

Abstract
Introduction:
The site of pathology in myasthenia gravis (MG) is the neuromuscular junction (NMJ). Our goal was to determine the ability to direct complement inhibition to the NMJ. Methods: A single-chain antibody directed against the alpha subunit of the acetylcholine receptor was synthesized (scFv-35) and coupled to decay-accelerating factor (DAF, scFv-35-DAF). scFv-35-DAF was tested in a passive model of experimentally acquired MG. Results: Administration of scFv-35-DAF to mice deficient in intrinsic complement inhibitors produced no weakness despite confirmation of its localization to the NMJ and no evidence of tissue destruction related to complement activation. Rats with experimentally acquired MG treated with scFV-35-DAF showed less weakness and a reduction of complement deposition.
Conclusions:
We demonstrate a method to effectively target a therapeutic agent to the NMJ. Muscle Nerve
Keywords: acetylcholine receptor, autoimmunity, complement, decay-accelerating factor, myasthenia gravis
Myasthenia gravis (MG) is a prototypic antibody-mediated autoimmune disease, which fulfills strict criteria that define autoimmunity.1,2 MG is caused primarily by antibodies directed against the skeletal muscle nicotinic acetylcholine receptor (AChR), which is concentrated on the postsynaptic surface of the neuromuscular junction. The antibodies produce a reduction in AChR number and damage the muscle endplate. This leads to failure of neuromuscular transmission and produces weakness that may be life-threatening. AChR antibodies of patients with MG are polyclonal and recognize a complex epitope repertoire that differs among individuals. The heterogeneity of AChR antibodies among patients is increased further by the variety of light-chain and heavy-chain use. The antibody synthesis is T-cell–dependent, and the epitope repertoire of AChR CD4+ cells in MG patients is complex and characteristic of the individual. Due to extreme diversity of the immune response, specific suppression of autoantibody production is an elusive goal.1,3
Therapies for MG fall into 2 broad categories4,5: those that focus on enhancing neuromuscular transmission by inhibition of cholinesterase, such as pyridostigmine; or agents that suppress or modulate the immune system, such as corticosteroids, azathioprine, tacrolimus, and mycophenolate mofetil. Acute exacerbations of weakness are usually treated with plasma exchange or intravenous immunoglobulins, which act primarily through removal of pathogenic antibody. All these treatments were developed after initial use in other disorders6 and produce systemic adverse effects. The immunotherapies are not aimed at production of the specific autoantibody in MG, but rather general moderation of all immune responses, whether this occurs through reduction of autoantibody levels directly or indirectly through suppression of B- and T-cell activity. The approach described here attempts inhibition of a final effector mechanism of pathogenic AChR antibody.
The AChR-specific antibodies induce disease by 3 mechanisms: (1) blocking binding and activation of the AChR; (2) accelerating degradation of AChR molecules cross-linked by antibodies (“antigenic modulation”); and (3) mobilizing complement at the NMJ.7–9 Autoantibodies are the only effectors of myasthenic pathology, as patients with MG do not have evidence of AChR-specific cell-mediated mechanisms of NMJ injury.1,10 Therefore, treatments focused on inhibiting antibody effector mechanisms are likely to work rapidly and be highly effective. Compromise of AChR function has not been found to be a major contributor to limitation of neuromuscular transmission.1,2 Antibodies from most MG patients accelerate the degradation rate of the AChR in whole muscle tissue culture and cultured muscle cells.11,12 However, not all AChR antibodies are capable of antigenic modulation, because the epitope location on the AChR surface restricts antibody ability to cross-link a second AChR molecule. Therefore, complement-mediated injury is the primary mechanism for pathology of the NMJ in MG.
The autoantibodies that bind to the AChR can activate the complement cascade and produce lysis of the NMJ through insertion of the membrane attack complex (MAC). The NMJs of skeletal muscle are protected from complement attack by specific cell-surface complement-regulatory proteins.13 Decay-accelerating factor (DAF or CD55), CD59, and membrane cofactor protein (complement receptor 1–related gene/protein y is the rodent analog) reduce the activation of complement through various pathways.14–18 The role of DAF as a complement regulator is to accelerate the decay of autologous C3 and C5 convertases. We assessed the ability to target DAF to the NMJ and thereby reduce the damage produced by complement activation. Concentrating DAF to the cell surface by a fusion protein including an antibody binding domain has been demonstrated to be effective in inhibiting complement-mediated lysis.19
Eculizumab, the only complement inhibitor available for human use, demonstrated signs of efficacy in a small, pilot study of patients with severe MG, and thus there is some support for complement inhibitor–based therapeutic approaches for MG.20 However, to date, all inhibitor strategies have led to systemic inhibition of complement. Here we describe an approach that couples the complement-inhibitory domains of the DAF molecule to a single-chain antibody that binds the AChR. We used the single-chain antibody as a control to assess whether blocking pathogenic antibody binding could contribute to a treatment effect. The expectation was that concentration of complement inhibition to the site of pathology could enhance efficacy without compromising neuromuscular transmission.
METHODS
Animals
Daf1−/− mice21 were provided by M.E. Medof (Case Western Reserve University) and CD59ab−/− mice were provided by J.A. Halperin (Harvard Medical School, Boston, MA). Knockout mouse strains were backcrossed 9 generations on the C57Bc/6 background. CD59ab−/−Daf1−/− knockouts were prepared by cross-breeding CD59ab−/− and Daf1−/− mice. Genotyping to assure knockout of the CD59 genes was performed as described previously.22 To confirm that at least 1 copy of the Daf deletion construct was present, polymerase chain reaction (PCR) was performed with specific primers (NeoFor: 5′TGC TCC TGC CGA GAA AGT AT 3′ NeoRev: 5′AAT ATC ACG GGT AGC CAA CG 3′); to assure that 2 copies of the deletion construct were present, quantitative polymerase chain reaction (PCR) was performed (NeoRT forward: 5′ AGACAATCGGCTGCTCTGAT 3′ NeoRT reverse: 5′ TCGTCC TGCAGTTCATTCAG 3′). Male mice, 6–8 weeks old, were used for experiments to determine localization of the scFv. Female Lewis rats (Harlan, Indianapolis, Indiana), 6–10 weeks old, were used for experimentally acquired MG (EAMG). All animals were maintained in the Saint Louis University or George Washington University animal facilities. Studies were carried out according to approved protocols by the local institutional animal care and use committees (IACUC).
Construction of Single-Chain Antibody Attached to Decay-Accelerating Factor
We utilized the TIB-175 hybridoma cell line (ATCC), which produces monoclonal antibody 35, an antibody known to bind the AChR,23,24 for isolation of RNA and then performed PCR amplification of variable heavy-chain (VH) and variable light-chain (VL) regions. We then performed overlapping PCR to generate VH–VL fragment with a linker (GGGGS)3 to produce the single-chain AChR antibody scFv-35 (Fig. 1). Monoclonal antibody 35 (MAb-35) is an antibody used commonly in EAMG studies.10,24,25 Constructs were produced with an IgG signal peptide. To attach the DAF domains, we utilized the C57Bl/6J for RNA and PCR to clone the 4 extracellular complement control protein modules of 255 amino acids and produced the scFv-35-Daf (Fig. 2). We cloned all constructs into the pIRES2-AcGFP1 vector (Clontech, Mountain View, California). This vector allows expression of 2 separate proteins, 1 green fluorescent protein (GFP) and the other protein of interest, rather than a single fusion protein, which limits concerns regarding the risk of loss of biological activity of the protein of interest.
Figure 1.
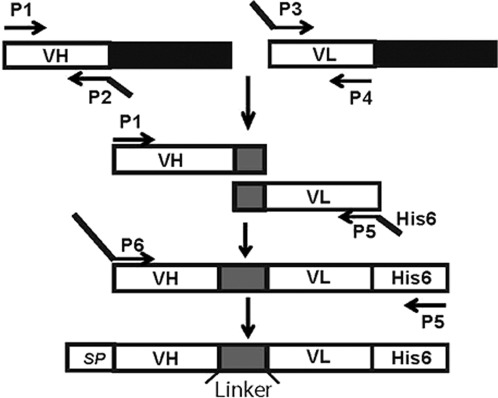
Schematic of cloning strategy to produce scFv-35. RNA from the TIB-175 hybridoma cell line was used to produce the VH and VL regions of a monoclonal antibody that targets the AChR.
Figure 2.

The 529-amino-acid sequence of the scFv-35-DAF construct. Each of the functional domains and corresponding sequences are indicated: IgGsp (signal peptide); VH (variable heavy chain); linker; VL (variable light chain); and DAF (decay-accelerating factor).
Transfection into BHK-21
BHK-21 cells, derived from P1 hamster kidneys, were grown in 100-mm dishes containing 10 ml of Eagle minimum essential medium supplemented with 10% fetal bovine serum and antibiotics (growth medium). Transfection was performed with Lipofectin (Invitrogen, Carlsbad, California), following the manufacturer’s protocol. Briefly, cells were plated at 1 × 105 on a 60-mm dish in growth medium, minus the antibiotics, the day before transfection. The following day, DNA was prepared by adding 2 μg of construct (IgG-scFv-35 and IgG-scFv-35-DAF) to Opt-MEM (Invitrogen) and combining it with the Lipofectin solution. The resulting solution was added to cells and incubated for 24 h at 37°C. Stable transfected cell lines were selected using G418 (200 μg/ml) in growth medium. Positive cells were detected by expression of GFP. Clones were selected and expanded in growth medium supplemented with G418. Cells were placed in serum-free and protein-free growth medium for collection of secreted recombinant protein. Collected medium was purified by Antibody Research (St. Charles, Missouri), and the resulting recombinant protein was analyzed with 12% sodium dodecylsulfate sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), followed by detection with Coomassie Blue G-250 (Bio-Rad, Hercules, California).
Complement Hemolytic Assay
Complement inhibition by scFv-35-DAF was assessed by hemolytic assay using sensitized sheep erythrocytes (Complement Technologies, Tyler, Texas) and rat serum, as a source of complement. Rat serum was diluted 1:200 and added to a concentration of scFv-35 or scFv-35-DAF ranging from 0.1 to 100 nM. Complement activity was initiated by addition of 100 μl of 2 × 108 sensitized sheep erythrocytes in Veronal buffer and incubated at 37°C for 1 h. After 60 min, sheep cells were removed by centrifugation, and complement hemolysis was measured at 412 nm (SpectraMax 340; Molecular Devices, Sunnyvale, California). Data are shown as a percentage of inhibition based on 100% lysis values.
Evaluation of scFv-35 and scFv-35–DAF into C57Bl/6 and CD59ab−/−Daf1−/− Mice
We utilized 2 groups of 4 mice on both C57Bl/6 and CD59ab−/−Daf1−/− strains. Mice were injected with 234 μg of scFv-35 or scFv-35-DAF and monitored for 24 h for weakness or signs of stress. Grip strengths were determined by ability to lift cage lids consecutive times. A generally accepted motor strength scale was used to assess for weakness: 0 = can grip and lift lid of the cage; 1 = can grip but cannot lift the lid of the cage; 2 = unable to grip cage lid; 3 = unable to grip plus hindlimb paralysis; and 4 = moribund.26 After 24 h, the mice were euthanized, and diaphragms were removed for analysis.
Induction of EAMG in Lewis Rats
To determine the efficacy of the scFv-35-DAF recombinant protein, 15 rats were grouped randomly and given scFv-35, scFv-35-DAF, or vehicle by intraperitoneal injection. One hour later, EAMG was induced by administration of rat anti-mouse muscle AChR MAb McAb-3 (gift of Vanda Lennon, Mayo Clinic27), similar to our previous investigations [50 μl of purified McAb-3 (4.6 mg/ml)].21,28,29 McAb-3 is an IgG2b isotype known to activate complement. Rats were kept under close observation and evaluated daily. A generally accepted motor strength scale was used to assess for weakness.26 Animals underwent euthanasia after 48 h, and diaphragms were removed for analysis.
Immunohistochemistry and Fluorescence Quantitation
For analysis of localization of scFv-35 to the neuromuscular junction, the diaphragms of mice were analyzed. Cryosections of diaphragm at 10 μm were stained with Alexa 488–labeled anti-rat IgG (Molecular Probes) and Alexa 594–labeled bungarotoxin (BTX) for identification of NMJs. For analysis of EAMG-induced rats treated with scFv-35 and scFv-35-DAF or PBS controls, diaphragm tissues were cryosectioned at 10 μm and stained for membrane attack complex (MAC) deposition. Briefly, rabbit polyclonal anti-complement 5b-9 (Calbiochem) diluted 1:100 was incubated on sections for 1 h. Alexa 488–labeled anti-rabbit IgG (Molecular Probes) was used to detect MAC and Alexa 594–labeled BTX was used to identify NMJs.
The sections were viewed with a fluorescence microscope (BX50; Olympus, Center Valley, Pennsylvania), and digital images were captured with a Spot camera (Diagnostic Instruments, Sterling Heights, Michigan) and analyzed with ImagePro software (Media Cybernetics, Silver Springs, Maryland). Adobe Photoshop and Illustrator (Adobe Systems, Inc., Seattle, Washington) software programs were used to prepare the figures. All BTX-stained junctions in 9–12 randomly selected fields were counted, and MAC immunoreactivity of these junctions was determined. Pixel density measurements were used to quantitate fluorescence at the neuromuscular junctions. A total of 80 NMJs were analyzed in each group. The percent of endplates with specific pixel density was expressed in a histogram.
Statistical Analysis
The data were analyzed and tested for statistical significance using paired Mann–Whitney U-tests. P < 0.05 was considered significant.
RESULTS
scFv-35 and scFv-35-DAF
The sequence of the combined scFv-35-DAF is shown in Figure 2 and confirms the identity of the construct. Recombinant protein was collected in serum-free and protein-free medium. SDS-PAGE gel was run to confirm size and purity of scFv-35-DAF (Fig. 3A). The recombinant protein is 529 amino acids with an expected size of 57.7 kDa.
Figure 3.
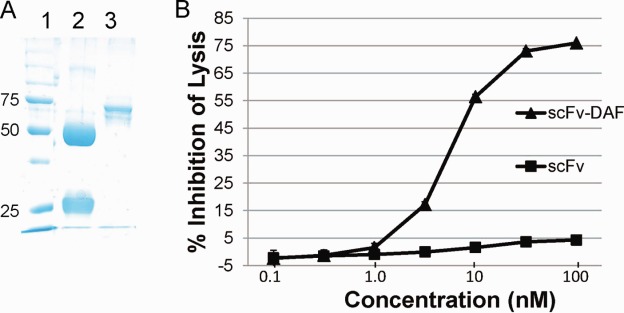
Expression and complement inhibition activity of scFv-35-DAF. (A) SDS-PAGE gel: lane 1—protein standards; lane 2—rat IgG; lane 3—scFv-35-DAF. (B) The scFv-35-DAF recombinant protein inhibits minimal cell lysis at a 5-nM concentration, a maximal 76.61 ± 3.89% inhibitory capacity at 100 nM, and an estimated CH50 of 9 nM. The scFV-35 demonstrates no native complement inhibitory potential.
Complement Inhibition of scFv-35-DAF
We performed a serial dilution to assess the ability of scFv-35-DAF to inhibit lysis of sensitized sheep red blood cells. The scFv-35 alone had no demonstrable effect on inhibition of cell lysis. The scFv-35-DAF inhibited cell lysis by 76.61 ± 3.89% at a concentration of 100 nM (Fig. 3B) and an estimated CH50 of 9 nM.
Evaluation of Toxicity of scFv-35-DAF
Administration of an antibody directed toward the AChR carries the risk of blocking neuromuscular transmission and potentially activating complement. We utilized C57Bl/6 mice and mice deficient in both CD59ab and Daf1, which have demonstrated severe, complement-mediated weakness within 24 h of EAMG induction.28,30,31 Injection of scFv-35 into C57Bl/6 and CD59ab−/−Daf1−/− mice showed no observable pathology. The mice maintained strength and mobility over the 24-h period. We were able to demonstrate that, at 24 h, scFv-35 ?A3B2 thyc?> localized to the NMJ in the C57Bl/6 and CD59ab−/−Daf1−/− mice (Fig. 4). Furthermore, the NMJs of CD59ab−/−Daf1−/− animals showed no evidence of MAC formation (Fig. 4).
Figure 4.
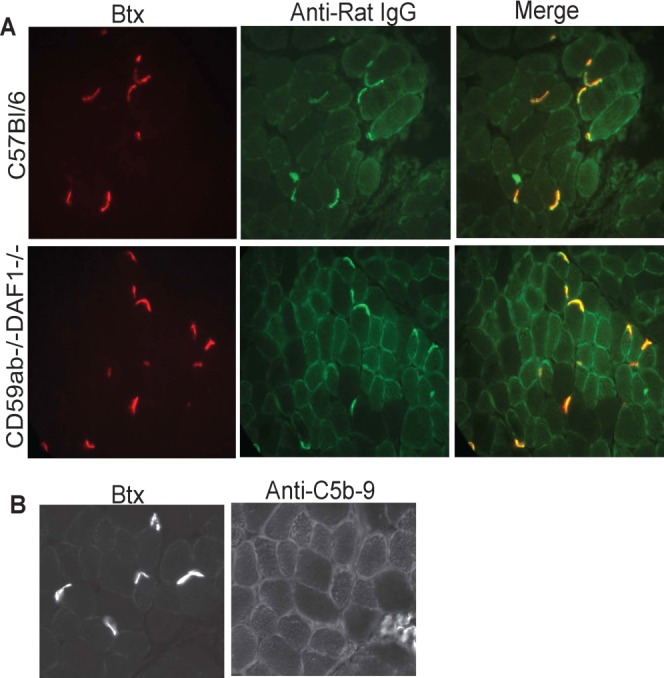
C57Bl/6 and CD59ab−/−Daf1−/− mice were injected with scFv-35 and killed 24 h later. Animals did not demonstrate weakness. (A) Diaphragm cryosections from scFv-35–injected c57Bl/6 and CD59ab−/−Daf1−/− mice were stained with Alexa 488–conjugated anti-rat IgG (green). Alexa 594–conjugated α-BTX (red) identifies the NMJs. (B) CD59ab−/−Daf1−/− mice demonstrated no evidence of MAC deposition. Diaphragms from scFv-35–injected CD59ab−/−Daf1−/− mice were stained with anti-MAC and detected with Alexa 488–conjugated anti-rabbit IgG. Alexa 594–conjugated α-BTX identifies the NMJs.
Therapeutic Effect of scFv-35 and scFv-35-DAF
After 24 h of EAMG induction, PBS-treated and scFv-35–treated rats began to show weakness, which was not evident in the scFv-35-DAF–treated rats (Fig. 5). By 48 h, vehicle- and scFv-35–treated rats had become profoundly weak (clinical scores of 3.4 ± 0.55 and 3.4 ± 0.89, respectively) to the point of requiring euthanasia, whereas scFv-35-DAF–treated rats had developed only moderate weakness (clinical score of 2 ± 0, P < 0.01 compared with both scFv-35 and PBS; Fig. 5). All animals were then killed for histological analysis. Quantitative analysis of complement deposition demonstrated significantly less MAC deposition at endplates of scFv-35-DAF–treated animals (mean 1226 ± 610 pixel density) compared with both vehicle- and scFv-35–treated rats (mean 1611 ± 644 pixel density and 1979 ± 453 pixel density, respectively; P < 0.01 compared with scFv-DAF treated), whereas scFv-35–treated rats had a marginally higher degree of complement deposition compared with vehicle-treated rats (Fig. 6). Consistent with the better clinical outcome, AChR density was significantly greater in the scFv-35-DAF–treated rats (mean 3208 ± 953 pixel density) than scFv-35–treated and vehicle-treated rats (means 1489 ± 865 pixel density and 1128 ± 716 pixel density, respectively; P < 0.001 compared with scFv-35-DAF).
Figure 5.
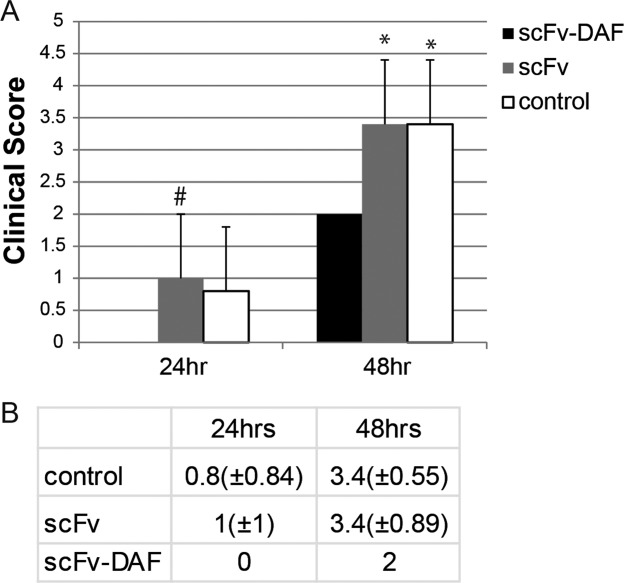
Clinical scores of EAMG-induced rats treated with scFv-35 (shaded bars), scFv-35-DAF (black bars), and PBS (white bars). (A) Rats were treated with scFv-35-DAF, scFv-35, or PBS control before being induced with MAb 3. The rats treated with scFv-35-DAF had no weakness at 24 h and minimal weakness at 48 h with vehicle-treated and scFv-35–treated rats (#P < 0.05 vs. scFv-35-DAF, *P < 0.01 vs. scFv-35-DAF). (B) Table of clinical scores for scFv-35, scFv-35-DAF, and PBS treatment at 24 h and 48 h.
Figure 6.
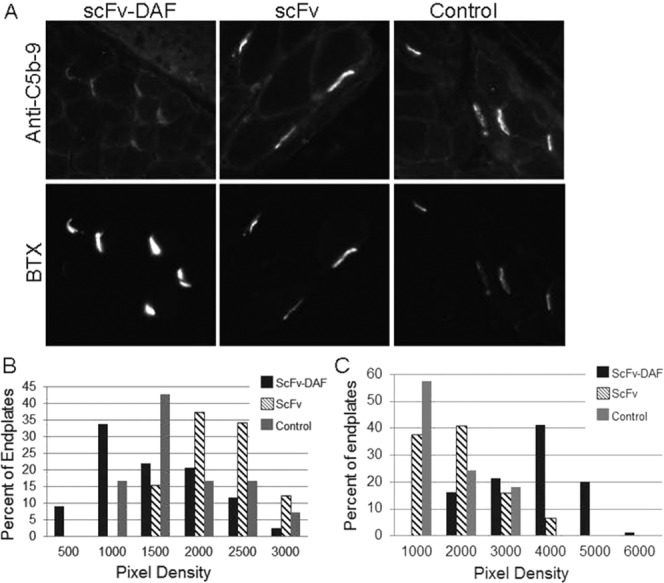
Membrane attack complex and AChR assessment after 48 h of induction of EAMG. (A) Diaphragms from EAMG-induced rats were cryosectioned and stained for MAC. Alexa 488–conjugated anti-rabbit IgG was used to detect MAC antibody. Alexa 594–conjugated α-BTX identified NMJs. (B) Fluorescent images were assessed for pixel density of MAC deposition at the NMJ. A distribution plot was constructed based on percent of endplates that expressed a specific pixel range (P < 0.01 for scFv-35-DAF vs. scFv-35 and control). (C) Alexa 594–conjugated α-BTX bound to AChR at the NMJs were assessed by pixel density. A distribution plot was constructed with percent NMJs identified within a range of pixel density.
DISCUSSION
In this study we have demonstrated the feasibility of concentrating a therapeutic agent at the NMJ without producing neuromuscular blockade or activation of complement. The scFv-35 was produced from the well-characterized McAb-35, which is known to bind the main immunogenic region on the extracellular surface of the rat α-subunit of the AChR. We confirmed that the scFv-35 localized to the NMJ and is stable for 24 h. Despite scFv-35 binding to the junction, c57Bl/6 and complement-deficient mice demonstrated no weakness. This indicates that the AChR can be used as a target to concentrate therapeutics to the NMJ (Fig. 7). We showed that the scFv-35-DAF constructed inhibited complement-mediated lysis of sheep red blood cells in vitro, whereas the scFv-35 itself had no such inhibitory activity. Despite using CD59ab−/−Daf1−/− mice, which are highly susceptible to complement activation by antibody,28,30,31 there was no immunohistochemical evidence of complement activation. The scFv-35 was engineered to lack the Fc portion, and we did not expect it to activate complement. In both mice deficient in intrinsic complement inhibitors (data not shown) and Lewis rats, we found the scFv-35-DAF moderated passive EAMG severity with reduced weakness, lessened MAC deposition, and preserved AChR density. Our findings are most likely explained by the complement-inhibitory effect of the scFv-35-DAF. We found the half-maximal inhibitory concentration (IC50) for scFv-35-DAF to be 9 nM, whereas scFv-35 alone did not inhibit complement. The IC50 compares favorably with that of eculizumab, the only complement inhibitor in clinical use, which is estimated to be in the range of 1–290 nM.33,34
Figure 7.
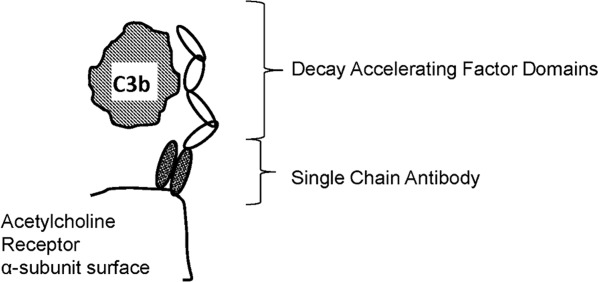
Schematic of mode of action of scFv-35-DAF. The figure illustrates the hypothesis of AChR being targeted by a single-chain antibody and concentrating DAF to the NMJ. Size differences among molecules are approximate.
We did not completely ablate the effects of EAMG, but only a single dose was evaluated. Optimization of dosing would be expected to improve efficacy, and, as with other therapeutics for human MG, repeat dosing would be required. Another limitation of our study is that we chose to treat animals with the scFv-35-DAF prior to EAMG induction to optimize the potential to detect a treatment effect. Further studies are required to assure that scFv-35-DAF can reduce EAMG severity after induction of disease.
Antigenic modulation, which occurs through cross-linking of AChR by the bivalent antibody and thereby increases internalization of receptor, contributes to reductions of AChR density and consequent development of weakness.1–4 Based on previous studies, monovalent AChR antibodies have not been demonstrated to induce disease and therefore there was an expectation that the scFv-35 would not induce weakness. As expected, the scFv-35 did not produce weakness in the CD59ab−/−Daf1−/− mice, but one may have expected in EAMG experiments that the scFv-35 would reduce severity of EAMG-induced weakness. Fab fragments produced from McAb-35 moderate the severity of EAMG induced by the whole antibody.35 We have limited evidence that any blockade of pathogenic antibody occurred in this experiment. The scFv-35 produced no moderation of disease severity and did not reduce complement activation, which could have been expected if the scFv-35 reduced pathogenic antibody binding with subsequent complement activation. The addition of the DAF domains to the scFv-35 could conceivably produce a blocking effect, but given the demonstration of reduced MAC deposition, it is most reasonable to consider the complement-inhibitory effect to be primary. Surprisingly, administration of scFv-35 to rats with EAMG produced a moderate increase in complement deposition and a reduced AChR density compared with EAMG rats given PBS alone. We are unaware of any antibody directed to the AChR that leads to activation of the ion channel,10 and there is none for the McAb-35 itself. Therefore, we do not believe svFc-35-DAF could have an agonist effect.
Blockade of AChR function could contribute to weakness,36 which led to significant concern that the scFv-35 could produce weakness. There was no evidence that the scFv-35 blocked channel function; however, we did not perform electrophysiological studies to assess subclinical compromise of neuromuscular transmission. Both mice and humans share the property of a high safety factor for neuromuscular transmission,37 and therefore small effects of scFv-35 and scFv-35-DAF on transmission would not have been identified by behavioral evaluations. However, even in animals with EAMG, scFv-35 and scFv-35-DAF did not worsen disease.
Although intravenous immunoglobulin treatment may moderate the severity of human MG and EAMG,38 it is unlikely that our findings can be explained only by infusion of immunoglobulin. First, scFv-35 had no effect on EAMG severity. Second, intravenous immunoglobulin appears to moderate EAMG by anti-idiotype mechanisms, which is unlikely to have occurred in our experimental paradigm.38,39 Finally, a study of intravenous immunoglobulin utilized a dose of 0.4 g/kg per rat, which is about 200 times the dose of scFv-35-DAF used in our study.40
As a complement inhibitor, we utilized the regulatory region of DAF, which is composed of 60-amino-acid-long repeating domains and acts efficiently on both the classical and alternative pathways.41,42 In DAF, 4 of these complement control repeats (CCPs) are suspended above the surface membrane. In a series of experiments, we and others have demonstrated that mice deficient in Daf and another cell surface complement inhibitor, CD59, are extremely susceptible to EAMG,28–31 which suggests that the DAF-inhibitory domain could function effectively in disease moderation, as demonstrated in this report. One investigation43 described construction of a single-chain antibody directed at the AChR coupled to DAF, which also was found to concentrate to the NMJ; however, the investigators did not evaluate for therapeutic efficacy in animals. Linking CCP domains of DAF to a viral vector has been successful in delivering DAF to the cell surface, leading to protection from complement-mediated injury.44 Delivery of DAF to the cell surface of erythrocytes protects them from complement-mediated lysis.45 Coupled with the present investigation, these studies support the use of DAF-based constructs as therapeutic complement inhibitors.
Complement inhibitors are in their developmental infancy,46 but they are a subject of intense interest.47 Presently, only 1 first-generation inhibitor, eculizumab, is FDA approved for paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Eculizumab also carries the distinction of being the most expensive medication on the market.48 Development of more advanced complement-based therapeutics will likely to be refined to focus inhibition on specific steps of the activation cascade tailored to specific pathology as well as enhancing potency. Here we have demonstrated the feasibility of concentrating complement inhibition to a site of pathology, which is an additional refinement needed for complement therapy development.
Glossary
- AChR
acetylcholine receptor
- CCP
complement control repeats
- DAF
decay-accelerating factor
- EAMG
experimentally acquired myasthenia gravis
- IC50
half-maximal inhibitory complex
- MAC
membrane attack complex
- McAb
monoclonal antibody
- MG
myasthenia gravis
- NMJ
neuromuscular junction
- PCR
polymerase chain reaction
- scFv
single-chain antibody
- SDS-PAGE
sodium dodecylsulfate–polyacrylamide gel electrophoresis
REFERENCES
- 1.Conti-Fine BM, Milani M, Kaminski HJ. Myasthenia gravis: past, present, and future. J Clin Invest. 2006;116:2843–2854. doi: 10.1172/JCI29894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vincent A. Autoimmune channelopathies: well-established and emerging immunotherapy-responsive diseases of the peripheral and central nervous systems. J Clin Immunol. 2010;30(suppl 1):S97–102. doi: 10.1007/s10875-010-9401-x. [DOI] [PubMed] [Google Scholar]
- 3.Drachman DB. Targeting T cells in myasthenia gravis. Ann Neurol. 1999;46:553–555. doi: 10.1002/1531-8249(199910)46:4<553::aid-ana1>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 4.Sanders DB, Evoli A. Immunosuppressive therapies in myasthenia gravis. Autoimmunity. 2010;43:428–435. doi: 10.3109/08916930903518107. [DOI] [PubMed] [Google Scholar]
- 5.Meriggioli MN, Sanders DB. Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurol. 2009;8:475–490. doi: 10.1016/S1474-4422(09)70063-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keesey JC. A history of treatments for myasthenia gravis. Semin Neurol. 2004;24:5–16. doi: 10.1055/s-2004-829584. [DOI] [PubMed] [Google Scholar]
- 7.Kusner LL, Kaminski HJ, Soltys J. Effect of complement and its regulation on myasthenia gravis pathogenesis. Exp Rev Clin Immunol. 2008;4:43–52. doi: 10.1586/1744666X.4.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tuzun E, Huda R, Christadoss P. Complement and cytokine based therapeutic strategies in myasthenia gravis. J Autoimmun. 2011;37:136–143. doi: 10.1016/j.jaut.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 9.Christadoss P, Tuzun E, Li J, Saini SS, Yang H. Classical complement pathway in experimental autoimmune myasthenia gravis pathogenesis. Ann NY Acad Sci. 2008;1132:210–219. doi: 10.1196/annals.1405.009. [DOI] [PubMed] [Google Scholar]
- 10.Lindstrom J. Acetylcholine receptors and myasthenia. Muscle Nerve. 2000;23:453–477. doi: 10.1002/(sici)1097-4598(200004)23:4<453::aid-mus3>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 11.Bevan S, Kullberg RW, Heinemann SF. Human myasthenic sera reduce acetylcholine sensitivity of human muscle cells in tissue culture. Nature. 1977;267:263–265. doi: 10.1038/267263a0. [DOI] [PubMed] [Google Scholar]
- 12.Drachman D, Angus CW, Adams RN, Kao I. Effect of myasthenic patients’ immunoglobulin on acetylcholine receptor turnover: selectivity of degradation process. Proc Natl Acad Sci USA. 1978;75:3422–3426. doi: 10.1073/pnas.75.7.3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kusner LL, Kaminski HJ, Soltys J. Effect of complement and its regulation on myasthenia gravis pathogenesis. Exp Rev Clin Immunol. 2008;4:43–52. doi: 10.1586/1744666X.4.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Medof ME, Kinoshita T, Nussenzweig V. Inhibition of complement activation on the surface of cells after incorporation of decay-accelerating factor (DAF) into their membranes. J Exp Med. 1984;160:1558–1578. doi: 10.1084/jem.160.5.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seya T, Turner J, Atkinson J. Purification and characterization of a membrane protein (gp45–70) that is a cofactor for cleavage of C3b and C4b. J Exp Med. 1986;163:837–855. doi: 10.1084/jem.163.4.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davies A, Simmons DL, Hale G, Harrison RA, Tighe H, Lachmann PJ, et al. CD59, an LY-6-like protein expressed in human lymphoid cells, regulates the action of the complement membrane attack complex on homologous cells. J Exp Med. 1989;170:637–654. doi: 10.1084/jem.170.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harada R, Okada N, Fujita T, Okada H. Purification of 1F5 antigen that prevents complement attack on homologous cell membranes. J Immunol. 1990;144:1823–1828. [PubMed] [Google Scholar]
- 18.Holguin MH, Fredrick LR, Bernshaw NJ, Wilcox LA, Parker CJ. Isolation and characterization of a membrane protein from normal human erythrocytes that inhibits reactive lysis of the erythrocytes of paroxysmal nocturnal hemoglobinuria. J Clin Invest. 1989;84:7–17. doi: 10.1172/JCI114172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang H, Lu S, Morrison SL, Tomlinson S. Targeting of functional antibody-decay-accelerating factor fusion proteins to a cell surface. J Biol Chem. 2001;276:27290–27295. doi: 10.1074/jbc.M100436200. [DOI] [PubMed] [Google Scholar]
- 20.Howard JF, Jr, Barohn R, Freimer M, Juel V, Mozaffar T, Mellion M, et al. Randomized, double-blind, placebo-controlled, crossover, multicenter, phase II study of eculizumab in patients with refractory generalized myasthenia gravis. Muscle Nerve. 2013;48:76–84. doi: 10.1002/mus.23839. [DOI] [PubMed] [Google Scholar]
- 21.Lin F, Emancipator SN, Salant DJ, Medof ME. Decay-accelerating factor confers protection against complement-mediated podocyte injury in acute nephrotoxic nephritis. Lab Invest. 2002;82:563–569. doi: 10.1038/labinvest.3780451. [DOI] [PubMed] [Google Scholar]
- 22.Qin X, Hu W, Song W, Grubissich L, Hu X, Wu G, et al. Generation and phenotyping of mCd59a and mCd59b double-knockout mice. Am J Hematol. 2009;84:65–70. doi: 10.1002/ajh.21319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tzartos SJ, Rand DE, Einarson BL, Lindstrom JM. Mapping of surface structures of electrophorus acetylcholine receptor using monoclonal antibodies. J Biol Chem. 1981;256:8635–8645. [PubMed] [Google Scholar]
- 24.Losen M, Stassen MH, Martinez-Martinez P, Machiels BM, Duimel H, Frederik P, et al. Increased expression of rapsyn in muscles prevents acetylcholine receptor loss in experimental autoimmune myasthenia gravis. Brain. 2005;128:2327–2337. doi: 10.1093/brain/awh612. [DOI] [PubMed] [Google Scholar]
- 25.Lindstrom J, Einarson B, Tzartos S. Production and assays of antibodies to acetylcholine receptors. Methods Enzymol. 1981;74:432–456. doi: 10.1016/0076-6879(81)74031-x. [DOI] [PubMed] [Google Scholar]
- 26.Piddlesden SJ, Jiang S, Levin JL, Vincent A, Morgan BP. Soluble complement receptor 1 (sCR1) protects against experimental autoimmune myasthenia gravis. J Neuroimmunol. 1996;71:173–177. doi: 10.1016/s0165-5728(96)00144-0. [DOI] [PubMed] [Google Scholar]
- 27.Lennon VA, Lambert EH. Myasthenia gravis induced by monoclonal antibodies to acetylcholine receptors. Nature. 1980;285:238–240. doi: 10.1038/285238a0. [DOI] [PubMed] [Google Scholar]
- 28.Kaminski HJ, Kusner LL, Richmonds C, Medof ME, Lin F. Deficiency of decay accelerating factor and CD59 leads to crisis in experimental myasthenia. Exp Neurol. 2006;202:287–293. doi: 10.1016/j.expneurol.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 29.Lin F, Kaminski HJ, Conti-Fine BM, Wang W, Richmonds C, Medof ME. Markedly enhanced susceptibility to experimental autoimmune myasthenia gravis in the absence of decay-accelerating factor protection. J Clin Invest. 2002;110:1269–1274. doi: 10.1172/JCI16086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morgan BP, Chamberlain-Banoub J, Neal JW, Song W, Mizuno M, Harris CL. The membrane attack pathway of complement drives pathology in passively induced experimental autoimmune myasthenia gravis in mice. Clin Exp Immunol. 2006;146:294–302. doi: 10.1111/j.1365-2249.2006.03205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kusner LL, Halperin JA, Kaminski HJ. Cell surface complement regulators moderate experimental myasthenia gravis pathology. Muscle Nerve. 2013;47:33–40. doi: 10.1002/mus.23448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Research CfDEa. Soliris (eculizumab) injectable I.V. Silver Spring, MD: Food and Drug Administration; 2007. [Google Scholar]
- 34.Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol. 2007;25:1256–1264. doi: 10.1038/nbt1344. [DOI] [PubMed] [Google Scholar]
- 35.Loutrari H, Kokla A, Tzartos SJ. Passive transfer of experimental myasthenia gravis via antigenic modulation of acetylcholine receptor. Eur J Immunol. 1992;22:2449–2452. doi: 10.1002/eji.1830220939. [DOI] [PubMed] [Google Scholar]
- 36.Poulas K, Tsouloufis T, Tzartos SJ. Treatment of passively transferred experimental autoimmune myasthenia gravis using papain. Clin Exp Immunol. 2000;120:363–368. doi: 10.1046/j.1365-2249.2000.01202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gomez CM, Richman DP. Anti-acetylcholine receptor antibodies directed against the alpha-bungarotoxin binding site induce a unique form of experimental myasthenia. Proc Natl Acad Sci USA. 1983;80:4089–4093. doi: 10.1073/pnas.80.13.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Slater CR. Reliability of neuromuscular transmission and how it is maintained. In: Vinken PJ, Bruyn GE, editors. Handbook of clinical neurology. 2008. pp. 27–101. [DOI] [PubMed] [Google Scholar]
- 39.Fuchs S, Feferman T, Meidler R, Margalit R, Sicsic C, Wang N, et al. A disease-specific fraction isolated from IVIG is essential for the immunosuppressive effect of IVIG in experimental autoimmune myasthenia gravis. J Neuroimmunol. 2008;194:89–96. doi: 10.1016/j.jneuroim.2007.11.020. [DOI] [PubMed] [Google Scholar]
- 40.Fuchs S, Feferman T, Meidler R, Margalit R, Sicsic C, Brenner T, et al. Immunosuppression of EAMG by IVIG is mediated by a disease-specific anti-immunoglobulin fraction. Ann NY Acad Sci. 2008;1132:244–248. doi: 10.1196/annals.1405.032. [DOI] [PubMed] [Google Scholar]
- 41.Fuchs S, Feferman T, Meidler R, Brenner T, Laub O, Souroujon MC. The disease-specific arm of the therapeutic effect of intravenous immunoglobulin in autoimmune diseases: experimental autoimmune myasthenia gravis as a model. Isr Med Assoc J. 2008;10:58–60. [PubMed] [Google Scholar]
- 42.Medof ME, Lublin DM, Holers VM, Ayers DJ, Getty RR, Leykam JF, et al. Cloning and characterization of cDNAs encoding the complete sequence of decay-accelerating factor of human complement. Proc Natl Acad Sci USA. 1987;84:2007–2011. doi: 10.1073/pnas.84.7.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Caras IW, Davitz MA, Rhee L, Weddell G, Martin DW, Jr, Nussenzweig V. Cloning of decay-accelerating factor suggests novel use of splicing to generate two proteins. Nature. 1987;325:545–549. doi: 10.1038/325545a0. [DOI] [PubMed] [Google Scholar]
- 44.Song C, Xu Z, Miao J, Xu J, Wu X, Zhang F, et al. Protective effect of scFv-DAF fusion protein on the complement attack to acetylcholine receptor: a possible option for treatment of myasthenia gravis. Muscle Nerve. 2012;45:668–675. doi: 10.1002/mus.23247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaname Y, Tani H, Kataoka C, Shiokawa M, Taguwa S, Abe T, et al. Acquisition of complement resistance through incorporation of CD55/decay-accelerating factor into viral particles bearing baculovirus GP64. J Virol. 2010;84:3210–3219. doi: 10.1128/JVI.02519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spitzer D, Unsinger J, Bessler M, Atkinson JP. ScFv-mediated in vivo targeting of DAF to erythrocytes inhibits lysis by complement. Mol Immunol. 2004;40:911–919. doi: 10.1016/j.molimm.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 47.Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat Biotechnol. 2007;25:1265–1275. doi: 10.1038/nbt1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang K, Deangelis RA, Reed JE, Ricklin D, Lambris JD. Complement in action: an analysis of patent trends from 1976 through 2011. Adv Exp Med Biol. 2013;734:301–313. doi: 10.1007/978-1-4614-4118-2_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herper M. The world’s most expensive drugs Forbes.com2010.