

Abstract
Normally, sonic hedgehog (Shh) signaling induces high levels of Patched 1 (Ptc1) and its associated transcription factor Gli1 with genesis of specific neuronal progeny. But their roles in the neural stem cells (NSCs), including glial precursor cells (GPCs), of Alzheimer's disease (AD) are unclear. Here, we show that Ptc1 and Gli1 are significantly deficits in the hippocampus of an aged AD transgenic mouse mode, whereas these two molecules are highly elevated at young ages. Our similar findings in autopsied AD brains validate the discovery in AD mouse models. To examine whether Aβ peptides, which are a main component of the amyloid plaques in AD brains, affected Ptc1-Gli1 signaling, we treated GPCs with Aβ peptides, we found that high dose of Aβ1–42 but not Aβ1–40 significantly decreased Ptc1-Gli1, while Shh itself was elevated in hippocampal NSCs/GPCs. Furthermore, we found that deficits of Ptc1-Gli1 signaling induced NSCs/GPCs into asymmetric division, which results in an increase in the number of dividing cells including transit-amplifying cells and neuroblasts. These precursor cells commit to apoptosis-like death under the toxic conditions. By this way, adult neural precursor cell pool is exhausted and defective neurogenesis happens in AD brains. Our findings suggest that Ptc1-Gli1 signaling deregulation resulting abnormal loss of GPCs may contribute to a cognition decline in AD brains. The novel findings elucidate a new molecular mechanism of adult NSCs/GPCs on neurogenesis and demonstrate a regulatory role for Ptc1-Gli1 in adult neural circuit integrity of the brain.
INTRODUCTION
Sonic hedgehog (Shh) binds to two Patched (Ptc) proteins, Ptc homolog 1 (Ptc1) and Ptc homolog 2 (Ptc2) with similar high-affinity and inhibits Smo acitivities (1,2). Smo triggers a signaling cascade (3) and regulates Gli zinc finger transcription factor, Gli1, Gli2 and Gi3, which have distinct and overlapping functions in response to Shh signaling (4,5). Gli1 and Gli2 are then translocated into the nucleus and activate gene transcription and Gli3 acts as a repressor (6). The balance of Gli functions influences expression of target genes (7).
Shh acts as a morphogen in embryonic neural development (8,9). Once development has been completed, the expression of Shh-mediated signaling molecules declines to low levels in normal healthy brains (10), maintains the neurogenic niches (11–13) and controls cell division of neural stem cells (NSCs) or glial precursor cells (GPCs) (14,15). The up-regulation of Shh signals is involved in brain stroke (16) and even multiple sclerosis (17).
The impairment of learning and memory is one of characteristics of Alzheimer's disease (AD), which is associated with the overproduction of amyloid β protein (Aβ) (18,19). There are arguments on the exploration of neurogenesis in AD brains (20–23). Hippocampal neurogenesis is closely associated with learning and memory. To our knowledge, however, there is still no investigation of Shh signaling and its relationship with neurogenesis in AD brains.
In the present study, we find an increased level of Shh signaling in the hippocampi of APP23 mice and AD patients. Soluble Aβ1–42 up-regulates Shh pathway of hippocampal progenitor cells. The elevation of Shh signaling accelerates NSCs or GPCs into division and differentiation, resulting in an increase in the number of immediate GPCs in response to elevated Shh level. Cyclopamine application inhibits an increase in cell proliferation induced by Shh signals. Rapidly increased NSCs or GPCs turn toward apoptosis-like death. Because of lacking potential of self-renewal, NSC and GPC pool is accelerated to be depleted by activated Shh signaling and finally results in an impaired neurogenesis, as well as astrogenesis in AD brains.
RESULTS
Levels of both Aβ1–40 and Aβ1–42 are elevated in the hippocampus of APP23 mice
Before we explore the effects of Aβ on GPCs, we used APP23 transgenic mice (24), a mouse model for AD with a plaque-predominant type, overproducing Aβ, Aβ1–40 and Aβ1–42. To characterize and ensure Aβ load in the hippocampus, immunostaining was performed with 6E10 antibody against Aβ1–17, results showed, in the hippocampus, intracellular Aβ-positive immunostaining without Aβ deposits at 3 months old, a few plaque deposits at 12 months old and a dense amyloid deposits in the molecular layer (Mol) at 24 months old APP23 mice (Fig. 1A). There are two main segments of Aβ peptides, Aβ1–40 and Aβ1–42. Due to different effects of the different forms of Aβ in AD brains (25), we measured by ELISA the soluble and insoluble Aβ of the hippocampus formation at different age stages. Similar to the previous report of Aβ measured in the neocortex of APP23 mice (26), the amount of both soluble Aβ1–40 and Aβ1–42 peptides in the hippocampus of APP23 mouse brains was constantly and significantly elevated at all ages when compared with age-matched WT mice (Fig. 1B). With age, levels of both Aβ1–42 and Aβ1–40 were slightly increased but not significantly different from 3 to 24 months old APP23 mice. The average levels of soluble Aβ1–42 were significant higher than those of soluble Aβ1–40 at all ages of APP23 mice. Within the hippocampus of WT, the average values of soluble Aβ1–40 and Aβ1–42, however, were much lower and similar at all ages (Fig. 1B). Moreover, we measured insoluble Aβ, main components as deposited plaques in AD and found that, in the hippocampus of APP23 mice, the amount of insoluble Aβ1–40 was maintained at a basal level in the first 9 months of the life span in APP23 mice (Fig. 1C). A significant increase in insoluble Aβ1–40 amount was observed until 12 months of age and then a sharp elevation at 18 and 24 months of age (Fig. 1C). Similarly, the levels of insoluble Aβ1–42 were also maintained at a basal level in the hippocampus of APP23 mice until the age of 12 months and then increased but significantly lower than Aβ1–40 levels by 18 and 24 months of age (Fig. 1C).
Figure 1.

Aβ is overexpressed and Shh levels are elevated in the hippocampus of APP23 mice. (A) Immunostaning of antibody clone 6E10 showed Aβ-positive reaction in the hippocampus of 3-, 12- and 24 months old APP23 mice. Intracellular Aβ-positive reaction in 3 months old, a few of Aβ deposit plaques in the subiculum of 12-month-old, lots of Aβ plaques, especially dense deposits in molecular layer in hippocampus were visualized in 24 months old APP23 mice. Scale bar: 50 µm. (B) The levels of soluble Aβ1–42 and Aβ1–40 levels were tested in the hippocampi of different age group of WT and APP23 mice (n = 5 in each group, Student t-test, **P < 0.01 of soluble Aβ1–42 and Aβ1–40 versus corresponding that of WT, respectively; ##P < 0.01 of soluble Aβ1–42 versus soluble Aβ1–40 amount in APP23 mice). (C) The levels of insoluble Aβ1–42 and Aβ1–40 were measured in the hippocampi of different age groups of WT and APP23 mice (n = 5 in each group, Student t-test, **P < 0.01 of insoluble Aβ1–42 and Aβ1–40 versus corresponding that of WT, respectively; ##P < 0.01 of insoluble Aβ1–42 versus insoluble Aβ1–40 levels in APP23 mice). (D) Representative micrographs showed Shh expression in the hippocampi of WT and APP23 mice. (E). A significant increase in the expression of ligand Shh with different molecular weights was observed in the hippocampus at 3, 12 and 24 months old APP23 mice (n = 10 in each group, Student's t-test, **P < 0.01 versus WT).
Shh levels are elevated in the hippocampus of APP23 mice
Shh signaling maintains NSC/GPC pool in the hippocampus (11,15). To test whether Shh protein levels in the hippocampus were changed in APP23 transgenic mice and if so, whether Shh expression was changed as aging. The hippocampi were dissected from WT and APP23 mice at ages of 3, 12 and 24 months old for Shh detection by western blot. We found three bands of the molecular weights: ∼50, ∼100 and ∼150 kDa in APP23 mouse brains and aged WT brains (Fig. 1D). Evidence has shown that ligand Shh is produced as an ∼20 kDa N-terminal signaling domain (ShhN) and modified by adding palmitoyl on N-terminal amino acids and cholesterol on carboxyl end (27,28). The high molecular weight Shh may be free diffusible and soluble multimeric forms of Shh with lipophilic modification(s-ShhNp) (28,29). By qualification analysis, we found that Shh expression levels at each molecular weight were significantly elevated at all ages of APP23 mice, compared with age-matched WT mice (Fig. 1E). Due to overdue accumulation of Aβ along with age in APP23 mice (24), the deregulation of Shh levels is probably associated with the effects of Aβ.
Expressions of Ptc-Gli signaling components are deficits in old AD transgenic mice where these signals are elevated at young ages
Shh-receiving cells exist in the hippocampal formation (11,15). It has been known that the application of Shh enhances the signaling component expressions of Shh-responding cells (30,31). Whether the elevated Shh level in the hippocampus of APP23 mice was accompanied with a corresponding up-regulation of Shh signaling component molecules was not clear. There are two Shh binding proteins, Patched homolog 1 (Ptc1) and 2 (Ptc2) (1,32). Ptc1 is highly expressed in adult hippocampus (10,33) as well as isolated NSCs/GPCs (11). To examine whether the Ptc1 expression level in APP23 mice was changed, immunostaining was first performed with antibody against Ptc1 at 12 months old mice. Results showed an increased expression in the granular cells of dentate gyrus, especially in the cells of hilus region (Fig. 2A). To further confirm the results from IHC, the hippocampus was dissected from APP23 mice and Ptc1 expression was probed. Western blot showed that the expression levels of Ptc1 had not been significantly elevated at 3 months old yet (Fig. 2B and C), but greatly increased in the hippocampus at 12 months old APP23 mice (Fig. 2D and E). Unexpectedly, the Ptc1 levels were reduced in the hippocampus of 24-month-old APP23 mice (Fig. 2F and G). Ptc2 has similar and overlapping functions of Ptc1 (1). Similar to Ptc1 expression pattern, we found that levels of Ptc2 were elevated at 12-month-old and reduced at 24-month-old APP23 mice (Fig. 2B–G). Smo membrane proteins, which is inhibited by Ptc (34), are expressed in the normal hippocampus (10,33). We tested Smo levels and found that Smo levels were significantly elevated at 3, 12 and 24 months old APP23 mice compared with corresponding WT controls (Fig. 2B–G). Gli proteins are three types of zinc finger transcription factors, Gli1, Gli2 and Gli3, which have distinct and overlapping functions (5). Gli1 is a canonical transcription factor as a read-out in response to Shh signals (32), which activates transcription and practice Shh signaling role in hippocampal NSCs (15). We measured Gli1 expression in the hippocampus and found a significant increase in 12- and 24-month-old APP23 mice (Fig. 2D–G), still without a significant increase at 3-month-old-APP23 mice (Fig. 2B and C). Gli2, an activator like as Gli1, is involved in initial Shh response (35,36). Similar to the Gli2 expression pattern, we found that expression levels of Gli2 were also greatly elevated in 24-month-old APP23 mice (Fig. 2F and G), but not be significantly increased at 3 and 12 months of age (Fig. 2B–E). Gli3, a predominant negative regulator for Shh signaling transcription, is upregulated in the absence of Shh signals (32). Gli3 was probed by western blot and results showed a significant decrease in expression levels in 24-month-old APP23 mice (Fig. 2F–G), but not significant changes at 3 and 12 months of age, compared with corresponding age-matched WT mice (Fig. 2B–E). Even if a decrease in Ptc levels is observed, the expression levels of Gli1 and Gli2 are still elevated. It indicates that the Shh signaling still play a role in the hippocampus of aged APP23 mice. These results show that, responsive to increased Shh levels, Shh-receiving cells in hippocampus at younger and aged stage have different responses in APP23 mice. The difference could stem from the changes of cell components in hippocampus at different age stage.
Figure 2.
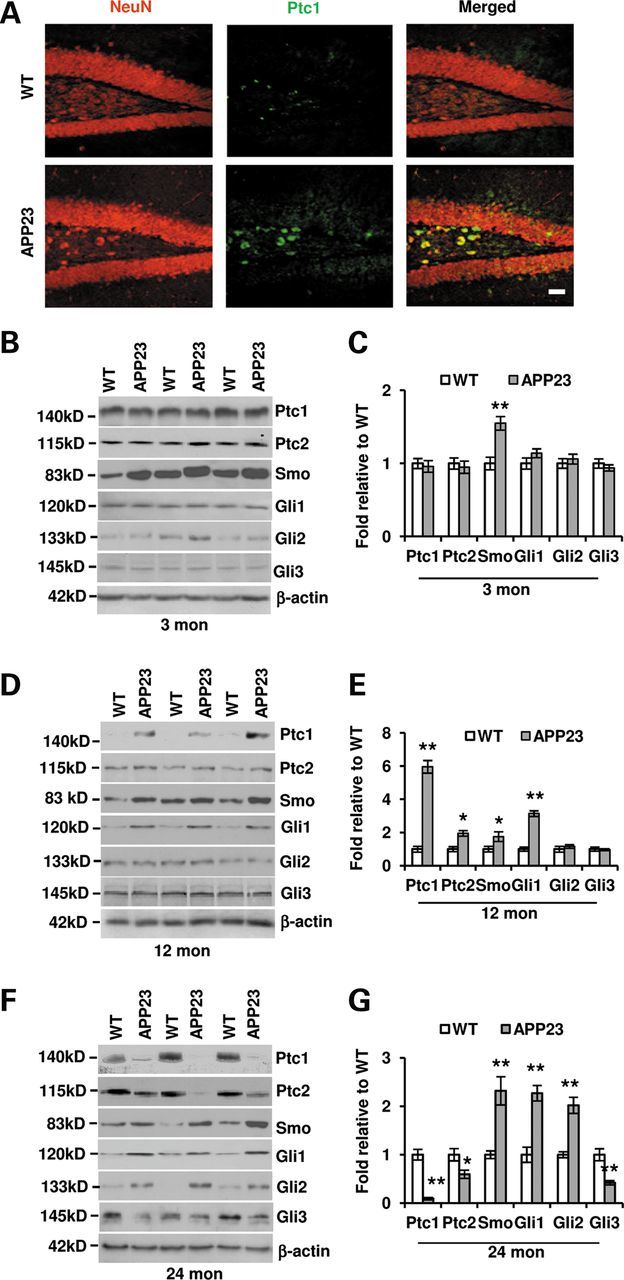

Shh signal components are altered in the hippocampus of both APP23 mice and AD patients. (A) Ptc1 expression (green) was visualized by immunostaining in the dentate gyrus with an increased Ptc1 expression in 12-month-old APP23 mice, especially in the cells of hilus region. Neurons were stained with red. Scale bar: 50 µm. (B) Western blot showed the protein expressions of Shh signaling components in the hippocampus of 3-month-old APP23 mice. (C) Statistical analysis showed that the Smo expression level is increased (n = 10, Student's t-test, **P < 0.01) in APP23 mice of 3 months old of age, whereas the levels of Ptc1, Ptc2, Gli1, Gli2 and Gli3 expressions were still not significantly changed. (D) Western blot showed the protein expressions of Shh signaling components in the hippocampus of 12-month-old APP23 mice. (E) Statistical analysis showed that the expression levels of Ptc1, Ptc2, Smo and Gli1 were increased (n = 10, Student's t-test, *P < 0.05, **P < 0.01 versus corresponding WT) in APP23 mice of 12 months old of age, whereas the levels of Gli2 and Gli3 expressions were not significantly changed. (F) Western blot showed the protein expressions of Shh signaling components in the hippocampus of 24-month-old APP23 mice. (G) Statistical analysis showed that the expression levels of Smo, Gli1 and Gli2 are increased (n = 10, Student's t-test, *P < 0.01 versus corresponding WT) in APP23 mice of 24 months old of age, whereas the levels of Ptc1, Ptc2 and Gli3 expressions were significantly reduced (n = 10, Student's t-test, *P < 0.05, **P < 0.01 versus corresponding WT). (H) Western blot showed that Shh signaling components were expressed in the hippocampus of isolated from human brains of HC and AD patients. (I) The protein levels of Shh, Smo, Gli1 and Gli2 expression were significantly increased, whereas the expression levels of Ptc1, Ptc2 and Gli3 were significantly reduced in comparison with corresponding health controls without dementia (n = 5, Student's t-test, *P < 0.05, **P < 0.01 versus HC).
Ptc-Gli signaling in human AD brains validates the findings from the transgenic mouse model
To examine whether the elevation of Shh signaling in the hippocampus of APP23 mice reflects the real pathological situation in AD brains, we harvested the hippocampal tissues from both AD (n = 5) and healthy control (HC) samples (n = 5) to examine the expression of Shh signaling. Western blotting study showed an apparent and significant increase in Shh expression levels in the AD hippocampus compared with the HC (Fig. 2H and I). Interestingly, different from mice with Shh molecular weights of ∼50, ∼100 and ∼150 kDa, only one obvious band with ∼50 kDa was observed in the hippocampus of human beings (Fig. 2H). Similarly, we found that the expression levels of Ptc1 and Ptc2 were reduced in AD (Fig. 2H and I), counterpart to the observation in 24-month-old APP23 mice. Next, we tested the expression of Smo and found significantly increased levels of Smo expression in the hippocampus of AD (Fig. 2H and I), also similar to the findings in 24 months old APP23 mice. Furthermore, we tested the transcription factor proteins Gli1, 2 and 3 and found that both Gli1 and Gli2 protein levels were significantly elevated, whereas Gli3 repressor levels were significantly decreased in AD brains (Fig. 2H and I). The results indicate that the alteration of Shh signaling in AD brains may be a counterpart of aged (24-month-old) APP23 mice, reflecting an activated Shh signaling in the hippocampus of AD brains.
Aβ1–42 decreases Ptc-Gli1 while it elevates Shh signaling in cultured GPCs
NSCs or GPCs isolated from mouse SVZ secrete Shh proteins in an autocrine fashion (37) and hippocampal GPCs express Shh mRNA (31). However, whether Shh proteins could be tested from isolated hippocampal GPCs was still not clear. To clarify Shh secretion from the hippocampal GPCs, we isolated GPCs from neonatal WT mice and cultured for 2 weeks. The spheres were dissociated and cultured for another 2 weeks. The conditioned media were collected and the secreted proteins were analyzed. Western blot showed that Shh molecules exist in the cultured media of hippocampal GPCs (Fig. 3A), suggesting a release of Shh from the cultured GPCs. Expectedly, Shh expression was found in the cell lysate of GPCs (Fig. 3A). It suggests that the hippocampal GPCs could produce Shh protein in an autocrine manner (31). It has been shown that Aβ1–42 peptide application increases new neuron production (38,39). To exam whether the enhanced differentiation by Aβ1–42 peptides would be associated with Shh signaling, the GPCs were treated with Aβ1–42 for 72 h. The conditioned medium and the cultured cells were harvested. Using western blot techniques, we found that the levels of Shh expression were elevated in the cultured medium with Aβ1–42 treatment (Fig. 3A) and in a dose-dependent manner (Fig. 3B). Similar findings of Shh expression were observed in the GPC lysate and Shh levels were expectedly elevated with Aβ1–42 incubation in dose-dependent manner. A little surprise, the different molecular weights between 37 and 150 kDa were observed (Fig. 3A), similar to the findings in the hippocampi of mice (Fig. 1D), suggesting the presence of Shh multimer molecule in isolated hippocampal GPCs. A fraction of GPCs is responsive to Shh signal (11,15). A previous report showed detectable mRNA levels of Shh signaling components in isolated hippocampal GPCs (31). Next, we examined the expressions of Shh pathway components. We found apparent increases in Ptc1, Ptc2, Smo, Gli1 and Gli2 expression levels but without significant changes of Gli3 levels in the presence of Aβ1–42 (Fig. 3C and D). However, the elevation of Ptc1 expression is blocked by Aβ1–42 application at the high concentration (100 µm) even if Shh levels are still elevated at this dose of Aβ1–42 (Fig. 3C and D). The result that the high dose of Aβ1–42 exposure reduces the molecule levels of Ptc1 expression is consistent with the change in the hippocampal formation at 24 months old APP23 mice (Fig. 2F and G) and in AD patient brains (Fig. 2H and I). It may be an effect of Aβ itself or other factors induced by excessive Aβ in the dysregulation.
Figure 3.
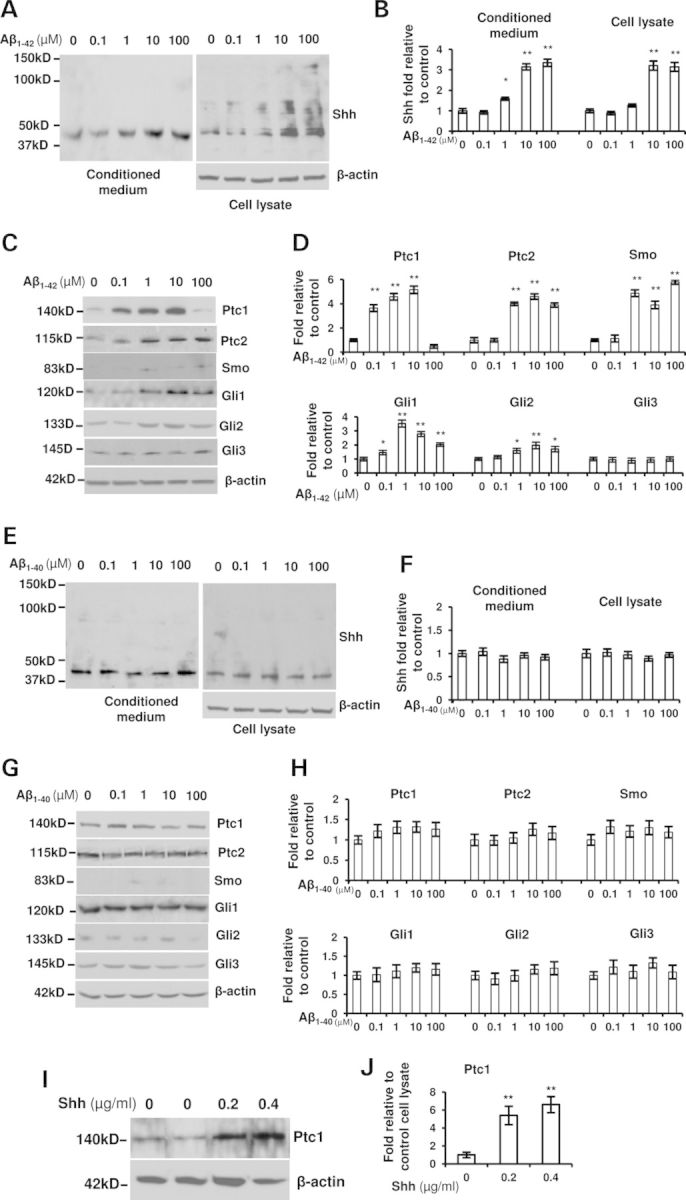
Aβ1–42 not Aβ1–40 increases the expressions of Shh signaling in hippocampal GPCs derived from WT mice. The suspended cells were incubated with the presence of 0, 0.1, 1, 10 or 100 µm of Aβ1–42 or Aβ1–40 for 3 days. (A) Shh expression with different molecular weights was shown by western blot from the cultured medium and the cells treated with Aβ1–42. (B) Shh levels were significantly increased in the conditioned medium (normalized to corresponding cell lysate) and cultured cells in a dose-dependent manner with Aβ1–42 treatment (ANOVA test, *P < 0.05, **P < 0.01). (C) The expression of Shh signaling components was shown in the cells with Aβ1–42 treatment. (D) The levels of Ptc1, Ptc2, Smo, Gli1 and Gli2 were significant elevated (ANOVA test, *P < 0.05, **P < 0.01) but not significant changes of Gli3 levels with the exposure of Aβ1–42 (ANOVA test, P > 0.05). (E) Shh expression was shown from the conditioned medium and GPCs treated with Aβ1–40. (F) Shh levels were not significantly increased in the conditioned medium (normalized to corresponding cell lysate) and the cultured GPCs with the Aβ1–40 treatment (ANOVA test, P > 0.05). (G) The expression of Shh signaling component molecules was shown in the GPC lysate with the Aβ1–40 treatment. (H) The levels of the components were not significantly changed with the Aβ1–40 treatment. (I) Western blot showed Ptc1 expression with the exogenous Shh application. (J) A statistical analysis showed a significant promotion of Ptc1 expression levels in WT hippocampus progenitor cells for 3 days of Shh incubation (ANOVA test, **P < 0.01). Experiments were repeated three times per condition.
It has also been reported that Aβ1–40 peptide enhances GPC proliferation in vitro (39,40). To test whether the increased proliferation is through Shh signaling, we treated the hippocampal GPCs with different doses of Aβ1–40 for 72 h. Results showed that there were not significant changes of Shh expression levels in the conditioned medium with various doses of Aβ1–40 incubation (Fig. 3E and F). Similarly, little changes of Shh levels were observed in the GPC lysate treated with Aβ1–40. By using the same approach, we tested the downstream molecules of Shh signaling and found no significant changes in protein levels in the GPCs exposed to Aβ1–40 (Fig. 3G and H). The results suggest that Aβ1–40-induced proliferation may not be associated with Shh signaling. It has been reported that Shh promotes Ptc1 expression levels (3,30,31). To further clarify that the increased expression of Shh pathway components is caused by Aβ1–42 itself or by Aβ1–42-triggered Shh, we treated the GPCs with Shh molecule for 72 h and found an increased level of Ptc1 expression (Fig. 3I and J), suggesting that Aβ-induced Shh level elevation is associated with the level increase of Shh signaling components instead of Aβ itself.
Cell proliferation is altered and Shh signaling blockage prevents an increase in the number of dividing cells in APP23 mice
It has been suggested an increased proliferation in the hippocampus of APP23 mice (20,40). To further clarify the possible change in cell proliferation happened in APP23 and WT mice at 3, 12 and 24 months old, they were injected with BrdU twice a day. The brain sections were analyzed within 24 h. BrdU-positive cells were visualized by immunostaining and the positive nuclei were primarily showed in SGZ (Fig. 4A). We found a significant increase of more than 2-folds in the number of BrdU-positive cells in SGZ of 3-month-old APP23 mice compared with corresponding WT (P < 0.01, Fig. 4B), but no significant differences in the hilus, granular cell layer (GCL) and Mol (P > 0.05). To analyze whether or not the increase of dividing cells at 3-month-old APP23 mice is mediated by activation of Shh signaling, we applied cyclopamine, a Smo inhibitor, which has been shown to block activation of Shh signal transduction pathway (34) and inhibit the proliferation of hippocampal dividing cells (11). WT and APP23 mice at age of 3 months old were injected with cyclopamine or vehicle alone as reported (11,34,41). One-week injection was followed by twice a day of BrdU injection. After 24 h last BrdU application, immunohistochemistry was performed with antibody against BrdU. The number of immunopositive nuclei was counted in SGZ. We found that about 40% of BrdU-positive cells were reduced along with cyclopamine application in the WT group (P < 0.05, Fig. 4C). However, ∼65% of BrdU+ cells were prevented with cyclopamine application, compared with APP23 mice in the vehicle group (P < 0.01, Fig. 4C). Meanwhile, we also observed the proliferation in 12 and 24 months old mice and found an ∼30% increase in the number of BrdU+ cells in SGZ at 12-month-old APP23 mice (P < 0.05, Fig. 4D). At the age of 24 months old, however, a significant decrease in BrdU-positive cell number was found in the SGZ (P < 0.01, Fig. 4E). The number of BrdU+ cells in the hilus, GCL and Mol did not have significant changes between WT and APP23 mice at this age (Fig. 4E). These results indicate that the elevation of Shh signaling levels contributes to the altered proliferation in the hippocampus of APP transgenic mice.
Figure 4.

Cell proliferation changes and Smo antagonist cyclopamine prevents Shh-induced cell dividing in APP23 mice. (A) Microphotographs showed BrdU-positive staining nuclei in the hippocampus of WT and APP23 mice at 3 months old. The dentate gyrus was counterstained with hemotaxylin. Scale bar: 50 µm. (B) At 3 months old of APP23 mice, the number of BrdU-positive cells in SGZ was significantly increased (n = 6 each group, Student's t-test, **P < 0.01) whereas there were not significant changes in the regions of Hilus, Mol and GCL, compared with age-matched WT controls. (C) With the application of cyclopamine for 7 days, the number of BrdU+ cells were significantly decreased in WT (n = 5 each group, Student's t-test, *P < 0.05) and APP23 (n = 5 each group, Student's t-test, **P < 0.01) mice of 3 months old, compared with corresponding controls. (D) At 12 months old of APP23 mice, the number of BrdU-positive cells in SGZ was counted and the total number per hippocampus was summed and shown as mean ± SEM per mice. BrdU-positive cells were significantly increased (n = 6 each group, Student's t-test, *P < 0.05) whereas there were not significant changes in the regions of Hilus, Mol and GCL. (E) At 24 months old of APP23 mice, however, the number of BrdU-positive cells in SGZ was significantly decreased (n = 6 each group, Student's t-test, **P < 0.01) whereas there was not significant changes in Hilus, Mol and GCL.
Survival of NSCs decreases and apoptotic cell number increases in APP23 mice
It is unknown whether the enhanced cell proliferation at 3-month-old APP23 mice could be in part associated with survival of dividing cells. The staining sections were analyzed 4 weeks after last BrdU injection to determine the survival of the newly dividing cells. We found that the number of BrdU-positive cells was reduced within 4 weeks. Within 24 h last BrdU injection, the number of BrdU+ cells was 2146 ± 192 in WT and 4214 ± 310 in APP23 mice. Four-week later, remaining BrdU+ cells were 677 ± 32 in WT and 450 ± 41 in APP23mice. About 32% in WT and ∼11% of BrdU+ cells in the hippocampus of APP23 mice survived 4 weeks, respectively (P < 0.01, Fig. 5A), indicating that about 70 and 90% newly generated cells disappear in 4 weeks in WT and APP23 mice. A part of surviving dividing cells could migrate toward and integrate into GCL. A few cells are still observed in SGZ. To understand the migration and integration of remaining cells, we calculated the remaining BrdU+ cells in the SGZ and GCL 4 weeks later. Results showed a significantly decreased number of BrdU+ cells in the SGZ of APP23 mice versus WT (P < 0.01, Fig. 5B). Similarly, the number of BrdU+ cells into GCL was also reduced in 3 months old APP23 mice (P < 0.05, Fig. 5B), suggesting that the loss of proliferating cells are accelerated in APP23 mice. Whether the great decrease in remaining BrdU-positive cells is a result of cell death remains to be verified. We performed transferase-mediated biotinylated UTP nick end labeling (TUNEL) staining for apoptotic cells in 3-month-old mice. Immunostaining revealed apoptosis-like nuclei in DG (Fig. 5C). Staining positive nuclei were counted and results showed a significant increase in the number of apoptosis-like nuclei in APP23 mice (P < 0.01, Fig. 5D).
Figure 5.

The survival of GPCs decrease and GPC apoptosis increases in APP23 mice. (A) Four weeks last BrdU injection the total number of remaining BrdU+ cells was significantly reduced (n = 6 in each group, Student's t-test, **P < 0.01) in APP23 mice. (B) Four weeks last BrdU application, the total number of surviving BrdU+ cells was counted and was shown as mean ± SEM per mice. Analysis showed a significant decrease in the remaining BrdU+ nuclei at SGZ and GCL of APP23 mice compared with WT (n = 6 in each group, Student's t-test, **P < 0.01, *P < 0.05) but no significant changes in hilus and Mol regions. (C) Representative micrographs of TUNEL staining showed TUNEL-positive nuclei (green) in the DG of WT and APP23 mice at age of 3 months old. Neurons were showed in red color with NeuN antibody. Arrowheads are for TUNEL-positive nuclei. Scale bar: 50 µm. (D) TUNEL-positive nuclei were counted and the average number per section was shown as mean ± SEM. Quantification of immunoreactive structures revealed a significant increase in the number of apoptotic nuclei in 3-month-old APP23 mice compared with age-matched WT (n = 6 in each group, Student's t-test, **P < 0.01). (E) GPC number declines and astrocyte number increases in the hippocampus of APP23 mice. Double labeling showed nestin-positive cells (green) and GFAP-positive cells (red). Closed white arrowhead was for GPCs (co-labeling of both GFAP and nestin); yellow arrowhead for transit-amplifying cells (nestin-positive and GFAP-negative), and white arrows for mature astrocytes (nestin-negative and GFAP-positive). Scale bar: 20 µm. (F) Nestin+GFAP+ expressing cells (NSCs), nestin+GFAP− cells (transit-amplifying cell) and GFAP+/nestin− cells (mature astrocyte) were counted and the average number per section was shown as mean ± SEM. Statistical analysis was significant difference (n = 6 in each group, Student's t-test, **P < 0.01 versus WT). (G) Double labeling showed BrdU+ cells (green) and GFAP+ cells (red) in the hilus of 12 months old WT and APP23 mice. Scale bar: 20 µm.
Numbers of GPCs were decreased and astrocytes were increased in APP23 mice
Hippocampal GPCs, a population of GPCs resided in the SGZ of hippocampus, have astroglia-like characteristics expressing GFAP, vimentin, nestin and produce new neurons. The cells, which lack S100β and DCX expression (42,43), extend small trees of dendritic-like processes perpendicular to the GCL into the inner molecular layer (44). A fraction of hippocampal GPCs expressing nestin respond to mitogen Shh (15). Whether the elevated Shh levels in the hippocampus could increase nestin-positive cells remains to be investigated. To observe the group of nestin+ GPCs, GFAP, another marker for radial glial-like GPCs (43), was immunolabeled together (Fig. 5E). We found a great reduction in GFAP+/nestin+ co-labeling NPCs in the DG of 3-month-old APP23 mice in comparison of WT controls (P < 0.01, Fig. 5F). As quiescent GPCs turn toward differentiation, GFAP expression is extinguished and cells adopt a migratory-like morphology. These cells represent a transient amplifying population of GPCs, referred to as fast-dividing transit-amplifying cells (43,44). We found the nestin-positive and GFAP-negative immunostaining cells with a short process localized in SGZ and hilus (Fig. 5E) and a significant decrease in the number of transit-amplifying cells (P < 0.01, Fig. 5F). Mature astrocytes, which are nestin-negative and GFAP-positive immunolabeling cells, were observed in hilus of DG (Fig. 5F). We found that the number of mature astrocytes (nestin−GFAP+) was significantly increased in 3 months old APP23 mice (P < 0.01, Fig. 5F). To confirm the astrocytes come from dividing GPCs, the double staining with antibodies against BrdU and GFAP are performed at 12 months old WT and APP23 mice. Results showed a few of double-labeling cells in the hilus (Fig. 5G), suggesting the increased number of astrocytes might be, at least in part, from dividing GPCs. In addition, the increased astrocyte number might be in part from the activation of dominant astrocytes in responsive to elevated Shh signaling (45) or inflammation by Aβ. However, we cannot perform statistical analysis between WT and APP23 mice due to few cells of co-labeling BrdU and GAFP.
Defective neurogensis in APP23 mice
After exiting the cell cycle, most of the nascent cells die, and the surviving cells then differentiate into neurons and glia. It has been shown that a fraction of psa-NCAM-positive cells (neuroblast) are Shh-responding cells (15). Whether the neuronal commitment of the dividing progenitor cells could be increased in response to the intensified Shh signaling is not clear. Doublecortin (DCX) is another marker for neuroblasts (46,47). Immunostaining was performed and results showed a significant increase in number of DCX+ cells at 3-month-old, whereas a significant decrease in the number at 12- and 24-month-old APP23 mice compared with age-matched WT (Fig. 6A and B). Whether the increased neuroblasts at 3-month-old APP23 mice are associated with dividing BrdU+ cells is not clear. Double staining of psa-NCAM and BrdU was applied. We found co-labeling cells of psa-NCAM and BrdU (Fig. 6C). Double labeling cells were counted and results showed that the average percentage of co-labeling cells to psa-NCAM-positive cells is 31.5 ± 7.2 and 12.8 ± 3.7 per section in APP23 and WT mice at age of 3 months old, respectively (Fig. 6D). Statistical analysis showed a significant increase in the psa-NCAM+/BrdU+ cells in 3 months old APP23 mice (P < 0.01, Fig. 6E), suggesting that the increase in the number of dividing cells in part is from the differentiating neuroblasts. Due to too few double-labeling cells, we did not perform statistical analysis. To observe the fate of the increased neuroblasts at 3-month-old APP23 mice, the mice were detected with double-labeling of BrdU and NeuN (mature neuron marker) after 4 weeks of BrdU application. The NeuN-positive nuclei with BrdU labeling were showed (Fig. 6E). Double labeling cells were counted and results showed that the average co-labeling cell number is 11.5 ± 1.9 and 5.2 ± 0.7 per section in WT and APP23mice, respectively. Statistical analysis showed a significant decrease in the co-labeling BrdU+NeuN+ cells in 3 months old APP23 mice (P < 0.01), suggesting a decrease in the number of neuroblasts to mature neurons due to apoptosis-like cell death (Fig. 6C–F).
Figure 6.
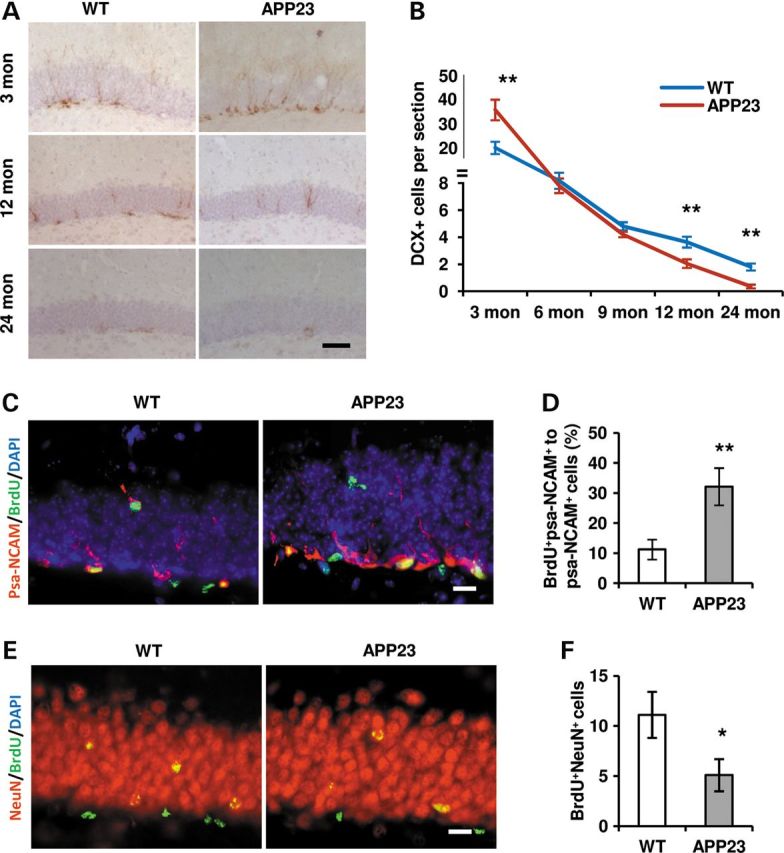
Neurogensis alters in the hippocampus of APP23 mice. (A) Representative micrographs showed DCX-expressing cells in the hippocampi of 3, 12 and 24 months old WT and APP23 mice. Scale bar: 50 µm. (B) The number of DCX+ cells was counted and the average number per section was shown at 3, 12 and 24 months old of WT and APP23 mice. The statistical differences were significant (n = 6 in each group, Student's t-test, **P < 0.01 versus WT). (C) Double labeling showed cells with BrdU+ (green) and psa-NCAM+ (red) in DG of 3 months of old WT and APP23 mice. Scale bar: 20 µm. (D) Immunostaining structures were accounted and the average number per section was shown as mean ± SEM. The ratio (%) of double-labeling cells with BrdU and psa-NCAM versus psa-NCAM expressing cells was significantly increased in 3-month-old APP23 mice (n = 6 in each group, Student's t-test, *P < 0.01 versus WT). (E) Double labeling showed cells with BrdU+ (green) and NeuN+ (red) in DG of 3 months old WT and APP23 mice after 4-weeks of BrdU application. Scale bar: 20 µm. (F) Co-labeling cells were accounted and the average number per section was shown as mean ± SEM. A differentiation to neurons with double-labeling of BrdU and NeuN was significantly decreased in 3-month-old APP23 mice after 4 weeks of BrdU injection (n = 6 in each group, Student's t-test, *P < 0.05 versus WT).
DISCUSSION
Shh-producing cells
In our current study, it is the first to discover that Shh signaling is greatly elevated in the hippocampal extracts of AD patients and APP23 mice (Fig. 1). As for the origin of Shh protein in hippocampus, it has been suggested that Shh-producing cells in the basal forebrains anterogradely transport Shh to the hippocampus via the axonal projection (10,11,33). Although failure to try immunolabeling Shh-expressing cells in vivo, we demonstrate Shh-expressing cells in cultured hippocampal progenitors, consistent with the reports of Shh mRNA expression in isolated hippocampal NSCs (31). Furthermore, we find that Aβ1–42 not Aβ1–40 up-regulates the Shh expression of the GPCs (Fig. 3). Due to the differences of cultured GPCs from that in vivo, Shh production from GPCs still need to be analyzed. Meanwhile, addition of Aβ1–42 in our study can elevate Shh signal. Whether other forms of Aβ peptides or inflammation factors play a similar role in the GPCs can regulate the shh-producing neurons in the forebrains also needs to be clarified. It is also reported that reactive astrocytes by freezing injuries in cortex secret Shh by the stimulation of pro-inflammatory cytokines (45). However, due to possible astrocyte diversity in response to Shh signaling (48), whether reactive astrocytes in hippocampus of AD-brains produce Shh signals still remains unclear. Shh levels can also be up-regulated in other pathological conditions, such as the brain stroke (16) and multiple sclerosis (17). It suggests that in addition to Aβ, other factors caused by injuries or disorders also contribute to Shh signaling up-regulation.
Shh-receiving cells
A fraction of NSCs and their immediate GPCs respond to Shh activity (11,15). Our results demonstrate that Shh signaling molecules, the Shh-Ptc-Smo-Gli axis, are universally up-regulated in the hippocampus of AD-like APP23 mice (Figs 2 and 7). And we find that isolated hippocampal NSCs express and up-regulate Shh pathway components in the presence of Shh signal (Fig. 7), which is consistent with a previous report (11,31). Here we demonstrated that, in addition to Shh signals by anterograde (10,33) or possible paracrine by activated astrocytes (45), Shh also plays a role in hippocampal GPCs in an autocrine fashion. In response to elevated amount of Shh, the expression levels of Shh signaling components change with along age differences, in which Ptc levels are greatly decreased even if Shh levels are elevated in the hippocampi of 24-month-old APP23 mice and AD brains (Fig. 2), controversial to Ptc level increase at a younger (3 and 12 months of age) stage. Shh and Ptc seem to change earlier (it increases most at 12 months old; Figs 1D and 2D) than that of insoluble Aβ at 24 months old (Fig. 1C). The reason of differences is possible relationship with a ratio change of cell types and/or cell reactive ability to Shh signaling, such as more GPCs at young stages and more reactive astrocytes or activated microglia at aged stages, etc, which may not be able to degrade or clear insoluble Aβ. Even so, lines of evidence have shown that Ptc1 may inhibit Smo in a non-stoichiometric, catalytic manner (16), e.g. the inhibition of Smo is less dependent on the dose of Ptc1 protein (49). Ptc expression levels decrease in the hippocampi of 24-month-old APP23 mice and AD patients may disclose a constitutive disinhibition of Smo by Ptc. In this case, Smo is no longer regulated by Ptc and can act as a constitutively activated receptor to propagate downstream signaling, including activation of Gli transcription gene expression (Fig. 7). All of these suggest an enhancing Shh signaling in aged AD brains. Recently, it has been reported that NSCs or GPCs isolated from the Down syndrome mouse model (Ts65Dn) highly express Ptc1 proteins, which impair cell proliferation in the absence of Shh molecules (50). It indicates Shh signaling changes under brain pathophysiological conditions.
Figure 7.
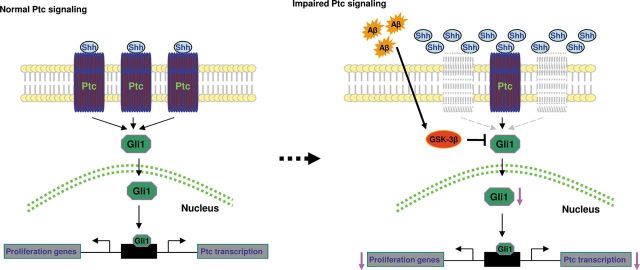
The schematic presentation shows Shh-Ptc1-Gli1 signaling pathway in normal (Left) condition and disease (right) conditions. In general, Shh binds Ptc, which activates and promotes the effecter Gli1. Gli1 enters the nucleus and binds to Ptc promoter and proliferative genes. Excessive Aβ in AD brains promotes and activates GSK-3β, which inhibits Gli1 and, in turn, decreased the transcription of Ptc and proliferative genes.
Cell proliferation and survival
In normal adult brains, responding to mitogens, such as Shh, NSCs exit Go state of cell cycle in a gradual manner and enter cell division (15,51). These dividing cells include a group of GFAP-expressing NSCs, fast-dividing transit-amplifying cells and psa-NCAM-expressing neuroblasts (immature neuronal precursors) (15). These cells can incorporate exogenous BrdU substrate. By BrdU labeling, we find an increased number of dividing cells in the hippocampus of APP23 transgenic mice at 3 and 12 months of age (Figs 4 and 5). Combining with the data of elevated Shh signaling in AD brains and Shh signaling inhibition by cyclopamine (Figs 2 and 3) (11), it indicates that Shh signaling may be associated with the regulation of GPCs proliferation above basal levels in APP23 brains at young stage. It seems to be consistent with a report that APP23 mice have more neurons until they develop amyloid plaques (52). Therefore, the increase in the number of dividing cells found at young stage of APP23 brains may present an acceleration of cell division of the resided GPCs in response to the elevation of Shh signaling (Fig. 7). A large population of post-mitotic cells commit to death (53) and a small group of differentiation cells can survive and replace dead neurons (Fig. 5). In AD brains, the existence of constant and high-level Aβ especially toxic soluble Aβ accelerates dividing cells to death (23,54). Our observation of increased apoptosis-like in DG of APP23 mice may also reflect more diving cells to death at aged APP23 mice (52). In addition, due to fast dividing of Shh-responding GPCs, it raises a possibility that the dilution of BrdU levels is beyond detection so that the decrease in the number of dividing cells is observed within 4 weeks (e.g. 90% of dividing cells disappear) in 3-month-old APP23 mice.
We notice a report that there is a discrepancy in cell proliferation in the hippocampus at age 5 and 25 months old of APP23 mice, respectively (20). The findings are different from our observation here with an increase or decrease in the dividing cell number at ages of 3 or 24 months old. It may suggest a different observation method. In that report, they applied BrdU for 7 days and immunostaining was performed 3 weeks after the last injection. It is similar with our observation of BrdU staining 4 weeks after the last BrdU application. It indicates a survival change in addition to proliferation.
Defective neurogenesis and increased gliogenesis
Hippocampal NSCs/GPCs are quiescent and can maintain in the state for lifetime (55,56). In normal adult brains, quiescent NSCs/GPCs are gradually released to exit cell cycle Go and enter the rounds of division at basic level of Shh signal stimulation (15). Different from the continuous self-renewal of hemapoietic GPCs (57), a recent report shows that adult hippocampal GPCs lose the capacity to self-renew and the cell number decreases in an age-dependent manner (Fig. 5) (51). Here, we find a great decrease in the number of radial glial-like stem cells in the hippocampus of 3-month-old APP23 mice (Figs 5 and 6). The GPC loss indicates that more quiescent GPCs enter dividing cycle and lose their characteristics of GPCs due to high levels of Shh signaling stimulation in AD brains. Still, we cannot exclude the possibility of Aβ toxicity to impair hippocampal quiescent GPCs. It has also been shown that, upon exiting quiescent state, adult hippocampal stem cells rapidly undergo a series of asymmetric divisions to produce dividing progeny destined to become neurons (51). If it is true in this case, increasing number of stem cells exiting quiescent state may be the reason of an increase in the number of fast-dividing transit-amplifying cells (GFAP-negative and nestin-positive cells) and neuroblasts (DCX- and psa-NCAM-positive cells) we observed here in the hippocampus of 3 months old APP23 mice (Fig. 6). In addition, transit-amplifying cells and neuroblasts self-responding to Shh (15) may also contribute to the dividing cell number observed here in 3-month-old APP23 brains. Meanwhile, the stem cells exiting quiescent state subsequently convert into mature astrocytes by division-coupled astrocytic differentiation (51). By this way, an elevated Shh signaling in AD brains contributes to an increase in the cell number of astrocytes originated from NSCs, a trend to gliogenesis. Unlike new neurons which are subject to death in response to a detrimental environment, newly generated astrocytes by asymmetric division survive brain disorders (23,54). In addition, it also is possible that a fraction of dormant astrocytes responding to elevated Shh levels (45,48,57) or Aβ (57) are activated and added the glial cell number in APP23 brains, i.e. a reactive astrogliosis.
In normal adult brains, NSCs store in hippocampus. Once injuries happen, mitogen signaling such as Shh is elevated (Fig. 7) (16). NSCs are inspired to divide and differentiation for potentially replacing dead cells. After the acute injuries stop, mitogen signaling is back to a basic level (45). However, in AD brains and other chronic brain disorders such as multiple sclerosis, Shh signaling maintains at high level (17). Because adult quiescent hippocampal NSCs lack self-renewal potential (51), high level of Shh signaling induced by pathogens such as soluble Aβ1–42 etc. accelerates loss of NSCs. In addition, excessive Aβ accumulation in AD brains is toxic to neural stem/progenitor cells (23,54). In conclusion, our studies provide the first evidence that a deregulation of Shh signaling in AD hippocampus, which induces the exit of NSCs toward proliferation and differentiation. By this way, hippocampal NSCs are lost and new neuron production decreases.
MATERIALS AND METHODS
Animals
All mice of APP23 transgenic (20 males and 20 females in each age group) and non-transgenic wild-type (20 males and 20 females in each age group) genotypes in our experiment are on the C57BL/6 background. APP23 transgenic mice were provided by Novartis Institute for Biomedical Research and the mice express mutated human βAPP (Swedish double mutation, KM670/671NL) under brain and neuron-specific murine Thy-1 promoter element. APP23 transgenic mice develop senile plaques in the cerebral cortex and hippocampus, along with neuronal loss that is most evident in the hippocampal CA1 region at 12–18 months of age (32). APP23 and non-transgenic wild-type mice were crossed and the progenies were genotyped and characterized as APP23 with PCR followed by western blot for brain APP protein.
Human hippocampal tissues
The hippocampal formation of human brains was harvested from rapidly autopsied geriatric patients (n = 5 HC, n = 5 AD) enrolled in the Brain Donation Program at Banner Sun Health Research Institute. The average postmortem interval was <3 h. The neuropathological characteristics were demonstrated (Table 1). The average ages of the HC and AD subjects were 82 ± 15 and 78 ± 7 years old, respectively.
Table 1.
The pathological profiles of human brains
| Case no. | Sex | Age (years) | Neuropathological diagnosis | Postmortem interval (h) | Disease duration (years) |
|---|---|---|---|---|---|
| 92-32 | F | 102 | HC* | 1.67 | |
| 95-26 | M | 63 | HC | 3.25 | |
| 97-50 | F | 88 | HC | 2.15 | |
| 98-33 | F | 82 | HC | 2.0 | |
| 03-31 | M | 75 | HC | 2.0 | |
| 97-20 | F | 83 | AD* | 3.0 | 38 |
| 98-28 | M | 88 | AD | 2.0 | 7 |
| 99-39 | M | 72 | AD | 2.5 | 12 |
| 00-47 | M | 72 | AD | 2.5 | 13 |
| 03-40 | F | 75 | AD | 2.25 | 6 |
HC, healthy control; AD, Alzheimer's disease.
5-Bromo-2-deoxyuridine (BrdU) incorporation
Mice were kept in a normal 12 h light 12 h dark cycle. 5-Bromo-2-deoxyuridine (BrdU) (B5002, Sigma) was prepared in a sterile stock solution of 10 mg/ml dissolved in 0.9% (w/v) NaCl solution. BrdU of 50 mg/kg body weight was intraperitoneally injected twice a day, interval of 4 h, at 3, 12 and 24 months old WT and APP23 mice (n = 6 each group). For proliferating analysis, after 24 h of last injection the mice were perfused and brain tissues were collected. For survival and differentiation observation, 3-month-old WT and APP23 mice (n = 6, respectively) were sacrificed and the brain tissues were harvested 4 weeks after last injection.
Cyclopamine injection
Cyclopamine specifically blocks Shh signaling by binding to Shh receptor complex Smo (34,58,59). WT and APP23 mice with age of 3 months old were injected intraperitoneally as described previously (11,45), with either vehicle [45% 2-hydroxypropyl-β-cyclodextrin (HBC) in sterile phosphate-buffered saline, Sigma] or cyclopamine (C4116, Sigma, 25 mg/kg/day, 1 mg/ml of w/v in 45% HBC) (38) for 7 consecutive days. The day following last injection, all animals were administered with the mitotic marker BrdU (100 mg/kg; Sigma) twice a day with interval of 4 h and were sacrificed 24 h later.
Glial precursor cell (GPC) culture
The hippocampus tissues from the neonatant WT mouse (P0–P3) were isolated under a stereomicroscope. As we previously reported (23), cells were dissociated using papain/DNase I (Vector Laboratories, CA, USA) and cultured in flasks that were pre-coated with 10 mg/ml poly(2-hydroxyethyl methacrylate) (P3932, Sigma) to avoid cell adhesion (31). Primary neurospheres were generated in the DMEM/F12 medium (growth medium) containing 10 ng/ml of bFGF (catalog: 13256-029, Invitrogen) and 10 ng/ml of EGF (Catalog: PMG8043, Invitrogen) for 14 days.
Shh or Aβ treatment on neural progenitor cells from WT hippocampus
To observe the effect of β amyloid peptides or Shh on the expression of Shh signaling, primary neurospheres were dissociated with papain and re-suspended in the identical growth medium. The free-floating cells were exposed to 0, 0.2 and 0.4 µg/ml of Shh (Catalog: 1845-SH, R&D), or 0, 0.1, 1, 10 and 100 µm of Aβ1–42 (Catalog: A9810, Sigma) or 0, 0.1, 1, 10 and 100 µm of Aβ1–40 (Catalog: H5568, Bachem) for 72 h. The medium and culture factors are purchased from Invitrogen Company. The experiment was repeated three times independently.
ELISA
Aβ ELISA was performed as described previously (23,26). In brief, wild-type and APP23 mice (n = 10 per group, 5 males and 5 females) were sacrificed at 3, 6, 9, 12, 18 and 24 months of age. The hippocampus formation of one hemisphere was isolated and homogenized in homogenization buffer. Protein concentrations were measured by protein assays (Bio-Rad Laboratories). The hippocampal homogenates were centrifuged at 14 000g for 1 h at 4°C. The supernatant including soluble Aβ fractions was collected for the assay of soluble Aβ1–40 and Aβ1–42. The pellet with insoluble Aβ was dissolved in 98% of formic acid and centrifuged for 30 min at 4°C. The supernatant from the pellet was collected for the assay of insoluble Aβ1–40 and Aβ1–42. The levels of Aβ1–40 and Aβ1–42 were measured with an Aβ1–40 and Aβ1–42 ELISA kit (KHB3481 and KHB3544, Invitrogen). The ELISA system has been extensively tested and no cross-reactivity between Aβ1–40 and Aβ1–42 was observed. The quantitation of soluble Aβ ELISA measurement was normalized to corresponding tissue protein concentration. Data are presented as means ± SD of four experiments.
Western blot
The hippocampus formation from mice (n = 12 each group, 6 males and 6 females), human brains (n = 5 each group) as well as the treated neural progenitor cells were lysed in cell lysis buffer (10 mm Tris–HCl pH 7.4, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 10% glycerol, 0.1 m Na3VO4, 0.5% Triton X-100). Twenty-five micrograms of protein were separated on 8% SDS–PAGE and transferred to PDVF membrane. To measure the levels of secreted protein Shh in the conditioned medium, the medium surrounding neural progenitor cells was collected and centrifuged. Equal volume of the media was mixed with 20% ice-cold trichloroacetic acid on ice for 30 min. The mix was centrifuged at 10 000g for 10 min. All supernatant was removed and the drops were dried. The samples were resuspended in SDS–PAGE loading buffer. Corresponding cultured cell lysates were for loading control. The blot was probed by rat anti-ShhN terminal protein (1:1000, MAB4641, R&D), rabbit anti-patched 1 (Ptc1, 1:2000, sc-9016, Santa Cruz Biotechnology), rabbit anti-patched 2 (Ptc2, PA1-46223, 1:1000, Thermo-Scientific), rabbit anti-smoothened (Smo, 1:2000, sc-13943, clone:488-787, Santa Cruz Biotechnology), rabbit anti-Gli1 (1:1000, AB3444, Millipore Bioscience Research Reagents), rabbit anti-Gli2 (Chip grade, ab26056, 1:1000, Abcam) and rabbit anti-Gli3 (ab69838, 1:1000, Abcam). The concentration of peroxidase-conjugated secondary antibodies was 1:50 000.
Immunostaining
Mice were perfused with 4% paraformaldehyde buffered with 0.1 m phosphate buffer. Serial sagittal sections (30 µm) were generated using a cryostat (Sigma). Sections were penetrated with 0.15% Triton X-100 and were blocked with 10% horse or goat serum. The primary antibody was applied against Aβ amino acid sequence 1–17 (clone 6E10, MAB1560, 1:2000, Millipore Bioscience Research Reagents), and rabbit anti-doublecortin (DCX, 1:1000, ab18723, Abcam) for neuroblast (immature neuronal precursor). Secondary antibodies were applied with horse anti-mouse or goat anti-rabbit IgG (1:1000, Vector Laboratories). For dividing progenitor cell detection, antigens were retrieved with 50% formamide in 2× BBS buffer for 60 min in 65°C, and 2 N HCl for 30 min in 37°C. BrdU-integrated cell nuclei were detected with a rabbit polyclonal antibody directing BrdU (1:10 000, Catalog: BP40250, Megabase). A secondary antibody with biotinylated goat anti-rabbit IgG (1:1000, BA-1000, Vector Laboratories) was applied and followed by Vestastain ABC Elit Kit and a DAB substrate (Vector Laboratories). Hematoxylin (Sigma) was used as counterstaining. For immunofluorescence, primary antibodies were used as follows, rabbit anti-patched 1 (1:200, sc-9016, Santa Cruz Biotechnology), mouse anti-BrdU (Catalog: 03-3900, 1:5000, Invitrogen), mouse anti-NeuN (MAB377, 1:400, Millipore Bioscience Research Reagents) and rabbit anti-mouse nestin (1:50, sc-20978, Santa Cruz Biotechnology). Secondary antibodies against rabbit IgG or mouse IgG were used (1:1000; Invitrogen) with fluorescent-labeling 488 (green) or 594 (red). DAPI (1:10 000, Santa Cruz Biotechnology) was used as counterstaining.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling
With the reference of our previous report (31), the method was briefed. The tissue sections of 3 months old WT and APP23 mice (n = 6 in each group) were treated with 0.5% Triton X-100 and blocked with 3% BSA for 30 min. The sections were then incubated with terminal deoxynucleotidyl TUNEL reaction mixture according to the manufacturer's procedures (Boehringer Mannheim). Negative controls were treated with Label Solution (without terminal transferase) instead of the TUNEL reaction mixture. Positive controls were incubated with DNase I to induce DNA strand breaks and were labeled with the TUNEL reaction mixture. Incubation was performed in a humidified atmosphere for 60 min at 37°C in dark. To show the DG structure, the sections were washed in PBS and incubated with antibody NeuN for 1 h in RT and secondary antibody coupled with fluorescence 594 for 30 min. Samples were analyzed using a fluorescent microscope. Normal nuclei, which contained only insignificant amounts of DNA 3′-OH ends, did not stain with this technique; however, cells with apoptotic morphology exhibited condensed nuclei (green). NeuN staining was visualized in red color.
Quantification of immunoreactive structures
Quantification of immunopositive structures was carried out by an experimenter blind to the study. Left hemisphere of mouse brains was sagittally sectioned with 30 µm by cryostat. All of the serial sections were harvested. The sections per interval of 400 µm were chosen as immunostaining. Counterstaining was performed with hematoxylin (0.1%, Catalogue: 03973, Sigma) for DAB substrate or DAPI for immunofluorescence to aid visualization of the dentate gyrus. A microscope (DMLS; Leica) with a 10× N PLAN and 20× and 40× PL FLUOTAR was used. Digitized images were captured with a DEI-470 digital camera (Optronics, Goleta, CA, USA) on a Leica microscope (Leica, Germany). MagnaFire software (version 2.1C; Optronics) was used. The immunopositive structures of each section were counted with same parameter. In general, 9–11 sections through the hippocampus formation per mouse were calculated (n = 10 mice in each group). The number of immune-positive cells or nuclei was totalized based on counting of subpopulation of each section, which was estimated as total number of immunopositive cells. The total number was divided by the number of sections and was expressed as immunopositive structure number per section.
Statistical analyses
Results were expressed as mean ± SD. All analyses were performed using a software program (SPSS version 11.5.1; SPSS). Differences between two groups were assessed using Student's t-tests. Differences between three or more groups in Aβ treatment on WT neural progenitor cells were evaluated by one-way analysis of variance models (ANOVA). The level of significance was P ≤ 0.05.
AUTHORS’ CONTRIBUTIONS
P.H. performed experiments, participated in data analyses and wrote the article draft. M.S. provided APP23 transgenic mice through collaboration. Y.S. and R.L. initiated the project and designed the experiments, and participated in data analyses and overviewed article drafting and finalized final version of the article. All authors have read and approved the final article.
FUNDING
This work was supported by NIHAGRO1 (8528888, 032441), ABRC 002 and Alzheimer Association (IIRG-09-61521 and IIRG-07-59510) and AHAF (G2006-118, A2008-642).
ACKNOWLEDGEMENTS
We sincerely thank Dr Fred H. Gage from the Laboratory of Genetics at the Salk Institute for Biological Studies for his critical reading and valuable comments on our early article and enthusiastic support.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Johnson R.L., Rothman A.L., Xie J., Goodrich L.V., Bare J.W., Bonifas J.M., Quinn A.G., Myers R.M., Cox D.R., Epstein E.H., Jr., Scott M.P. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668–1671. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- 2.Motoyama J., Takabatake T., Takeshima K., Hui C. Ptch2, a second mouse Patched gene is co-expressed with Sonic hedgehog. Nat. Genet. 1998;18:104–106. doi: 10.1038/ng0298-104. [DOI] [PubMed] [Google Scholar]
- 3.Chen Y., Struhl G. Dual roles for patched in sequestering and transducing Hedgehog. Cell. 1996;87:553–563. doi: 10.1016/s0092-8674(00)81374-4. [DOI] [PubMed] [Google Scholar]
- 4.Kinzler K.W., Ruppert J.M., Bigner S.H., Vogelstein B. The GLI gene is a member of the Kruppel family of zinc finger proteins. Nature. 1988;332:371–374. doi: 10.1038/332371a0. [DOI] [PubMed] [Google Scholar]
- 5.Ruiz I, Altaba A., Nguyen V., Palma V. The emergent design of the neural tube: prepattern, SHH morphogen and GLI code. Curr. Opin. Genet. Dev. 2003;13:513–521. doi: 10.1016/j.gde.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 6.Ruiz I., Altaba A., Palma V., Dahmane N. Hedgehog-Gli signalling and the growth of the brain. Nat. Rev. Neurosci. 2002;3:24–33. doi: 10.1038/nrn704. [DOI] [PubMed] [Google Scholar]
- 7.Methot N., Basler K. Hedgehog controls limb development by regulating the activities of distinct transcriptional activator and repressor forms of Cubitus interruptus. Cell. 1999;96:819–831. doi: 10.1016/s0092-8674(00)80592-9. [DOI] [PubMed] [Google Scholar]
- 8.Echelard Y., Epstein D.J., St-Jacques B., Shen L., Mohler J., McMahon J.A., McMahon A.P. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75:1417–1430. doi: 10.1016/0092-8674(93)90627-3. [DOI] [PubMed] [Google Scholar]
- 9.Roelink H., Augsburger A., Heemskerk J., Korzh V., Norlin S., Ruiz I., Altaba A., Tanabe Y., Placzek M., Edlund T., Jessell T.M., et al. Floor plate and motor neuron induction by vhh-1, a vertebrate homolog of hedgehog expressed by the notochord. Cell. 1994;76:761–775. doi: 10.1016/0092-8674(94)90514-2. [DOI] [PubMed] [Google Scholar]
- 10.Traiffort E., Angot E., Ruat M. Sonic Hedgehog signaling in the mammalian brain. J. Neurochem. 2010;113:576–590. doi: 10.1111/j.1471-4159.2010.06642.x. [DOI] [PubMed] [Google Scholar]
- 11.Lai K., Kaspar B.K., Gage F.H., Schaffer D.V. Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nat. Neurosci. 2003;6:21–27. doi: 10.1038/nn983. [DOI] [PubMed] [Google Scholar]
- 12.Balordi F., Fishell G. Hedgehog signaling in the subventricular zone is required for both the maintenance of stem cells and the migration of newborn neurons. J. Neurosci. 2007;27:5936–5947. doi: 10.1523/JNEUROSCI.1040-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han Y.G., Spassky N., Romaguera-Ros M., Garcia-Verdugo J.M., Aguilar A., Schneider-Maunoury S., Alvarez-Buylla A. Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat. Neurosci. 2008;11:277–284. doi: 10.1038/nn2059. [DOI] [PubMed] [Google Scholar]
- 14.Machold R., Hayashi S., Rutlin M., Muzumdar M.D., Nery S., Corbin J.G., Gritli-Linde A., Dellovade T., Porter J.A., Rubin L.L., et al. Sonic hedgehog is required for progenitor cell maintenance in telencephalic stem cell niches. Neuron. 2003;39:937–950. doi: 10.1016/s0896-6273(03)00561-0. [DOI] [PubMed] [Google Scholar]
- 15.Ahn S., Joyner A.L. In vivo analysis of quiescent adult neural stem cells responding to Sonic hedgehog. Nature. 2005;437:894–897. doi: 10.1038/nature03994. [DOI] [PubMed] [Google Scholar]
- 16.Sims J.R., Lee S.W., Topalkara K., Qiu J., Xu J., Zhou Z., Moskowitz M.A. Sonic hedgehog regulates ischemia/hypoxia-induced neural progenitor proliferation. Stroke. 2009;40:3618–3626. doi: 10.1161/STROKEAHA.109.561951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y., Imitola J., Rasmussen S., O'Connor K.C., Khoury S.J. Paradoxical dysregulation of the neural stem cell pathway sonic hedgehog-Gli1 in autoimmune encephalomyelitis and multiple sclerosis. Ann. Neurol. 2008;64:417–427. doi: 10.1002/ana.21457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walsh D.M., Selkoe D.J. Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron. 2004;44:181–193. doi: 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 19.Kapogiannis D., Mattson M.P. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer's disease. Lancet Neurol. 2011;10:187–198. doi: 10.1016/S1474-4422(10)70277-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ermini F.V., Grathwohl S., Radde R., Yamaguchi M., Staufenbiel M., Palmer T.D., Jucker M. Neurogenesis and alterations of neural stem cells in mouse models of cerebral amyloidosis. Am. J. Pathol. 2008;172:1520–1528. doi: 10.2353/ajpath.2008.060520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin K., Peel A.L., Mao X.O., Xie L., Cottrell B.A., Henshall D.C., Greenberg D.A. Increased hippocampal neurogenesis in Alzheimer's disease. Proc. Natl Acad. Sci. USA. 2004;101:343–347. doi: 10.1073/pnas.2634794100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li B., Yamamori H., Tatebayashi Y., Shafit-Zagardo B., Tanimukai H., Chen S., Iqbal K., Grundke-Iqbal I. Failure of neuronal maturation in Alzheimer disease dentate gyrus. J. Neuropathol. Exp. Neurol. 2008;67:78–84. doi: 10.1097/nen.0b013e318160c5db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He P., Shen Y. Interruption of beta-catenin signaling reduces neurogenesis in Alzheimer's disease. J. Neurosci. 2009;29:6545–6557. doi: 10.1523/JNEUROSCI.0421-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sturchler-Pierrat C., Abramowski D., Duke M., Wiederhold K.H., Mistl C., Rothacher S., Ledermann B., Burki K., Frey P., Paganetti P.A., et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl Acad. Sci. USA. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McGowan E., Pickford F., Kim J., Onstead L., Eriksen J., Yu C., Skipper L., Murphy M.P., Beard J., Das P., et al. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He P., Zhong Z., Lindholm K., Berning L., Lee W., Lemere C., Staufenbiel M., Li R., Shen Y. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer's mice. J. Cell Biol. 2007;178:829–841. doi: 10.1083/jcb.200705042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Porter J.A., Young K.E., Beachy P.A. Cholesterol modification of hedgehog signaling proteins in animal development. Science. 1996;274:255–259. doi: 10.1126/science.274.5285.255. [DOI] [PubMed] [Google Scholar]
- 28.Zeng X., Goetz J.A., Suber L.M., Scott W.J., Jr., Schreiner C.M., Robbins D.J. A freely diffusible form of Sonic hedgehog mediates long-range signalling. Nature. 2001;411:716–720. doi: 10.1038/35079648. [DOI] [PubMed] [Google Scholar]
- 29.Goetz J.A., Singh S., Suber L.M., Kull F.J., Robbins D.J. A highly conserved amino-terminal region of sonic hedgehog is required for the formation of its freely diffusible multimeric form. J. Biol. Chem. 2006;281:4087–4093. doi: 10.1074/jbc.M511427200. [DOI] [PubMed] [Google Scholar]
- 30.Charytoniuk D., Traiffort E., Hantraye P., Hermel J.M., Galdes A., Ruat M. Intrastriatal sonic hedgehog injection increases Patched transcript levels in the adult rat subventricular zone. Eur. J Neurosci. 2002;16:2351–2357. doi: 10.1046/j.1460-9568.2002.02412.x. [DOI] [PubMed] [Google Scholar]
- 31.Galvin K.E., Ye H., Wetmore C. Differential gene induction by genetic and ligand-mediated activation of the Sonic hedgehog pathway in neural stem cells. Dev. Biol. 2007;308:331–342. doi: 10.1016/j.ydbio.2007.05.031. [DOI] [PubMed] [Google Scholar]
- 32.Marigo V., Davey R.A., Zuo Y., Cunningham J.M., Tabin C.J. Biochemical evidence that patched is the Hedgehog receptor. Nature. 1996b;384:176–179. doi: 10.1038/384176a0. [DOI] [PubMed] [Google Scholar]
- 33.Traiffort E., Charytoniuk D., Watroba L., Faure H., Sales N., Ruat M. Discrete localizations of hedgehog signalling components in the developing and adult rat nervous system. Eur. J. Neurosci. 1999;11:3199–3214. doi: 10.1046/j.1460-9568.1999.00777.x. [DOI] [PubMed] [Google Scholar]
- 34.Taipale J., Chen J.K., Cooper M.K., Wang B., Mann R.K., Milenkovic L., Scott M.P., Beachy P.A. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406:1005–1009. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- 35.Chen J.K., Taipale J., Cooper M.K., Beachy P.A. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002;16:2743–2748. doi: 10.1101/gad.1025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bai C.B., Auerbach W., Lee J.S., Stephen D., Joyner A.L. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development. 2002;129:4753–4761. doi: 10.1242/dev.129.20.4753. [DOI] [PubMed] [Google Scholar]
- 37.Rafuse V.F., Soundararajan P., Leopold C., Robertson H.A. Neuroprotective properties of cultured neural progenitor cells are associated with the production of sonic hedgehog. Neuroscience. 2005;131:899–916. doi: 10.1016/j.neuroscience.2004.11.048. [DOI] [PubMed] [Google Scholar]
- 38.Lopez-Toledano M.A., Shelanski M.L. Neurogenic effect of beta-amyloid peptide in the development of neural stem cells. J. Neurosci. 2004;24:5439–5444. doi: 10.1523/JNEUROSCI.0974-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heo C., Chang K.A., Choi H.S., Kim H.S., Kim S., Liew H., Kim J.A., Yu E., Ma J., Suh Y.H. Effects of the monomeric, oligomeric, and fibrillar Abeta42 peptides on the proliferation and differentiation of adult neural stem cells from subventricular zone. J. Neurochem. 2007;102:493–500. doi: 10.1111/j.1471-4159.2007.04499.x. [DOI] [PubMed] [Google Scholar]
- 40.Mirochnic S., Wolf S., Staufenbiel M., Kempermann G. Age effects on the regulation of adult hippocampal neurogenesis by physical activity and environmental enrichment in the APP23 mouse model of Alzheimer disease. Hippocampus. 2009;19:1008–1018. doi: 10.1002/hipo.20560. [DOI] [PubMed] [Google Scholar]
- 41.Berman D.M., Karhadkar S.S., Hallahan A.R., Pritchard J.I., Eberhart C.G., Watkins D.N., Chen J.K., Cooper M.K., Taipale J., Olson J.M., Beachy P.A. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science. 2002;297:1559–1561. doi: 10.1126/science.1073733. [DOI] [PubMed] [Google Scholar]
- 42.Doetsch F., Caille I., Lim D.A., Garcia-Verdugo J.M., Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- 43.Encinas J.M., Vaahtokari A., Enikolopov G. Fluoxetine targets early progenitor cells in the adult brain. Proc. Natl Acad. Sci. USA. 2006;103:8233–8238. doi: 10.1073/pnas.0601992103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao C., Teng E.M., Summers R.G., Jr., Ming G.L., Gage F.H. Distinct morphological stages of dentate granule neuron maturation in the adult mouse hippocampus. J. Neurosci. 2006;26:3–11. doi: 10.1523/JNEUROSCI.3648-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeWitt D.A., Perry G., Cohen M., Doller C., Silver J. Astrocytes regulate microglial phagocytosis of senile plaque cores of Alzheimer's disease. Exp. Neurol. 1998;149:329–340. doi: 10.1006/exnr.1997.6738. [DOI] [PubMed] [Google Scholar]
- 46.Rao M.S., Shetty A.K. Efficacy of doublecortin as a marker to analyse the absolute number and dendritic growth of newly generated neurons in the adult dentate gyrus. Eur. J. Neurosci. 2004;19:234–246. doi: 10.1111/j.0953-816x.2003.03123.x. [DOI] [PubMed] [Google Scholar]
- 47.Couillard-Despres S., Winner B., Schaubeck S., Aigner R., Vroemen M., Weidner N., Bogdahn U., Winkler J., Kuhn H.G., Aigner L. Doublecortin expression levels in adult brain reflect neurogenesis. Eur. J. Neurosci. 2005;21:1–14. doi: 10.1111/j.1460-9568.2004.03813.x. [DOI] [PubMed] [Google Scholar]
- 48.Garcia A.D., Petrova R., Eng L., Joyner A.L. Sonic hedgehog regulates discrete populations of astrocytes in the adult mouse forebrain. J. Neurosci. 2010;30:13597–13608. doi: 10.1523/JNEUROSCI.0830-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taipale J., Cooper M.K., Maiti T., Beachy P.A. Patched acts catalytically to suppress the activity of Smoothened. Nature. 2002;418:892–897. doi: 10.1038/nature00989. [DOI] [PubMed] [Google Scholar]
- 50.Trazzi S., Mitrugno V.M., Valli E., Fuchs C., Rizzi S., Guidi S., Perini G., Bartesaghi R., Ciani E. APP-dependent up-regulation of Ptch1 underlies proliferation impairment of neural precursors in Down syndrome. Hum. Mol. Genet. 2011;20:1560–1573. doi: 10.1093/hmg/ddr033. [DOI] [PubMed] [Google Scholar]
- 51.Encinas J.M., Michurina T.V., Peunova N., Park J.H., Tordo J., Peterson D.A., Fishell G., Koulakov A., Enikolopov G. Division-coupled astrocytic differentiation and age-related depletion of neural stem cells in the adult hippocampus. Cell Stem Cell. 2011;8:566–579. doi: 10.1016/j.stem.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bondolfi L., Calhoun M., Ermini F., Kuhn H.G., Wiederhold K.H., Walker L., Staufenbiel M., Jucker M. Amyloid-associated neuron loss and gliogenesis in the neocortex of amyloid precursor protein transgenic mice. J. Neurosci. 2002;22:515–522. doi: 10.1523/JNEUROSCI.22-02-00515.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Emsley J.G., Mitchell B.D., Kempermann G., Macklis J.D. Adult neurogenesis and repair of the adult CNS with neural progenitors, precursors, and stem cells. Prog. Neurobiol. 2005;75:321–341. doi: 10.1016/j.pneurobio.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 54.Verret L., Jankowsky J.L., Xu G.M., Borchelt D.R., Rampon C. Alzheimer's-type amyloidosis in transgenic mice impairs survival of newborn neurons derived from adult hippocampal neurogenesis. J. Neurosci. 2007;27:6771–6780. doi: 10.1523/JNEUROSCI.5564-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morrison S.J., Spradling A.C. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132:598–611. doi: 10.1016/j.cell.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li L., Clevers H. Coexistence of quiescent and active adult stem cells in mammals. Science. 2010;327:542–545. doi: 10.1126/science.1180794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kiel M.J., He S., Ashkenazi R., Gentry S.N., Teta M., Kushner J.A., Jackson T.L., Morrison S.J. Haematopoietic stem cells do not asymmetrically segregate chromosomes or retain BrdU. Nature. 2007;449:238–242. doi: 10.1038/nature06115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van den Brink G.R., Hardwick J.C., Tytgat G.N., Brink M.A., Ten Kate F.J., Van Deventer S.J., Peppelenbosch M.P. Sonic hedgehog regulates gastric gland morphogenesis in man and mouse. Gastroenterology. 2001;121:317–328. doi: 10.1053/gast.2001.26261. [DOI] [PubMed] [Google Scholar]
- 59.Cooper M.K., Porter J.A., Young K.E., Beachy P.A. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science. 1998;280:1603–1607. doi: 10.1126/science.280.5369.1603. [DOI] [PubMed] [Google Scholar]