

Abstract
Summary: We present a tool, diCal-IBD, for detecting identity-by-descent (IBD) tracts between pairs of genomic sequences. Our method builds on a recent demographic inference method based on the coalescent with recombination, and is able to incorporate demographic information as a prior. Simulation study shows that diCal-IBD has significantly higher recall and precision than that of existing single-nucleotide polymorphism–based IBD detection methods, while retaining reasonable accuracy for IBD tracts as small as 0.1 cM.
Availability: http://sourceforge.net/projects/dical-ibd
Contact: yss@eecs.berkeley.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The notion of identity-by-descent (IBD) between distantly related individuals is playing an increasing role in a variety of genetic analyses, including association mapping (Browning and Thompson, 2012), inferring past demographic history (Palamara et al., 2012; Ralph and Coop, 2013) and detecting signals of natural selection (Albrechtsen et al., 2010). Currently there exist several useful methods for detecting IBD tracts. These methods are based on characterizing similar haplotypes [e.g. GERMLINE (Gusev et al., 2009)] or considering patterns of linkage disequilibrium [e.g. fastIBD, Refined IBD and IBDseq (Browning and Browning, 2011, 2013a, b)], but they do not explicitly model genealogical relationships between genomic sequences. Here, we present a new IBD detection tool, diCal-IBD, which is based on a well-used genealogical process in population genetics, namely, the coalescent with recombination. Another feature that distinguishes our method is that we can incorporate demographic information as a prior.
There seems to be no universally accepted definition of IBD. The definition we adopt is the same as that in Palamara et al. (2012) and Ralph and Coop (2013). Specifically, an IBD tract is defined as a maximally contiguous genomic region that is wholly descended from a common ancestor without any recombination occurring within the region. In contrast to other methods, we allow IBD tracts to contain point mutations, which are likely to occur in humans due to comparable mutation and recombination rates.
diCal-IBD is able to detect IBD tracts with high accuracy in unrelated individuals, between whom the vast majority of shared tracts are <1 cM. Single-nucleotide polymorphism (SNP)-based methods are successful in detecting tracts >2 cM, but have low power for shorter tracts, whereas sequence-based methdods, such as diCal-IBD and IBDseq, maintain reasonable accuracy for tracts as small as 0.1 cM.
2 METHOD
diCal-IBD uses a recently developed demographic inference method called diCal (Sheehan et al., 2013). diCal is formulated as a hidden Markov model, a decoding of which returns the time to the most recent common ancestor (TMRCA) for each site when analyzing only a pair of sequences. A change in TMRCA requires a recombination event and diCal-IBD uses the posterior decoding of TMRCA to call IBD tracts above a user-specified length, optionally trimming the ends of the tracts that have low posterior probabilities.
diCal requires discretizing time by partitioning it into non-overlapping intervals. The user has the option of specifying any discretization scheme. The default setting implemented in diCal-IBD distributes the pairwise coalescence probability uniformly over the intervals, similarly as in PSMC (Li and Durbin, 2011), under a constant population size model. An alternative scheme concentrates the intervals in the period that is most likely to give rise to tracts that are long enough to be detected accurately. These schemes are detailed in the Supplementary Information. Given a variable population size history, we approximate it with a piecewise constant population size.
As an application of IBD prediction, we provide a framework for detecting natural selection. Using the average IBD sharing and posterior probability along the sequence, diCal-IBD identifies regions that exhibit high sharing relative to the background average, indicating possible influence of positive selection.
We refer the reader to the online Supplementary Information for details on data processing, options used in calling diCal, post-processing of posterior decoding and identification of selection.
3 IMPLEMENTATION
diCal-IBD is written in Python 2.7, is platform independent and has a command line interface that allows the user to completely specify its behavior. The implementation allows for parallel runs of diCal on different sequence pairs. diCal-IBD provides a visualization of the predicted tracts, their posterior probabilities and the corresponding TMRCAs and sequence-wide average IBD sharing and posterior probability. Accuracy information is also provided if the true IBD tracts are known.
4 PERFORMANCE
We carried out a simulation study to compare diCal-IBD with the state-of-the-art IBD detection methods. We used ms (Hudson, 2002) to simulate full ancestral recombination graphs (ARGs) for 50 sequences of 10 Mb each. We used a constant recombination rate of , and the African and European demographic histories inferred by Tennessen et al. (2012). We simulated perfectly phased sequence data on the ARGs with a constant mutation rate of per base per generation. From the simulated ARGs, we reconstructed the true pairwise IBD tracts by finding maximally consecutive sites that have the same TMRCA for the pair in question. We only considered tracts of length >0.1 cM.
To run SNP-based methods, we generated SNP data with ∼1 marker per 0.2 kb. For further details on running existing tools, see Supplementary Information.
Figure 1 shows the recall (percentage of true tracts that were correctly recovered), precision (percentage of predicted tracts that were correctly predicted) and F-score (harmonic mean of recall and precision) for each method. See Supplementary Information for other measures of accuracy as a function of the true tract length, as well as the effects of errors in the data, demography, discretization and trimming based on posterior probabilities. As the figure shows, diCal-IBD was able to recall significantly more tracts with greater precision than could SNP-based methods, leading to a much higher F-score. diCal-IBD was run assuming a constant population size, but its accuracy performance for the examples considered did not seem to be affected much by using this incorrect prior. This suggests that the posterior distribution inferred by diCal is robust to mis-specification of population sizes; whether this trend persists for more complex demographies deserves further investigation.
Fig. 1.
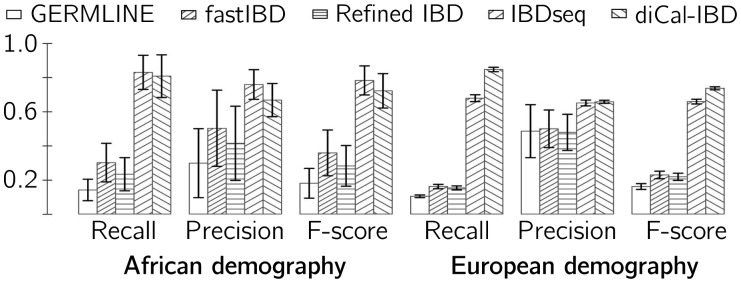
diCal-IBD was run with bin size 100, constant population size, time discretized in 10 intervals according to unconditional coalescence probability, minimum tract length of 0.1 cM and trimming of tracts with a threshold of 0.2. Error bars show the variance
The precision and recall performance of diCal-IBD was comparable with that of IBDseq, with neither one strictly dominating the other on all population size histories. See Figure 1. A strength of diCal-IBD is its ability to explicitly incorporate demographic information. diCal, on which diCal-IBD is based, was originally developed for inferring variable effective population sizes, but it is being extended to handle more complex demographic models, incorporating multiple populations, population splits, migration and admixture. diCal-IBD will be updated in parallel with diCal and hence will be able to use a complex demographic model as a prior.
Figure 2 illustrates the potential of applying diCal-IBD to identify regions under selection (Albrechtsen et al., 2010). We refer the reader to the Supplementary Information for further details.
Fig. 2.
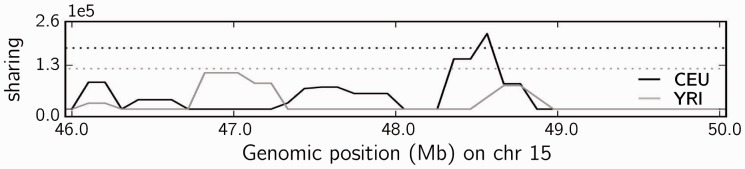
Detection of high sharing using diCal-IBD, on a 4 Mb genomic segment (46.0–50.0 Mb) on chromosome 15 from Complete Genomics data (Drmanac et al., 2010). This region contains a gene thought to be under positive selection in the European population (CEU), but not in the African population (YRI), located at 48.41–48.43 Mb, corresponding with the observed peak in the plot. Dotted lines indicate the thresholds for considering that a region exhibits high sharing
Supplementary Material
ACKNOWLEDGEMENTS
We thank Sara Sheehan, Jack Kamm, Matthias Steinrücken and other members of the Song group for helpful discussions.
Funding: This research is supported in part by an NSF IGERT grant (J.A.N.) from CiBER at UC Berkeley, an NIH grant R01-GM094402 (Y.S.S.) and a Packard Fellowship for Science and Engineering (Y.S.S.).
Conflict of interest: none declared.
REFERENCES
- Albrechtsen A, et al. Natural selection and the distribution of identity-by-descent in the human genome. Genetics. 2010;186:295–308. doi: 10.1534/genetics.110.113977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning BL, Browning SR. A fast, powerful method for detecting identity by descent. Am. J. Hum. Genet. 2011;88:173–182. doi: 10.1016/j.ajhg.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning BL, Browning SR. Detecting identity by descent and estimating genotype error rates in sequence data. Am. J. Hum. Genet. 2013a;93:840–851. doi: 10.1016/j.ajhg.2013.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning BL, Browning SR. Improving the accuracy and efficiency of identity-by-descent detection in population data. Genetics. 2013b;194:459–471. doi: 10.1534/genetics.113.150029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning SR, Thompson EA. Detecting rare variant associations by identity-by-descent mapping in case-control studies. Genetics. 2012;190:1521–1531. doi: 10.1534/genetics.111.136937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drmanac R, et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science. 2010;327:78–81. doi: 10.1126/science.1181498. [DOI] [PubMed] [Google Scholar]
- Gusev A, et al. Whole population, genome-wide mapping of hidden relatedness. Genome Res. 2009;19:318–326. doi: 10.1101/gr.081398.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493–496. doi: 10.1038/nature10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palamara PF, et al. Length distributions of identity by descent reveal fine-scale demographic history. Am. J. Hum. Genet. 2012;91:809–822. doi: 10.1016/j.ajhg.2012.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph P, Coop G. The geography of recent genetic ancestry across Europe. PLoS Biol. 2013;11:e1001555. doi: 10.1371/journal.pbio.1001555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan S, et al. Estimating variable effective population sizes from multiple genomes: a sequentially Markov conditional sampling distribution approach. Genetics. 2013;194:647–662. doi: 10.1534/genetics.112.149096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen JA, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.