaMKII) and calcineurin (CaN) as indexed by nuclear factor of activated T-cell-luciferase activity. Loss of AKAP5 interfered with the recycling of cardiac β1-ARs, which was mediated in part by CaN binding to AKAP5. Carvedilol reversed cardiac hypertrophy and haemodynamic deficiencies in AKAP5−/− mice by normalizing the activities of cardiac CaN and CaMKII.
Conclusions
These findings identify a novel cardioprotective role for AKAP5 that is mediated by regulating the activities of cardiac CaN and CaMKII and highlight a significant role for cardiac β-ARs in this phenomenon.
Keywords: AKAP, Cardiac hypertrophy, β-Adrenergic receptors, Calcineurin, Calmodulin kinase II
1. Introduction
Activation of cardiac β-adrenergic receptors (β-ARs) by catecholamines promotes acute elevation of cyclic AMP, which results in chronotropic and inotropic effects on the heart muscle. A-kinase anchoring proteins (AKAPs) bind protein kinase A (PKA) and other proteins and compartmentalize them into signalling complexes.1 Among the more than 50 AKAPs that have been identified, AKAP5 has attracted a significant attention. AKAP5 is widely expressed in the periphery and plays a major role in forming discrete signalling networks. AKAP5 binds to PKA, protein kinase C and calcineurin (CaN), and other signalling molecules, such as adenylyl cyclase (AC) 5/6, voltage-gated l-type calcium channels (LTCCs), and post-synaptic density forming proteins such as SAP97.2,3 A relationship between cardiac β1-ARs and AKAP5 was determined by immunohistochemical methods that revealed close association between post-synaptic β1-AR and AKAP5.4,5
Studies in cardiac myocytes derived from AKAP5−/− mice indicated that AKAP5 exerted effects on the regulation of Ca2+ signalling and trafficking of the β1-AR.6,7 Nichols et al.7 determined that β-AR-dependent increase in [Ca2+]i and β-AR-mediated regulation of excitation–contraction coupling were eliminated in AKAP5−/− myocytes. The defect in β-AR-mediated activation of [Ca2+]i was eventually traced to a subpopulation of LTCCs that were associated simultaneously with AKAP5 and cardiac caveolin 3.7 Li et al.8 determined that signalling by β1-AR to cyclic AMP was more pronounced in neonatal AKAP5−/− neonatal mouse ventricular myocytes (NMVM). Sensitization of β1-AR signalling in AKAP5−/− cardiac myocytes resulted in increased cardiomyocyte contractility and size in response to β1-agonists. In addition, intracellular trafficking of the β1-AR was impaired in AKAP5−/− cardiac myocytes and restored by the expression of AKAP5. These findings indicate that AKAP5 was a major AKAP involved in regulating the physiology and trafficking of cardiac β1-AR.6–8
In this study, we determined cardiac parameters in AKAP5−/− vs. wild-type (WT) AKAP5+/+ mice to determine the effect of global ablation of AKAP5 on cardiac function. Our results indicate that AKAP5 was actually cardioprotective, because its deletion was associated with significant cardiac hypertrophy and insufficiency that were reversed by the mixed α- and β-blocker carvedilol. These results are the first to indicate a clear role for an AKAP in cardiac remodelling and in regulating cardiac haemodynamics.
2. Methods
2.1. Mice and rats
AKAP5+/+ and AKAP5−/− mice have previously been described.7,8 Animal experiments were performed according to protocols that were approved by the University of Tennessee-HSC Institutional Animal Care and Use Committee. Studies conformed to the ‘Guide for Care and Use of Laboratory Animals' published by the US National Institutes of Health (NIH publication No. 85-23, revised 1996). All mice were in the C57Bl6/J background and backcrossed >10 generations. Spargue–Dawley rats were used as the source of neonatal cardiac myocytes. Mice or rats were sacrificed by barbiturate overdose (pentobarbital, 150 mg/kg, i.p.).
2.2. Drug administration
One hour prior to placement of the Alzet mini-pump, each mouse received 0.125 mg/g of carprofen analgesic, i.p. Four-month-old mice were anaesthetized by inhalation of 2% isofluorane and the back area slightly posterior to the scapulae was shaved and wiped in betadine. A 0.5 cm skin incision was made and an Alzet model 2002 mini-pump that was filled with buffer (1% ascorbic acid in PBS) or isoprenaline (40 mg/kg/d) was inserted subcutaneously. The wound was closed with a 7 mm clip, followed by the application of triple antibiotic ointment. Pumps were removed 24 h before echocardiography following the same procedure used to implant them. Mouse chow containing placebo or carvedilol 1500 mg/kg was provided ad libitum to 4-month-old AKAP+/+ or AKAP5−/− mice for 10 days.9,10 Cyclosporine A (CsA, 10 mg/kg) was injected i.p. daily for 2 weeks to matched littermates of 4-month-old AKAP5+/+ or AKAP5−/− mice of either sex.
2.3. Echocardiography and histology
Mice were anaesthetized with isofluorane (2%) and placed on the recording stage of a Vevo 2100 ultrasound and imaged using an 18–32 MHz MS400 transducer. The anterior chest was shaved and ultrasonic gel applied. Two-dimensional images were recorded in parasternal long- and short-axis projections with guided M-mode recordings at the midventricular level in both views. M-mode echocardiographic measurements were obtained before and after each treatment.
2.4. Immunohistochemistry
After echocardiography, mice or rats were sacrificed by barbiturate overdose. Hearts were excised, cleaned, and embedded in tissue freezing medium at −40°C and sectioned. Eight-micron slices were adhered to silanated glass slides and stained with haematoxylin–eosin or Masson trichrome. Other tissue sections were permeabilized in 0.2% Triton X-100 in PBS for 10 min at 4°C, incubated with anti-sarcomeric α-actinin and then incubated with a 1 : 100 dilution of rhodamine-conjugated goat anti-mouse (EMD Biosciences, San Diego, CA, USA), and analysed by immunocytochemistry.
2.5. Confocal recycling microscopy protocol
Cardiac myocytes were prepared from 1-day-old Sprague–Dawley rats and cultured on collagen-coated cover slips as described.5,6,8 Myocytes were infected for 6 h with adenovirus expressing WT FLAG-tagged β1-AR, WT FLAG-tagged β2-AR, or activated CaN (JMAd-03, Seven Hills Bioreagents, Cincinnati, OH, USA) at multiplicity of infection (m.o.i.) of 100. Myocytes were washed and cultured for an additional 24–36 h before use. Cardiomyocytes were incubated with Cy3-labelled anti-FLAG IgG (5–10 µg/mL) and with 1 µM of FK-506 or buffer (1% DMSO) for 1 h at 37°C. HEK-293 cells stably expressing 1.2 pmol/mg protein of FLAG-tagged β1-AR were transiently transfected with pEGFP-N1, AKAP79(Δ321–360) in pEGFP-N1, or PVIVIT in pEGFP-N1 for 2 days and then used.8,11,12 Internalization and recycling were conducted as described,5,6,8 followed by confocal microscopy using Cy3 (pseudo-red, for β-AR), DAPI (pseudo-violet, for nuclei), and GFP (pseudo-green) laser settings of the Olympus FluoView™ FV-1000 confocal microscope.
2.6. Measurement of the activity of luciferase-linked to nuclear factor of activated T-cells
AKAP5−/− mice were bred with mice that express luciferase upon the activation of nuclear NFAT (nuclear factor of activated T-cells; Jackson Laboratories, stock 013781). Hearts from AKAP5+/+/NFAT-luc or AKAP5−/−/NFAT-luc were excised from age-matched mice and fresh frozen with liquid N2. Hearts were powdered and then extracted in 1× luciferase lysis buffer (Promega) with protease inhibitors by three freeze/thaw cycles. Clarified cardiac lysates were used to measure the enzymatic activity of luciferase by a commercial kit (Luciferase Assay System, Promega Corp.). Light intensity was measured with a Turner-20 luminometer over 10 s and expressed as relative light units (RLUs) over 10 s/mg protein.
2.7. CaN activity assay
Phosphatase activity was measured using the CaN cellular activity kit (Enzo Life sciences), according to manufacturer's instructions. Cardiac extracts from either three AKAP5+/+ or three AKAP5−/− mice were prepared, and CaN activity was measured as the dephosphorylation rate of a synthetic phosphopeptide substrate (RII peptide). The amount of PO4 released was determined colorimetrically with the BIOMOL GREEN™ reagent.
2.8. Measurement of the activity of cardiac calmodulin kinase II
Calmodulin kinase II (CaMKII) activity in cardiac homogenates was assayed in a total volume of 25 µL with the use of a biotinylated CaMKII peptide (SignaTect™ CaMKII kit, Promega). Hearts were powdered and extracted by three freeze/thaw cycles in extraction buffer [20 mM Tris–HCL, (pH 8.0), 2 mM EDTA and 2 mM EGTA, and 2 mM dithiothreitol] with protease inhibitors. The assay contained 50 mM Tris (pH 7.4), 20 µM biotinylated CaMKII substrate, 0.1 mM ATP ([γ-32P]ATP = 1 µCi per assay), 10 mM magnesium acetate, and 0.5 mM dithiothreitol. CaMKII activity was measured in the presence of ±1 mM CaCl2 and ±1 µM calmodulin. The reaction was initiated by adding 10 µg of the homogenate, followed by incubation for 3 min at 30°C. Incorporation of 32P in the peptide substrate was determined following the manufacturer's instructions. Cardiac homogenates prepared from age-matched AKAP5+/+ or AKAP5−/− mice were subjected to immunoblotting with anti-δCaMKII (Santa Cruz) or anti-phospho (Thr286) CaMKII (Cell Signaling Technology), followed by densitometric scanning of each band to determine optical density ratios.
2.9. Immunoprecipitation
CaNα in pcDNA3.0 myc/His was ectopically expressed in HEK-293 cells with either empty p-EGFP-N1 or with AKAP5(321–360)-GFP.11 Cells were lysed and equal amounts of protein were subjected to affinity-purification on anti-AKAP5-IgG or pre-immune IgG beads. Then, equal volumes of eluate were probed for co-immunoprecipitation (IP) of CaNα by western blotting using anti-Myc (9E10) antibody (Santa Cruz Biotechnology) as described.
2.10. Statistical analysis
All data are expressed as mean ± SE. The significance of differences between baseline and drug treatment within same genetic species and between pre-carvedilol and post-carvedilol echocardiographic values within the group was evaluated by Student's t-test. To evaluate differences in haemodynamic parameters among groups of transgenic mice, multigroup comparisons were made by two-way ANOVA followed by the Bonferroni post hoc test, using the Graph Pad Prism™ software. In addition, two-way ANOVA with Bonferroni correction for multiple comparisons was used to evaluate the morphometric measurements and enzymatic activities. Statistical significance was defined as P < 0.05.
3. Results
3.1. Ablation of AKAP5 leads to ventricular hypertrophy
AKAP5−/− mice did not display obvious anatomical, growth, or productive changes.7 However, deletion of AKAP5 resulted in progressive ventricular hypertrophy (Figure 1A and B). Histological sections confirmed enlarged left ventricular (LV) chambers in 3-month-old AKAP5−/− compared with AKAP5+/+ mice and the enlargement progressed with age (Figure 1A). Body weights and left tibial lengths were not different, but there was a ∼16% increase (P < 0.05) in the ratio of heart-to-body weight between AKAP5−/− and AKAP5+/+ mice (Figure 1B). These differences were apparent when heart weights were normalized to body weight showing a 16% increase in the ratios at 4 months (P < 0.05). Cross-sectional area of cardiac cells in AKAP5−/− mice at 4 months (679 ± 100 µm2) was significantly larger than the corresponding parameter (439 ± 103 µm2) in AKAP5+/+ littermates (Figure 1C and D). In addition, hearts from AKAP5−/− mice manifested ventricular myocyte interstitial fibrosis and increased collagen deposition (Figure 1E and F). Deletion of AKAP5 induced marked up-regulation in the expression of molecular markers of hypertrophy,13 such as atrial and brain natriuretic peptides, and the ratio of β- to α-MHC (Figure 1G).
Figure 1.
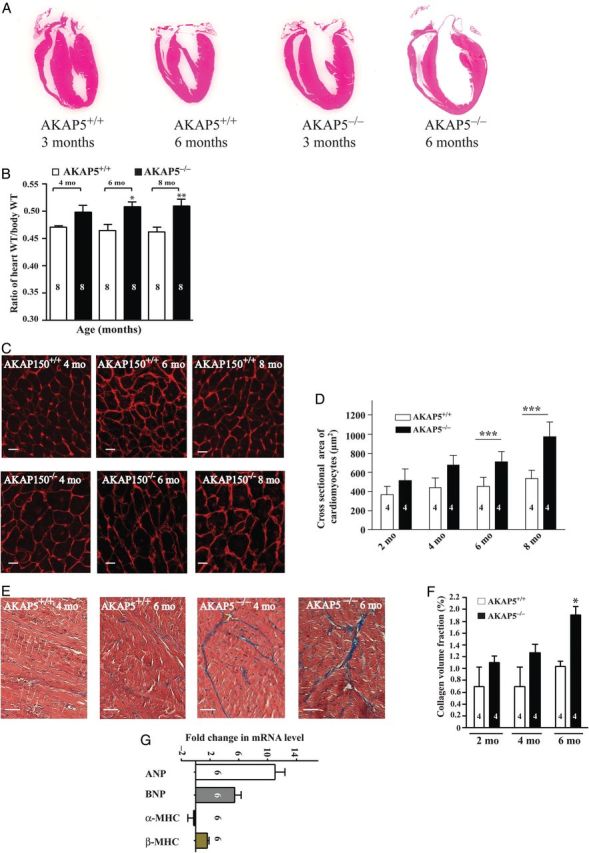
AKAP5−/− mice display a hypertrophic phenotype. (A) Histological sections of WT or AKAP5−/− hearts from 3- or 6-month-old mice. (B) Means ± SE of heart-to-body weight ratios between WT and AKAP5−/− mice at 4, 6, and 8 months of age. (C and D) Cell membrane staining (wheat germ agglutinin) and quantification of cross-sectional areas of heart cells from 4-, 6-, or 8-month-old WT and AKAP5−/− mice. Scale bars represent 25 µm. (E and F) Trichrome staining (fibrosis in blue) and quantification of fibrosis between 4- and 6-month-old WT and AKAP5−/− mice. Scale bars represent 20 µm. (G) RT-PCR analysis of hypertrophic marker gene expression in WT vs. AKAP5−/− mice at 4 months of age. Data were plotted as means ± SE. *P < 0.05, **P < 0.01, and ***P < 0.001 for either base in WT vs. base in knockout (KO) mice.
3.2. Loss of AKAP5 leads to compromised cardiac performance
Cardiac function was monitored by echocardiography in anaesthetized mice (Figure 2A). Loss of AKAP5 led to compromised cardiac function (Figure 2B–F). There was an age-dependent proportional decrease in systolic performance assessed as per cent ejection fraction from 65% in 4-month-old AKAP5+/+ animals to 41% in matched AKAP5−/− littermates (P < 0.01). In addition, stroke volumes were significantly reduced (P < 0.05) in AKAP5−/− mice when compared with age-matched AKAP5+/+ littermates (Figure 2E). Declines in cardiac function were associated with ventricular dilatation and a progressive increase in LV end-systolic and -diastolic dimensions (Figure 2C and D). An echocardiographic estimate of LV volume and necroscopic evaluation of heart-to-body weight indicated that loss of AKAP5 was associated with eccentric hypertrophy and dilatation.14
Figure 2.
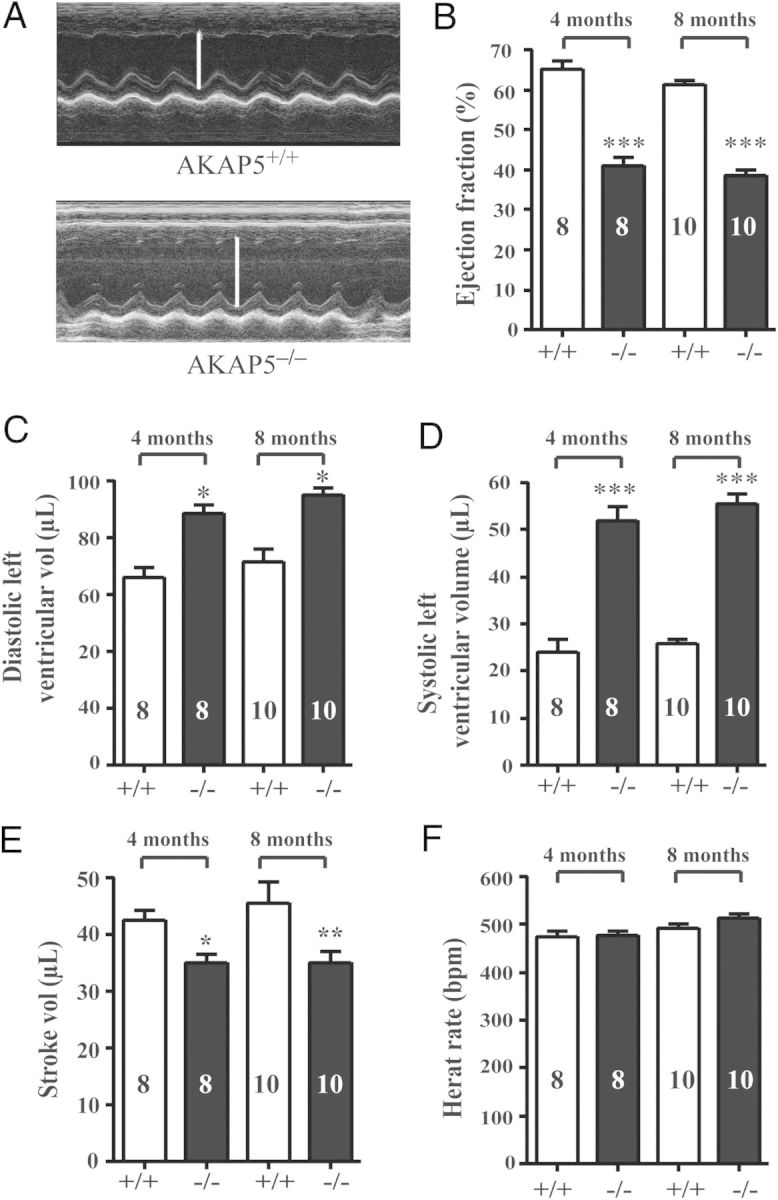
Cardiac echocardiography between WT and AKAP5−/− mice. (A) M-mode echocardiographic analysis of hearts from WT and AKAP5−/− mice. (B–F) Parameters of cardiac haemodynamic function in WT and AKAP5−/− mice. (B) Ejection fraction in %; (C) diastolic LV volume in µL; (D) systolic LV volume in µL; (E) stroke volume in µL and heart rate in bpm. *P < 0.05, **P < 0.01, and ***P < 0.001 represent the P-value for either base in WT vs. base in KO mice.
3.3. AKAP5 mice are resistant to stress-induced cardiac hypertrophy elicited by isoprenaline
Infusions of isoprenaline produce significant haemodynamic insufficiency, cardiac hypertrophy, and remodelling that is associated with significant apoptosis.15,16 To determine the role of AKAP5 in β-AR-induced cardiomyopathy, isoprenaline was infused for 10 days into AKAP5−/− and AKAP5+/+ mice littermates (Figure 3A). In WT AKAP5+/+ mice, isoprenaline led to −20 ± 8% reduction in the ejection fraction (P < 0.05) and significantly increased systolic LV volume by 24 ± 6% without affecting heart rate (Figure 3A).17 In contrast, isoprenaline-mediated changes in cardiac function in AKAP5−/− mice were not statistically significant (Figure 3A). However, isoprenaline increased in the ratio of heart-to-body weight in AKAP5+/+ or in AKAP5−/− mice by 1.5 ± 0.18-fold (P < 0.05 compared with no drug treatment; Figure 3B). Isoprenaline also increased the expression of hypertrophic markers, such as β-MHC, collagen-1a, and collagen-3a, by more than five-fold in AKAP5+/+ and AKAP5−/− mice (Figure 3C).
Figure 3.
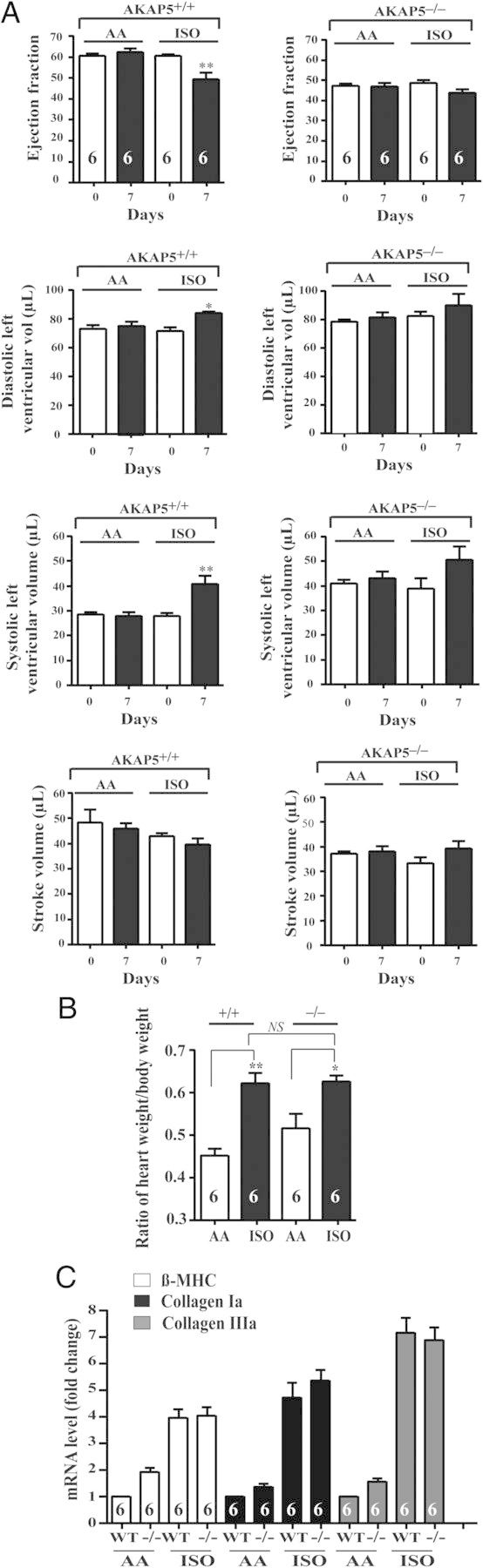
Effect of isoprenaline on cardiac haemodynamics and size in WT vs. AKAP5−/− mice. (A) Two-month-old WT and AKAP5−/− were echoed before the placement of the Alzet mini-osmotic pump. Buffer (1% ascorbic acid in PBS) or isoprenaline (100 mg/mL of buffer) was infused by a model 2002 mini-osmotic pump at a rate of 0.5 µL/h for 10 days. Then, the pumps were removed and the mice were echoed the next day. The data were plotted as means ± SE and then compared among different groups. (B) After the second echocardiography, mice were weighed and the heart was excised, weighed, and followed by extraction of RNA. (C) mRNA expression levels of hypertrophy markers were determined by quantitative RT-PCR. *P < 0.05 and **P < 0.01 represent the P-value of either base in WT vs. base in KO mice or isoprenaline in WT vs. isoprenaline in KO.
3.4. Role of Ca2+/CaM downstream signalling to CaN and CaMKII in haemodynamic dysfunction of AKAP5−/− mice
Nichols et al.7 reported that β-AR-mediated activation of [Ca2+]i via LTCC was impaired in AKAP5−/− cardiac myocytes. Aberrant handling of Ca2+ in cardiomyocytes affects the activity of the Ca2+-binding protein CaM, which regulates the activities of Ca2+/CaM-activated protein phosphatase 2B or CaN and CaMKII.18,19 Hyperactive CaN or CaMKII is associated with cardiac hypertrophy.18–20 To determine the effect of gene deletion of AKAP5 on the activity of cardiac CaN, AKAP5−/− mice were crossed with mice that expressed luciferase under the control of the NFAT nuclear transcription factor.21 This approach is used to determine the activity of CaN in vivo because CaN dephosphorylates and activates cytosolic NFAT.22 The activity of luciferase in cardiac extract from 4-month-old AKAP5+/+-NFAT-LucTg/? mice was 620 ± 80 RLU/min/mg (Figure 4A). In age- and sex-matched AKAP5 knockout mice (AKAP5−/−-NFAT-LucTg/? mice), the activity of CaN as indexed by luciferase increased by an additional three-fold (P < 0.001). Similarly, total CaN phosphatase activity (okadaic acid-resistant and EGTA-sensitive) in AKAP5−/− cardiac extracts was significantly higher (P < 0.05) than in AKAP5+/+ extracts (0.40 ± 0.08 vs. 0.27 ± 0.06 pmol PO4 released/µg protein, respectively; Figure 4B).
Figure 4.
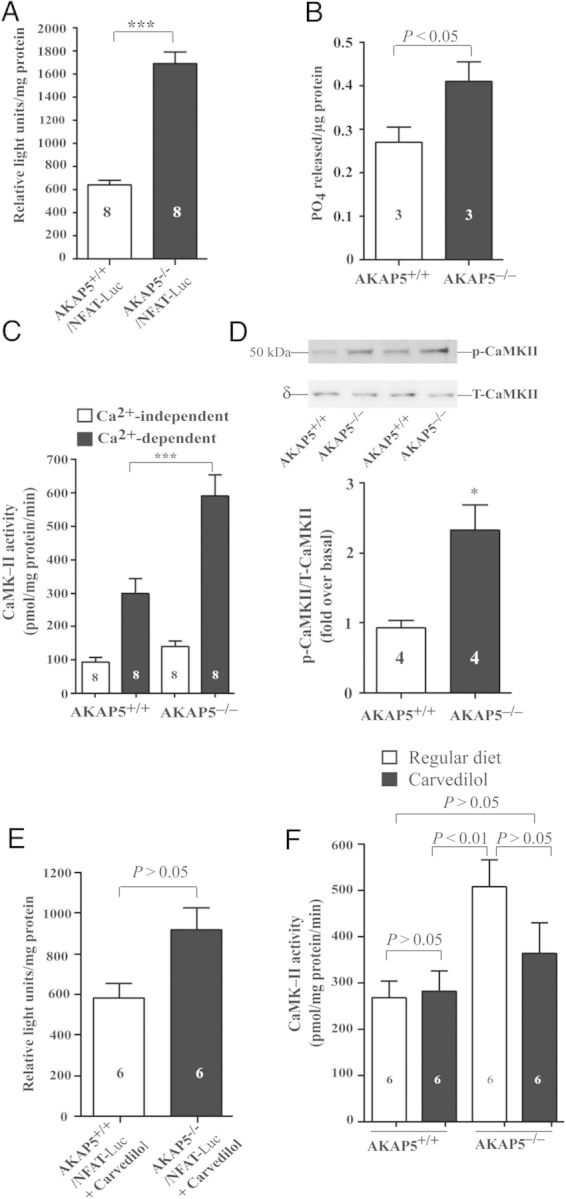
Comparison of the activities of CaN and CaMKII in hearts of transgenic mice that were fed regular chow and chow containing carvedilol. (A) Relative luc levels from cardiac protein extracts from 4-month-old mice containing NFAT-luc, in a WT AKAP5 (AKAP5+/+) background or in a homozygous AKAP5 (AKAP5−/−) background. All data were shown relative to background (no luc transgene), which were set a value of 1. (B) Comparison of the activity of CaN in cardiac extracts from control or AKAP5−/− mice. (C) CaMKII activity assayed in heart homogenate of 4-month-old (AKAP5+/+) or AKAP5−/− mice in the absence of CaM/Ca2+ (open rectangle) or the presence of CaM/Ca2+ (closed rectangle). (D) Ratios of phospho-CaMKII (Thr286)/total δ-CaMKII in cardiac extracts prepared from WT or KO mice. (E) Four-month-old mice containing NFAT-luc, in a WT AKAP5 (AKAP5+/+) background or in a homozygous AKAP5 (AKAP5−/−) background, were fed regular diet or diet containing carvedilol for 10 days. Cardiac extracts were prepared and assayed for the activity of luciferase. (F) CaMKII activity was assayed in the presence of CaM/Ca2+ in heart homogenates prepared from 4-month-old WT or KO mice that were fed regular chow (open square) or chow containing carvedilol (closed square). In (A–E), the P-value represents WT vs. KO. In (E), the P-value represents treatment with carvedilol vs. without carvedilol for each group.
Extracts from hearts of age- and sex-matched 4-month-old AKAP5+/+ and AKAP5−/− mice were compared for activity of CaMKII in the absence or presence of CaM and Ca2+ (Figure 4C). Ca2+ and CaM increased the enzymatic activity of CaMKII by 3 ± 0.5-fold in cardiac extracts from AKAP5+/+ mice and by 6 ± 1.5-fold in cardiac extracts from AKAP5−/− mice (P < 0.001). Activity of CaMKII,21 as indexed by the ratio of anti-phospho-CaMKII (Thr286) over δCaMKII, was elevated in cardiac homogenates prepared from ventricles of AKAP5−/− mice vs. control mice (Figure 4D). Therefore, activities of CaN and CaMKII were both increased in hearts of AKAP5−/− mice.
3.5. Role of CaN and the CaN-binding site in AKAP5 on trafficking of β-AR in rodent cardiac myocytes
β1-AR and β2-AR are internalized in response to isoprenaline and recycle back into the cell membrane of cardiac myocytes or other mammalian cells.5,8 These results were replicated in rat cardiac myocytes that expressed either the β1-AR or the β2-AR (Figure 5A and B). WT β1-AR or WT β2-AR (pseudo-red colour) was expressed on the surface of the cardiomyocyte and then internalized in response to isoprenaline (Figure 5A, a and b, and B, p–q). Removal of isoprenaline promoted the recycling of internal β1-AR or β2-AR back into the myocardial cell membrane (Figure 5A, c–e and B, r–t). Since the activity of CaN was increased in hearts of AKAP5−/− mice, we wondered whether altered CaN activity somehow affected the trafficking of these β-AR. Therefore, the effect of enhancing or suppressing the activity of CaN on trafficking of the β1-AR vs. the β2-AR was determined (Figure 5A and B). In cardiac myocytes that expressed constitutively active CaN, WT β1-AR or β2-AR was internalized and recycled normally (Figure 5A, f–j, and B, u–y). Inhibition of CaN with FK-506 did not affect the internalization of WT β1-AR in response to isoprenaline, but inhibited its recycling (Figure 5A, m–o). In contrast, FK-506 had no effect on the internalization and recycling of WT β2-AR in rat cardiomyocytes (Figure 5B, a′–e′).
Figure 5.
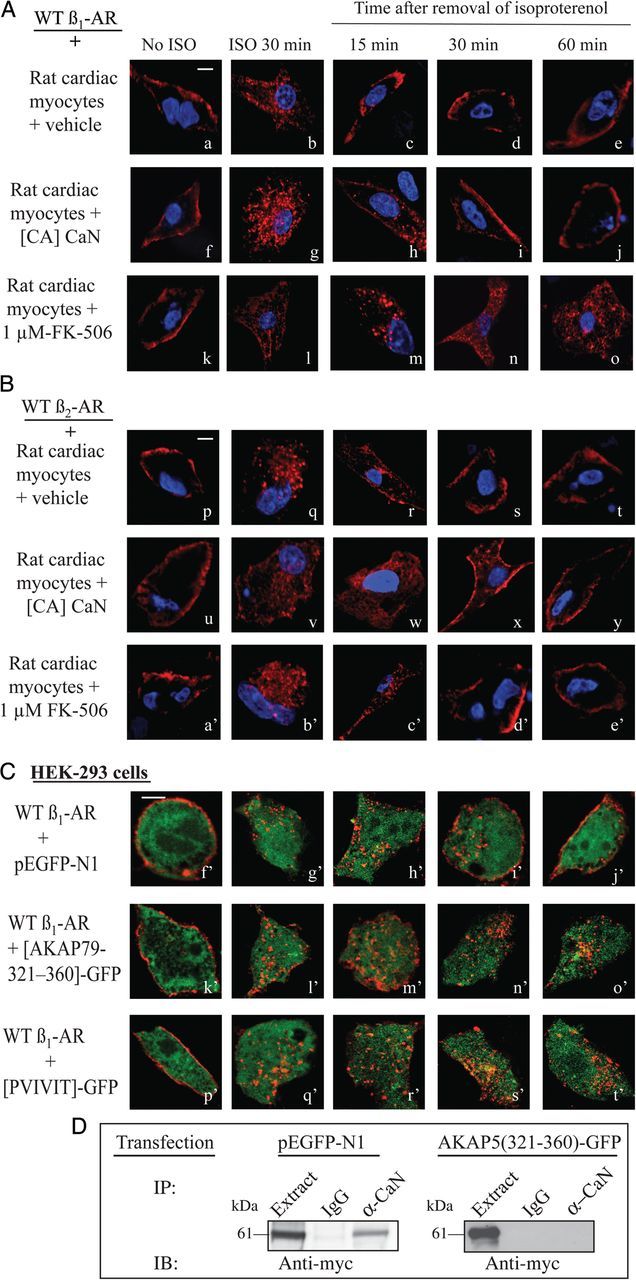
Effect of CaN on trafficking of the β1-AR in cardiac myocytes. (A and B) Rat neonatal cardiac myocytes were infected with 100 m.o.i. with adenoviruses harbouring the FLAG-tagged β1-AR (a–e and k–o) or FLAG-tagged β2-AR (p–t and a′–e′). Another set of cells was infected with 50 m.o.i. of β1-AR adenovirus and 50 m.o.i. of constitutively active (CA) CaN adenovirus (f–j and u–y). Cells infected with the β1-AR or β2-AR adenovirus were exposed to buffer or 1 µM FK-506 and then labelled Cy3-anti-FLAG IgG. Cells were washed, fixed (a, f, k, p, u, and a′), or exposed to 10 µM isoprenaline for 30 min and then fixed (b, g, l, q, v, and b′). The remaining slides were incubated with 100 µM alprenolol and subjected to recycling conditions for the indicated time period and then fixed. Shown is pseudo-colour ‘red’ for the β1-AR and ‘violet’ for nuclei. (C) HEK-293 cells stably expressing FLAG β1-AR were transiently transfected with the empty pEGFP-N1 (f′–j′), AKAP5 (321–360)-GFP (k′–o′), or with PVIVIT-GFP (p′–t′) vector then processed for trafficking as described above. Scale bars represent 5 µm. (D) Extracts from HEK-293 cells expressing AKAP5 and either pEGFP-N1 or AKAP5 (312–360)-GFP peptide were IP’s on anti-AKAP5 or pre-immune resin. Then extracts (2% of total) or eluate were subjected to western blotting and probed with anti-myc antibody to detect α-CaN.
These findings are the first to show that inhibition of CaN differentially interfered with the trafficking of the β1-AR, but how this might occur is unknown. As outlined earlier, deletion of AKAP5 also interfered with recycling of the WT β1-AR in AKAP5−/− NMVM without affecting the trafficking of WT β2-AR in these cells.8 Therefore, we wondered whether CaN bound to AKAP5 was primarily responsible for this selectivity. We addressed this question by repeating the recycling experiments in cells that expressed the WT β1-AR along with peptides that competed with endogenous CaN in binding to AKAP5 (Figure 5C). Plasmids harbouring the cDNA for a GFP-tagged peptide corresponding to the CaN-binding site between amino acids 321–360 of AKAP5 and another that expressed the core VIVIT sequence that binds avidly to CaN were used.11,12 Binding to AKAP5 was verified by co-IP's, where by ectopic expression of AKAP5 (312–360)-GFP interfered with AKAP5 binding to CaN, while soluble GFP did not (Figure 4D). Expression of the empty GFP vector ‘pseudo-green colour’ did not interfere with the membranous localization, agonist-mediated internalization, or recycling of internal WT β1-AR ‘pseudo-red colour’ (Figure 5C, f′–j′). However, recycling of the agonist-internalized β1-AR was inhibited in cells that expressed the CaN-binding peptides (Figure 5C, k′–o′ and p′–t′). These results along with the findings of Li et al.8 indicate that CaN bound to AKAP5 was critical for recycling of cardiac β1-AR.
3.6. Reversal of the AKAP5−/− cardiac phenotype with carvedilol but not with CsA
To find out if the compromised cardiac performance associated with loss of AKAP5 bears similarities to cardiac insufficiency and failure in humans, we determined the effect of carvedilol on these parameters in age-matched AKAP5+/+ vs. AKAP5−/− mice. Carvedilol is a non-selective β-blocker as well as an α1-AR blocker that is widely used in the management of heart failure in humans.10 Carvedilol was administered to 4-month-old mice in chow at a concentration of 1500 p.p.m. for 10 days.11 Echocardiography of WT AKAP5+/+ mice before and after carvedilol indicated that this compound has no effect on haemodynamic parameters (Figure 6). Echocardiography of AKAP5−/− littermates, however, showed a significant improvement in cardiac performance in both male and female mice (Figure 6). Carvedilol increased the ejection fraction by 26 ± 4%, markedly reduced systolic LV volumes by 43 ± 8% in AKAP5−/− mice, and produced a 30 ± 6% increase in the stroke volume (Figure 6A). In aggregate, these results indicate that haemodynamic parameters in AKAP5−/− after carvedilol were not significantly different from those in untreated AKAK5+/+ mice (P > 0.05, n = 8).
Figure 6.
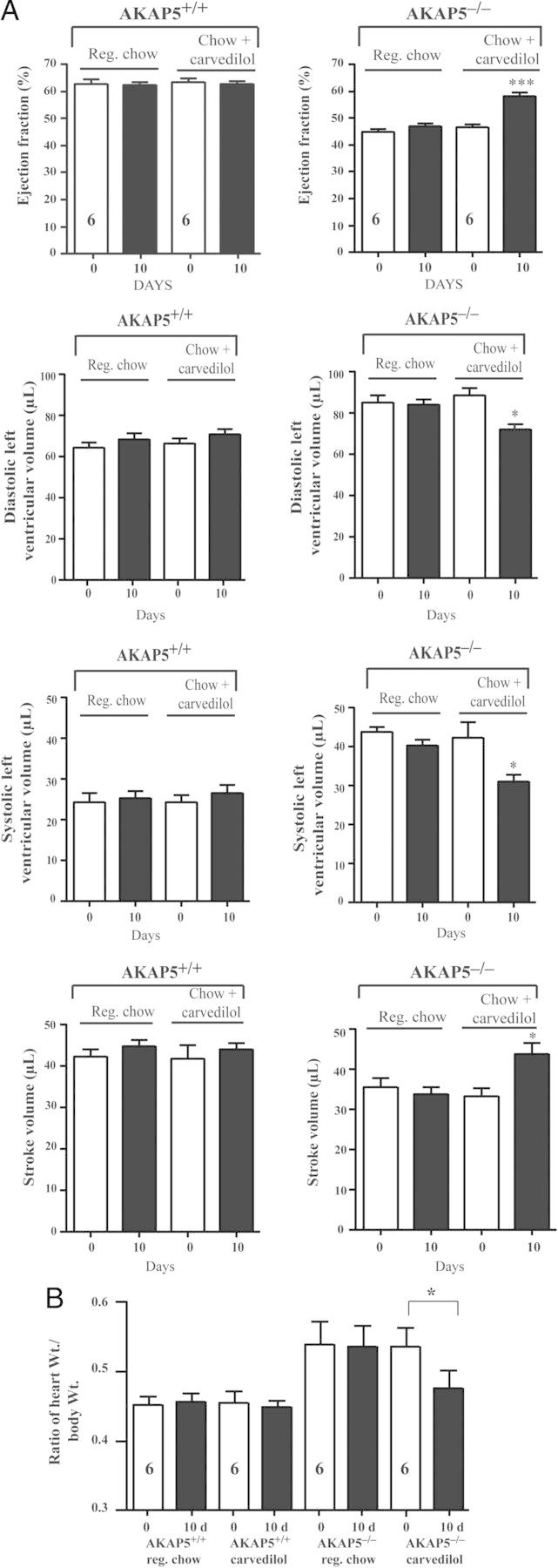
Effect of carvedilol on cardiac haemodynamics in AKAP5+/+ vs. AKAP5−/− mice. (A) Four-month-old AKAP5+/+ or AKAP5−/− mice were echoed. Then, fed control chow or chow with 1500 p.p.m. of carvedilol for 10 days and echoed again. (B). Ratios of heart-to-body weight. The data were plotted as means ± SE and then compared among different groups. n, represents the number of mice in each condition. In (A), *P < 0.05 and ***P < 0.001 represent the P-value of either base in WT vs. base in KO mice or carvedilol in WT vs. carvedilol in KO. In (B), the P-value represents either base in WT vs. base in KO or carvedilol in WT vs carvedilol in KO mice.
Since ablation of the AKAP5 locus resulted in significant activation of CaN, we evaluated whether inhibition of CaN with CsA would affect the cardiac phenotype of AKAP5−/− mice (Table 1). A 14-day regimen of subcutaneous administration of 10 mg/kg/day of CsA did not produce a statistically significant effect on cardiac haemodynamics either in AKAP5+/+ or in AKAP5−/− mice.
Table 1.
Effect of CsA on echocardiographic parameters in WT vs. AKAP5 KO littermates
| Solvent AKAP5+/+ |
Cyclosporine A AKAP5+/+ |
Solvent AKAP5−/− |
Cyclosporine A AKAP5−/− |
|||||
|---|---|---|---|---|---|---|---|---|
| Before | After | Before | After | Before | After | Before | After | |
| (n = 8) | (n = 8) | (n = 8) | (n = 8) | |||||
| EF (%) | 63.1 ± 4.6 | 63.6 ± 3.6 | 62.5 ± 2.9 | 60.6 ± 3.1 | 46.9 ± 2.5 | 46.6 ± 3.4 | 46.7 ± 2.7 | 50.5 ± 3.6 |
| FS (%) | 43.8 ± 2.2 | 44.0 ± 2.4 | 43.4 ± 2.3 | 42.8 ± 2.4 | 33.4 ± 1.5 | 33.0 ± 2.7 | 33.7 ± 3.3 | 35.5 ± 3.0 |
| LV Vold (µL) | 64.1 ± 4.6 | 65.0 ± 5.3 | 65.9 ± 4.3 | 59.7 ± 5.7 | 79.2 ± 6.3 | 79.3 ± 6.9 | 78.4 ± 6.1 | 76.4 ± 6.3 |
| LV Vols (µL) | 23.7 ± 3.4 | 23.8 ± 3.7 | 24.9 ± 2.8 | 22.7 ± 3.2 | 42.9 ± 4.6 | 42.5 ± 3.5 | 41.6 ± 5.5 | 38.2 ± 4.4 |
| SV (µL) | 40.4 ± 4.1 | 41.3 ± 4.3 | 41.0 ± 4.0 | 36.9 ± 4.4 | 37.2 ± 4.8 | 36.8 ± 4.3 | 36.7 ± 3.8 | 38.2 ± 4.2 |
| CO/BW | 0.74 | 0.75 | 1.02 | 0.93 | 0.77 | 0.71 | 0.69 | 0.74 |
Echocardiography was before and after the administration of 10 mg CsA /kg/day or solvent for 14 days to 4-month-old mice.
EF, ejection fraction; FS, Fractional sgortening; LV Vold, diastolic left ventricular volume; LV Vols, systolic left ventricular volume; SV, stroke volume; CO/BW, ratio of cardiac output/min/body weight in grams.
Next, we determined if improvement in cardiac function of AKAP5−/− mice by carvedilol was mediated, in part, by modulating the activities of CaN and CaMKII. Four-month-old AKAP5+/+-NFAT-LucTg/? mice and AKAP5−/−-NFAT-LucTg/? mice were fed chow supplemented with carvedilol for 10 days (Figure 4E). Activities of luciferase in cardiac extract from carvedilol-fed AKAP5+/+-NFAT-LucTg/? mice or AKAP5−/−-NFAT-LucTg/? mice were 555 ± 50 and 919 ± 104 RLU/min/mg, respectively (P > 0.05).
The effect of carvedilol on CaMKII was determined in 4-month-old AKAP5+/+ vs. AKAP5−/− mice that were fed chow or chow containing carvedilol for 10 days (Figure 4F). Activities of CaMKII in heart homogenates of AKAP5+/+ mice on regular chow or chow with carvedilol were 269 ± 36 and 283 ± 44 pmol/mg protein/min, respectively (P> 0.05). Activities of CaMKII in heart homogenates of AKAP5−/− mice on regular chow or chow containing carvedilol were 509 ± 58 and 364 ± 66 pmol/mg protein/min, respectively (P > 0.05). Therefore, the activity of CaMKII in carvedilol-fed AKAP5−/− mice was not significantly different from that of CaMKII in AKAP5+/+ without or with carvedilol (P > 0.05). In addition, carvedilol normalized the activity of CaMKII (Figure 4D) and the ratio of heart-to-body weight (Figure 6B) in AKAP5−/− mice to its levels in AKAP5+/+ mice. These results indicate that AKAP5 is cardioprotective by virtue of its inhibition of cardiac CaN and CaMKII signalling cascades and highlight a link between AKAP5 and cardiac β1-AR in this phenomenon.
4. Discussion
4.1. Role of AKAPs in cardiac physiology and pathology
Several approaches have been used to investigate the role of the more than 14 cardiac AKAPs in regulating a variety of cardiac events. Knockdown of muscle AKAPβ with an siRNA in cardiac myocytes inhibited α- and β-adrenergic agonist-induced hypertrophy.23 siRNA-mediated knockdown of cardiac Yotiao (AKAP9) indicated that this AKAP was involved in regulating the activity of cardiac potassium channels and β-adrenergic signalling.24 The gene deletion approach was used in characterizing the physiological function of AKAPlbc (AKAP13), AKAP5, and AKAP15/18 (AKAP7). Ablation of AKAP13 was embryonically lethal, indicating that this AKAP was involved in genetic programming.25 Deletion of the various isoforms of AKAP7 did not affect the responsiveness of cardiomyocytes to adrenergic stimulation.26 Global deletion of AKAP5 interfered with β-agonist-mediated regulation of [Ca2+]i and phosphorylation of substrates involved in Ca2+ handling in cardiomyocytes derived from adult AKAP5−/− mice.7 However, isoprenaline elicited [Ca2+]i were maintained in cardiomyocytes derived from AKAP5 D36 mice, which express an AKAP5 construct that lacks the PKA-binding site.7 Another study indicated that β1-AR-mediated increase in cyclic AMP was ∼40% higher in AKAP5−/− cardiac myocytes than in AKAPP5+/+ myocytes.8 These results imply that AKAP domains other than the PKA-binding domain were involved in cardiac effects of AKAPs.
4.2. Roles of enhanced signalling by the β1-AR or defective trafficking of cardiac β1-AR in genesis of the cardiac phenotype of AKAP5−/− mice
Ablation of AKAP5 might lead to cardiac hypertrophy and insufficiency by two independent mechanisms. First, myocardial AKAP5 interacts with AC isoforms V and VI and facilitates their phosphorylation by AKAP5-anchored PKA to inhibit cyclic AMP formation.2,3 AKAP5-anchored PKA is selectively activated by the β-AR signalling pathway to blunt excessive signalling by the receptor.2,3 In this view, ablation of AKAP5 in NMVM augmented β1-AR signalling to cyclic AMP by alleviating the inhibitory effect of AKAP5-anchored PKA on AC-V and VI. Enhanced β1-AR signalling to cyclic AMP and subsequently to downstream hypertrophic pathways such as Akt, exchange protein directly activated by cyclic AMP (Epac), CaMKII and many other kinases might explain the cardiac phenotype observed in vitro in NMVM and in vivo in the heart (Figure 1).17,22,27,28 Secondly, ablation of AKAP5 interfered with recycling of the agonist-internalized β1-AR in NMVM.8 We believe that altered trafficking of the myocardial β1-AR and the role of CaN bound to AKAP5 in this phenomenon also plays a significant role in hypertrophy of AKAP5−/− mice. There is ample evidence that internalized β2-AR and other GPCR still signal intracellularly.29,30 Thus, retention of the agonist-internalized β1-AR might prolong the duration of Gs-signalling in the heart of AKAP5−/− mice. Moreover, internalization was required for β1-AR-induced cardiac hypertrophy.31 Consequently, enhanced hypertrophic signalling either from membrane-bound or internalized β1-AR could potentially contribute to hypertrophy in hearts of AKAP5−/− mice.
4.3. Differential resistance of AKAP5−/− mice to isoprenaline
Indiscriminate inhibition of PKA by global expression of a PKA inhibitory peptide or by delocalization of PKA from AKAP by the Ht-31 peptide blunted signalling by the β-AR.15,32,33 Similarly, isoprenaline-mediated increase in [Ca2+]i was markedly reduced in AKAP5−/− cardiomyocytes.7 Nichols et al.7 attributed uncoupling between excitation and contraction in response to isoprenaline in AKAP5−/− cardiomyocytes to loss of PKA-dependent phosphorylation of ryanodine2 receptors, phospholamban, and LTCC that were associated with caveolin 3. We also determined that chronic isoprenaline infusions failed to further diminish cardiac haemodynamic in AKAP5−/− mice (Figure 3), presumably by similar mechanisms to those alluded to by Nichols et al.7
4.4. Role of PKA-independent up-regulation of downstream signalling by Ca2+/CaM in genesis of the cardiac phenotype of AKAP5−/− mice
Ablation of AKAP5 did not alter the expression of the immunoreactive catalytic αA subunit of CaN,7 so what is the cause of the marked elevation of CaN activity in AKAP5−/− mice? We speculate that loss of AKAP5 was involved in activating CaN because expression of AKAP5 markedly reduced the activity of CaN and myocardial hypertrophy after isoprenaline infusion or pressure overload.11,34,35 A role for enhanced CaN activity in cardiac hypertrophy is well established, particularly that inhibition of CaN with CsA (15 mg/kg/days) or FK506 has been shown to prevent cardiac hypertrophy in transgenic mice.36,37 However, in our hands, these doses resulted in unacceptable side effects, such as chewing of extremities and early death in WT and in AKAP5−/− mice. Assuming that our dose of CsA was sufficient, we interpret the inability of CsA to reverse the AKAP5−/− phenotype as an indication for the preponderance of other signalling or transcriptional pathways in imparting the AKAP5−/− cardiac phenotype. One such other pathway is the cardiac CaMKII pathway, which is activated by Ca2+ and by the β1-AR via PKA-independent mechanisms involving altered Epac, β-arrestin signalling, and other kinases and phosphatases.17,22,27,28,38
4.5. Reversal of the AKAP5−/− phenotype with carvedilol
The effects of AKAP5 in the heart were mediated by several mechanisms. Therefore, we used carvedilol, a third generation β-blocker, that acts by multiple mechanisms to ameliorate heart failure in humans.10 These mechanisms involve blockade of α1- and β-AR, inhibition of reactive oxygen species, and biased agonism involving the activation of G protein receptor kinase/β-arrestin signalling.10,39 The mechanism underlying the beneficial effects of carvedilol in AKAP5−/− mice is not known, but might involve its ability to inhibit aberrant β1-AR, G protein-dependent (Gs-mediated), G protein-independent (Epac/β-arrestin), and numerous other kinases/phosphatases and their downstream signalling effectors in the heart.15,22,27–31,39 Our results indicate that carvedilol normalized the activities of CaN and CaMKII in AKAP5−/− mice to their baseline levels in WT mice.
This study indicates that hypertrophy and insufficiency in AKAP5−/− mice bear similarities to these conditions in humans in that they are progressive and mediated by aberrant GPCR and Ca2+ signalling. These results emphasize an unexpected cardioprotective role for AKAP5, which is exerted on β1-AR, CaN, and CaMKII signalling that is worthy of further translational investigation.
Funding
This work was supported by the National Institutes of Health grant (HL-085848 to S.W.B.).
Acknowledgements
We thank G. Stanley McKnight, University of Washington Health Sciences Center, for providing heterozygous AKAP5 mice and Marc Dell'Acqua, University of Colorado-Denver Medical Center, for providing CaNα in pcDNA3.0 myc/His, AKAP79(Δ321–360)-pEGFPN1, and PVIVIT-pEGFP-N1 plasmids.
Conflict of interest: none declared.
References
- 1.Diviani D, Dodge-Kafka KL, Li J, Kapiloff MS. A-kinase anchoring proteins: scaffolding proteins in the heart. Am J Physiol Heart Circ Physiol. 2011;301:H1742–H1753. doi: 10.1152/ajpheart.00569.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Efendiev R, Samelson BK, Nguyen BT, Phatarpekar PV, Baameur F, Scott JD, Dessauer CW. AKAP79 interacts with multiple adenylyl cyclase (AC) isoforms and scaffolds AC5 and -6 to alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptors. J Biol Chem. 2010;285:14450–14458. doi: 10.1074/jbc.M110.109769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bauman AL, Soughayer J, Nguyen BT, Willoughby D, Carnegie GK, Wong W, Hoshi N, Langeberg LK, Cooper DM, Dessauer CW, Scott JD. Dynamic regulation of cAMP synthesis through anchored PKA-adenylyl cyclase V/VI complexes. Mol Cell. 2006;23:925–931. doi: 10.1016/j.molcel.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shcherbakova OG, Hurt CM, Xiang Y, Dell'Acqua ML, Zhang Q, Tsien RW, Kobilka BK. Organization of β-adrenoceptor signaling compartments by sympathetic innervation of cardiac myocytes. J Cell Biol. 2007;176:521–533. doi: 10.1083/jcb.200604167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gardner LA, Naren AP, Bahouth SW. Assembly of a SAP97-AKAP79-PKA scaffold at the type-1 PDZ motif of the human β1-adrenergic receptor generates a receptosome involved in receptor recycling and networking. J Biol Chem. 2007;282:5085–5099. doi: 10.1074/jbc.M608871200. [DOI] [PubMed] [Google Scholar]
- 6.Gardner LA, Tavalin SJ, Goehring AS, Scott JD, Bahouth SW. AKAP79-mediated targeting of the cyclic AMP-dependent protein kinase to the β1-adrenergic receptor promotes recycling and functional resensitization of the receptor. J Biol Chem. 2006;281:33537–33553. doi: 10.1074/jbc.M601809200. [DOI] [PubMed] [Google Scholar]
- 7.Nichols CB, Rossow CF, Navedo MF, Westenbroek RE, Catterall WA, Santana LF, McKnight GS. Sympathetic stimulation of adult cardiomyocytes requires association of AKAP5 with a subpopulation of l-type calcium channels. Circ Res. 2010;107:747–756. doi: 10.1161/CIRCRESAHA.109.216127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li X, Nooh MM, Bahouth SW. Role of the AKAP79/150 protein in β1-adrenergic receptor trafficking and signaling in mammalian cells. J Biol Chem. 2013;288:33797–33812. doi: 10.1074/jbc.M113.470559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feuerstein GZ, Ruffolo RR. Carvedilol, a novel vasodilating beta-blocker with the potential for cardiovascular organ protection. Eur Heart J. 1996;17(Suppl B):24–29. doi: 10.1093/eurheartj/17.suppl_b.24. [DOI] [PubMed] [Google Scholar]
- 10.Frishman WH. Carvedilol. N Engl J Med. 1998;339:1759–1765. doi: 10.1056/NEJM199812103392407. [DOI] [PubMed] [Google Scholar]
- 11.Dell'Acqua ML, Dodge-Kafka KL, Tavalin SJ, Scott JD. Mapping the protein phosphatase-2B anchoring site on AKAP79. J Biol Chem. 2002;277:48796–48802. doi: 10.1074/jbc.M207833200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nystoriak MA, Nieves-Cintrón M, Nygren PJ, Hinke SA, Nichols CB, Chen CY, Puglisi JL, Izu LT, Bers DM, Dell'Acqua ML, Scott JD, Santana LF, Navedo MF. AKAP150 contributes to enhanced vascular tone by facilitating large-conductance Ca2+-activated K+ channel remodeling in hyperglycemia and diabetes mellitus. Circ Res. 2014;114:607–615. doi: 10.1161/CIRCRESAHA.114.302168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Izumo S, Nadal-Ginard B, Mahdavi V. Protooncogene induction and reprogramming of cardiac gene expression produced by pressure overload. Proc Natl Acad Sci USA. 1988;85:339–343. doi: 10.1073/pnas.85.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signaling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 15.Zhang X, Szeto C, Gao E, Tang M, Jin J, Fu Q, Makarewich C, Ai X, Li Y, Tang A, Wang J, Gao H, Wang F, Ge XJ, Kunapuli SP, Zhou L, Zeng C, Xiang KY, Chen X. Cardiotoxic and cardioprotective features of chronic β-adrenergic signaling. Circ Res. 2013;112:498–509. doi: 10.1161/CIRCRESAHA.112.273896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oudit GY, Crackower MA, Eriksson U, Sarao R, Kozieradzki I, Sasaki T, Irie-Sasaki J, Gidrewicz D, Rybin VO, Wada T, Steinberg SF, Backx PH, Penninger JM. Phosphoinositide 3-kinase gamma-deficient mice are protected from isoproterenol-induced heart failure. Circulation. 2003;108:2147–2152. doi: 10.1161/01.CIR.0000091403.62293.2B. [DOI] [PubMed] [Google Scholar]
- 17.Zhu WZ, Wang SQ, Chakir K, Yang D, Zhang T, Heller-Brown J, Devic E, Kobilka BK, Cheng H, Xiao RP. Linkage of β1-adrenergic receptor stimulation to apoptotic heart cell death through protein kinase A-independent activation of Ca2+/calmodulin kinase II. J Clin Invest. 2003;111:617–625. doi: 10.1172/JCI16326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maier LS. Role of CaMKII for signaling and regulation in the heart. Front Biosci. 2009;14:486–496. doi: 10.2741/3257. [DOI] [PubMed] [Google Scholar]
- 20.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Jr, Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409–417. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 21.Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR, Molkentin JD. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 22.Mangmool S, Shukla AK, Rockman HA. Beta-arrestin-dependent activation of Ca 2+ /calmodulin kinase II after β1-adrenergic receptor stimulation. J Cell Biol. 2010;189:573–587. doi: 10.1083/jcb.200911047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pare GC, Bauman AL, McHenry M, Michel JJ, Dodge-Kafka KL, Kapiloff MS. The mAKAP complex participates in the induction of cardiac myocyte hypertrophy by adrenergic receptor signaling. J Cell Sci. 2005;118:5637–5646. doi: 10.1242/jcs.02675. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Chen L, Kass RS, Dessauer CW. The A-kinase anchoring protein Yotiao facilitates complex formation between adenylyl cyclase type 9 and the IKs potassium channel in heart. J Biol Chem. 2012;287:29815–29824. doi: 10.1074/jbc.M112.380568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mayers CM, Wadell J, McLean K, Venere M, Malik M, Shibata T, Driggers PH, Kino T, Guo XC, Koide H, Gorivodsky M, Grinberg A, Mukhopadhyay M, Abu-Asab M, Westphal H, Segars JH. The Rho guanine nucleotide exchange factor AKAP13 (BRX) is essential for cardiac development in mice. J Biol Chem. 2010;285:12344–12354. doi: 10.1074/jbc.M110.106856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones BW, Brunet S, Gilbert ML, Nichols CB, Su T, Westenbroek RE, Scott JD, Catterall WA, McKnight GS. Cardiomyocytes from AKAP7 knockout mice respond normally to adrenergic stimulation. Proc Natl Acad Sci USA. 2012;109:17099–17104. doi: 10.1073/pnas.1215219109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oestreich EA, Malik S, Goonasekera SA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV. Epac and phospholipase Cε regulate Ca2+ release in the heart by activation of protein kinase Cε and calcium-calmodulin kinase II. J Biol Chem. 2009;284:1514–1522. doi: 10.1074/jbc.M806994200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lundby A, Andersen MN, Steffensen AB, Horn H, Kelstrup CD, Francavilla C, Jensen LJ, Schmitt N, Thomsen MB, Olsen JV. In vivo phosphoproteomics analysis reveals the cardiac targets of β-adrenergic receptor signaling. Sci Signal. 2013;6:rs11. doi: 10.1126/scisignal.2003506. [DOI] [PubMed] [Google Scholar]
- 29.Calebiro D, Nikolaev VO, Persani L, Lohse MJ. Signaling by internalized G-protein-coupled receptors. Trends Pharmacol Sci. 2010;31:221–228. doi: 10.1016/j.tips.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 30.Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, Rasmussen SG, Sunahara RK, El-Samad H, Huang B, von Zastrow M. Conformational biosensors reveal GPCR signalling from endosomes. Nature. 2013;2495:534–538. doi: 10.1038/nature12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morisco C, Marrone C, Galeotti J, Shao D, Vatner DE, Vatner SF, Sadoshima J. Endocytosis machinery is required for β1-adrenergic receptor-induced hypertrophy in neonatal rat cardiac myocytes. Cardiovasc Res. 2006;78:36–44. doi: 10.1093/cvr/cvn008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patel HH, Hamuro LL, Chun BJ, Kawaraguchi Y, Quick A, Rebolledo B, Pennypacker J, Thurston J, Rodriguez-Pinto N, Self C, Olson G, Insel PA, Giles WR, Taylor SS, Roth DM. Disruption of protein kinase A localization using a trans-activator of transcription (TAT)-conjugated A-kinase-anchoring peptide reduces cardiac function. J Biol Chem. 2010;285:27632–27640. doi: 10.1074/jbc.M110.146589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McConnell BK, Popovic Z, Mal N, Lee K, Bautista J, Forudi F, Schwartzman R, Jin JP, Penn M, Bond M. Disruption of protein kinase A interaction with A-kinase-anchoring proteins in the heart in vivo: effects on cardiac contractility, protein kinase A phosphorylation, and troponin I proteolysis. J Biol Chem. 2009;284:1583–1592. doi: 10.1074/jbc.M806321200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Pink MD, Murphy JG, Stein A, Dell'Acqua ML, Hogan PG. Balanced interactions of calcineurin with AKAP79 regulate Ca2+-calcineurin-NFAT signaling. Nature Struct Mol Biol. 2012;19:337–345. doi: 10.1038/nsmb.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kashishian A, Howard M, Loh C, Gallatin WM, Hoekstra MF, Lai Y. AKAP79 inhibits calcineurin through a site distinct from the immunophilin-binding region. J Biol Chem. 1998;273:27412–27419. doi: 10.1074/jbc.273.42.27412. [DOI] [PubMed] [Google Scholar]
- 36.De Windt LJ, Lim HW, Bueno OF, Liang Q, Delling U, Braz JC, Glascock BJ, Kimball TF, del Monte F, Hajjar RJ, Molkentin JD. Targeted inhibition of calcineurin attenuates cardiac hypertrophy in vivo. Proc Natl Acad Sci USA. 2001;98:3322–3327. doi: 10.1073/pnas.031371998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sussman MA, Lim HW, Gude N, Taigen T, Olson EN, Robbins J, Colbert MC, Wieczorek DF, Molkentin JD. Prevention of cardiac hypertrophy in mice by calcineurin inhibition. Science. 1998;281:1690–1693. doi: 10.1126/science.281.5383.1690. [DOI] [PubMed] [Google Scholar]
- 38.Wang W, Zhu W, Wang S, Yang D, Crow MT, Xia R-P, Cheng H. Sustained β1-adrenergic stimulation modulates cardiac contractility by Ca2+/calmodulin kinase signaling pathway. Circ Res. 2004;95:798–806. doi: 10.1161/01.RES.0000145361.50017.aa. [DOI] [PubMed] [Google Scholar]
- 39.Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, Shenoy SK, Lefkowitz RJ. A unique mechanism of β-blocker action: carvedilol stimulates β-arrestin signaling. Proc Natl Acad Sci USA. 2007;104:16557–16662. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]