

SUMMARY
Cells contain multiple F-actin assembly pathways including the Arp2/3 complex, formins, and Ena/VASP, which have largely been analyzed separately. They collectively generate the bulk of F-actin from a common pool of G-actin; however, the interplay/competition between these pathways remains poorly understood. Using fibroblast lines derived from an Arpc2 conditional knockout mouse, we established matched-pair cells with and without the Arp2/3 complex. Arpc2−/− cells lack lamellipodia and migrate slower than WT cells, but have F-actin levels indistinguishable from controls. Actin assembly in Arpc2−/− cells was resistant to cytochalasin-D and was highly dependent on profilin-1 and Ena/VASP, but not formins. Profilin-1 depletion in WT cells increased F-actin and Arp2/3 complex in lamellipodia. Conversely, addition of exogenous profilin-1 inhibited Arp2/3 complex actin nucleation in vitro and in vivo. These observations suggest that antagonism of the Arp2/3 complex by profilin-1 in cells maintains actin homeostasis by balancing Arp2/3 complex-dependent and independent actin assembly pathways.
INTRODUCTION
Actin assembly is critical for many cellular processes including migration, vesicular trafficking and adhesion (Campellone and Welch, 2010). F-actin can form spontaneously in vitro, but de novo filament nucleation is energetically and kinetically disfavored and requires additional factors to efficiently polymerize both in vitro and in vivo (Campellone and Welch, 2010). The factors responsible for assembling F-actin networks include the Arp2/3 complex, which forms branched actin filaments, and formin and Ena/VASP proteins which form long, unbranched actin filaments. Each of these classes of actin assembly factors polymerizes F-actin at specific subcellular locations, leading to various cellular responses.
The seven subunit Arp2/3 complex localizes to endocytic and phagocytic structures, adherens junctions, invadopodia, and to the lamellipodia, where it generates the branched actin network under the control of Nucleation Promoting Factors (NPFs) (Rotty et al., 2013). Ena/VASP localizes to the distal tip of the lamellipodium where it regulates the density and length of Arp2/3 complex-generated branches through its antagonistic relationship with capping protein, while also incorporating G-actin to growing barbed ends (Bear et al., 2002; Hansen and Mullins, 2010; Winkelman et al., 2014). However, Ena/VASP proteins also localize to both focal adhesions and filopodia, and directly contribute to forming the unbranched, bundled F-actin of the latter (Lanier et al., 1999; Reinhard et al., 1992; Svitkina et al., 2003). Formins are multidomain proteins encoded by 15 distinct genes in mammals that assemble actin in filopodia and stress fibers, and contribute to lamellipodial dynamics, vesicular transport, cytokinesis, and phagocytosis (Breitsprecher and Goode, 2013). Though much is known about these pathways individually, both in vitro and in cells, we lack a systematic understanding of the collaboration and competition between these pathways in cells.
All of these pathways are thought to share a common pool of G-actin, which must be divided among distinct F-actin assembly factors at various subcellular locations (Chesarone and Goode, 2009; Gao and Bretscher, 2008). In yeast, which lack Ena/VASP proteins and have only two (budding yeast) or three formins (fission yeast), the Arp2/3 complex is known to generate actin patches involved in endocytosis (Winter et al., 1999) while formin isoforms generate a completely distinct network of actin cables that polarize cells for division, and form the contractile ring (Evangelista et al., 2002; Sagot et al., 2002). Recent studies revealed that inhibition of the Arp2/3 complex leads to compensatory F-actin assembly by formins in fission yeast (Burke et al., 2014). Arp2/3 complex-dependent and –independent assembly pathways show a similar compensation in mammalian cells, although until now the mechanism remained obscure (Hotulainen and Lappalainen, 2006; Steffen et al., 2006; Suraneni et al., 2012; Wu et al., 2012).
Here we report a detailed analysis of the F-actin network structure, dynamics and content of fibroblasts genetically null for the p34 subunit of the Arp2/3 complex. We find that Ena/VASP and profilin maintain F-actin levels in the absence of Arp2/3 complex function in mammalian cells. We also find evidence for an inhibitory relationship between profilin and the Arp2/3 complex. Our findings in mouse fibroblasts, together with the work of Suarez et al using fission yeast and in vitro single molecule imaging techniques (see accompanying paper), suggest that profilin preferentially delivers actin monomers to Ena/VASP and formin pathways and inhibits Arp2/3 complex-based nucleation. The profilin-dependent interplay between these pathways creates a homeostatic balance that allows each pathway to function side-by-side in a common cytoplasmic compartment in order to drive higher order cellular processes like lamellipodial protrusion, endocytosis and cell division that depend on complex and varied actin networks.
RESULTS
Generation and characterization of Arpc2−/− fibroblast lines
Based on our recent finding that cells depleted of Arp2/3 complex by RNAi are viable in the Ink4a/Arf−/− genetic background and proliferate in culture (Wu et al., 2012), we crossed mice containing a conditional Arpc2 allele (consisting of LoxP sites flanking exon 8 of the gene encoding the p34 subunit of the Arp2/3 complex) into the Ink4a/Arf−/− background. Cells derived from these mice lack both Arf and p16INK4a and proliferate readily in culture before and after the deletion of the Arpc2 gene. Fibroblasts were isolated from both embryonic (MEFs) and adult tail (MTFs) tissue of these mice, stably transduced with CreER, and grown up as clonal lines. Based on initial validation of multiple clonal lines, we proceeded with one MEF line (MEF 10-4) and one MTF line (MTF24) for subsequent experiments. Treatment of clonal lines with tamoxifen (4-OHT) to activate the Cre recombinase activity generated matched pair cell lines with and without the complete loss of p34 protein (referred to as Arpc2−/− and WT throughout) and led to the loss of other Arp2/3 complex subunits as well (Fig. 1A,B). These Arpc2−/− lines lack lamellipodia and are dominated instead by filopodial protrusions, as well as containing abundant stress fibers (Figs. 1C, S1A). Stable reintroduction of p34-GFP via lentivirus restored Arp2/3 complex protein levels and lamellipodia (Fig. 1D,E). Both Arpc2−/− lines showed severe defects in single cell motility that were rescued by the re-introduction of p34-GFP (Fig. 1F).
Figure 1. Generation and characterization of Arpc2−/− fibroblast cell lines.
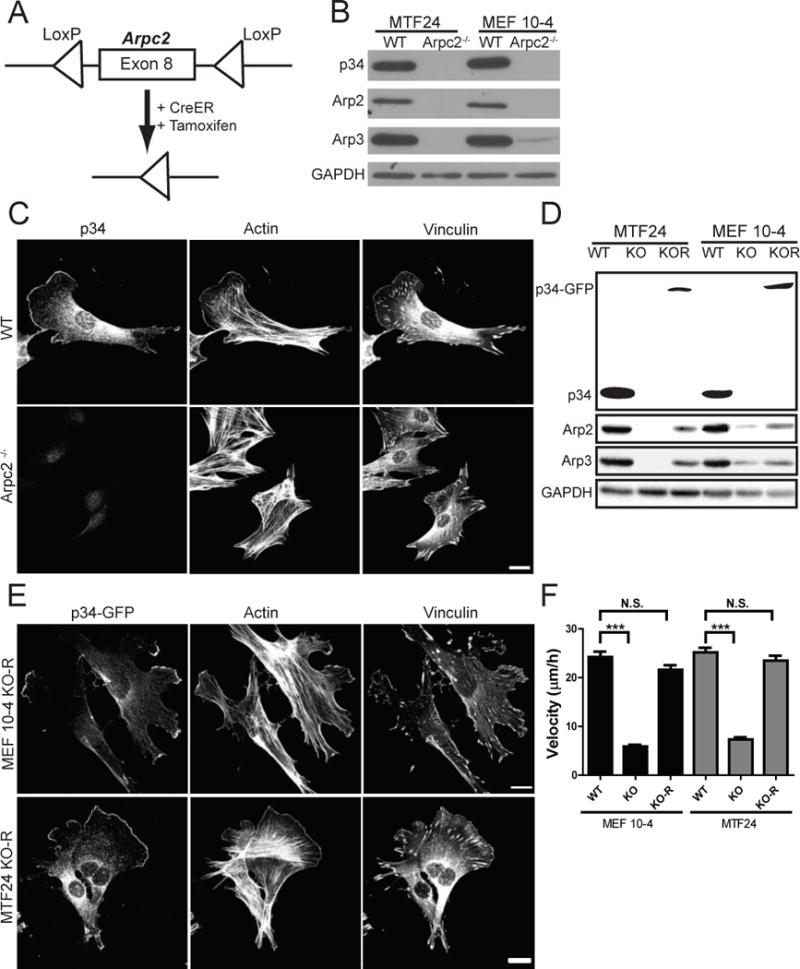
A) Schematic representation of tamoxifen-inducible CreER activation and Arpc2 (p34) deletion. B) Blot analysis of two mouse fibroblast cell lines without (WT) or with (Arpc2−/−) tamoxifen treatment. C) Staining of MTF24 WT and Arpc2−/− fibroblasts; scale bar = 20 microns. D) Blot analysis of cell lines without (WT), with (KO) tamoxifen treatment, or KO cells stably rescued with p34-GFP (KO-R). E) Staining of MEF 10-4 KO-R and MTF24 KO-R fibroblasts; GFP indicates p34-GFP; scale bars = 20 microns. F) Random migration velocity of WT, KO, and KO-R MEF 10-4 (black bars) and MTF24 (grey bars) fibroblasts; N = at least 54 cells per condition; error bars represent standard error of the mean; ***p-value < 0.0001. See also Figure S1.
Loss of Arp2/3 complex affects F-actin structure and dynamics, but not total F-actin levels
The genetic ablation of Arp2/3 complex in mammalian fibroblasts provides an opportunity to analyze the actin cytoskeleton in the absence of one of its major regulators. Actin filaments are considerably less dynamic in Arpc2−/− cells than WT controls as shown by imaging the actin probe LifeAct (LA), leading to less dynamic protrusion and retraction (Fig. 2A, S1B, Movies S1, S2), consistent with their slow migration. We used cryo-shadowing EM (Wu et al., 2012) to confirm the absence of a dense network of lamellipodial actin in Arpc2−/− cells with F-actin organized instead into parallel bundles of actin within filopodial protrusions (Fig. 2B). Although the organization of F-actin is strikingly different in these cells, we tested whether the balance of F- vs G-actin was affected by the loss of Arp2/3 complex.
Figure 2. Comparison of actin structure and dynamics in WT and Arpc2−/− cells.
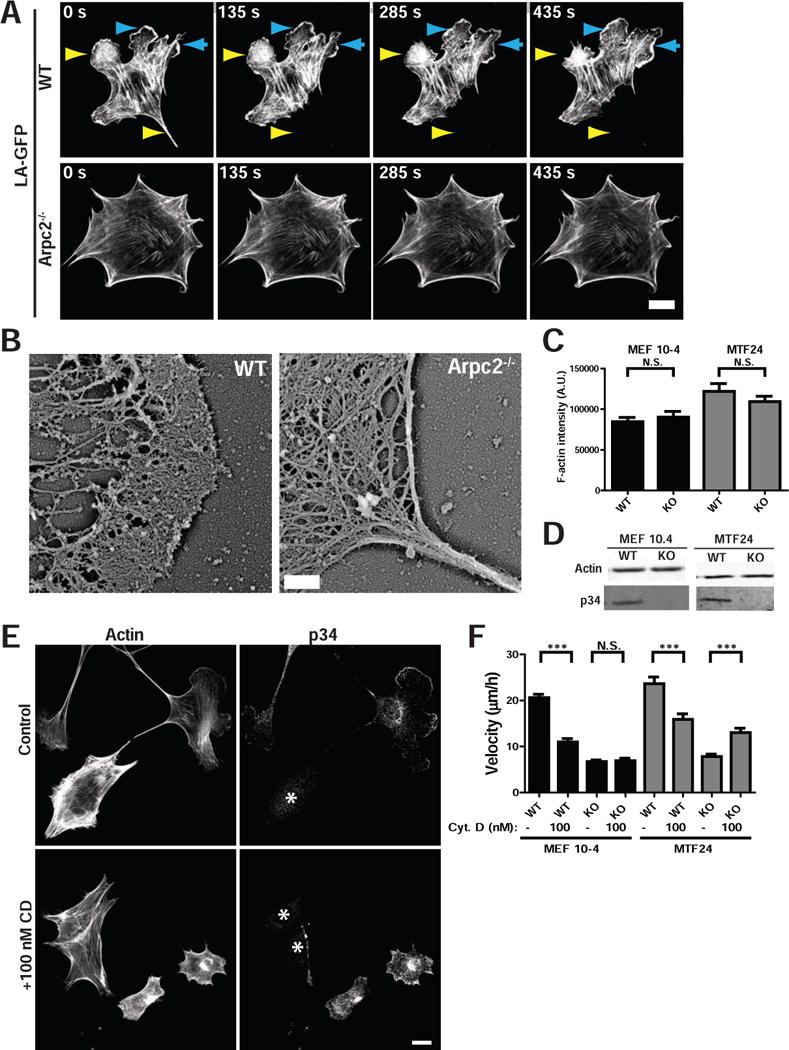
A) Still frames from live cell imaging of MEF 10-4 WT and Arpc2−/− cells stably transfected with the live cell actin probe LifeAct (LA)-GFP showing dynamic F-actin behavior in each cell type. Cyan arrowheads denote protrusion, yellow arrowheads denote retraction; scale bar = 20 microns. B) Cryo-shadowing EM of F-actin networks in MTF24 WT and Arpc2−/− cellular protrusions; scale bar = 500 nm. C) Integrated pixel density of phalloidin staining in fixed WT and KO cells from both lines plotted as average F-actin intensity/cell. N = 100 cells per condition; error bars represent standard error of the mean; N.S. = Not Significant. D) Blots of whole cell lysates loaded by cell equivalents for both lines. E) Staining of MTF24 WT and Arpc2−/− fibroblasts in mixed culture (KO cells marked with *) after addition of 100 nM cytochalasin D (CD) for 2h; scale bar = 20 microns. F) Random migration velocity of MEF 10-4 (black bars) and MTF24 (grey bars) WT and KO control (−) cells or cells treated with 100 nM CD (+); N = at least 30 cells per condition; error bars represent standard error of the mean; ***p-value ≤ 0.0001. See also Figure S1.
Total F-actin levels were analyzed with fluorescent phalloidin in fixed cells plated in mixed populations (i.e. WT and Arpc2−/− cells plated side by side; Fig. S1C). Low magnification epifluorescent imaging captured fluorescent signal from all focal planes, and integrated pixel density of phalloidin fluorescence was used to calculate relative F-actin content. Whole cell lysates of matched cell numbers were prepared to analyze total actin levels via immunoblot. WT and Arpc2−/− cells had similar levels of total actin and, surprisingly, similar levels of F-actin (Fig. 2C–D, S1C). Thus other polymerization mechanisms compensate for loss of Arp2/3 complex activity and maintain actin filament levels.
Both WT and Arpc2−/− F-actin was equally susceptible to 500 nM Latrunculin B (LatB) (Fig. S1D), but F-actin in Arpc2−/− lines was more resistant to 100 nM Cytochalasin D (CD) than WT cells when examined side by side (Fig. 2E; S1E). Furthermore, WT cell motility was significantly reduced by CD while Arpc2−/− cell motility was either not affected by the drug or enhanced, depending on the cell line (Fig. 2F; confirmed in Arp2/3 complex-depleted 2xKD cells in Fig. S1F). LatB predominately sequesters actin monomers (Spector et al., 1989) while 100 nM CD predominantly binds actin filament barbed ends rather than actin monomers (Cooper, 1987). Thus, actin polymerization in cells without Arp2/3 complex depends on actin assembly factors that are resistant to CD and generate unbranched actin filaments.
Actin assembly in Arpc2−/− cells is profilin-1 dependent
Based on the difference in susceptibility to the barbed end toxin CD, we used barbed end labeling assays to localize and quantify actin assembly in situ using labeled actin monomers in cells with or without Arp2/3 complex (Symons and Mitchison, 1991). To our surprise, monomer incorporation in both Arpc2−/− and Arp2/3 complex-depleted cells (2xKD) was reduced compared to control cells (Fig. 3A–B, S2A). Several non-Arp2/3 complex based actin assembly pathways preferentially utilize profilin-actin complexes (Hansen and Mullins, 2010; Romero et al., 2004) and transcriptome profiling indicates that profilin-1 is by far the dominant profilin isoform expressed in fibroblasts (>100 fold at the mRNA level than profilin-2) (Wu et al., 2013). Therefore, we repeated the barbed end incorporation with a mixture of labeled actin and purified profilin-1, and found that the presence of profilin rescued actin monomer incorporation in Arpc2−/− and 2xKD cells (Fig. 3A–B, S2A). Interestingly, barbed end labeling of WT cells was reduced overall (Fig. 3B) as well as being significantly reduced at the leading edge (Fig. 3C) in the presence of profilin-1. Based on these data, we postulated that Arpc2−/− cells would be more sensitive to the loss of profilin-1 than WT cells.
Figure 3. Actin assembly in Arpc2−/− cells is highly dependent upon profilin.
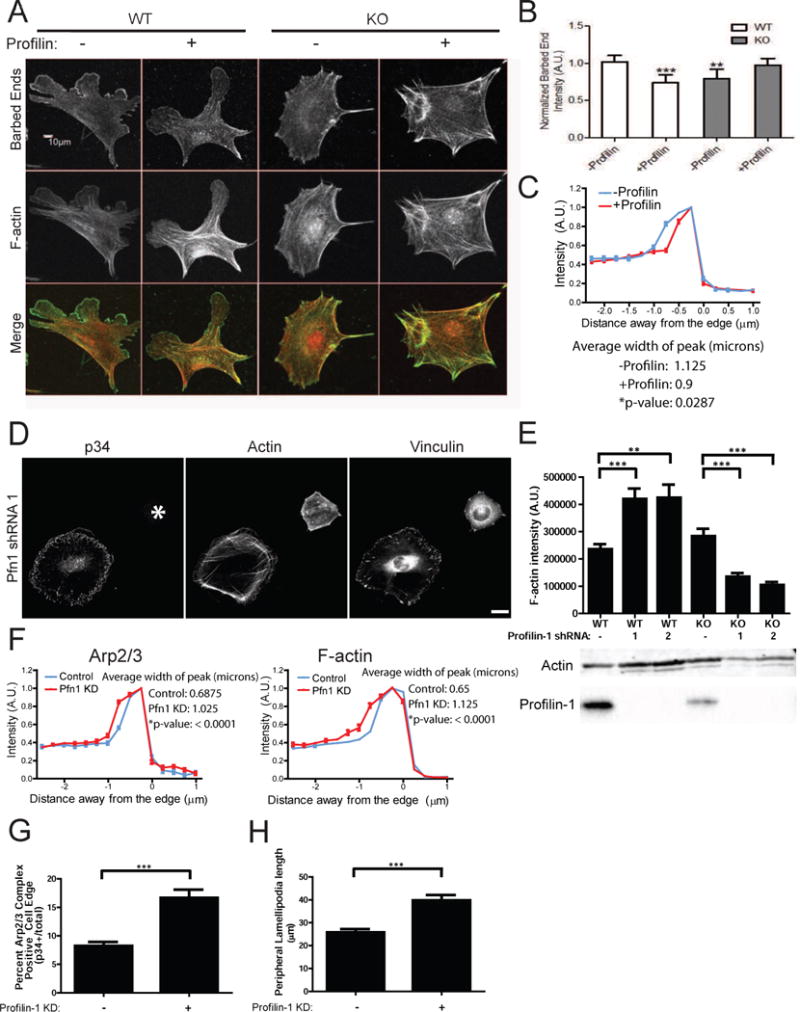
A) Barbed end assay relating the distribution of labeled barbed ends to total F-actin in MEF 10-4 WT or KO cells in the absence (−) or presence (+) of profilin; scale bar = 10 microns. B) Quantification of barbed end staining. Barbed end fluorescence intensity normalized to F-actin, with each condition plotted relative to control cells (WT minus profilin). **p-value = 0.0036, ***p-value = 0.0001. C) Barbed end distribution at the periphery of WT MEF 10-4 cells in the presence (+) or absence (−) of profilin in barbed end assay (cell edge = 0, negative values = intracellular distance from edge). Plotted as mean ± SEM. The mean width of the peak intensity was also quantified, analyzed by T-test and is presented numerically alongside the graph. D) Staining of MEF 10-4 WT and Arpc2−/− Profilin-1 KD fibroblasts (Pfn1 KD) in mixed culture (KO cell marked with *); scale bar = 20 microns. E) Integrated pixel density of phalloidin staining in fixed MEF 10-4 WT and KO cells ± Pfn1 KD plotted as average F-actin intensity/cell, with SEM. N = 50 cells per condition, ***p-value < 0.0001, **p-value = 0.0003. Blots of whole cell lysate matched by cell number directly below. F) Distribution of p34 and F-actin at the periphery of control or Pfn1 KD MEF 10-4 cells (cell edge = 0, negative values = intracellular distance from edge). Plotted as mean ± SEM. The mean width of the peak intensity was also quantified, analyzed by T-test and is presented numerically alongside the graph. G) Comparison of Arp2/3 positive edge in MEF 10-4 control or Pfn1 KD cells. High Arp2/3 complex signal in a narrow band along the perimeter was detected and divided by total cell perimeter to yield Arp2/3 complex enriched edge, plotted as average percent Arp2/3 complex positive edge with SEM. N = 29 for WT, 24 for WT Pfn1 KD cells, ***p < 0.0001. H) Peripheral lamellipodia length. The length of p34 positive edge was determined by outlining the periphery of each protrusion in ImageJ to yield the peripheral length in microns. N = at least 128 lamellipodia. ***p < 0.0001. See also Figure S2, S3.
Consistent with our hypothesis, depletion of profilin-1 in Arpc2−/− cells (via two distinct shRNAs) leads to decreased F-actin levels (Fig. 3D,E; S2B–D). These Arpc2−/−, profilin-depleted cells have a severely compromised ability to spread and highly disorganized F-actin, with a decrease in the appearance and length of filopodia (Fig. 3D; S2C,E). These data confirm that compensatory actin assembly in Arpc2−/− cells requires profilin-bound actin monomers.
Profilin-1 inhibits Arp2/3 complex function
Based on our in situ cell staining results with profilin-1, we hypothesized that its depletion would lead to cellular effects consistent with enhanced Arp2/3 complex activity. Indeed, depletion of profilin-1 in cells with intact Arp2/3 complex (WT) leads to strikingly different effects than depletion in Arpc2−/− cells. In WT cells, profilin-depletion led to increased levels of F-actin (Fig. 3E, S2D) and alterations in cell morphology and actin organization (Fig. 3D, S2C). WT cells depleted of profilin-1 have broad lamellipodia that contain abundant Arp2/3 complex (Fig. 3D, S2C). These cells have increased actin arcs, bundled actin structures that run parallel to the cell edge that are known to be Arp2/3 complex derived (Fig. 3D, S2C) (Hotulainen and Lappalainen, 2006). Unlike Arpc2−/− cells depleted of profilin-1, these cells had increased spread area relative to controls (Fig. S2E). To quantify the changes in Arp2/3 complex distribution in the WT (Arp2/3+); profilin-depleted cells, we used previously established edge mapping techniques (Cai et al., 2007). The width of both the Arp2/3 complex and F-actin bands at the cell periphery is significantly increased in profilin-1 depleted cells (Fig. 3F). Furthermore, a greater fraction of the cell edge is positive for Arp2/3 complex and the average peripheral length of lamellipodia is greater in profilin-1 depleted cells compared to WT controls (Fig. 3G,H, S3A,B). Profilin-1 depletion enhances Arp2/3 complex localization to the leading edge, broadens the lamellipodial F-actin band and increases lamellipodia size, suggesting enhanced branch nucleation. These findings led us to directly test whether profilin-1 inhibits Arp2/3 complex branch nucleation.
We used in vitro TIRF microscopy-based actin polymerization assays to re-visit the observation that profilin inhibits Arp2/3 complex nucleation in vitro (Machesky et al., 1999). We found that profilin-1 inhibits Arp2/3 complex branch generation ~6-fold (Fig. 4A,B). We utilized profilin-1 mutants to further probe the underlying mechanism. The inhibitory activity of profilin-1 is dependent on its ability to bind to actin monomers, as the G-actin binding mutant (R88E) has no inhibitory effect. Conversely, the poly-proline binding mutant (Y6D) inhibits Arp2/3 complex-dependent branch formation comparably to WT profilin-1.
Figure 4. Profilin inhibits Arp2/3 complex actin nucleation, disrupts Arp2/3 complex leading edge localization and impedes lamellipodia generation.
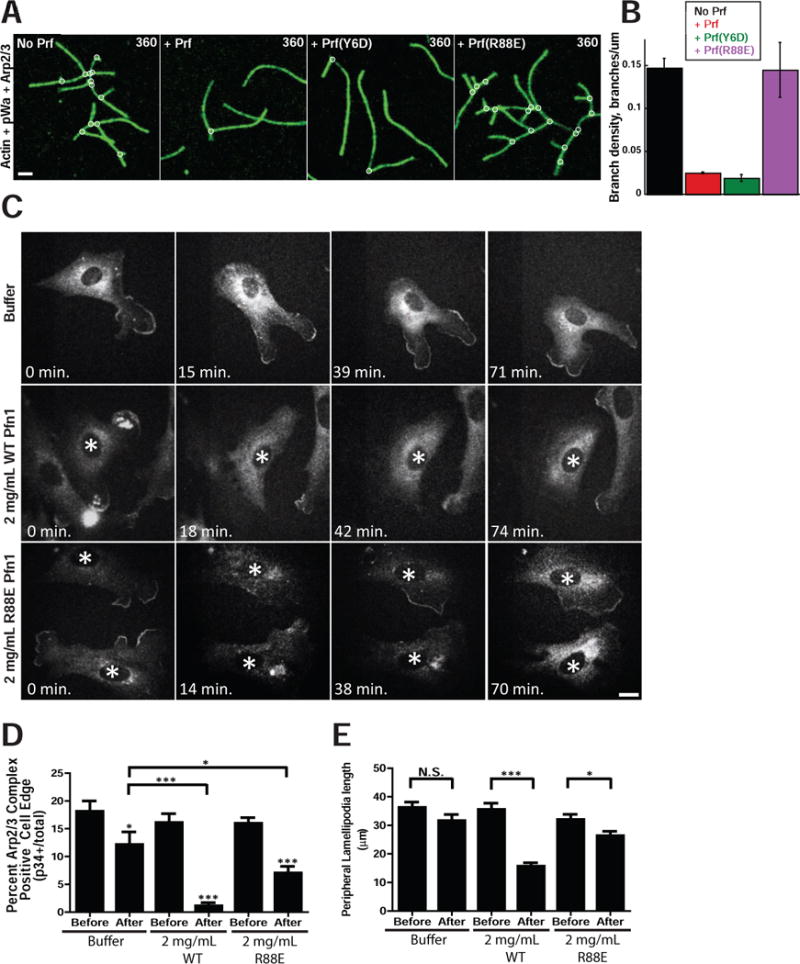
A) Time-lapse TIRF microscopy of 1.5 μM Oregon green-labeled actin polymerized in the presence of 40 nM Arp2/3 complex, 150 nM pWA in the absence (No Prf) or presence of either 5 μM WT (+ Prf), Y6D, or R88E hProfilin-1. Scale bar = 2 microns. B) Effect of WT, Y6D or R88E hProfilin-1 on branch density, quantified from time-lapse TIRF experiments in A. Plotted as mean plus SEM. C) Representative images of p34-GFP localization before (0 min) and at various times after microinjection of buffer, 2 mg/mL WT hProfilin-1, or 2 mg/mL R88E hProfilin-1. Scale bar = 20 microns. Asterisks denote microinjected cells in images with multiple cells. D) Percent of p34-GFP positive edge. Quantified as positive edge/total edge × 100% based on measurements done by hand in ImageJ. Measurements were made before and directly after microinjection for each condition, plotted as mean with SEM. N = at least 31 cells per condition. ***p-value < 0.0001, *p-value < 0.05; p-values for each post-injection mean are to pre-injected cells of same condition, unless explicitly noted otherwise. E) Peripheral lamellipodia length. The length of p34-GFP positive edge was determined by outlining the periphery of each protrusion in ImageJ to yield the peripheral length in microns, plotted as mean with SEM. N = at least 66 lamellipodia in pre-injection for each condition; N = 67 for buffer post-injection, 58 for R88E post-injection and 11 for WT post-injection. ***p-value = 0.0006, *p-value = 0.0426. See also Figure S3.
To test the inhibitory effect of profilin-1 on Arp2/3 complex activity in cells, we performed the counter-experiment to our profilin-depletion studies by elevating profilin levels. We were unable to achieve satisfactory genetic profilin-1 overexpression in our lines, so instead we turned to profilin-1 microinjection to directly test for an inhibitory effect on Arp2/3 complex function. Precedence exists in the literature for disruption of lamellipodia after profilin microinjection (Cao et al., 1992), but these observations were limited to morphological analysis and did not include an investigation of the molecular processes involved. For our experiments we used our Arpc2−/− p34-GFP rescue line which also stably expressed LifeAct-RFP (LA-RFP) to monitor both Arp2/3 complex and F-actin in the same cell before and after microinjection of ~0.5pL of 2 mg/mL (133 μM) human profilin-1. Although the precise volume injected and the starting volume of the cell vary by ~2-fold, this corresponds approximately to a step increase in cytoplasmic profilin-1 concentration from 29 μM to 39 μM or 36% (see Experimental Procedures). Cy5-dextran was used in the mixture to mark injected cells (Fig. S3C). Pre-injection images (0 min) were taken immediately before microinjection, after which the saved stage positions were revisited and imaged cells were microinjected. The exact time that postinjection images were acquired varied slightly from experiment to experiment based on the length of time required for microinjection. Buffer alone-injected cells typically maintained lamellipodia and Arp2/3 complex localization at the periphery, whereas microinjection of 2 mg/mL WT profilin led to acute and persistent disruption of Arp2/3 complex edge localization and the disappearance of lamellipodia (Fig. 4C). To test the requirement for G-actin binding by profilin in this response, we microinjected R88E mutant profilin-1, which failed to inhibit the Arp2/3 complex in our TIRF experiments. With this mutant profilin-1, we observed an intermediate phenotype with cells retaining some peripheral Arp2/3 localization and morphological lamellipodia, albeit to a reduced extent relative to buffer alone injections (Fig. 4C). Unfortunately, the Y6D mutant profilin-1 proved unsuitable for microinjection due to needle clogging. To quantify the effects of profilin microinjection, we measured the percent of p34 positive edge and the average length of lamellipodia before and after microinjection, and measured changes consistent with our visual impressions (Fig. 4D,E). These data suggest that profilin antagonizes the Arp2/3 complex and that its ability to bind G-actin plays a significant role in this activity.
In addition to the Arp2/3 complex, we concomitantly observed the F-actin network via LA-RFP labelling in these cells. At early time points after WT profilin microinjection, the F-actin network was disrupted in protrusive lamellipodia as well as in stress fibers, while cells responded later by re-polymerizing actin into stress fibers but not lamellipodia (Fig. 5A, quantified as shown in Fig. S3D). Neither buffer nor R88E-injected cells demonstrated the same level of F-actin disruption (Fig. 5A), suggesting that actin monomer binding was important for WT profilin’s acute effect on F-actin. We tested whether Arpc2−/− cells expressing LA-RFP were similarly affected by WT profilin. In these cells, we saw no significant disruption of F-actin in Arpc2−/− cells regardless of microinjection condition (Fig. 5B), aside from a modest effect with R88E profilin reflecting a possible dominant negative effect toward endogenous profilin-dependent pathways. The Arpc2−/− cells generate no F-actin via the Arp2/3 complex, are highly dependent upon profilin for maintaining F-actin homeostasis (Fig. 3E; Fig. S2D) and are much less dynamic than WT cells (Fig. 1F; Fig. 2A), leading to lower levels of filament turnover and less free G-actin available for profilin binding. These factors together likely contribute to Arpc2−/− resistance to exogenous profilin.
Figure 5. Profilin affects overall F-actin structure in cells with functional Arp2/3 complex.
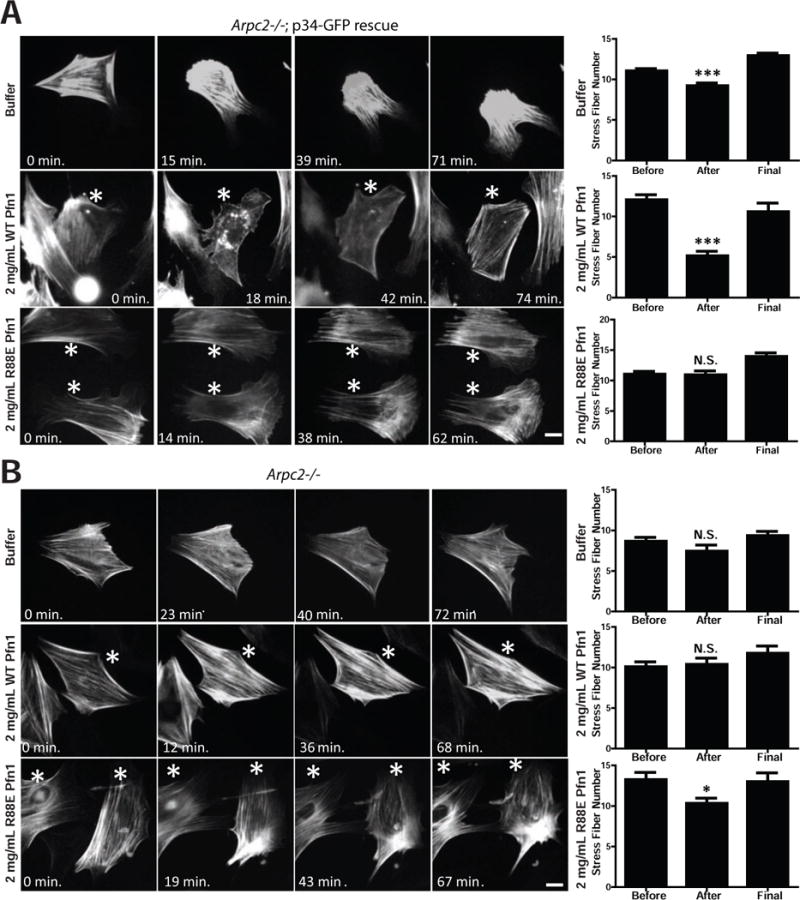
A) Representative images of Lifeact-RFP labeling in p34 knockout-rescue cells (Arpc2−/−; p34-GFP rescue) before (0 min.) and at various times after microinjection of buffer, 2 mg/mL WT hProfilin-1, or 2 mg/mL R88E hProfilin-1. Scale bar = 20 microns. Asterisks denote microinjected cells in images with multiple cells. Right: Quantification of stress fiber number from images before, after and at the end (‘final’) of the post-injection time course. Counted as number of stress fibers across a line drawn perpendicular to the predominant stress fiber orientation, plotted as mean with SEM; N = 198 measurements from 66 buffer-injected cells, 78 measurements from 26 WT hProfilin-1 injected cells, 60 measurements from 20 R88E hProfilin-1 injected cells. ***p-value < 0.0001. B) Representative images of Lifeact-RFP labeling in Arpc2−/− cells before (0 min.) and at various times after microinjection of buffer, 2 mg/mL WT hProfilin-1, or 2 mg/mL R88E hProfilin-1. Scale bar = 20 microns. Asterisks denote microinjected cells in images with multiple cells. Right: Quantification of stress fiber number, plotted as mean with SEM; N = 36 measurements from 12 buffer-injected cells, 60 measurements from 20 WT hProfilin-1 injected cells, or 42 measurements from 14 R88E hProfilin-1 injected cells. *p-value = 0.0103. See also Figure S3.
Ena/VASP proteins, but not formins, maintain Arpc2−/− F-actin levels
We assessed the contribution of formins to the F-actin network in WT and Arpc2−/− cells. Formins are profilin-dependent actin assembly factors that nucleate actin filaments, and increase the F-actin barbed end elongation rate up to 10-fold (Kovar et al., 2006). The mammalian formin family contains isoforms encoded by fifteen genes, of which eleven are expressed in mouse fibroblasts (Wu et al., 2013). Since all formins contain an FH2 domain, we used the recently characterized formin inhibitor SMIFH2 that directly inhibits the FH2 domain (Rizvi et al., 2009), and should act as a pan-formin inhibitor though the drug’s efficacy toward every member of the formin family has not been carefully tested. WT cell motility is decreased by SMIFH2 while Arpc2−/− cell motility is unaffected or slightly enhanced by the drug, depending on the cell line (Fig. 6A). F-actin levels are not significantly altered in WT cells treated with 15 μM SMIFH2 compared to untreated controls (Fig. 6B); and cells retain lamellipodia and stress fiber staining in the presence of the drug (Fig. S4A–B). Surprisingly, Arpc2−/− cells are largely resistant to the effects of SMIFH2 as measured by F-actin levels and F-actin organization does not significantly differ from untreated Arpc2−/− cells (Fig. 6B, Fig. S4A–B). Together, these data indicate that formins clearly contribute to generating F-actin for cell motility in cells with Arp2/3 complex, but may not be as important in the absence of Arp2/3 complex, at least in mammalian cells.
Figure 6. Formin and Ena/VASP differentially affect actin homeostasis depending on cellular Arp2/3 complex status.
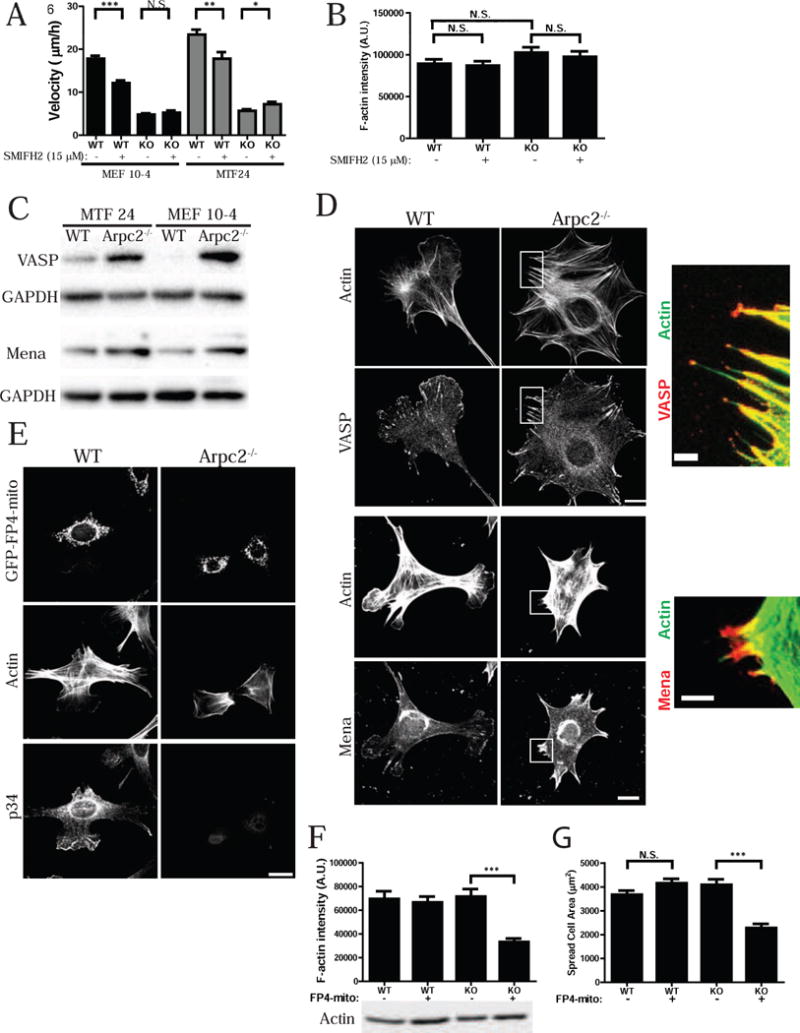
A) Random migration velocity of MEF 10-4 (black bars) and MTF24 (grey bars) WT and KO control (−) cells or cells treated with 15 μM SMIFH2 (+), plotted as mean and SEM; N = at least 48 cells per condition. ***p-value < 0.0001, **p-value = 0.0040, *p-value = 0.0219, N.S. = Not Significant. B) Integrated pixel density of phalloidin staining in fixed MEF 10-4 WT and KO cells in the presence or absence of 15 μM SMIFH2, plotted as average F-actin intensity/cell, with SEM. N = at least 120 cells per condition. C) Blots of WT and Arpc2−/− cells. D) Staining of MEF 10-4 WT and Arpc2−/− fibroblasts; scale bar = 20 microns. Boxed regions are magnified and merged with VASP or Mena in red and F-actin in green; scale bar of magnified image is 5 microns. E) Staining of MEF 10-4 WT and Arpc2−/− cells expressing GFP-FP4-mito; scale bar = 20 microns. F) Integrated pixel density of phalloidin staining in fixed MEF 10-4 WT and KO cells, or WT/KO cells stably expressing GFP-FP4-mito (FP4-mito +), plotted as average F-actin intensity/cell, with standard error of the mean. N = at least 38 cells per condition; ***p-value < 0.0001. Blots of whole cell lysate matched by cell number directly below. G) Spread cell area in sq. microns of MEF 10-4 WT, WT FP4-mito, KO and KO FP4-mito cells plotted as average area/cell with SEM. N = at least 140 cells per condition; ***p-value < 0.0001. See also Figures S4–S6.
Ena/VASP proteins are profilin-binding, actin assembly proteins that bind to the barbed ends of actin filaments, block capping protein binding and enhance barbed end growth (Bear et al., 2002; Hansen and Mullins, 2010; Winkelman et al., 2014). Profilin-1 hemizygous mice bred into a Mena−/− genetic background die in utero due to severe neurulation defects, indicating dosage-dependent genetic interactions (Lanier et al., 1999). A puzzling aspect of Ena/VASP function is its inhibition of Arp2/3 complex branching in favor of filament elongation or ‘anti-branching’ (Bear et al., 2002; Skoble et al., 2001). TIRF microscopy assays using mammalian proteins reveal that VASP alone is not sufficient to block Arp2/3 complex nucleation (Fig. S5A). However, inclusion of profilin in the reaction led to a significant decrease in actin branches (Fig. S5A, B). Control experiments reveal that VASP was active in these assay conditions as it was able to enhance the elongation rate of actin filaments ~2-fold (Fig. S5C). These data argue that the ‘anti-branching’ effect observed in previous studies may be due to preferential usage of profilin-actin by Ena/VASP proteins to elongate rather than nucleate actin filaments.
We tested the role that Ena/VASP proteins play in the actin assembly occurring in the Arpc2−/− cells. Interestingly, both VASP and Mena are overexpressed in our Arpc2−/− cell lines relative to their WT counterparts (Fig. 6C; 4-fold for VASP and 1.74-fold for Mena on average). The staining pattern of VASP and Mena in WT cells is consistent with the known localization of Ena/VASP proteins to lamellipodia, focal adhesions, and filopodial tips (Fig. 6D). In Arpc2−/− cells, VASP and Mena localize to filopodial tips and to focal adhesions that form at the base of these filopodia (Fig. 6D, and insets), which is similar to the barbed end staining pattern in Arpc2−/− cells incubated with profilin (Fig. 3A). Depletion of profilin-1 does not affect VASP localization in cells with intact Arp2/3 complex, however in the absence of Arp2/3 complex and depletion of profilin-1, VASP is largely restricted to small focal adhesions at the periphery (Fig. S5D). To functionally address the role of Ena/VASP proteins in Arp2/3 complex-independent actin assembly and homeostasis, we stably transduced WT or Arpc2−/− cells with a previously characterized dominant interfering construct (termed GFP-FP4-mito, or FP4-mito) that sequesters all Ena/VASP family proteins to mitochondria (Fig. S5E) (Bear et al., 2000). Disruption of Ena/VASP activity in WT fibroblasts (via FP4-mito expression or genetic null lines) alters actin organization in lamellipodia and enhances cell motility, but does not block lamellipodia generation (Bear et al., 2000). As expected, total F-actin levels are unchanged in WT (Arp2/3+) FP4-mito cells (Fig. 6E–F, Fig. S6A–C). However, the combination of Arp2/3 complex deficiency and Ena/VASP sequestration significantly decreases spread cell area and reduces cellular protrusions (i.e. bundled filopodia of Arpc2−/−cells) (Fig. 6E,G Fig. S6A,D), as well as overall F-actin levels (Fig. 6F, Fig. S6C). Thus, compromising Ena/VASP activity in Arpc2−/− cells phenocopies key effects of profilin-1 depletion in the same cells. Together, these data suggest mammalian cells maintain F-actin homeostasis in the absence of Arp2/3 complex by activating profilin-1 and Ena/VASP-dependent actin assembly.
DISCUSSION
In this work, we utilized new cell lines isolated from a conditional knockout mouse in the Arpc2 gene encoding the p34 subunit of the Arp2/3 complex to interrogate F-actin dynamics, organization, and homeostasis in the presence or absence of the Arp2/3 complex. We found, as in previous studies (Suraneni et al., 2012; Wu et al., 2012), that cells lacking the Arp2/3 complex possessed filopodial protrusions containing bundled actin filaments, lacked lamellipodia, migrated slowly and had less dynamic actin networks than control cells. Surprisingly, we found that Arpc2−/− cells could compensate for loss of Arp2/3 complex and maintain overall F-actin levels similar to control cells. In addition, Arpc2−/− cells are resistant to low doses of cytochalasin D and require profilin-1 and Ena/VASP, but not formins, to maintain proper levels of F-actin. Finally, consistent with previous biochemical studies, our data reveal an important role for profilin-1 in inhibiting Arp2/3 complex function in mammalian cells. Together, these observations suggest that profilin-1 plays a major gatekeeper role in actin assembly by directing actin monomers towards formin and Ena/VASP pathways and away from Arp2/3-based actin assembly.
Profilin-actin is thought to be the major form of monomeric actin in cells (Kaiser et al., 1999). Our data identify a critical role for profilin-1 in maintaining F-actin levels in the absence of Arp2/3 complex. Formins and Ena/VASP associate with filament barbed ends and act as actin polymerases that elongate actin filaments while protecting barbed ends from capping (Bear et al., 2002; Breitsprecher and Goode, 2013; Hansen and Mullins, 2010; Winkelman et al., 2014). Our experiments yielded the surprising finding that formins did not appear to contribute significantly to global F-actin maintenance in Arpc2−/− cells. The fact that formin inhibition did not affect the F-actin organization and content of the Arpc2−/− cells is surprising given the key role that formins play in compensating for the loss of Arp2/3 complex activity in fission yeast (Burke et al., 2014). However, yeast cells do not have proteins homologous to Ena/VASP and recent data in insect cells indicate that Ena/VASP proteins and formins do not merely substitute for each other functionally, but have a more complex interaction than previously suspected (Bilancia et al., 2014). Our data in mammalian cells indicate that Ena/VASP proteins play a major role in compensating for the loss of Arp2/3 complex when it comes to maintaining overall F-actin levels. It will be important to revisit the issue of the relative contribution of Ena/VASP and formins to both specific actin structures, as well as overall F-actin levels as reagents to perturb formins in mammalian systems continue to improve.
In addition to being described as anti-capping factors, Ena/VASP proteins have also been described as ‘anti-branching’ factors (Bear and Gertler, 2009). FP4-mito expression, which blocks Ena/VASP function, leads to increased actin filament branching and decreased filament length at the leading edge, whereas targeting of Ena/VASP proteins to the membrane gives rise to a converse phenotype, implying Arp2/3 complex inhibition by Ena/VASP (Bear et al., 2002). Additionally, ActA-induced Arp2/3 complex nucleation is inhibited in the presence of VASP (Skoble et al., 2001). Two possible mechanisms for the proposed ‘anti-branching’ activity of Ena/VASP are: a) direct inhibition of Arp2/3 complex by Ena/VASP and b) indirect inhibition via competition for actin monomers. We find in TIRF microscopy assays that the presence of VASP alone does not affect Arp2/3 complex nucleation, arguing against direct inhibition. Ena/VASP’s ‘anti-branching’ activity in cells may relate to its recruitment of profilin-actin that cannot be used by the Arp2/3 complex for branch nucleation. It is already known that profilin enhances but is not required for Ena/VASP’s anti-capping activity (Barzik et al., 2005). Future experiments will be required to clarify the relationship between Ena/VASP, profilin-1 and the Arp2/3 complex and whether ‘anti-branching’ is a distinct mechanism from anti-capping. However, this matter may be even more complicated given the recent finding that VASP can bind to and positively regulate the WAVE Regulatory Complex via Abi-1 and, therefore, Arp2/3 complex activity (Chen et al., 2014).
One of the most important findings in this work is the inhibitory role that profilin-1 plays in Arp2/3-dependent actin assembly pathways in cells. Decreasing profilin-1 (via RNAi) and increasing profilin-1 (via microinjection) levels led to reciprocal changes in Arp2/3 complex activity, as evidenced by leading edge incorporation and lamellipodia formation. In fact, the relationship between the Arp2/3 complex and profilin-1 appears to be precisely balanced, a balance that can be shifted even by modest changes in their relative levels. While these findings establish an inhibitory activity for profilin-1 toward the Arp2/3 complex in mammalian cells, prior biochemical studies showed profilin inhibition of Arp2/3 complex nucleation in vitro (Machesky et al., 1999; Rodal et al., 2003). Direct observation of Arp2/3 complex branch formation in vitro using purified mammalian proteins yielded the important observation that profilin decreased Arp2/3 complex branching while increasing filament length (Blanchoin et al., 2000), similar to experiments reported in our study as well as in the accompanying paper from Suarez et al.
Our data confirms and extends these in vitro findings, but there are several mechanistic possibilities that are worth considering about the inhibitory effect of profilin towards Arp2/3 complex, which could be either direct or indirect. The Arp2/3 complex was initially discovered and characterized as a profilin-binding complex from Acanthamoeba (Machesky et al., 1994; Mullins et al., 1998). Though the affinity of the interaction is relatively low (KD = 7 μM) (Mullins et al., 1998), a direct protein-protein interaction could allosterically inhibit Arp2/3 complex activity or block recruitment of NPFs to the complex. The structure/function and mutagenesis experiments required to test this interesting possibility conclusively lie outside the scope of the present effort.
Both our work and the accompanying paper from Suarez and colleagues support the idea of indirect inhibition of Arp2/3 complex by profilin through competition for actin monomers. Both groups show that profilin mutants (R88E or K81E, respectively) with greatly reduced affinity for G-actin do not inhibit Arp2/3 complex branch generation in vitro. Our microinjection studies also show reduced ability of this mutant to inhibit Arp2/3-containing lamellipodia, although the effect is less clear-cut than the in vitro studies. One possible explanation for this partial effect of the R88E mutant in vivo is that this mutant may act as a dominant negative mutant for the formin and Ena/VASP pathways, which are known to contribute to lamellipodia formation (Yang et al., 2007). Furthermore, Suarez et al directly demonstrate competition between fission yeast profilin and WASP (VCA) for actin monomers in vitro. In cells, according to this model, profilin-actin preferentially delivers actin monomers to Ena/VASP and formins, whereas NPFs deliver actin monomers to the Arp2/3 complex for nucleation of daughter filaments. It is known that the affinity of VCA and profilin for G-actin is similar (Marchand et al., 2001). Despite the high cellular concentrations of profilin (Kaiser et al., 1999; Tseng et al., 1984), it is also known that nanomolar VCA/NPF concentrations can maintain lower but observable Arp2/3 complex activity in the presence of saturating (μM) profilin concentrations (Blanchoin et al., 2000; Machesky et al., 1999; Rodal et al., 2003). In future studies, it will be interesting to test whether known profilin regulatory mechanisms such as PIP2 binding and/or tyrosine phosphorylation (Ding et al., 2012; Fan et al., 2012) can tip the balance between Arp2/3-dependent and –independent actin assembly pathways. Our results suggest that disruption of any aspect of actin regulation will lead to a resulting compensation (or defect) that reveals functional connections and interplay occurring in normal cells. Physiological cues and cellular context will certainly help determine the regulatory interplay between these pathways during cellular events such as vesicular trafficking, migration and adhesion.
EXPERIMENTAL PROCEDURES
A complete description of all experimental procedures can be found in the supplementary materials.
Mouse strains
C57BL/6 mice with conditional Arpc2 alleles were ordered from the EUCOMM consortium, mated to FLP recombinase mice, and were then crossed with previously established Ink4a/Arf−/− mice (in a mixed genetic background; mice were null for both Ink4a and Arf) (Serrano et al., 1996) and homozygosed to generate p34FL/FL; Ink4a/Arf−/− mice in a mixed strain background. All mouse experiments were reviewed and approved by the Institutional Animal Care and Use Committee and were provided with food and water ad libitum.
F-actin and total actin quantitation
After processing coverslips according to standard lab protocol (reported in depth in supplemental experimental procedures), images were taken on an Olympus IX81 microscope with a 0.30 N.A. 10X objective and an iXon+ front-illuminated EMCCD camera (Andor Technology) and controlled by Metamorph software (Molecular Devices). Images of fluorescent phalloidin were used to determine F-actin content. Briefly, image files were imported into ImageJ and background was subtracted by the program. Cells were carefully outlined by hand and integrated pixel density was measured on a per cell basis to generate average F-actin content per cell. Measurements of the area of outlined cells were generated, and subsequently reported as average spread cell area. As cells were plated for F-actin visualization, a subset of trypsinized cells of the same population (matched at 25,000 cells per condition) were spun down at 1,000xg for 3 minutes at room temperature and whole cell lysates were prepared from pelleted cells. Ten μL of WCL was used per sample on SDS-PAGE gels. Actin and p34 signal from WCL was simultaneously detected with Li-Cor fluorescent antibodies on an Odyssey detection system (Li-Cor).
Single Molecule TIRF
Full experimental conditions can be found in the supplementary experimental procedures. TIRF microscopy images were collected at 5s intervals with an iXon EMCCD camera (Andor Technology) using an Olympus IX-71 microscope equipped with through-the-objective TIRFM illumination. Elongation rates were measured using imageJ, branch densities were calculated by counting the number of branch points and dividing by the total filament length.
Microinjection
Cells were plated overnight in low CO2 adjustment media on Delta-T dishes (Bioptechs) and sealed with parafilm. Cells were removed the next morning and placed into a heated insert for imaging on an Olympus IX81 microscope with a 1.05 N.A. 30X silicon oil objective and an iXon+ front-illuminated EMCCD camera (Andor Technology) and controlled by Metamorph software (Molecular Devices). Several cells were imaged in the GFP, RFP and Cy5 channels prior to microinjection and stage positions were saved. Saved positions were then revisited for microinjection. Microinjection with Femtotip needles was accomplished with the FemtoJet injection controller and InjectMan NI 2 system (Eppendorf). Needles were loaded with 2 mg/mL (133 μM) WT or R88E human profilin in profilin buffer (20 mM Tris, pH 7.5; 150 mM KCl; 0.2 mM DTT) plus 0.67 μg/mL Cy5-conjugated dextran (Sigma), or buffer and dextran alone. Using a standard curve of purified His-profilin-1 (Cytoskeleton), we determined the average level of profilin-1 in 2,500 cells to be ~5 ng. The corresponding level per cell is 2 pg or 133.33 amol profilin-1. We imaged cells in suspension and found them to have an average cytoplasmic volume of 4.6 pL. Based on these values, we calculated a cellular profilin-1 concentration of ~29 μM, in line with published values in mammalian cells that range from 8.4–50 μM (Finkel et al., 1994; Goldschmidt-Clermont et al., 1991). Though microinjected volume likely differed from cell to cell, the manufacturer’s data regarding their femtotip needle reported an injection range of 0.1–0.5 pL. As 0.5 pL is ~10% of cell volume we find it reasonable as an upper bound for injection volume. Given this and the needle concentration of profilin, we could deliver 1 pg (66.67 amol) of profilin. The post-injection concentration of profilin-1 would be 39 μM, an immediate 36% increase in profilin-1 levels. After microinjection cells were allowed to re-equilibrate until Z-drift was minimized. Cells at each saved position were imaged every 8 minutes on GFP, RFP and Cy5 channels.
Image analysis
Actin and Arp2/3 complex edge detection
The ImageJ Macro Edgeratio (http://www.unc.edu/~cail/code/EdgeRatio.txt) (Cai et al., 2007) was used to measure the distribution of F-actin and p34Arc at the cell edge. A full description of this analysis method can be found in the supplemental experimental procedures.
In situ actin assembly quantification
F-actin and Alexa 488-Actin signal was background subtracted, cells were outlined, and fluorescence of both channels was quantified in ImageJ. Barbed end intensity was normalized to total F-actin. Averages of normalized values are reported relative to control WT cells minus profilin.
Arp2/3 complex edge ratio
Confocal stacks of cells imaged on an Olympus FV1000 microscope at 40x were analyzed using MATLAB (MathWorks). Cells were automatically segmented from background via k-means clustering. High Arp2/3 complex signal (designated as intensity greater than 1.2 standard deviations above the mean signal for the whole cell) was analyzed within a 5 pixel band along the cell perimeter. Perimeter area covered by high Arp2/3 complex signal was divided by the total perimeter area of the cell to achieve the final fraction of Arp2/3 complex enriched edge.
Actin stress fiber quantification
Analysis was done similarly to already established approaches (Wei et al., 2011). A full description can be found in the supplemental experimental procedures.
Peripheral lamellipodia length
p34-positive lamellipodia length was analyzed in ImageJ by hand drawing a curved line on top of p34-positive lamellipodial regions. The length of the curved line in microns was measured using the ImageJ software.
Statistical analysis
All means are graphed with standard error of the mean, with the exception of barbed end quantification in which error bars represent 95% confidence intervals. Statistical significance was assessed using unpaired two-tailed t tests, with p-values < 0.05 being considered significant. All statistical tests of raw data were done in GraphPad Prism (GraphPad Software).
Supplementary Material
Supplemental Figure 1. Additional characterization of fibroblast cell lines: morphology, F-actin character, response to drug treatment. A) Staining of fixed MEF 10-4 WT and Arpc2−/− fibroblasts; scale bar = 20 microns. B) Still frames from live cell timecourse of MTF24 WT and Arpc2−/− cells stably transfected with the live cell actin probe lifeact (LA)-GFP showing dynamic F-actin behavior in each cell type. Cyan arrowheads depict areas of active protrusion; scale bar = 20 microns. C) Representative 10X images of phalloidin (Actin) and p34 used to generate F-actin integrated pixel density measurements from both lines, with WT and KO cells plated in mixed culture for side by side comparison (KO cells denoted by *); scale bar = 100 microns. D) Indirect immunofluorescence of fixed MEF 10-4 and MTF24 cells in mixed culture plus 500 nM Latrunculin B (KO cells denoted with *); scale bar = 20 microns. E) Staining of fixed MEF 10-4 WT and Arpc2−/− fibroblasts in mixed culture (KO cells marked with *) after addition of 100 nM cytochalasin D (CD) for two hours; scale bar = 20 microns. F) Random migration velocity of control (NS) IA32 cells or IA32 cells stably depleted of p34 and Arp2 (2xKD) in the presence or absence of CD. Means are plotted with 95% confidence intervals; N = at least 49 cells per condition, ****p-value < 0.0001. Relates to Figures 1 and 2.
Supplemental Figure 2. Additional characterization of profilin-1 activity in WT and Arp2/3-depleted lines. A) Barbed end assay relating the distribution of labeled barbed ends to total F-actin in control (NS) or Arp2/3 complex depleted (2xKD) IA32 cells in the absence (−) or presence (+) of profilin; scale bar = 10 microns. B) Representative 10X images of Pfn1 KD WT and Arpc2−/− cells in mixed culture stained for phalloidin (Actin) and p34 (KO cells denoted by *) used to generate F-actin integrated pixel density measurements from both lines; scale bar = 100 microns. C) Indirect immunofluorescence of fixed MTF24 WT and Arpc2−/− Profilin-1 KD fibroblasts; scale bar = 20 microns. D) Integrated pixel density of phalloidin staining in fixed MTF24 WT and KO cells ± Pfn1 KD plotted as average F-actin intensity/cell, with SEM. N = 50 cells per condition, ***p-value < 0.0001, **p-value < 0.001. Blots of whole cell lysate matched by cell number directly below. E) Spread cell area in sq. microns for MEF 10-4 (black bars) and MTF24 (grey bars) WT and KO cells ± Pfn1 KD, plotted as average area/cell, with SEM. N = 50 cells per condition. ****p-value < 0.0001, ***p-value < 0.0053, **p-value = 0.0124, *p-value = 0.0366. Relates to Figure 3.
Supplemental Figure 3. Image quantification and analysis approaches used for data generation. A) Automated p34 edge detection workflow. Parental image is imported into MATLAB and single cells are detected by the edge analysis program. Steps 2–4 are repeated for each cell detected in the initial analysis step. After step 4, the program calculates the ratio of high p34 edge compared to total cell edge. Scale bar = 20 microns. B) Images used for edge detection were also imported into ImageJ and peripheral lamellipodia length in microns was measured for each lamellipodia (outlined in red). Measurements for each example lamellipodia is reported to the right of the image. Scale bar = 20 microns. C) Example of Cy5-dextran signal in Arpc2−/−; p34-GFP rescue cell from Figs. 4C and 5A, before (0 min) and after microinjection of WT hProfilin-1 at indicated time points. Scale bar = 20 microns. D) LA-RFP signal in Arpc2−/−; p34-GFP cell. Scale bar = 20 microns. Individual images were background subtracted in ImageJ and straight lines were drawn through the cell perpendicular to the predominant stress fiber orientation (as shown by white line). When signal was detectable above background in ImageJ line traces (example to the right of image), independent peaks of LA-RFP signal were counted (S = stress fiber). Line trace reports Absolute Grey Value vs. distance across the line (in microns), with the section related to intracellular space marked. Relates to Figures 3, 4 and 5.
Supplemental Figure 4. Effect of SMIFH2 on cell morphology, actin organization and F-actin levels. A) Indirect immunofluorescence of fixed MEF 10-4 and MTF24 WT and Arpc2−/− cells plus or minus SMIFH2; scale bar = 20 microns. B) Representative 10X images of WT and Arpc2−/− cells + SMIFH2 in mixed culture stained for phalloidin (Actin) and p34 (KO cells denoted by *) used to generate F-actin integrated pixel density measurements from both lines; scale bar = 100 microns. Relates to Figure 6.
Supplemental Figure 5. Characterization of Ena/VASP interplay with the Arp2/3 complex and confirmation of FP4-mito approach. A) Time-lapse TIRF microscopy of 1.5 μM Oregon green-labeled actin polymerized in the presence of 50 nM Arp2/3 complex, 50 nM pWA, and 20 nM SNAP-549-hVASP without (top) or with (bottom) 5 μM hPRF1. B) Effect of Profilin on branch density (branches per micron) in the presence of hVASP and Arp2/3 complex, quantified from time-lapse TIRF experiments in A. Plotted as mean with SEM. C) Actin filament elongation rate (subunits/s) in the absence of VASP (−VASP), with VASP alone (+VASP) or with VASP and hProfilin-1 (+VASP/+Prf). Plotted as mean with SEM. D) Indirect immunofluorescence of fixed profilin-1 depleted MEF 10-4 and MTF24 WT and Arpc2−/− cells for F-actin and VASP localization; scale bar = 20 microns. E) Indirect immunofluorescence of fixed MEF 10-4 WT and Arpc2−/− fibroblasts, GFP-FP4-mito detected by direct fluorescence rather than antibody. Note VASP localization to mitochondria in FP4-mito (+) cells. Scale bar = 20 microns. Relates to Figure 6.
Supplemental Figure 6. Additional characterization of Ena/VASP involvement in F-actin homeostasis. A) Indirect immunofluorescence of fixed MTF24 WT and Arpc2−/− cells transfected with GFP-FP4-mito construct. GFP signal is direct fluorescence, rather than antibody; scale bar = 20 microns. B) Representative 10X images of GFP-FP4-mito expressing WT and Arpc2−/− cells in mixed culture stained for phalloidin (Actin) and p34 (KO cells denoted by *) used to generate F-actin integrated pixel density measurements from both lines; scale bar = 100 microns. C) Integrated pixel density of phalloidin staining in fixed MTF24 WT and KO cells, or WT/KO cells stably expressing FP4-mito-GFP (FP4-mito +), plotted as average F-actin intensity/cell, with SEM. N = 50 cells per condition; ***p-value < 0.0001. Western blots of whole cell lysate matched by cell number directly below. D) Spread cell area in sq. microns of MTF24 WT, WT FP4-mito, KO and KO FP4-mito cells plotted as average area/cell with SEM. N = at least 50 cells per condition; ***p-value < 0.0001. Relates to Figure 6.
Movie S1. Time lapse imaging of LA-GFP labeled actin in MEF 10-4 WT or Arpc2−/− cells. Time lapse imaging of Lifeact (LA)-GFP labeled actin in MEF 10-4 WT (left) or Arpc2−/− (right) cells corresponding to Figure 2D still frames. Images were taken every 15 seconds for 435 seconds. Imaging was done at 37°C with 40X magnification in 5% CO2. Scale bar = 20 microns. Relates to Figure 2.
Movie S2. Time lapse imaging of LA-GFP labeled actin in MTF24 WT or Arpc2−/− cells. Time lapse imaging of LA-GFP labeled actin in MTF24 WT (left) or Arpc2−/− (right) cells corresponding to Figure S1C still frames. Images were taken every 15 seconds for 435 seconds. Imaging was done at 37°C with 40X magnification in 5% CO2. Scale bar = 20 microns. Relates to Figure S1.
Acknowledgments
This work was supported by NIH grants to JEB (GM111557) and DRK (GM079265), NSF grant to JMH (1133476), and support from HHMI to JEB. The data reported in this paper are presented in the main text and supplementary materials. We thank Jack D. Griffith and the Lineberger Electron Microscopy core for assistance with cryoshadowing EM experiments, and Tao Bo for technical assistance with mouse husbandry and dissection, and members of our laboratories for critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTION:
J.D.R. and C.W. designed and performed experiments, analyzed data, and wrote the paper; E.M.H. analyzed data; J.D.W and C.S. performed and analyzed TIRF experiments and wrote associated methodological text; H.E.J. and J.M.H. developed the edge ratio MATLAB application; D.R.K. proposed and directed TIRF experiments; J.E.B. proposed and designed experiments, analyzed data and wrote the paper. All authors reviewed and made comments on the manuscript before publication.
LITERATURE CITED
- Barzik M, Kotova TI, Higgs HN, Hazelwood L, Hanein D, Gertler FB, Schafer DA. Ena/VASP proteins enhance actin polymerization in the presence of barbed end capping proteins. J Biol Chem. 2005;280:28653–28662. doi: 10.1074/jbc.M503957200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear J, Loureiro J, Libova I, Fassler R, Wehland J, Gertler F. Negative regulation of fibroblast motility by Ena/VASP proteins. Cell. 2000;101:717–728. doi: 10.1016/s0092-8674(00)80884-3. [DOI] [PubMed] [Google Scholar]
- Bear JE, Gertler FB. Ena/VASP: towards resolving a pointed controversy at the barbed end. J Cell Sci. 2009;122:1947–1953. doi: 10.1242/jcs.038125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear JE, Svitkina TM, Krause M, Schafer DA, Loureiro JJ, Strasser GA, Maly IV, Chaga OY, Cooper JA, Borisy GG, et al. Antagonism between Ena/VASP Proteins and Actin Filament Capping Regulates Fibroblast Motility. Cell. 2002;109:509–521. doi: 10.1016/s0092-8674(02)00731-6. [DOI] [PubMed] [Google Scholar]
- Bilancia CG, Winkelman JD, Tsygankov D, Nowotarski SH, Sees JA, Comber K, Evans I, Lakhani V, Wood W, Elston TC, et al. Enabled negatively regulates diaphanous-driven actin dynamics in vitro and in vivo. Dev Cell. 2014;28:394–408. doi: 10.1016/j.devcel.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchoin L, Amann KJ, Higgs HN, Marchand JB, Kaiser DA, Pollard TD. Direct observation of dendritic actin filament networks nucleated by Arp2/3 complex and WASP/Scar proteins. Nature. 2000;404:1007–1011. doi: 10.1038/35010008. [DOI] [PubMed] [Google Scholar]
- Breitsprecher D, Goode BL. Formins at a glance. J Cell Sci. 2013;126:1–7. doi: 10.1242/jcs.107250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke TA, Christensen JR, Barone E, Suarez C, Sirotkin V, Kovar DR. Homeostatic Actin Cytoskeleton Networks Are Regulated by Assembly Factor Competition for Monomers. Curr Biol. 2014 doi: 10.1016/j.cub.2014.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Marshall TW, Uetrecht AC, Schafer DA, Bear JE. Coronin 1B coordinates Arp2/3 complex and cofilin activities at the leading edge. Cell. 2007;128:915–929. doi: 10.1016/j.cell.2007.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campellone KG, Welch MD. A nucleator arms race: cellular control of actin assembly. Nat Rev Mol Cell Biol. 2010;11:237–251. doi: 10.1038/nrm2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao LG, Babcock GG, Rubenstein PA, Wang YL. Effects of profilin and profilactin on actin structure and function in living cells. J Cell Biol. 1992;117:1023–1029. doi: 10.1083/jcb.117.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XJ, Squarr AJ, Stephan R, Chen B, Higgins TE, Barry DJ, Martin MC, Rosen MK, Bogdan S, Way M. Ena/VASP Proteins Cooperate with the WAVE Complex to Regulate the Actin Cytoskeleton. Dev Cell. 2014;30:569–584. doi: 10.1016/j.devcel.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesarone MA, Goode BL. Actin nucleation and elongation factors: mechanisms and interplay. Curr Opin Cell Biol. 2009;21:28–37. doi: 10.1016/j.ceb.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JA. Effects of cytochalasin and phalloidin on actin. J Cell Biol. 1987;105:1473–1478. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Z, Bae YH, Roy P. Molecular insights on context-specific role of profilin-1 in cell migration. Cell Adh Migr. 2012;6:442–449. doi: 10.4161/cam.21832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelista M, Pruyne D, Amberg DC, Boone C, Bretscher A. Formins direct Arp2/3-independent actin filament assembly to polarize cell growth in yeast. Nat Cell Biol. 2002;4:260–269. doi: 10.1038/ncb770. [DOI] [PubMed] [Google Scholar]
- Fan Y, Arif A, Gong Y, Jia J, Eswarappa SM, Willard B, Horowitz A, Graham LM, Penn MS, Fox PL. Stimulus-dependent phosphorylation of profilin-1 in angiogenesis. Nat Cell Biol. 2012;14:1046–1056. doi: 10.1038/ncb2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, Theriot JA, Dise KR, Tomaselli GF, Goldschmidt-Clermont PJ. Dynamic actin structures stabilized by profilin. Proc Natl Acad Sci U S A. 1994;91:1510–1514. doi: 10.1073/pnas.91.4.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Bretscher A. Analysis of unregulated formin activity reveals how yeast can balance F-actin assembly between different microfilament-based organizations. Molecular biology of the cell. 2008;19:1474–1484. doi: 10.1091/mbc.E07-05-0520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt-Clermont PJ, Machesky LM, Doberstein SK, Pollard TD. Mechanism of the interaction of human platelet profilin with actin. J Cell Biol. 1991;113:1081–1089. doi: 10.1083/jcb.113.5.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SD, Mullins RD. VASP is a processive actin polymerase that requires monomeric actin for barbed end association. J Cell Biol. 2010;191:571–584. doi: 10.1083/jcb.201003014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotulainen P, Lappalainen P. Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. J Cell Biol. 2006;173:383–394. doi: 10.1083/jcb.200511093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser DA, Vinson VK, Murphy DB, Pollard TD. Profilin is predominantly associated with monomeric actin in Acanthamoeba. J Cell Sci. 1999;112(Pt 21):3779–3790. doi: 10.1242/jcs.112.21.3779. [DOI] [PubMed] [Google Scholar]
- Kovar DR, Harris ES, Mahaffy R, Higgs HN, Pollard TD. Control of the assembly of ATP- and ADP-actin by formins and profilin. Cell. 2006;124:423–435. doi: 10.1016/j.cell.2005.11.038. [DOI] [PubMed] [Google Scholar]
- Lanier LM, Gates MA, Witke W, Menzies AS, Wehman AM, Macklis JD, Kwiatkowski D, Soriano P, Gertler FB. Mena is required for neurulation and commissure formation. Neuron. 1999;22:313–325. doi: 10.1016/s0896-6273(00)81092-2. [DOI] [PubMed] [Google Scholar]
- Machesky LM, Atkinson SJ, Ampe C, Vandekerckhove J, Pollard TD. Purification of a cortical complex containing two unconventional actins from Acanthamoeba by affinity chromatography on profilin-agarose. J Cell Biol. 1994;127:107–115. doi: 10.1083/jcb.127.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machesky LM, Mullins RD, Higgs HN, Kaiser DA, Blanchoin L, May RC, Hall ME, Pollard TD. Scar, a WASp-related protein, activates nucleation of actin filaments by the Arp2/3 complex. Proc Natl Acad Sci U S A. 1999;96:3739–3744. doi: 10.1073/pnas.96.7.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand JB, Kaiser DA, Pollard TD, Higgs HN. Interaction of WASP/Scar proteins with actin and vertebrate Arp2/3 complex. Nat Cell Biol. 2001;3:76–82. doi: 10.1038/35050590. [DOI] [PubMed] [Google Scholar]
- Mullins RD, Kelleher JF, Xu J, Pollard TD. Arp2/3 complex from Acanthamoeba binds profilin and cross-links actin filaments. Molecular biology of the cell. 1998;9:841–852. doi: 10.1091/mbc.9.4.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard M, Halbrugge M, Scheer U, Wiegand C, Jockusch BM, Walter U. The 46/50 kDa phosphoprotein VASP purified from human platelets is a novel protein associated with actin filaments and focal contacts. EMBO J. 1992;11:2063–2070. doi: 10.1002/j.1460-2075.1992.tb05264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi SA, Neidt EM, Cui J, Feiger Z, Skau CT, Gardel ML, Kozmin SA, Kovar DR. Identification and characterization of a small molecule inhibitor of formin-mediated actin assembly. Chem Biol. 2009;16:1158–1168. doi: 10.1016/j.chembiol.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodal AA, Manning AL, Goode BL, Drubin DG. Negative regulation of yeast WASp by two SH3 domain-containing proteins. Curr Biol. 2003;13:1000–1008. doi: 10.1016/s0960-9822(03)00383-x. [DOI] [PubMed] [Google Scholar]
- Romero S, Le Clainche C, Didry D, Egile C, Pantaloni D, Carlier MF. Formin is a processive motor that requires profilin to accelerate actin assembly and associated ATP hydrolysis. Cell. 2004;119:419–429. doi: 10.1016/j.cell.2004.09.039. [DOI] [PubMed] [Google Scholar]
- Rotty JD, Wu C, Bear JE. New insights into the regulation and cellular functions of the ARP2/3 complex. Nat Rev Mol Cell Biol. 2013;14:7–12. doi: 10.1038/nrm3492. [DOI] [PubMed] [Google Scholar]
- Sagot I, Rodal AA, Moseley J, Goode BL, Pellman D. An actin nucleation mechanism mediated by Bni1 and profilin. Nat Cell Biol. 2002;4:626–631. doi: 10.1038/ncb834. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- Skoble J, Auerbuch V, Goley ED, Welch MD, Portnoy DA. Pivotal role of VASP in Arp2/3 complex-mediated actin nucleation, actin branch-formation, and Listeria monocytogenes motility. J Cell Biol. 2001;155:89–100. doi: 10.1083/jcb.200106061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector I, Shochet NR, Blasberger D, Kashman Y. Latrunculins–novel marine macrolides that disrupt microfilament organization and affect cell growth: I. Comparison with cytochalasin D. Cell Motil Cytoskeleton. 1989;13:127–144. doi: 10.1002/cm.970130302. [DOI] [PubMed] [Google Scholar]
- Steffen A, Faix J, Resch GP, Linkner J, Wehland J, Small JV, Rottner K, Stradal TE. Filopodia formation in the absence of functional WAVE- and Arp2/3-complexes. Molecular biology of the cell. 2006;17:2581–2591. doi: 10.1091/mbc.E05-11-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suraneni P, Rubinstein B, Unruh JR, Durnin M, Hanein D, Li R. The Arp2/3 complex is required for lamellipodia extension and directional fibroblast cell migration. J Cell Biol. 2012;197:239–251. doi: 10.1083/jcb.201112113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina TM, Bulanova EA, Chaga OY, Vignjevic DM, Kojima S, Vasiliev JM, Borisy GG. Mechanism of filopodia initiation by reorganization of a dendritic network. J Cell Biol. 2003;160:409–421. doi: 10.1083/jcb.200210174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons MH, Mitchison TJ. Control of actin polymerization in live and permeabilized fibroblasts. J Cell Biol. 1991;114:503–513. doi: 10.1083/jcb.114.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng PC, Runge MS, Cooper JA, Williams RC, Jr, Pollard TD. Physical, immunochemical, and functional properties of Acanthamoeba profilin. J Cell Biol. 1984;98:214–221. doi: 10.1083/jcb.98.1.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S, Gao X, Du J, Su J, Xu Z. Angiogenin enhances cell migration by regulating stress fiber assembly and focal adhesion dynamics. PLoS One. 2011;6:e28797. doi: 10.1371/journal.pone.0028797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkelman JD, Bilancia CG, Peifer M, Kovar DR. Ena/VASP Enabled is a highly processive actin polymerase tailored to self-assemble parallel-bundled F-actin networks with Fascin. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1322093111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter DC, Choe EY, Li R. Genetic dissection of the budding yeast Arp2/3 complex: a comparison of the in vivo and structural roles of individual subunits. Proc Natl Acad Sci U S A. 1999;96:7288–7293. doi: 10.1073/pnas.96.13.7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Asokan SB, Berginski ME, Haynes EM, Sharpless NE, Griffith JD, Gomez SM, Bear JE. Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell. 2012;148:973–987. doi: 10.1016/j.cell.2011.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Haynes EM, Asokan SB, Simon JM, Sharpless NE, Baldwin AS, Davis IJ, Johnson GL, Bear JE. Loss of Arp2/3 induces an NF-kappaB-dependent, nonautonomous effect on chemotactic signaling. J Cell Biol. 2013;203:907–916. doi: 10.1083/jcb.201306032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Czech L, Gerboth S, Kojima S, Scita G, Svitkina T. Novel roles of formin mDia2 in lamellipodia and filopodia formation in motile cells. PLoS Biol. 2007;5:e317. doi: 10.1371/journal.pbio.0050317. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Additional characterization of fibroblast cell lines: morphology, F-actin character, response to drug treatment. A) Staining of fixed MEF 10-4 WT and Arpc2−/− fibroblasts; scale bar = 20 microns. B) Still frames from live cell timecourse of MTF24 WT and Arpc2−/− cells stably transfected with the live cell actin probe lifeact (LA)-GFP showing dynamic F-actin behavior in each cell type. Cyan arrowheads depict areas of active protrusion; scale bar = 20 microns. C) Representative 10X images of phalloidin (Actin) and p34 used to generate F-actin integrated pixel density measurements from both lines, with WT and KO cells plated in mixed culture for side by side comparison (KO cells denoted by *); scale bar = 100 microns. D) Indirect immunofluorescence of fixed MEF 10-4 and MTF24 cells in mixed culture plus 500 nM Latrunculin B (KO cells denoted with *); scale bar = 20 microns. E) Staining of fixed MEF 10-4 WT and Arpc2−/− fibroblasts in mixed culture (KO cells marked with *) after addition of 100 nM cytochalasin D (CD) for two hours; scale bar = 20 microns. F) Random migration velocity of control (NS) IA32 cells or IA32 cells stably depleted of p34 and Arp2 (2xKD) in the presence or absence of CD. Means are plotted with 95% confidence intervals; N = at least 49 cells per condition, ****p-value < 0.0001. Relates to Figures 1 and 2.
Supplemental Figure 2. Additional characterization of profilin-1 activity in WT and Arp2/3-depleted lines. A) Barbed end assay relating the distribution of labeled barbed ends to total F-actin in control (NS) or Arp2/3 complex depleted (2xKD) IA32 cells in the absence (−) or presence (+) of profilin; scale bar = 10 microns. B) Representative 10X images of Pfn1 KD WT and Arpc2−/− cells in mixed culture stained for phalloidin (Actin) and p34 (KO cells denoted by *) used to generate F-actin integrated pixel density measurements from both lines; scale bar = 100 microns. C) Indirect immunofluorescence of fixed MTF24 WT and Arpc2−/− Profilin-1 KD fibroblasts; scale bar = 20 microns. D) Integrated pixel density of phalloidin staining in fixed MTF24 WT and KO cells ± Pfn1 KD plotted as average F-actin intensity/cell, with SEM. N = 50 cells per condition, ***p-value < 0.0001, **p-value < 0.001. Blots of whole cell lysate matched by cell number directly below. E) Spread cell area in sq. microns for MEF 10-4 (black bars) and MTF24 (grey bars) WT and KO cells ± Pfn1 KD, plotted as average area/cell, with SEM. N = 50 cells per condition. ****p-value < 0.0001, ***p-value < 0.0053, **p-value = 0.0124, *p-value = 0.0366. Relates to Figure 3.
Supplemental Figure 3. Image quantification and analysis approaches used for data generation. A) Automated p34 edge detection workflow. Parental image is imported into MATLAB and single cells are detected by the edge analysis program. Steps 2–4 are repeated for each cell detected in the initial analysis step. After step 4, the program calculates the ratio of high p34 edge compared to total cell edge. Scale bar = 20 microns. B) Images used for edge detection were also imported into ImageJ and peripheral lamellipodia length in microns was measured for each lamellipodia (outlined in red). Measurements for each example lamellipodia is reported to the right of the image. Scale bar = 20 microns. C) Example of Cy5-dextran signal in Arpc2−/−; p34-GFP rescue cell from Figs. 4C and 5A, before (0 min) and after microinjection of WT hProfilin-1 at indicated time points. Scale bar = 20 microns. D) LA-RFP signal in Arpc2−/−; p34-GFP cell. Scale bar = 20 microns. Individual images were background subtracted in ImageJ and straight lines were drawn through the cell perpendicular to the predominant stress fiber orientation (as shown by white line). When signal was detectable above background in ImageJ line traces (example to the right of image), independent peaks of LA-RFP signal were counted (S = stress fiber). Line trace reports Absolute Grey Value vs. distance across the line (in microns), with the section related to intracellular space marked. Relates to Figures 3, 4 and 5.
Supplemental Figure 4. Effect of SMIFH2 on cell morphology, actin organization and F-actin levels. A) Indirect immunofluorescence of fixed MEF 10-4 and MTF24 WT and Arpc2−/− cells plus or minus SMIFH2; scale bar = 20 microns. B) Representative 10X images of WT and Arpc2−/− cells + SMIFH2 in mixed culture stained for phalloidin (Actin) and p34 (KO cells denoted by *) used to generate F-actin integrated pixel density measurements from both lines; scale bar = 100 microns. Relates to Figure 6.
Supplemental Figure 5. Characterization of Ena/VASP interplay with the Arp2/3 complex and confirmation of FP4-mito approach. A) Time-lapse TIRF microscopy of 1.5 μM Oregon green-labeled actin polymerized in the presence of 50 nM Arp2/3 complex, 50 nM pWA, and 20 nM SNAP-549-hVASP without (top) or with (bottom) 5 μM hPRF1. B) Effect of Profilin on branch density (branches per micron) in the presence of hVASP and Arp2/3 complex, quantified from time-lapse TIRF experiments in A. Plotted as mean with SEM. C) Actin filament elongation rate (subunits/s) in the absence of VASP (−VASP), with VASP alone (+VASP) or with VASP and hProfilin-1 (+VASP/+Prf). Plotted as mean with SEM. D) Indirect immunofluorescence of fixed profilin-1 depleted MEF 10-4 and MTF24 WT and Arpc2−/− cells for F-actin and VASP localization; scale bar = 20 microns. E) Indirect immunofluorescence of fixed MEF 10-4 WT and Arpc2−/− fibroblasts, GFP-FP4-mito detected by direct fluorescence rather than antibody. Note VASP localization to mitochondria in FP4-mito (+) cells. Scale bar = 20 microns. Relates to Figure 6.
Supplemental Figure 6. Additional characterization of Ena/VASP involvement in F-actin homeostasis. A) Indirect immunofluorescence of fixed MTF24 WT and Arpc2−/− cells transfected with GFP-FP4-mito construct. GFP signal is direct fluorescence, rather than antibody; scale bar = 20 microns. B) Representative 10X images of GFP-FP4-mito expressing WT and Arpc2−/− cells in mixed culture stained for phalloidin (Actin) and p34 (KO cells denoted by *) used to generate F-actin integrated pixel density measurements from both lines; scale bar = 100 microns. C) Integrated pixel density of phalloidin staining in fixed MTF24 WT and KO cells, or WT/KO cells stably expressing FP4-mito-GFP (FP4-mito +), plotted as average F-actin intensity/cell, with SEM. N = 50 cells per condition; ***p-value < 0.0001. Western blots of whole cell lysate matched by cell number directly below. D) Spread cell area in sq. microns of MTF24 WT, WT FP4-mito, KO and KO FP4-mito cells plotted as average area/cell with SEM. N = at least 50 cells per condition; ***p-value < 0.0001. Relates to Figure 6.
Movie S1. Time lapse imaging of LA-GFP labeled actin in MEF 10-4 WT or Arpc2−/− cells. Time lapse imaging of Lifeact (LA)-GFP labeled actin in MEF 10-4 WT (left) or Arpc2−/− (right) cells corresponding to Figure 2D still frames. Images were taken every 15 seconds for 435 seconds. Imaging was done at 37°C with 40X magnification in 5% CO2. Scale bar = 20 microns. Relates to Figure 2.
Movie S2. Time lapse imaging of LA-GFP labeled actin in MTF24 WT or Arpc2−/− cells. Time lapse imaging of LA-GFP labeled actin in MTF24 WT (left) or Arpc2−/− (right) cells corresponding to Figure S1C still frames. Images were taken every 15 seconds for 435 seconds. Imaging was done at 37°C with 40X magnification in 5% CO2. Scale bar = 20 microns. Relates to Figure S1.