

Abstract
Aims
The majority of patients diagnosed with arrhythmogenic right ventricular cardiomyopathy (ARVC) have mutations in genes encoding desmosomal proteins, raising the possibility that abnormal intercellular adhesion plays an important role in disease pathogenesis. We characterize cell mechanical properties and molecular responses to oscillatory shear stress in cardiac myocytes expressing mutant forms of the desmosomal proteins, plakoglobin and plakophilin, which are linked to ARVC in patients.
Methods and results
Cells expressing mutant plakoglobin or plakophilin showed no differences in cell–cell adhesion relative to controls, while knocking down these proteins weakened cell–cell adhesion. However, cells expressing mutant plakoglobin failed to increase the amount of immunoreactive signal for plakoglobin or N-cadherin at cell–cell junctions in response to shear stress, as seen in control cells. Cells expressing mutant plakophilin exhibited a similar attenuation in the shear-induced increase in junctional plakoglobin immunoreactive signal in response to shear stress, suggesting that the phenotype is independent of the type of mutant protein being expressed. Cells expressing mutant plakoglobin also showed greater myocyte apoptosis compared with controls. Apoptosis rates increased greatly in response to shear stress in cells expressing mutant plakoglobin, but not in controls. Abnormal responses to shear stress in cells expressing either mutant plakoglobin or plakophilin could be reversed by SB216763, a GSK3β inhibitor.
Conclusions
Desmosomal mutations linked to ARVC do not significantly affect cell mechanical properties, but cause myocytes to respond abnormally to mechanical stress through a mechanism involving GSK3β. These results may help explain why patients with ARVC experience disease exacerbations following strenuous exercise.
Keywords: Arrhythmia, Cardiomyopathy, Desmosome, Plakoglobin, Shear stress
1. Introduction
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a familial heart muscle disease characterized by myocardial degeneration with fibrofatty replacement, a high incidence of arrhythmias, and an increased risk of sudden cardiac death.1–4 More than half of affected individuals exhibit one or more mutations in genes encoding desmosomal proteins.5 This has led many investigators to suggest that ARVC is a ‘disease of the desmosome’ in which defective cell–cell adhesion plays a critical pathogenic role. In support of this widely held hypothesis are EM studies of patient endomyocardial biopsies and mouse models of human disease showing decreased numbers of desmosomes in the ventricular myocardium and apparent clefts or widening of various components within the intercalated disk.6–8 Delmar and associates9 have shown that knockdown of plakophilin-2 in neonatal rat cardiac myocytes in vitro greatly reduces cell–cell adhesion strength, and we have previously shown that, when expressed in HEK cells, mutant forms of plakoglobin linked to ARVC can alter cell stiffness or cause a similar marked loss of cell–cell adhesion strength.10 There is also clinical evidence implicating abnormal biomechanical factors in ARVC. For example, recent findings have revealed that ARVC progression may be exacerbated by strenuous exercise.11 The RV apex, an area preferentially affected in ARVC, exhibits heterogeneous fibre orientation and elevated incremental strains, potentially creating a mechanically weak spot in the heart.12 ARVC may be part of a cardiocutaneous syndrome in which the skin phenotype appears to be directly related to mechanical stress. One such condition is Naxos disease which, in addition to ARVC, is characterized by keratoderma of the palms and soles arising after birth once these regions are subjected to mechanical stresses during normal daily activities. However, while these observations all suggest that mechanical stress plays a critical role in promoting the ARVC phenotype, the underlying mechanisms remain poorly defined and there is no direct evidence of weakened cell–cell adhesion in the hearts of patients with ARVC.
Previous studies of mechanotransduction in cardiac myocytes have characterized responses of monolayers of cultured myocytes to stretch, but responses to shear stress may also be particularly pertinent. In vivo, cardiac myocytes are organized into a laminar sheet-like architecture with loose collagenous coupling between sheets. Relative movement of these laminar sheets during the cardiac cycle may cause direct shearing between adjacent sheets or shear from interstitial fluid motion.13,14 Canine studies, for example, have identified myocardial shear as a significant factor in wall thickening.15,16 Additionally, short-term fluid shear stresses of 90–600 mdyn/cm2 regulate expression of a wide variety of cardiac markers in rat cardiac myocytes, including the contractile proteins, such as α-sarcomeric actin and troponin-T, the gap junction protein connexin43, and the cell–cell adhesion protein N-cadherin without affecting phosphorylation of p38, an established marker of cell apoptosis.17,18
Accordingly, to gain greater insights into potential mechanisms by which desmosomal mutations cause ARVC, we characterized biomechanical properties and responses to shear stress in neonatal rat ventricular myocytes (NRVMs) expressing two distinct mutant forms of the desmosomal protein plakoglobin, which have been linked to ARVC in patients. We used atomic force microscopy (AFM) to measure cell stiffness and two different assays to measure cell–cell adhesion strength. We also measured changes in junctional protein distribution and molecular markers associated with hypertrophy and apoptosis in myocytes subjected to oscillatory shear stress. Furthermore, we repeated key experiments in cells expressing a mutant form of plakophilin-2, to determine whether our observations are dependent on the type of mutant desmosomal protein being expressed. We observed no differences in cell–cell adhesion resulting from mutant protein expression. Instead, myocytes expressing either mutant plakoglobin or plakophilin responded similarly, exhibiting abnormal responses to mechanical stress that were corrected by SB216763, which is annotated as a GSK3β inhibitor.19
2. Methods
2.1. Culture and transfection of neonatal rat ventricular myocytes
Primary cultures of neonatal rat ventricular myocytes (NRVM) were prepared from ventricles excised from 1-day-old Wistar rat pups following decapitation (Charles River, IN, USA) as previously described.20 Cells were seeded at a density of 310K cells/cm2. Experiments were performed on fully confluent monolayers 24 h after viral transfection. See Supplementary material online for a detailed description of cell culture, adenoviral transfection, and siRNA knockdown methods. All animal work was performed in accordance with the NIH guidelines and the Institutional Animal Care and Use Committee of Columbia University.
2.2. Cell–cell adhesion assays
Cell–cell adhesion strength was measured using dispase assays and cell aggregate assays. Briefly, dispase assays were performed using monolayers of transfected NRVM on collagen-coated glass chamber slides. Intact cell sheets that had lifted off the culture dish were agitated for 20 min in a rotary shaker at 70 rpm, and the number of fragments was quantified using ImageJ.9,10 Cell aggregate assays were performed by seeding 20 µL of hanging drops of culture media with 10 000 cardiac myocytes. Three days later, cell aggregates were agitated by pipetting, and the number of resulting fragments was quantified using Metamorph and ImageJ. See Supplementary material online for a detailed description.
2.3. Immunoblotting
Immunoblotting was performed as previously described.21 Briefly, cultured NRVMs were lysed in standard RIPA lysis buffer. Samples were solubilized, and soluble fractions containing equal amounts of total protein were separated using SDS–PAGE and transferred onto PVDF membranes (Millipore, MA, USA). Immunoblotting was performed using mouse anti-human antibodies and a horseradish peroxidase-conjugated secondary antibody. Blots were developed with ECL reagents (Perkin Elmer, MA, USA) and imaged using a FUJI imaging unit (Fujifilm, CT, USA). See Supplementary material online for a detailed description.
2.4. Atomic force microscopy
AFM was performed using a BioScope Catalyst AFM mounted on an Olympus IX-81 inverted light microscope (Olympus, PA, USA). A silicon-nitride DNP probe (Bruker, CA, USA) was used to characterize the stiffness of control myocytes and cells expressing mutant forms of plakoglobin. Indentations were made on the surface of the cells in a confluent monolayer, and the resulting force–deflection plot was used to extract the elastic modulus at a depth of 400 nm. Readings were acquired between consecutive contractions (which were easily visible on the AFM real-time readout) without further treatment. Five measurements from each cell were acquired and averaged to yield a ‘whole-cell’ stiffness.
2.5. Shear flow
NRVMs were sheared under oscillatory flow conditions in a parallel-plate shear chamber at 0.06 Pascal (Pa) for 4 h at 37°C. Thereafter, the cells were either immunostained for plakoglobin/N-cadherin or lysed for immunoblotting measurements of plakoglobin, V5, and phosphorylated or total ERK. The shear level was estimated by (i) using a parallel plate model to simulate the shear induced by adjacent myocardial sheets18 and (ii) using a range of shear stresses that have been previously shown to induce ERK phosphorylation without activation of the p38 pathway.17 Minimal shear controls were performed by shearing control myocytes at the lowest possible shear setting (≤0.01 Pa). In some experiments, cells were incubated with SB216763 (5 µM) for 24 h before shearing and throughout the duration of shear to determine whether this compound, identified in a previous chemical screen in a zebrafish model of ARVC,22 could reverse changes in cell mechanical and mechanotransduction properties caused by the expression of mutant plakoglobin or plakophilin.
2.6. Immunofluorescence
NRVMs were immunostained using established protocols.23 Cells were simultaneously blocked and permeabilized for 45 min in 3% normal goat serum, 1% w/v BSA, and 0.15% Triton X-100 in PBS, followed by an overnight incubation at 4°C with either mouse-monoclonal anti-plakoglobin (Sigma; 1 : 1500 in blocking buffer), anti-plakophilin (Fitzgerald, 1 : 50), anti-V5 (Invitrogen, 1 : 500), or anti-N-cadherin (Sigma, 1 : 1500) antibodies. Samples were then incubated with secondary antibody (Invitrogen; 1 : 1500) for 2 h at room temperature. The immunostained preparations were imaged at ×40 (1.3 NA) using an Olympus IX-81 inverted confocal microscope. Each image was subsequently masked to isolate cell junctions, allowing for discrete quantification of junctional immunoreactive signal. Images were analysed using quantitative confocal microscopy as previously described.10,24
2.7. TUNEL assay
Terminal deoxynucleotidyl transferase dUTP nick-end labelling (TUNEL) assays were performed using an in situ cell death detection kit (Roche Diagnostics, Basel, Switzerland) as previously described.21
2.8. Statistical analysis
Data sets with more than three independent groups were analysed using the non-parametric Kruskal–Wallis analysis of variance (ANOVA) test in Prism. Data showing significance (P ≤ 0.05) were further analysed using Dunn's post hoc test for multiple comparisons. For data sets involving fewer than three independent groups, data were analysed using the Mann–Whitney test. Differences were considered to be statistically significant and indicated with * when P ≤ 0.05 or ** when P ≤ 0.01. Error bars are plotted as the S.D. of the data.
3. Results
3.1. Expression of mutant plakoglobin and plakophilin in NRVM
Immunostaining with an antibody against the V5 epitope tag in myocytes transfected to express epitope-tagged wild-type (WT PG), Naxos (2057del2 PG, 2057del2 plakoglobin mutation), or S39_K40insS [InsS PG, S39_K40insS plakoglobin mutation (linked to autosomal dominant ARVC)] confirmed the localization of exogenous plakoglobin at cell junctions, and in the case of 2057del2 PG transfected cells, also within the nucleus (Figure 1A). Only background levels of signal were seen in non-transfected control cells (Figure 1A). Immunostaining with an antibody against an N-terminal epitope in plakoglobin showed a similar pattern of signal distribution (Figure 1B). Quantification of immunoblots of transfected cardiac myocyte lysates with an anti-plakoglobin antibody confirmed the expression of exogenous plakoglobin at levels comparable with that of the endogenous protein (Figure 1C). All lysates contained endogenous plakoglobin, whereas lysates from WT PG and InsS PG transfected myocytes also showed a second, more slowly migrating band corresponding to the exogenous form of plakoglobin containing the V5 tag. As expected, the 2057del2 PG protein was smaller than the endogenous protein because the Naxos mutation results in a premature stop codon and truncation of the C-terminus. Similar to cells transfected with plakoglobin constructs, immunostaining with an antibody against the V5 epitope tag in non-transfected (control) and wild-type plakophilin (WT PKP) transfected myocytes confirmed the localization of exogenous plakophilin-2 at cell junctions (Figure 1D). Mutant plakophilin-2 (1851del123 PKP, mutant plakophilin-2 protein with a deletion of Exon 10) failed to localize at cell junctions. Immunostaining with an antibody against a C-terminal epitope of plakophilin-2 was used to determine the localization of both exogenous and endogenous plakophilin-2 (Figure 1E). A V5 antibody was used to detect the presence of exogenous plakophilin-2, as the similar sizes of exogenous and endogenous plakophilin-2 prevented the detection of discrete bands using the anti-plakophilin antibody (Figure 1F). Transfection efficiency was determined in cells incubated with a virus containing a GFP construct and was calculated as the proportion of total nuclei contained within cellular regions showing green fluorescence (see Supplementary material online, Figure S1). Using this technique, the viral efficiency was determined to be 98.9 ± 1.1% (Figure 1G) and was used to determine the multiplicity of infection for viral constructs.
Figure 1.

ARVC models express transgenic proteins at endogenous levels: (A) V5 immunostaining of control (non-transfected) myocytes (C) and myocytes transfected to express wild-type plakoglobin (WT PG), S39_K40insS (InsS PG), or 2057del2 PG. No signal is seen in non-transfected control cells (thus demonstrating the absence of non-specific binding). Normal junctional localization of V5 labelled protein is seen in cells transfected to express WT PG, InsS PG, and 2057del2 PG mutants. Cells transfected with 2057del2 PG also show nuclear localization of truncated 2057del2 plakoglobin. (B) Immunostaining with an anti-plakoglobin antibody against an N-terminal epitope (which recognizes both endogenous and transfected forms of WT PG, InsS PG, and 2057del2 PG) shows a similar pattern. (C) Immunoblot (left) using an N-terminal anti-plakoglobin antibody, with quantification (right) showing roughly equivalent amounts of endogenous plakoglobin and transgene plakoglobin in WT PG, InsS PG, and 2057del2 PG cells (n = 4). While the control lane shows a single band corresponding to endogenous plakoglobin, the WT PG, InsS PG, and 2057del2 PG lanes exhibit a second band, corresponding to the transgenic plakoglobin. The larger mol. wt of transgenic plakoglobin observed in the WT PG and InsS PG lanes is explained by the addition of a C-terminal V5 tag, connected using a linker peptide. The lower mol. wt transgenic plakoglobin band observed in the 2057del2 PG lane is explained by the truncation of the C-terminal end of plakoglobin—a result of the premature stop codon generated by the frame-shift 2057del2 mutation. (D) V5 stain on control (C), wild-type plakophilin (WT PKP), and mutant plakophilin (1851del123 PKP) transfected myocytes. Normal junctional localization is seen in WT PKP transfected cells. No junctional signal is observed in cells expressing 1851del123 PKP. (E) Immunostaining with an anti-plakophilin-2 antibody against a C-terminal epitope (which recognizes both endogenous and transfected forms of WT PKP and 1851del123 PKP. (F) Immunoblots using anti-V5 (top), anti-C-terminal plakophilin (middle), and anti-GAPDH (bottom) antibodies. V5 bands in the WT PKP and 1851del123 PKP lysates confirm successful transfection. The relatively similar size between exogenous and endogenous plakophilin prevented detection of discrete bands using the anti-C-terminal plakophilin antibody. (G) Quantification of cells transfected with a GFP virus to confirm 98.9 ± 1.1% transfection efficiency (n = 5). Scale bars = 20 μm.
3.2. Cell–cell adhesion strength is unaffected by mutant plakoglobin or plakophilin expression
Dispase and cell aggregate assays were used to determine whether expression of mutant desmosomal proteins affected the strength of cell–cell adhesion. As shown in Figure 2A and B, the number of fragments resulting from mechanical agitation of monolayers in the dispase assay (Figure 2A) or cell clusters in the cell aggregate assay (Figure 2B) was not significantly different among the control myocytes and myocytes expressing either mutant form of plakoglobin. To further validate the dispase assay, control monolayers were exposed to 1 µM cytochalasin-D and then lifted from the culture dish and subjected to rotary agitation. As shown in Figure 2C, treatment with cytochalasin-D caused 3.0- and 3.5-fold increases in monolayer fragmentation after 1 and 3 min of agitation, respectively, thus providing independent confirmation that the dispase assay sensitively detects changes in cell–cell adhesion strength.
Figure 2.

Cell–cell adhesion strength is unaffected by mutant plakoglobin or plakophilin expression: (A) quantification of control and mutant plakoglobin-transfected cardiac myocyte monolayer fragmentation in dispase assays before and after 1 or 3 min of agitation. Cardiac myocytes expressing mutant plakoglobin show no differences in cell–cell adhesion relative to WT PG (n = 5). (B) Quantification of cluster sizes in control and transfected cardiac myocytes in cell aggregate assays (n = 3). Cardiac myocyte aggregates expressing mutant plakoglobin show no differences in aggregate fragmentation relative to WT PG. (C) Dispase assays in normal (non-transfected) cardiac myocytes showing a significant reduction in cell–cell adhesion in cells exposed to 1 µM cytochalasin-D (**P < 0.01 after 1 and 3 min agitation; n = 5). (D) Dispase assays in normal (non-transfected) cardiac myocytes showing a significant reduction in cell–cell adhesion in cells following siRNA knockdown of endogenous plakoglobin (*P < 0.05 after 1 or 3 min of agitation, n = 5). Plakoglobin knockdown was confirmed by immunoblotting (inset), and quantified (right). (E) Quantification of control and mutant plakophilin transfected cardiac myocyte monolayer fragmentation in dispase assays before and after 1 or 3 min of agitation. Similar to myocytes expressing mutant plakoglobin, cardiac myocytes expressing mutant plakophilin-2 (n = 6) also show no differences in cell–cell adhesion relative to WT PKP (n = 4), suggesting the effect does not depend on which desmosomal protein is being mutated. (F) Dispase assays in normal (non-transfected) cardiac myocytes showing a significant reduction in cell–cell adhesion in cells following siRNA knockdown of endogenous plakophilin-2 (*P < 0.05 after 1 or 3 min of agitation, n = 5). Plakophilin-2 knockdown was confirmed by immunoblotting (inset) and quantified (right).
The apparent lack of effect of mutant plakoglobin on adhesion is different than previous studies,9 reporting reduced adhesion in cardiac myocytes following siRNA knockdown of expression on the desmosomal protein plakophilin-2 in which dominant mutations are often found in ARVC patients. To determine whether the effects on cell adhesion are different following knockdown of a desmosomal protein vs. expression of a disease-causing mutation, we treated non-transfected myocytes with two different specific siRNAs against plakoglobin. As shown in Figure 2D, treated cells exhibited a marked reduction in plakoglobin expression. Both siRNAs led to between 2.0- and 2.7-fold increases in monolayer fragmentation relative to time-matched controls after 1 or 3 min of agitation, whereas treatment with a negative siRNA had no effect on cell–cell adhesion. Similarly, cells transfected to express mutant plakophilin-2 showed no difference in fragmentation following agitation (Figure 2E), but cells treated with either of two specific siRNAs against plakophilin showed between 1.9- and 3.5-fold increases in monolayer fragmentation relative to time-matched controls after 1 or 3 min of agitation (Figure 2F). Suppression of N-cadherin showed a similar reduction in cell–cell adhesion strength, suggesting that this effect is not limited to the suppression of desmosomal proteins, but other adhesion proteins as well (see Supplementary material online, Figure S2). Knockdown efficiency was quantified by western blot, and TUNEL assays were used to exclude reduced cell viability as a possible cause of increased sheet fragmentation (see Supplementary material online, Figure S3). Taken together, these results indicate that expression of mutant forms of plakoglobin and plakophilin-2 at levels roughly equivalent to that of the endogenous protein does not affect cell–cell adhesion strength, whereas knockdown of the endogenous protein has a marked effect in reducing adhesion.
3.3. Fluid shear induces junctional remodelling
A custom oscillatory shear device and parallel plate flow chamber were used to shear control and transfected myocytes. In response to oscillatory fluid shear at 0.06 Pa, control and WT PG transfected myocytes exhibited between 2.3 and 3.3-fold increases in immunoreactive signal for junctional plakoglobin (Figure 3A and B) compared with unsheared cells. In contrast, myocytes expressing the 2057del2 PG or InsS PG mutants failed to increase junctional plakoglobin signal following shear. A considerable amount of the plakoglobin in myocytes expressing the 2057del2 PG mutation was distributed within cells in a distinct perinuclear pattern, but there was little if any change in either the amount or distribution of plakoglobin signal in response to shear. Similar to control and WT PG transfected cells, WT PKP transfected cells exhibited a two-fold increase in immunoreactive signal for junctional plakoglobin (Figure 3C and see Supplementary material online, Figure S4), whereas myocytes expressing the 1851del123 PKP mutant protein were unable to increase junctional plakoglobin signal following shear. To determine whether SB216763 could restore the control-level response to shear in cells expressing mutant plakoglobin, we repeated experiments in cultures exposed to 5 µM SB216763 for 24 h. As shown in Figure 3D and E, cells expressing either InsS PG, 2057del2 PG, or 1851del123 PKP exhibited robust increases in junctional signal for plakoglobin in response to oscillatory shear equivalent to the response in control cells and cells expressing WT proteins (2.0-, 2.2-, and 2.6-fold increases in 2057del2 PG, InsS PG, and 1851del123 PKP cells, respectively). Thus, failure to increase junctional signal for plakoglobin in response to shear can be normalized by SB216763 in cells expressing either mutant plakoglobin or plakophilin-2.
Figure 3.
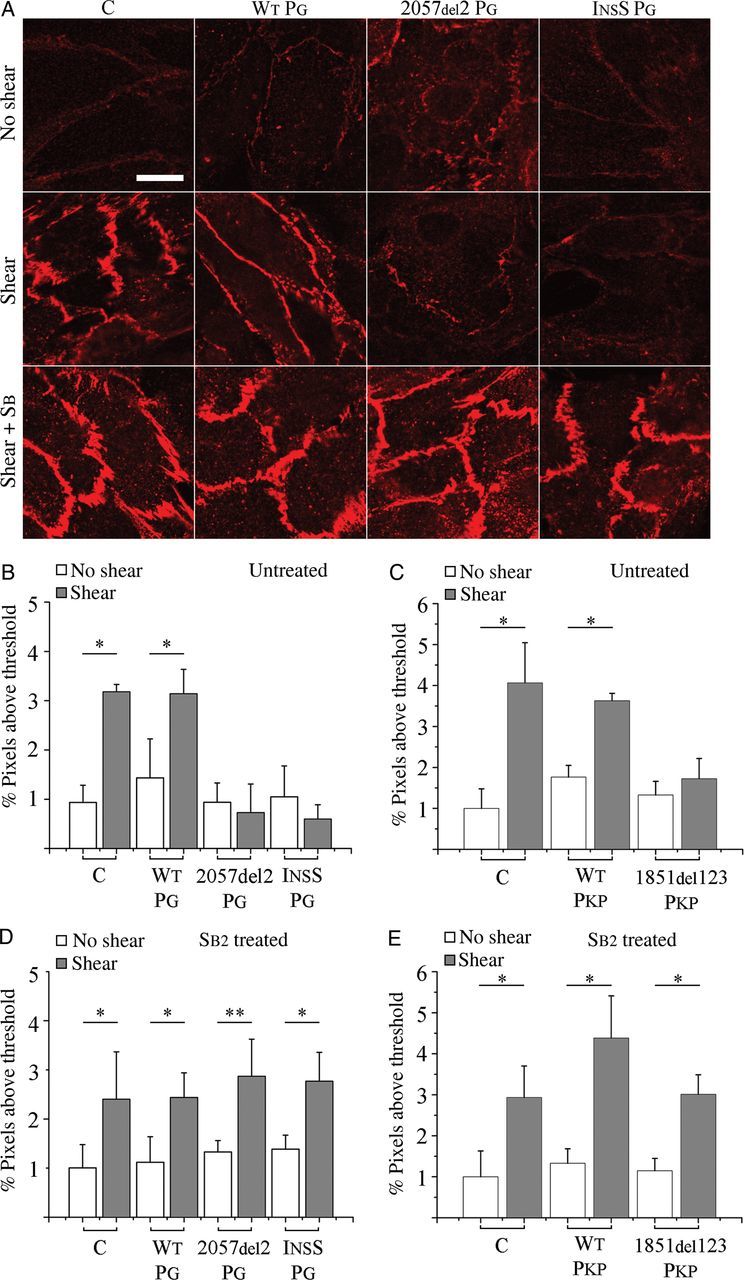
Shear-induced plakoglobin remodelling is reversibly altered in ARVC models: (A) immunofluorescence images showing the distribution of plakoglobin (N-terminal antibody) in control (C) and transfected cardiac myocytes before (top) and after (middle) exposure to shear, and after treatment with SB216763 (SB2; bottom). Control and WT PG cells show an increased immunoreactive signal with junctional localization after shear, whereas cells expressing InsS PG or 2057del2 PG show no apparent increase in junctional signal after shear. Treatment with the drug restored the normal shear-induced increase in junctional signal in cells expressing mutant plakoglobin or plakophilin. (B and C) Quantification of junctional plakoglobin signal before and after shear in (B) plakoglobin (n = 5) or (C) plakophilin (n = 4) transfected cells. (D and E) Quantification of junctional plakoglobin signal before and after shear in (D) plakoglobin (n = 6) or (E) plakophilin (n = 4) transfected cells treated with SB216763. Scale bars = 20 μm. *P < 0.05; **P < 0.01.
Control and WT PG cells also showed 2- and 1.6-fold increases in junctional signal for N-cadherin, respectively, following oscillatory shear (Figure 4A and B). In contrast, cells expressing either mutant form of plakoglobin failed to increase junctional N-cadherin signal in response to shear (Figure 4A and B). As was seen for plakoglobin, SB216763 restored the ability of cells expressing mutant plakoglobin to increase junctional N-cadherin signal in response to shear (Figure 4C; 1.9- and 1.7-fold increases in 2057del2 PG and InsS PG cells, respectively, compared with 1.6- and 2.1-fold increases in control and WT PG cells). SB216763 also reversed Cx43 gap junction remodelling in myocytes expressing mutant forms of plakoglobin and plakophilin-2 (see Supplementary material online, Figure S5) and, as previously reported,21 increased total Cx43 expression in control (non-transfected) myocytes and in cells expressing mutant desmosomal proteins.
Figure 4.
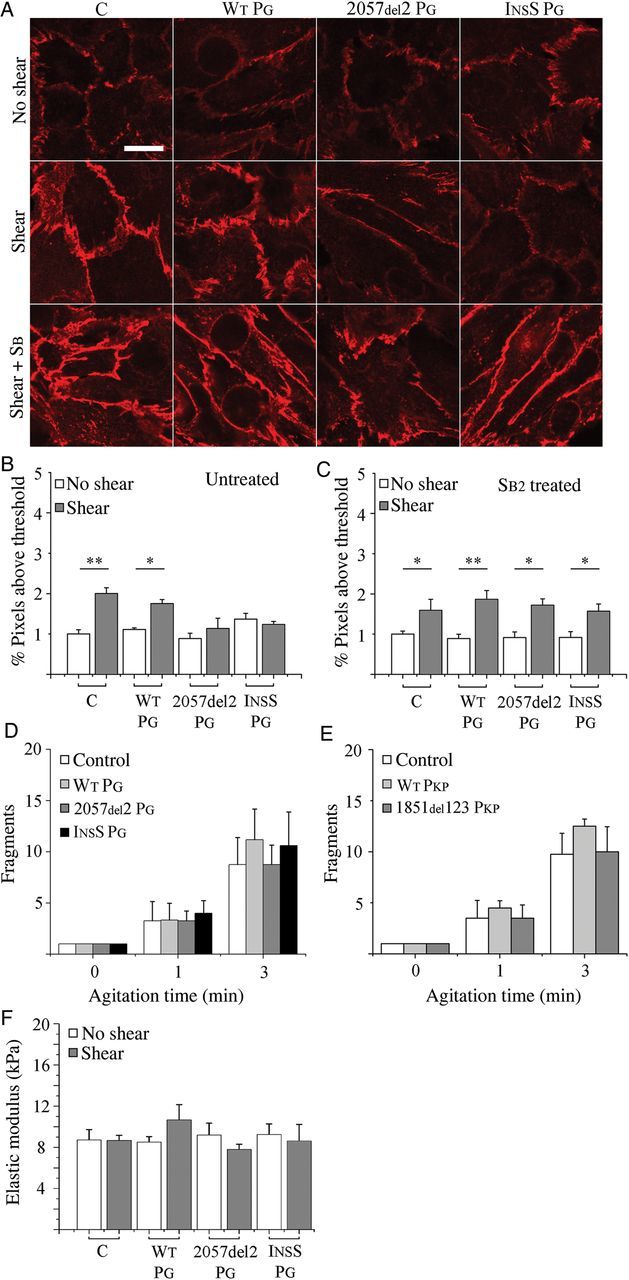
Shear-induced N-cadherin remodelling is reversibly altered in ARVC models: (A) N-cadherin IF images showing the distribution of N-cadherin in control (C) and transfected cardiac myocytes before (top) and after (middle) exposure to shear, and after treatment with SB216763 (bottom). As seen in Figure 3, control (non-transfected) cells and cells transfected to express wild-type plakoglobin (WT PG) show an increased junctional signal for N-cadherin after shear, whereas no shear-induced increase was seen in cells expressing mutant forms of plakoglobin. Treatment with the drug restored the normal shear-induced increase in junctional signal in cells expressing mutant plakoglobin. (B) Quantification of junctional N-cadherin signal before and after shear (n = 5). (C) Quantification of junctional N-cadherin signal before and after shear in cardiac myocytes treated with SB216763 (n = 5). SB216763 restored the normal shear-induced increase in junctional N-cadherin signal. (D and E) Dispase assay measurements in control cells (C) and cells expressing (D) transgenic plakoglobin (n = 5) or (E) transgenic plakophilin (n = 4) after exposure to mechanical shear stress. Cardiac myocytes expressing mutant plakoglobin or plakophilin-2 show no differences in cell–cell adhesion when compared with control cells. (F) Cell stiffness measurements in control cells (C) and cells expressing WT or mutant plakoglobin before and after exposure to mechanical shear stress (n = 4). Cardiac myocytes expressing mutant plakoglobin show no differences in cell stiffness compared with control cells. *P < 0.05; **P < 0.01.
In view of the increased signals for junctional plakoglobin and N-cadherin in control and WT PG cells following shear, we repeated studies of cell mechanical properties to determine whether there was a correlation between the amount of junctional protein signals and measures of cell–cell adhesion strength. As shown in Figure 4D and E, there was no significant change in cell–cell adhesion strength following shear in control myocytes or myocytes expressing mutant plakoglobin or plakophilin-2. Thus, within the limits of these assays, there appears to be no apparent relationship between the amount of junctional immunoreactive signals for plakoglobin or N-cadherin and cell–cell adhesion.
We next used AFM to measure cell stiffness.25 Whereas our previous studies have shown a change in cell stiffness in HEK epithelial cells expressing the InsS PG (but not the 2057del2 PG) mutation in plakoglobin,10 we found no such change in stiffness in cardiac myocytes expressing either mutant form of plakoglobin (Figure 4F).
3.4. Apoptosis is increased following shear in myocytes expressing mutant plakoglobin
Apoptosis is increased in the myocardium of patients with ARVC,26 and myocytes expressing the 2057del2 PG mutant form of plakoglobin exhibit increased apoptosis rates under basal condition with even greater apoptosis in response to cyclical stretch.22 To determine whether a similar response occurs in oscillatory shear, we measured apoptosis by TUNEL assays. As shown in Figure 5A, myocytes expressing 2057del2 PG showed elevated levels of apoptosis in response to shear stress, relative to unsheared cells. In contrast, myocytes expressing the InsS PG variant of plakoglobin exhibited no changes in apoptosis with exposure to oscillatory shear, although the mean level of apoptosis was elevated relative to control cells. Treatment with SB216763 returned apoptosis levels to control levels in myocytes expressing 2057del2 PG (Figure 5B).
Figure 5.

Cell viability, but not ERK phosphorylation, is altered by mutant plakoglobin expression: (A) quantification of apoptosis (percent TUNEL-positive nuclei) in control (C) cells and cells transfected to express WT or mutant forms of plakoglobin before and after exposure to shear (n = 4). Mechanical shear stress caused an increase in apoptosis in cells expressing 2057del2 PG. (B) Quantification of apoptosis (percent TUNEL-positive nuclei) in cells treated with SB216763 (SB2) before and after shear (n = 3). SB216763 prevented the shear-induced increase in apoptosis in cells expressing 2057del2 PG. (C) Representative immunoblot and quantitative data showing equivalent levels of phospho-ERK activation (P-ERK), with total ERK (T-ERK) as a loading control, in control cells (C) or cells transfected to express WT or mutant forms of plakoglobin (n = 3). *P < 0.05.
ERK phosphorylation, which is involved in hypertrophic response in cardiac myocytes, is responsive to shear stress. Given that hypertrophy may, in turn, be modulated in part by desmosomal integrity in cardiac myocytes,27,28 and based on the altered mechanoresponse of our ARVC cell model, we next assessed whether ERK phosphorylation may be disrupted by ARVC mutant plakoglobin. Immunoblotting performed on myocytes exposed to oscillatory shear flow confirmed an increase in the amount of phosphorylated ERK (p-ERK) in response to shear for control and WT PG myocytes, as well as cells expressing mutant forms of plakoglobin (Figure 5C). After 10 min, control, WT PG, 2057del2 PG, and insS PG transfected myocytes exposed to oscillatory shear exhibited 3.3-, 3.2-, 4.2-, and 3.5-fold increases in p-ERK relative to unsheared controls, respectively.
4. Discussion
The key observation in this study is that expression of mutant desmosomal proteins in ventricular myocytes in amounts roughly equivalent to that of the native protein has no apparent effect on cell mechanical properties per se, but instead has a marked effect on how cells respond to mechanical stimulation. This is an important distinction that has significant implications about how mutations in desmosomal proteins cause the complex disease phenotypes seen in patients with ARVC. It is widely believed that defective intercellular adhesion plays an important pathogenic role in ARVC patients with desmosomal mutations, but direct evidence for this hypothesis has been lacking. Previously, it has been shown that when expression of plakophilin-2 (the most frequently mutated desmosomal protein in ARVC) is knocked down in NRVM, there is a dramatic decrease in cell–cell adhesion strength.9 To reconcile the apparent discrepancy between this observation and the present study, we used two separate siRNAs targeting different regions to knockdown the expression of plakoglobin or plakophilin, and in both cases observed a similar marked decrease in cell–cell adhesion strength. This emphasizes important differences in the pathophysiological consequences of suppression of a desmosomal protein vs. expression of a mutant form of a desmosomal protein. In this regard, it appears that mutant desmosomal proteins are expressed in the myocardium of patients with ARVC. This is certainly true in Naxos disease which, although inherited as a recessive trait, is not related to loss of gene expression but rather to expression of mutant plakoglobin with a gene-dosage effect. Indeed, germline deletions of plakoglobin, desmoplakin, and plakophilin all cause embryonic lethal phenotypes in mice,29–32 providing further evidence that ARVC is related to expression of mutant proteins rather than haploinsufficiency. While it should not be surprising that knockdown of a desmosomal gene leads to loss of cell–cell adhesion, our results show that expression of a mutant desmosomal gene has a different effect which is more likely to reflect the pathophysiology of the disease in patients.
Another interesting related observation from our study is the apparent lack of correlation between the amount of immunoreactive signal for plakoglobin at cell–cell junctions and the strength of cell–cell adhesion. While there is likely a positive correlation between desmosome number and immunoreactive signal intensity, many factors (i.e.—variations in antibody-binding specificity, macromolecular crowding, and epitope accessibility) can make the exact relationship difficult to determine. Our data are consistent with the idea that redistribution of plakoglobin from junctional to intracellular/intranuclear sites, a feature seen in both patients and experimental models of ARVC, has no major effect on cell–cell adhesion strength, but rather leads to abnormal responses to mechanical stimuli that appear to be a fundamental part of the disease pathway in ARVC. Finally, we have previously reported that when HEK cells overexpress the 2057del2 PG mutation, there is a marked reduction in cell–cell adhesion strength without a change in cell stiffness, whereas overexpression of the InsS PG mutation in HEK cells has the opposite effects. The discrepancy between previous results and those presented in this study may result from differences in cell type, and the disparate roles of desmosomes in cardiac myocytes and HEK cells. It is possible that the robust actin–myosin network present in cardiac myocytes, but not in HEK cells, may mask any minor alterations in cell stiffness that result from the expression of mutant desmosomal proteins. The results of the present study reinforce the important idea that epithelial cells are not necessarily good surrogates for cardiac myocytes, and changes in HEK cells expressing cardiomyopathy-related disease mutations must be interpreted with caution.
4.1. Cell viability, but not ERK phosphorylation, is altered by mutant plakoglobin expression
The lack of differences in cell–cell adhesion between control and myocytes expressing mutant plakoglobin or plakophilin-2 motivated our search for a molecular mechanism underlying the pathogenesis of ARVC. Thus, we chose to investigate (i) ERK phosphorylation, a documented promoter of myocyte hypertrophy in vitro and in vivo,17,33,34 and (ii) apoptosis. While ERK phosphorylation (before and after shear) remained unaffected by the expression of ARVC-causing mutant plakoglobin, apoptosis was elevated in myocytes expressing 2057del2 PG and insS PG. Noting the nuclear localization of plakoglobin in these cells, elevated levels of apoptosis are consistent with work by Garcia-Gras et al.35 that suggests that nuclear localization of plakoglobin can recapitulate the ARVC phenotype (i.e.—adipogenesis, fibrogenesis, and myocyte apoptosis).
4.2. SB216763 treatment has restorative effects in ARVC models
Treatment of myocytes expressing mutant plakoglobin with SB216763 attenuated apoptosis back to control levels in myocytes expressing the 2057del2 PG and InsS PG mutations. Furthermore, SB216763 treatment recovered junctional remodelling of plakoglobin and N-cadherin in cells expressing 2057del2 PG, InsS PG, and 1851del123 PKP mutant proteins. The rapidity with which plakoglobin changes its subcellular distribution in response to SB216763 (24 h following drug exposure) further supports the hypothesis of underlying defects in protein trafficking.22 Although not within the scope of this study, attenuation of apoptosis may be explained by a decrease in the suppression of canonical wnt/beta-catenin signalling by nuclear plakoglobin, as evidenced by the loss of the nuclear localization of plakoglobin.
4.3. Limitations and conclusion
The cell–cell adhesion data do not necessarily represent in vivo tissue cohesion, particularly because neonatal cells do not possess the anisotropic characteristics of adult myocytes, which may affect cell–cell adhesion. However, it was unexpected, given all the previous work, that there was no significant alteration in cell–cell adhesion. While shear responses are consistent with ARVC, we do not claim that ARVC development is attributed primarily to shear response; likely stretch and non-mechanical pathways are also involved.
We note that the use of viral transduction of ARVC mutant plakoglobin did not lead to significant overexpression of the ARVC plakoglobin proteins, which increases confidence that our results are due to the effects of the transgene and not protein overexpression. Since Naxos disease is recessive, it is possible that our model cells express sufficient endogeneous plakoglobin to attenuate the effects of the 2057del2 PG mutation. However, the insS PG mutation is dominant and no significant biomechanical changes were observed, while both mutant plakoglobin and plakophilin variants led to molecular changes. Thus, we have some confidence that molecular changes likely dominate in the manifestation of ARVC. We did not repeat all of the experiments using mutant plakophilin-2, because our previous findings suggested that plakoglobin re-distribution is a key event in the final common disease pathway regardless of the underlying pathogenic mutation, and partially because the 1851del123 PKP mutation is not directly linked to ARVC cases, but rather a deletion of a region that is prone to ARVC mutation.
We conclude that ARVC-causing mutations in desmosomal proteins lead to altered cellular distribution of plakoglobin and N-cadherin, as well as increased apoptosis, without alterations in cell mechanical properties or certain early signalling pathways.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported in part by the National Institutes of Health (R01HL102361 and S10RR027943-01) and National Science Foundation (CMMI-1130376 and Graduate Research Fellowship). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or National Science Foundation.
Acknowledgements
The authors thank Qi Wei for helpful discussions, and Keith Yeager for assistance in designing the shear flow devices.
Conflict of interest: A patent application has been filed for the use of SB216763 in arrhythmogenic cardiomyopathy and related heart diseases.
References
- 1.Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, Grosgogeat Y. Right ventricular dysplasia—a report of 24 adult cases. Circulation. 1982;65:384–398. doi: 10.1161/01.cir.65.2.384. [DOI] [PubMed] [Google Scholar]
- 2.Mckenna WJ, Thiene G, Nava A, Fontaliran F, Blomstromlundqvist C, Fontaine G, Camerini F. Diagnosis of arrhythmogenic right-ventricular dysplasia/cardiomyopathy. Br Heart J. 1994;71:215–218. doi: 10.1136/hrt.71.3.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Norman MW, McKenna WJ. Arrhythmogenic right ventricular cardiomyopathy: perspectives on disease. Z Kardiol. 1999;88:550–554. doi: 10.1007/s003920050324. [DOI] [PubMed] [Google Scholar]
- 4.Sen-Chowdhry S, Morgan RD, Chambers JC, McKenna WJ. Arrhythmogenic cardiomyopathy: etiology, diagnosis, and treatment. Annu Rev Med. 2010;61:233–253. doi: 10.1146/annurev.med.052208.130419. [DOI] [PubMed] [Google Scholar]
- 5.Sen-Chowdhry S, Syrris P, McKenna WJ. Role of genetic analysis in the management of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2007;50:1813–1821. doi: 10.1016/j.jacc.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 6.Basso C, Czarnowska E, Della Barbera M, Bauce B, Beffagna G, Wlodarska EK, Pilichou K, Ramondo A, Lorenzon A, Wozniek O, Corrado D, Datiento L, Danieli GA, Valente M, Nava A, Thiene G, Rampazzo A. Ultrastructural evidence of intercalated disc remodelling in arrhythmogenic right ventricular cardiomyopathy: an electron microscopy investigation on endomyocardial biopsies. Eur Heart J. 2006;27:1847–1854. doi: 10.1093/eurheartj/ehl095. [DOI] [PubMed] [Google Scholar]
- 7.Fujita S, Terasaki F, Otsuka K, Katashima T, Kanzaki Y, Kawamura K, Tanaka T, Kitaura Y. Markedly increased intracellular lipid droplets and disruption of intercellular junctions in biopsied myocardium from a patient with arrhythmogenic right ventricular cardiomyopathy. Heart Vessels. 2008;23:440–444. doi: 10.1007/s00380-008-1079-0. [DOI] [PubMed] [Google Scholar]
- 8.Rizzo S, Lodder EM, Verkerk AO, Wolswinkel R, Beekman L, Pilichou K, Basso C, Remme CA, Thiene G, Bezzina CR. Intercalated disc abnormalities, reduced Na+ current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc Res. 2012;95:409–418. doi: 10.1093/cvr/cvs219. [DOI] [PubMed] [Google Scholar]
- 9.Sato PY, Coombs W, Lin XM, Nekrasova O, Green KJ, Isom LL, Taffet SM, Delmar M. Interactions between ankyrin-G, plakophilin-2, and connexin43 at the cardiac intercalated disc. Circ Res. 2011;109:193–201. doi: 10.1161/CIRCRESAHA.111.247023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang H, Asimaki A, Lo D, McKenna W, Saffitz J. Disparate effects of different mutations in plakoglobin on cell mechanical behavior. Cell Motil Cytoskel. 2008;65:964–978. doi: 10.1002/cm.20319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corrado D, Basso C, Pavei A, Michieli P, Schiavon M, Thiene G. Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. JAMA. 2006;296:1593–1601. doi: 10.1001/jama.296.13.1593. [DOI] [PubMed] [Google Scholar]
- 12.Hariharan V, Provost J, Shah S, Konofagou E, Huang H. Elevated strain and structural disarray occur at the right ventricular apex. Cardiovasc Eng Technol. 2012;3:52–61. doi: 10.1007/s13239-011-0081-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caulfield JB, Borg TK. Collagen network of the heart. Lab Invest. 1979;40:364–372. [PubMed] [Google Scholar]
- 14.Dou JG, Tseng WYI, Reese TG, Wedeen VJ. Combined diffusion and strain MRI reveals structure and function of human myocardial laminar sheets in vivo. Magn Reson Med. 2003;50:107–113. doi: 10.1002/mrm.10482. [DOI] [PubMed] [Google Scholar]
- 15.Legrice IJ, Takayama Y, Covell JW. Transverse-shear along myocardial cleavage planes provides a mechanism for normal systolic wall thickening. Circ Res. 1995;77:182–193. doi: 10.1161/01.res.77.1.182. [DOI] [PubMed] [Google Scholar]
- 16.Costa KD, Takayama Y, McCulloch AD, Covell JW. Laminar fiber architecture and three-dimensional systolic mechanics in canine ventricular myocardium. Am J Physiol Heart Circ. 1999;276:H595–H607. doi: 10.1152/ajpheart.1999.276.2.H595. [DOI] [PubMed] [Google Scholar]
- 17.Dvir T, Levy O, Shachar M, Granot Y, Cohen S. Activation of the ERK1/2 cascade via pulsatile interstitial fluid flow promotes cardiac tissue assembly. Tissue Eng. 2007;13:2185–2193. doi: 10.1089/ten.2006.0364. [DOI] [PubMed] [Google Scholar]
- 18.Lorenzen-Schmidt I, Schmid-Schonbein GW, Giles WR, McCulloch AD, Chien S, Omens JH. Chronotropic response of cultured neonatal rat ventricular myocytes to short term fluid shear. Cell Biochem Biophys. 2006;46:113–122. doi: 10.1385/CBB:46:2:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eldar-Finkelman H, Martinez A. GSK-3 inhibitors: preclinical and clinical focus on CNS. Front Mol Neurosci. 2011;4:1–18. doi: 10.3389/fnmol.2011.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Asimaki A, Tandri H, Duffy ER, Winterfield JR, Mackey-Bojack S, Picken MM, Cooper LT, Wilber DJ, Marcus FI, Basso C, Thiene G, Tsatsopoulou A, Protonotarios N, Stevenson WG, McKenna WJ, Gautam S, Remick DG, Calkins H, Saffitz JE. Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol. 2011;4:743–752. doi: 10.1161/CIRCEP.111.964890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei Q, Hariharan V, Huang H. Cell-cell contact preserves cell viability via plakoglobin. PLoS ONE. 2011;6:1–9. doi: 10.1371/journal.pone.0027064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Asimaki A, Kapoor S, Plovie E, Arndt AK, Adams E, Liu Z, James CA, Judge DP, Calkins H, Churko J, Wu JC, MacRae CA, Kleber AG, Saffitz JE. Identification of a new modulator of the intercalated disc in a zebrafish model of arrhythmogenic cardiomyopathy. Sci Transl Med. 2014;6:240ra274. doi: 10.1126/scitranslmed.3008008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwong KF, Schuessler RB, Green KG, Laing JG, Beyer EC, Boineau JP, Saffitz JE. Differential expression of gap junction proteins in the canine sinus node. Circ Res. 1998;82:604–612. doi: 10.1161/01.res.82.5.604. [DOI] [PubMed] [Google Scholar]
- 24.Saffitz JE, Green KG, Kraft WJ, Schechtman KB, Yamada KA. Effects of diminished expression of connexin43 on gap junction number and size in ventricular myocardium. Am J Physiol Heart Circ. 2000;278:H1662–H1670. doi: 10.1152/ajpheart.2000.278.5.H1662. [DOI] [PubMed] [Google Scholar]
- 25.Ingber DE. Tensegrity I. Cell structure and hierarchical systems biology. J Cell Sci. 2003;116:1157–1173. doi: 10.1242/jcs.00359. [DOI] [PubMed] [Google Scholar]
- 26.Valente M, Calabrese F, Thiene G, Angelini A, Basso C, Nava A, Rossi L. In vivo evidence of apoptosis in arrhythmogenic right ventricular cardiomyopathy. Am J Pathol. 1998;152:479–484. [PMC free article] [PubMed] [Google Scholar]
- 27.Yang Z, Bowles NE, Scherer SE, Taylor MD, Kearney DL, Ge SP, Nadvoretskiy VV, DeFreitas G, Carabello B, Brandon LI, Godsel LM, Green KJ, Saffitz JE, Li H, Danieli GA, Calkins H, Marcus F, Towbin JA. Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Res. 2006;99:646–655. doi: 10.1161/01.RES.0000241482.19382.c6. [DOI] [PubMed] [Google Scholar]
- 28.Li J, Swope D, Raess N, Cheng L, Muller EJ, Radice GL. Cardiac tissue-restricted deletion of plakoglobin results in progressive cardiomyopathy and activation of {beta}-catenin signaling. Mol Cell Biol. 2011;31:1134–1144. doi: 10.1128/MCB.01025-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bierkamp C, McLaughlin KJ, Schwarz H, Huber O, Kemler R. Embryonic heart and skin defects in mice lacking plakoglobin. Dev Biol. 1996;180:780–785. doi: 10.1006/dbio.1996.0346. [DOI] [PubMed] [Google Scholar]
- 30.Ruiz P, Brinkmann V, Ledermann B, Behrend M, Grund C, Thalhammer C, Vogel F, Birchmeier C, Gunthert U, Franke WW, Birchmeier W. Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. J Cell Biol. 1996;135:215–225. doi: 10.1083/jcb.135.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gallicano GI, Kouklis P, Bauer C, Yin M, Vasioukhin V, Degenstein L, Fuchs E. Desmoplakin is required early in development for assembly of desmosomes and cytoskeletal linkage. J Cell Biol. 1998;143:2009–2022. doi: 10.1083/jcb.143.7.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grossmann KS, Grund C, Huelsken J, Behrend M, Erdmann B, Franke WW, Birchmeier W. Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J Cell Biol. 2004;167:149–160. doi: 10.1083/jcb.200402096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wakatsuki T, Schlessinger J, Elson EL. The biochemical response of the heart to hypertension and exercise. Trends Biochem Sci. 2004;29:609–617. doi: 10.1016/j.tibs.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 34.Hausenloy DJ, Yellon DM. Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc Res. 2006;70:240–253. doi: 10.1016/j.cardiores.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 35.Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, Marian AJ. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest. 2006;116:2012–2021. doi: 10.1172/JCI27751. [DOI] [PMC free article] [PubMed] [Google Scholar]