

Abstract
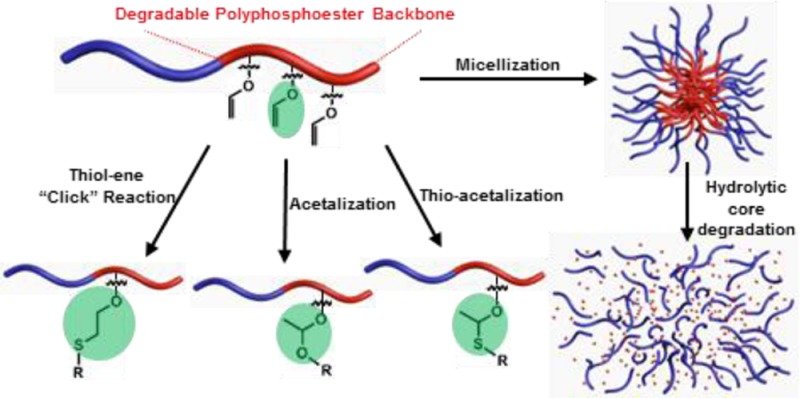
A novel polyphosphoester (PPE) with vinyl ether side chain functionality was developed as a versatile template for postpolymerization modifications, and its degradability and biocompatibility were evaluated. An organocatalyzed ring-opening polymerization of ethylene glycol vinyl ether-pendant cyclic phosphotriester monomer allowed for construction of poly(ethylene glycol vinyl ether phosphotriester) (PEVEP). This vinyl ether-functionalized PPE scaffold was coupled with hydroxyl- or thiol-containing model small molecules via three different types of conjugation chemistries—thiol–ene “click” reaction, acetalization, or thio-acetalization reaction—to afford modified polymers that accommodated either stable thio–ether or hydrolytically labile acetal or thio–acetal linkages. Amphiphilic diblock copolymers of poly(ethylene glycol) and PEVEP formed well-defined micelles with a narrow and monomodal size distribution in water, as confirmed by dynamic light scattering (DLS), transmission electron microscopy, and atomic force microscopy. The stability of the micelles and the hydrolytic degradability of the backbone and side chains of the PEVEP block segment were assessed by DLS and nuclear magnetic resonance spectroscopy (1H and 31P), respectively, in aqueous buffer solutions at pH values of 5.0 and 7.4 and at temperatures of 25 and 37 °C. The hydrolytic degradation products of the PEVEP segments of the block copolymers were then identified by electrospray ionization, gas chromatography, and matrix-assisted laser desorption/ionization mass spectrometry. The parent micelles and their degradation products were found to be non-cytotoxic at concentrations up to 3 mg/mL, when evaluated with RAW 264.7 mouse macrophages and OVCAR-3 human ovarian adenocarcinoma cells.
Introduction
Although ring-opening polymerizations (ROPs) provide a method to transform cyclic monomers into well-defined, functional, and degradable polymers,1−3 it has been challenging, in general, with some exceptions,4,5 to overcome difficulties that are often experienced in the attempt to introduce functional moieties within the cyclic monomer structures and with incompatibilities of these desired functionalities during the polymerizations. Alternatively, “click” chemistries such as azide–alkyne Huisgen cycloaddition,6 Diels–Alder reaction,7 and radical-mediated thiol–ene/yne reactions8,9 have revolutionized polymer chemistry, allowing for efficient and reliable routes toward postpolymerization modifications in the design of complex and functional polymeric materials.10−13
Combined with the extraordinary efficiency in the generation of stable covalent linkages by “click”-type reactions in biomaterials,14 acid-labile functionalities, such as acetals,15−17 ortho esters,18,19 and hydrazones,20,21 are also of major interest in contemporary materials design, especially toward biomedical applications.22 In spite of the wide utilization of acetals/thio–acetals in synthetic organic chemistry (e.g., as hydroxyl protecting groups), they have received less attention in polymer chemistry. The most intriguing characteristic of acetal/thio–acetal functionalities originates from their unique property of behaving as a “dynamic covalent bond”, i.e., being cleavable upon exposure to acidic conditions such as those found in the gastrointestinal tract, tissue at sites of inflammation and in tumors, and in endosomal and lysosomal compartments,23−25 while being relatively stable at the normal physiological pH.16,17,26−28 In this context, acetals/thio–acetals are appealing for their incorporation into biomacromolecules, where the dynamic covalent linkages can bridge to biologically active molecules and, ultimately, release them at a targeted site in a controlled manner. Furthermore, introduction of hydroxyl functionalities by controlled cleavage of acetals/thio–acetals would improve the hydrophilicity of a given polymer, which has been limited conventionally due to their incompatibility with the polymerization processes.29
The inherent acid-labile property of acetals has been introduced to the backbone or the side chains of functional polymers or cross-linkers in the preparation of cross-linked nanoparticles. For instance, among several elegant works, reports on acetal-linked linear polymers or dendrimers have elucidated the cleavability as well as the chemical stability of acetal linkages in a broad spectrum of polymeric systems, e.g., degradable, biocompatible dextrans, by Frechet et al.;15−17,26,28 poly(ethylene oxides) connected with cleavable acetals by Hawker et al.;30 hydroxyl-group protection during anionic polymerization and hyperbranched polyethers with acetal backbone linkages by Frey et al.;29,31 acetal-based degradable shell cross-linked polymeric micelles by Wooley et al.;32 introduction of vinyl ether side chain moieties along poly(ethylene glycol) backbones by Wurm et al.33 and Brocchini et al.;34 and acetal-linked backbone-cleavable ABA-type triblock copolymers by Ni et al.35 Here, our interest is the application of acid-labile acetal/thio–acetal linkages to the side chains in degradable polymeric systems for their potential use as drug delivery carriers. Recently, acetal-linked prodrug micellar nanoparticles and their pH-triggered controlled drug release from the side chains have been highlighted by Zhang et al.36 and Zhong et al.37 However, the polymeric nanoconstructs used in these studies were hydrocarbon-based, nondegradable backbone systems—polymethacrylates and poly(acrylic acid), respectively—which limit their application as potential biomaterials because of the possible long-term accumulation in the human body.
A major effort of our group has been to develop polyphosphoesters (PPEs) tailored by manipulation of pendant functional groups on the pentavalent phosphorus atoms and to integrate the PPE system into new classes of degradable, biocompatible polymeric nanomaterials.38−42 Particularly, introduction of reactive vinyl ether moieties to degradable, biocompatible PPEs43 and their utilization to conjugate a library of hydroxyl- or thiol-containing biologically active molecules via multiple types of conjugation chemistries are envisioned to expand the breadth of this chemical approach in the development of functional biomaterials.
Herein, we report the organocatalyzed ROP of ethylene glycol vinyl ether-containing cyclic phosphotriester monomer, as an initial fundamental study toward unique degradable, functional polymer materials. After demonstrating the well-controlled homopolymerization of the monomer with predetermined molecular weights and narrow molecular weight distributions, the resulting vinyl ether-functionalized PPE scaffold was conjugated with hydroxyl- or thiol-containing model small molecules via three different types of conjugation chemistries—thiol–ene “click” reaction, acetalization, or thio–acetalization reaction—revealing efficient routes for postpolymerization modifications of functional polymers. Furthermore, amphiphilic diblock copolymers containing the ethylene glycol vinyl ether-functionalized PPE as a hydrophobic block segment, also prepared by ROP, afforded well-defined micelles that showed a pH-dependent hydrolytic core degradability of both backbone and side chains in aqueous solutions. The degradation products, as identified by mass spectrometry, were found to be nontoxic toward two standard cell lines: RAW 264.7 mouse macrophages and OVCAR-3 human ovarian adenocarcinoma cells. Hence, this polymeric system can have potential applications in the development of nanomedical devices for a variety of biomedical applications.
Results and Discussion
Monomer Design and Synthesis
A novel ethylene glycol vinyl ether-functionalized cyclic phosphotriester monomer, 2-ethylene glycol vinyl ether-1,3,2-dioxaphospholane 2-oxide (EVEP), 1, was synthesized by following a typical condensation method: coupling of ethylene glycol vinyl ether (EVE) to 2-chloro-3-oxo-1,3,2-dioxaphospholane (COP) in the presence of triethylamine (TEA) in dichloromethane (DCM) at 4 °C (Scheme 1). It was critical to purify the monomer rigorously, by removing the residual starting materials completely, because of the potential for undesired dual initiation by residual EVE and the possible formation of random/branched (co)polymers by remaining reactive COP during the polymerization process. Multiple purification methods, including silica gel column chromatography, (vacuum) distillation, extraction, and precipitation, were attempted, but decomposition of the reactive cyclic monomer and/or incomplete removal of the impurities were inevitable in all cases. Therefore, use of an exact stoichiometric equivalence of reagents was attempted, and then the purification process was applied for removal of the TEA salts by a series of recrystallization in DCM, precipitation in diethyl ether, and filtration. Even though tetrahydrofuran (THF) could be used as a solvent, DCM was more efficient for recrystallization of the TEA salts. The quantitative conversion of EVE and COP to 1 was supported by 1H, 13C, and 31P NMR spectroscopy (Figure S1). By 13C NMR spectroscopy, the methylene carbon of the hydroxymethyl group of EVE resonating as a singlet (1H-decoupled) at 61.2 ppm disappeared upon coupling to COP to result in a doublet (J(31P,13C)) having a coupling constant of 10.2 Hz at 66.1 ppm for the methylene carbon of the new phosphoester linkage. In addition, consumption of the COP was observed by 31P NMR spectroscopy as replacement of the COP phosphorus resonance at 23.20 ppm with that for the monomer at 17.91 ppm. Although the conversion to monomer appeared to be complete, 1H NMR analysis indicated that there were residual solvents and TEA salts that were not removed (∼96% purity). However, those impurities did not impact the polymerization.
Scheme 1. Synthetic Route for the Preparation of Ethylene Glycol Vinyl Ether-Functionalized Cyclic Phosphotriester Monomer 1 and Homopolymer 2 by ROP Followed by Postpolymerization Modifications via Three Different Types of Conjugation Chemistries: Thiol–Ene “Click” Reaction, Acetalization, or Thio–Acetalization Reaction.

Homopolymerization of the Monomer 1 by Using an Organocatalyst, DBU
The ROP of 1 was performed in a glovebox at ambient temperature. For the kinetic study, 1 and benzyl alcohol (BnOH) (molar ratio of 100:1) were mixed in DCM, and the polymerization began with the addition of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (molar ratio to initiator of 3:1). After being stirred for a predetermined period of time, an aliquot of the reaction mixture was collected, immediately quenched by adding a solution of excess benzoic acid in DCM, and then analyzed by 31P NMR spectroscopy. A portion of the collected samples was precipitated into diethyl ether prior to injection into the gel permeation chromatography (GPC) instrument (Figure S2). Although routine decoupled 31P NMR spectroscopy has complications that may limit the quantitative value, the distinct resonance frequencies of the phosphorus nuclei of the monomer vs the polymer provided the best opportunity (relative to 1H or 13C NMR spectroscopy) to monitor the polymerization. Therefore, the conversion was estimated from 31P NMR spectroscopy by comparing the integral ratio of two distinct peaks of monomer 1 at 17.91 ppm and homopolymer, PEVEP, at −0.68 ppm, on crude polymerization aliquots. Once the polymerization was quenched and worked up, 1H NMR spectroscopy end-group analysis of the degree of polymerization was found to be in agreement with the monomer conversion calculated from the 31P NMR data. Both the molecular weight and its distribution were determined by GPC. The kinetic study displayed a rapid initial polymerization rate, in which the monomer conversion reached at 23% within the beginning 3 min. However, the maintenance of linearity of Mn vs monomer conversion suggested a living ROP up to 79% conversion (Figure S2b). The consistent low PDIs (≤1.04) until the monomer conversion reached at 79% indicated that there was minimal adverse transesterification of the PPE backbone during the polymerization process. A kinetic plot of ln([M]0/[M]) vs polymerization time illustrated pseudo-first-order kinetics (Figure S2a), which is a typical characteristic of ROP.
The same molar ratio used for the kinetic study was applied for a scaled-up production of 2. The purification of 2 by precipitation in diethyl ether was insufficient to remove the residual starting materials and benzoic acid completely. Silica gel column chromatography was also attempted, but instability of the PPE backbone was problematic. Dialysis (MWCO 6–8 kDa) of the reaction mixture against organic solvents, switching from MeOH to DCM, was the most assured purification method among those tested. Dialysis in basic aqueous solutions (e.g., carbonate buffer at pH 7–8) was also feasible, but the complete removal of water was challenging, which is imperative for the following postpolymerization modification reactions. Being consistent with the kinetic study, quenching the polymerization at a predetermined time (9 min), targeting at 50% conversion, yielded the predicted molecular weight and PDI. The degree of polymerization (DPn) calculated based on 31P NMR spectroscopy-determined monomer conversion was in agreement with that calculated from chain-end analysis by 1H NMR spectroscopy, i.e., by comparisons of the integrals of proton resonances of the benzyl group (7.43–7.34 or 5.08 ppm, labeled as a or b, respectively, in Figure 1a) of the initiated chain end to those of the distinct double bonds (6.49 or 4.06 ppm, labeled as f or g2, respectively, in Figure 1a) or β protons on the substituents to the phosphorus atom (3.94–3.86 ppm, labeled as e in Figure 1a) of 2, which was indicative of retention of the vinyl groups (Figure 1a). In addition, one distinct 31P resonance confirmed the stability of the degradable PPE backbone during ROP of 1 and the work-up process of 2. GPC analysis of 2 showed a monomodal peak with PDI of 1.05.
Figure 1.

1H (300 MHz, CD2Cl2, ppm) and 31P (121 MHz, CD2Cl2, ppm, inset) NMR spectra of (a) 2, (b) 3, (c) 4, and (d) 5.
Thiol–Ene “Click” Reaction of the Vinyl Ether Side Chain Moieties of PEVEP502 with 2-(2-Methoxyethoxy)ethanethiol
The vinyl ether side chain moieties of 2 were functionalized by thiol–ene “click” reaction with thiol-containing model small molecule, 2-(2-methoxyethoxy)ethanethiol. The radical-mediated thiol–ene “click” chemistry is a robust and versatile method that tolerates a variety of functional groups in achieving a high degree of functionalization on vinyl groups.8,9 Herein, this efficient chemistry was applied to demonstrate the presence and chemical availability of vinyl groups on 2. To verify the integrity of the PPE backbone in the presence of radicals during UV irradiation, a mixture of 2 and 2,2-dimethoxy-2-phenylacetophenone (DMPA) in methanol-d4 (MeOD4) was irradiated under UV light (365 nm, 6 W) for several hours, as a preliminary control reaction. 31P NMR spectroscopy confirmed that the polymer backbone was intact under these conditions. Accordingly, an excess of 2-(2-methoxyethoxy)ethanethiol relative to the vinyl ether bonds was employed with DMPA in MeOH and UV irradiation for 1 h to ensure a high coupling efficiency and to avoid undesired cross-linking reactions between the double bonds along the backbone. The purified products were obtained by conducting precipitation in diethyl ether followed by sequential dialysis (MWCO 6–8 kDa) against MeOH and DCM. Comparison of the 1H NMR spectra of 1 and 2 before and after thiol–ene “click” reaction, as shown in Figures 1a and 1b, respectively, verified the disappearance of the vinyl proton resonance (6.49 ppm, labeled as f in Figure 1a) and the corresponding appearance of two distinct proton resonances of methoxy (3.33 ppm, labeled as f in Figure 1b) and α-protons adjacent to the sulfur atom (2.78–2.69 ppm, labeled as g1 and g2 in Figure 1b) in the thio–ether functional group. GPC analysis of 3 clearly showed a peak shift to shorter elution time, relative to 2, with a monomodal peak having a PDI of 1.07 after thiol–ene “click” reaction (Figure S3).
Acetalization of the Vinyl Ether Side Chain Moieties of PEVEP502 with 4-Methylbenzyl Alcohol
Using commercially available 4-methylbenzyl alcohol and a catalytic amount of p-toluenesulfonic acid (PTSA), the presence and chemical availability of vinyl ether moieties of 2 were demonstrated via acetalization. Initially, to confirm the integrity of the PPE backbone in the presence of alcohols and strong acidic catalyst, PTSA (pKa ∼ −2.8 (water)), a mixture of 2 and 2 equiv of 4-methylbenzyl alcohol (relative to the absolute number of vinyl ether bonds) or that of 2 and 0.2 equiv of PTSA (in relation to the absolute number of vinyl ether bonds) or that of 2, 4-methylbenzyl alcohol, and PTSA in N,N-dimethylformamide-d7 (DMF-d7) was allowed to stir for a period of time, as a preliminary control reaction. An aliquot of each reaction solution was collected at a predetermined time, quenched by the addition of an excess of TEA, and then evaluated by 31P NMR spectroscopy. 31P NMR spectra confirmed the intact PPE backbone in the presence of either 4-methylbenzyl alcohol or PTSA for several hours. However, the combination of 2, 4-methylbenzyl alcohol, and PTSA in the mixture solution was accompanied by undesired degradation/transesterification along the PPE backbone structure after 6 min of reaction, as confirmed by 31P NMR spectroscopy. Accordingly, a scaled-up production of 4 was conducted using the same molar ratio as used for the preliminary control reaction, and the acetalization reaction was quenched at 5 min by the addition of an excess of TEA to ensure the intact PPE backbone structure. With respect to the purification, Hawker et al. employed an extraction method to purify their acetal-linked linear poly(ethylene glycol) by using water;30 however, this extraction method was not compatible with the instability of the PPE backbone in our system. In addition, the complete removal of PTSA and/or 4-methylbenzyl alcohol was not achieved by precipitation of the desired polymer in organic solvent, such as diethyl ether. More importantly, this precipitation method was not desirable, especially for acetal-bearing polymers as a result of the possible cross-linking side reactions by trans-acetalization in the presence of a trace amount of PTSA.33 Thus, sequential dialysis (MWCO 6–8 kDa) against two different organic solvents, DMF and DCM, was conducted for purification. Finally, the desired product, 4, was obtained after removal of the organic solvents, as confirmed by 1H and 31P NMR spectroscopy (Figure 1c).
The conversion percentage of vinyl ethers to acetals was calculated from chain-end group analysis by 1H NMR spectroscopy, i.e., by comparison of the integrals of proton resonances of the benzyl group (7.44–7.32 ppm, labeled as a in Figure 1c) of the initiated chain end to those of two distinct acetal linkages, methyl (1.33 ppm, labeled as j in Figure 1c) and methylene (4.87–4.75 ppm, labeled as i in Figure 1c), or 4-methyl protons on the benzyl substituents (2.32 ppm, labeled as m in Figure 1c). According to the chain-end analysis by 1H NMR spectroscopy, approximately 18% of the initial vinyl ether groups were converted into the acetal linkages, ca. 50% remained intact, and ca. 32% of the repeat units carried hydroxyl groups as a result of hydrolysis. The hydrolysis side reaction was probably attributed to a trace of water present in PTSA and/or highly viscous PPE. It is noteworthy that the complete disappearance of vinyl proton resonances was observed with prolonged reaction time (1–2 h), which could have enhanced the acetal conversion percentage. However, quenching the reaction at the optimal time, 5 min according to the preliminary reaction, was essential for the integrity of the PPE backbone. This backbone stability was verified by the existence of one distinct 31P resonance peak at −0.68 ppm, observed by 31P NMR spectroscopy. GPC analysis of 4 was not available in THF because of its polar nature, conferred by the newly formed hydroxyl groups on the side chains.
Thio–Acetalization of the Vinyl Ether Side Chain Moieties of PEVEP502 with 4-Methylbenzyl Mercaptan
The same reaction protocol for the acetalization reaction was applied to the thio–acetalization of 2. The 1H NMR spectrum of thio–acetal-bearing PEVEP50, 5, is shown in Figure 1d. The conversion percentage of vinyl ethers to thio–acetals was calculated from chain-end group analysis by 1H NMR spectroscopy, i.e., by comparisons of the integrals of proton resonances of the benzyl group (7.44–7.32 ppm, labeled as a in Figure 1d) of the initiated chain end to those of the two distinct thio–acetal linkages, methyl (1.51 ppm, labeled as j in Figure 1d), methylene (4.71 ppm, labeled as i in Figure 1d), or 4-methylbenzyl protons (2.31 ppm, labeled as m in Figure 1d). By 1H NMR chain-end analysis, approximately 8% of the initial vinyl groups were found to be converted into the thio–acetal linkages, ca. 56% of them remained intact, and ca. 36% of the repeat units underwent hydrolysis of the side chains to present hydroxyl groups. The intact PPE backbone was affirmed by the presence of one distinct 31P resonance peak at −0.67 ppm, as measured by 31P NMR spectroscopy. Similar to the acetal linkage-bearing polymer, 4, the polar nature endowed by the newly formed hydroxyl groups on the side chains did not allow for GPC analysis.
Synthesis of the Amphiphilic Diblock Copolymer, α-Methoxy Poly(ethylene glycol)44-block-poly(ethylene glycol vinyl ether phosphotriester)33 (mPEG44-b-PEVEP33), 6
Based on the kinetic study of 2, amphiphilic diblock copolymer, mPEG44-b-PEVEP33, 6, was prepared by ROP using the molar ratio 100:1:3 of 1, α-methoxy-ω-hydroxy poly(ethylene glycol) 2000 Da (mPEG44-OH), and DBU, respectively (Scheme 2). The polymerization was quenched at 6 min by the addition of a solution of excess of benzoic acid in DCM, and the desired diblock copolymer product was obtained after sequential dialysis (MWCO 6–8 kDa) against organic solvents, MeOH and DCM. The DPn calculated based on 31P NMR spectroscopy-determined monomer conversions was in agreement with that calculated from chain-end analysis by 1H NMR spectroscopy, i.e., by comparisons of the integrals of the proton resonance of the methyl group of the initiated chain end (3.33 ppm, labeled as a in Figure 2a) to that of the double bond (6.49 or 4.06 ppm, labeled as f or g2, respectively, in Figure 2a) or α-protons to the vinyl ether oxygen atom (3.94–3.86 ppm, labeled as e in Figure 2a) of the PEVEP block segment. Additionally, one distinct 31P resonance peak at −0.71 ppm confirmed the intact PEVEP block backbone structure during the chain extension by ROP of 1 and the work-up process of 6. GPC analysis of 6 showed a distinct peak shift to lower elution time from that of the macroinitiator, mPEG44-OH, after polymerization, with a monomodal peak with a PDI of 1.09 (Figure 2b).
Scheme 2. Synthetic Route for the Preparation of Amphiphilic Diblock Copolymer 6 and Schematic Illustration of the Assembly of 6 into Micelles 7 by Direct Dissolution in Water Followed by the Hydrolytic Core Degradation of 7.
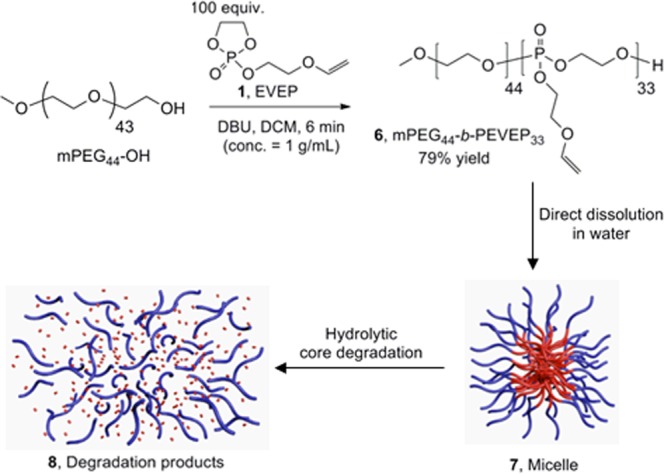
Figure 2.
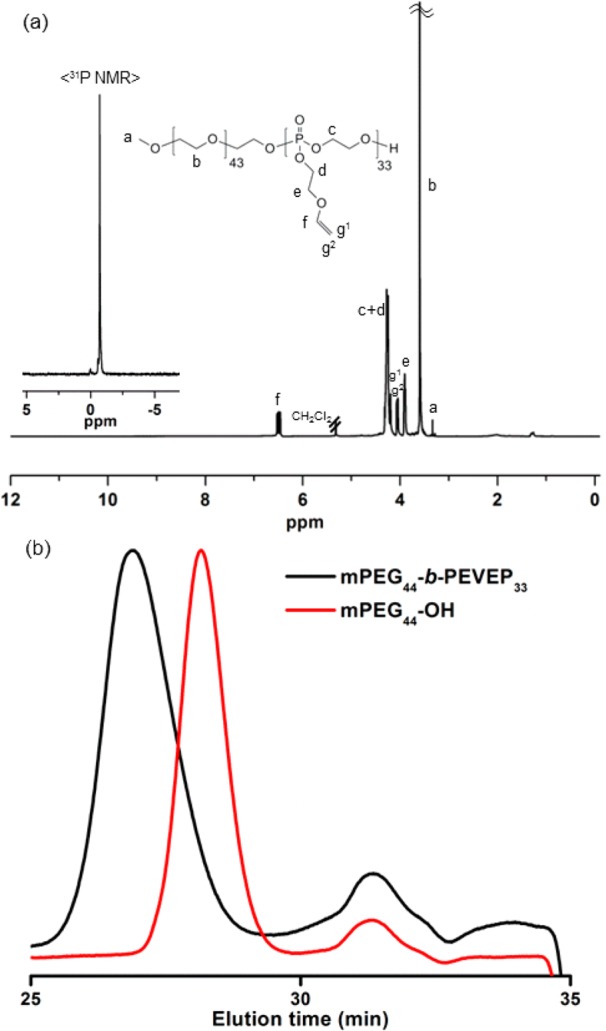
(a) 1H (300 MHz, CD2Cl2, ppm) and 31P (121 MHz, CD2Cl2, ppm, inset) NMR spectra of 6. (b) GPC traces of macroinitiator, mPEG44-OH, and diblock copolymer 6, mPEG44-b-PEVEP33, as a function of elution time (min).
Thermal Properties
The Tg values of the prepared polymers varied, as measured by differential scanning calorimetry (DSC), depending on the side chain substituents (Table 1). The conjugation of the 2-(2-methoxyethoxy)ethanethioether groups onto the side chains induced a decrease of the Tg value from −39 °C of 2 to −64 °C of 3 after the thiol–ene “click” reaction. The slight increase of Tg value of 4 and 5, −27 and −31 °C, respectively, as compared with that of 2, −39 °C, was ascribed to the rigidity and π–π interactions of the aromatic rings. Meanwhile, the Tg value of the diblock copolymer with the extended ethylene glycol backbone units, 6, was complicated to analyze but appeared to give only a single Tg value, similar to that of 2, −38 °C. In addition, no Tm for the PEG block segment was observed for the diblock system; therefore, an extensive investigation against the homopolymers, mPEG44-OH and PEVEP50, and a mPEG44-OH/PEVEP50 blend was conducted. These studies confirmed that the PEVEP block fully suppresses PEG crystallinity in the diblock system, while the physical blend does little to suppress PEG crystallization (full DSC traces for all systems upon heating and cooling are presented with detailed discussions in the Supporting Information (Figure S9)).
Table 1. Comparisons of Glass Transition Temperatures, Tg, of the Prepared Polymers, As Measured by Differential Scanning Calorimetry (DSC).
| polymer | Tg (°C) |
|---|---|
| PEVEP50, 2 | –39 |
| PEVEP50 after thiol–ene “click” reaction, 3 | –64 |
| PEVEP50 after acetalization reaction, 4 | –27 |
| PEVEP50 after thio-acetalization reaction, 5 | –31 |
| mPEG44-b-PEVEP33, 6 | –38 |
Self-Assembly of the Amphiphilic Diblock Copolymer, mPEG44-b-PEVEP336
The self-assembly behavior of the amphiphilic diblock copolymer, mPEG44-b-PEVEP336, was studied by direct dissolution in nanopure water. A high concentration of 6 could be dispersed in nanopure water or buffer (>15 mg/mL) without a significant turbidity or precipitation, which allowed for analyses to be performed across a broad range of concentrations and also serves as a promising criterion for its use as a drug delivery carrier. The morphology and surface charge of the resulting micellar nanoparticles, 7, were characterized by dynamic light scattering (DLS), transmission electron microscopy (TEM), atomic force microscopy (AFM), and ζ-potential measurements (Figure 3 and Figure S4). DLS and ζ-potential analyses indicated narrow and monomodal size distributions (PDI = 0.114) with almost neutral charges and a negligible difference at pH 5.0 and 7.4, −4.52 and −7.64 mV, respectively. The number-averaged hydrodynamic diameter of 7 by DLS was ca. 44 ± 6 nm. The TEM images also revealed uniform nanoparticles with an average diameter of 39 ± 5 nm. Although the hydrodynamic and dry-state diameters measured by DLS and TEM, respectively, were in agreement, AFM indicated significant deformation of the micelles upon deposition and drying on the mica substrate. The ca. 3 nm height and 40 ± 7 nm diameter indicate flattening of the micelles, which is predicted to occur based upon the fluid shell and core components, each being composed of a highly viscous polymer, PEG (Tg = −17 °C) and PEVEP (Tg = −39 °C, Table 1 and Figure S9). The AFM data are useful qualitatively; however, further quantitative analysis is complicated by the presence of substantial amounts of polymer debris and agglomerations of 7 across the substrate (Figure 3c,d), which are also indicators of the fluidity of the micellar assemblies.
Figure 3.

Self-assembly results of micelle 7 in water. (a) DLS results of 7: Dh(intensity) = 49 ± 7 nm, Dh(volume) = 46 ± 7 nm, and Dh(number) = 44 ± 6 nm (PDI = 0.114). (b) TEM image of 7: Dav = 39 ± 5 nm, after counting more than 150 nanoparticles. AFM height image (c) and three-dimensional image (d) of 7: Dav = 40 ± 7 nm, after counting more than 100 nanoparticles.
Degradation Study
The phosphoester linkages of PPEs can be cleaved by spontaneous hydrolysis and/or enzymatic degradation; the hydrolysis rate of the phosphoester linkages is known to be highly pH-dependent.44−49 In this study, we postulated that the exposure of micelles, 7, in acidic aqueous environment would induce a dual degradation of the phosphoester backbone linkages and the vinyl ether side chain moieties of the PEVEP block segment. Hence, the stability of the phosphoester backbone linkage and the vinyl ether functionality of 7 were monitored in deuterated buffer solutions by 31P and 1H NMR spectroscopies, respectively, in parallel to measuring the micelle size and the intensity by DLS. Furthermore, the collected degradation products were identified using electrospray ionization (ESI), gas chromatography (GC), and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry.
Degradability of the PEVEP Backbone in Aqueous Solutions As Monitored by 31P NMR Spectroscopy
The backbone stability of the PEVEP segment in D2O at pH values of 5.0 and 7.4 and temperature of 37 °C was monitored by 31P NMR spectroscopy (Figure 4). The integral ratio of the 31P resonance of the intact PEVEP segment at −0.71 ppm to that of the newly emerging peaks was analyzed. The pH-dependent degradation of the PEVEP backbone linkages became apparent by the disappearance of 31P resonance corresponding to the intact PEVEP backbone, coincident with the appearance of new 31P resonances as a result of hydrolytic degradation at pH 5.0, whereas there was no significant change in the PEVEP 31P resonance when the samples were incubated at pH 7.4.
Figure 4.
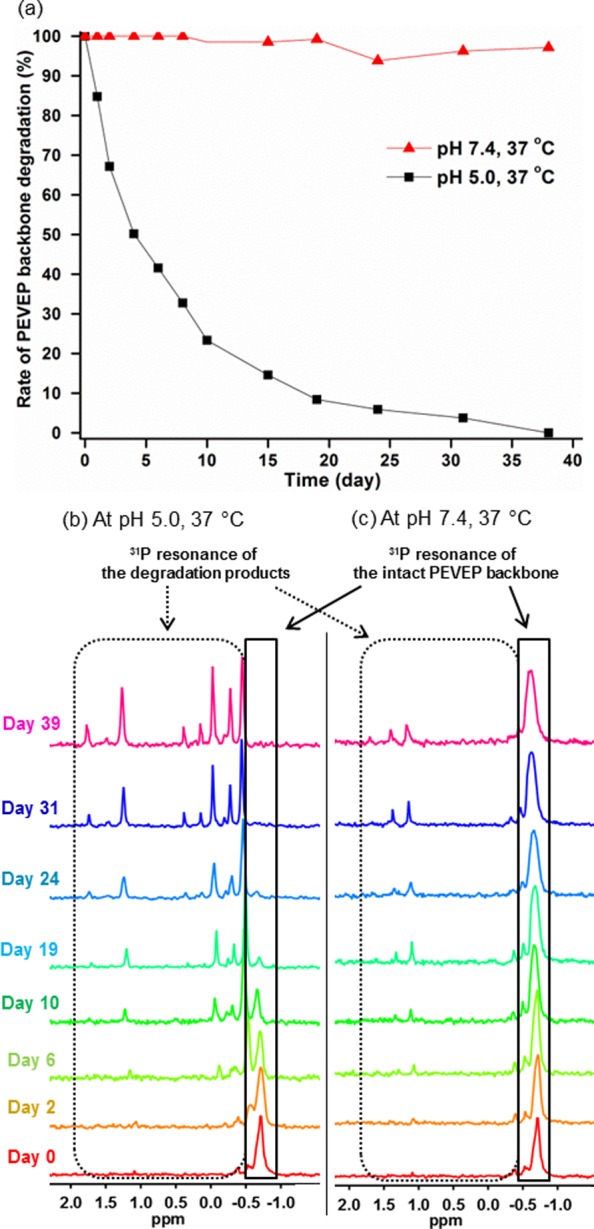
(a) Rate of the PEVEP backbone degradation of 7 at pH 5.0 (black line) or pH 7.4 (red line) at 37 °C as a function of time, as measured by a comparison of the integrals of initial to the newly appeared 31P NMR resonances. Changes in the 31P NMR resonance of PEVEP backbone of 7 at pH 5.0 (b) and 7.4 (c) at 37 °C over a period of time.
Stability of the Vinyl Ether Side Chain Moieties in Aqueous Solutions As Monitored by 1H NMR Spectroscopy
To compensate for potential complications in the detection of vinylic proton resonances of the micelle core, we evaluated the pH-dependent hydrolytic reactivity of the vinyl ether functionalities by observing both the polymer side chain functionalities and the small molecule hydrolysis product using 1H NMR spectroscopy. To determine the reactivity of the acid-labile vinyl ether functionality, the integral ratio of a distinct vinyl proton resonance of the micelles at 6.49 ppm to the newly appeared proton resonance of acetaldehyde, one of the hydrolysis products of the vinyl ether functionality, at 9.60 ppm was compared in D2O at pH values of 5.0 and 7.4 at 37 °C over a period of time (Figure 5). For the micellar nanoparticles in pH 5.0 aqueous solution at 37 °C, both the gradual disappearance of vinyl proton resonance intensity and the appearance of an acetaldehyde proton resonance signal were clearly observed within 1 day, and the equal ratio of these two different proton resonances was reached after 14 days. By contrast, the vinyl proton resonance of the micelles in pH 7.4 aqueous solution at 37 °C remained consistent without any observable generation of acetaldehyde over 39 days. Indeed, this observation bolstered our hypothesis that the spontaneous cleavage of the vinyl ether moieties occurred in pH 5.0 aqueous solution simultaneously with the PPE backbone degradation, but not at pH 7.4.
Figure 5.
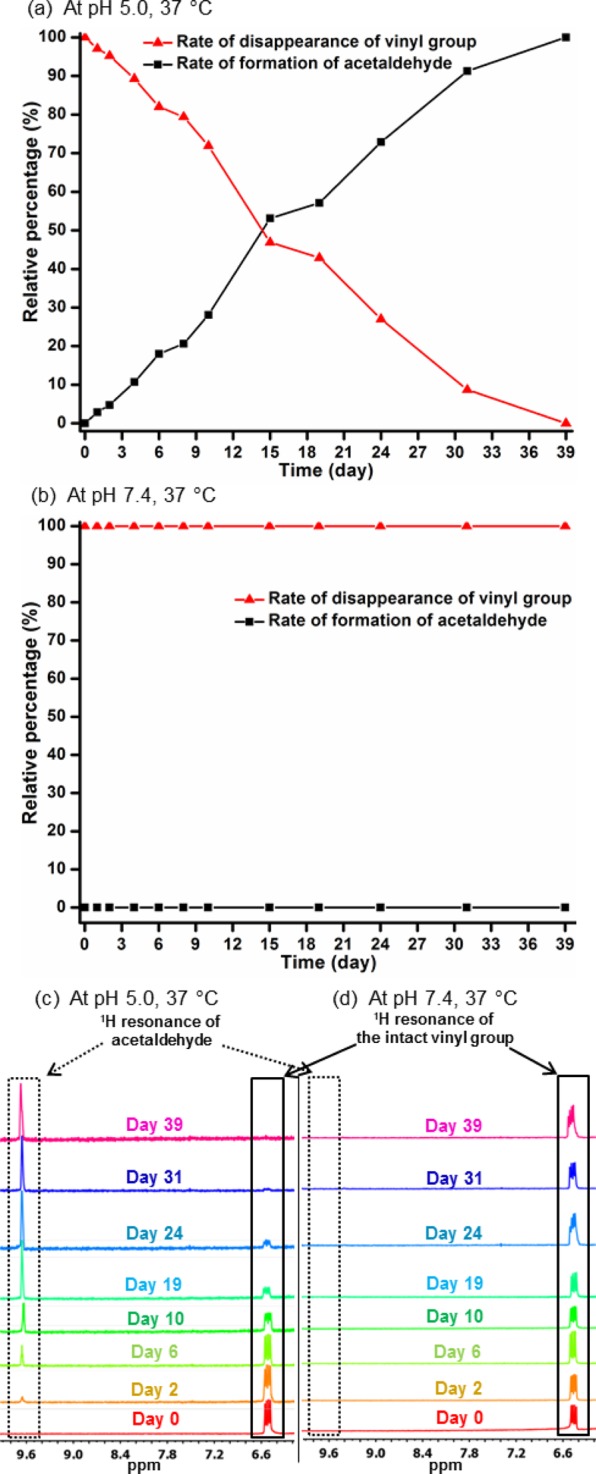
Rate of formation of acetaldehyde or disappearance of vinyl proton resonance at pH 5.0 (a) and pH 7.4 (b) and at 37 °C as a function of time, as measured by a comparison of the integrals of vinyl and acetaldehyde proton resonance. Transition of proton resonances of acetaldehyde and vinyl groups at pH 5.0 (c) or pH 7.4 (d) and at 37 °C over a period of time.
Hydrolytic Micelle Stability in Water by Monitoring the Changes in (1) Hydrodynamic Diameter and (2) the Intensity of Light, Scattered by Micelles 7, As Measured by DLS
It was hypothesized that hydrolysis of the PPE backbone would decrease the proportion of the hydrophobic:hydrophilic block segment ratio and that hydrolysis of the side chain vinyl ether groups would increase the hydrophilicity of the PPE backbone; thereby, each would weaken the micelle assemblies. In order to demonstrate the effects of pH-dependent hydrolytic degradation of the polymer on the behavior and stability of the micelles, 7, aqueous buffer solutions containing 6 at pH 5.0 and 7.4 were incubated at 25 and 37 °C, and their degradation profiles were assessed by measuring the changes in hydrodynamic diameter and the intensity of light, scattered by micelles, when measured by DLS over a period of time (Figure 6). Overall, as predicted from the NMR degradation studies, the micelle assemblies in pH 5.0 aqueous solutions, at both 25 and 37 °C, became unstable within 1 day, and the nanoparticles were undetectable within 7 and 2 days, respectively. The swelling behavior of the micelles upon hydrolysis of PEVEP segment was accounted for by the diffusion of water into the core region (Figure 6a). Interestingly, the unstable and dissociated micelles did not cause the formation of visible precipitates, and thus, the micelle solutions remained clear during monitoring. Also, in agreement with the lack of backbone or side chain hydrolysis observed by NMR spectroscopy, the nanoparticle sizes in pH 7.4 aqueous solutions at 25 °C remained consistent over 39 days (Figure 6b). Surprisingly, when the samples were incubated at pH 7.4 and 37 °C, large aggregates formed after 20 days, which persisted until day 43 when particles were no longer detectable. The lack of changes in the NMR data over the same period of time and conditions suggests that transesterification reactions may be a possible chemical change that produced subsequent morphological changes. In accordance with these observations, the intensity of light, scattered by the nanoparticles, as measured by DLS, was also dependent on the pH of water and temperature (Figure 6c). In the case of nanoparticles in pH 5.0 aqueous solutions, both at 25 and 37 °C, the signal intensities became weaker rapidly, reaching below 20% as compared to the initial intensity, and finally were undetectable within a week. The signal intensity of light scattered by micelles in pH 7.4 aqueous solution at 37 °C decreased gradually over a period of month. Meanwhile, there was no significant change in the signal intensity for the micelles in pH 7.4 aqueous solution at 25 °C throughout the monitoring period. Taken together, the acidity of solutions was the primary determinant of the micelle stability.
Figure 6.
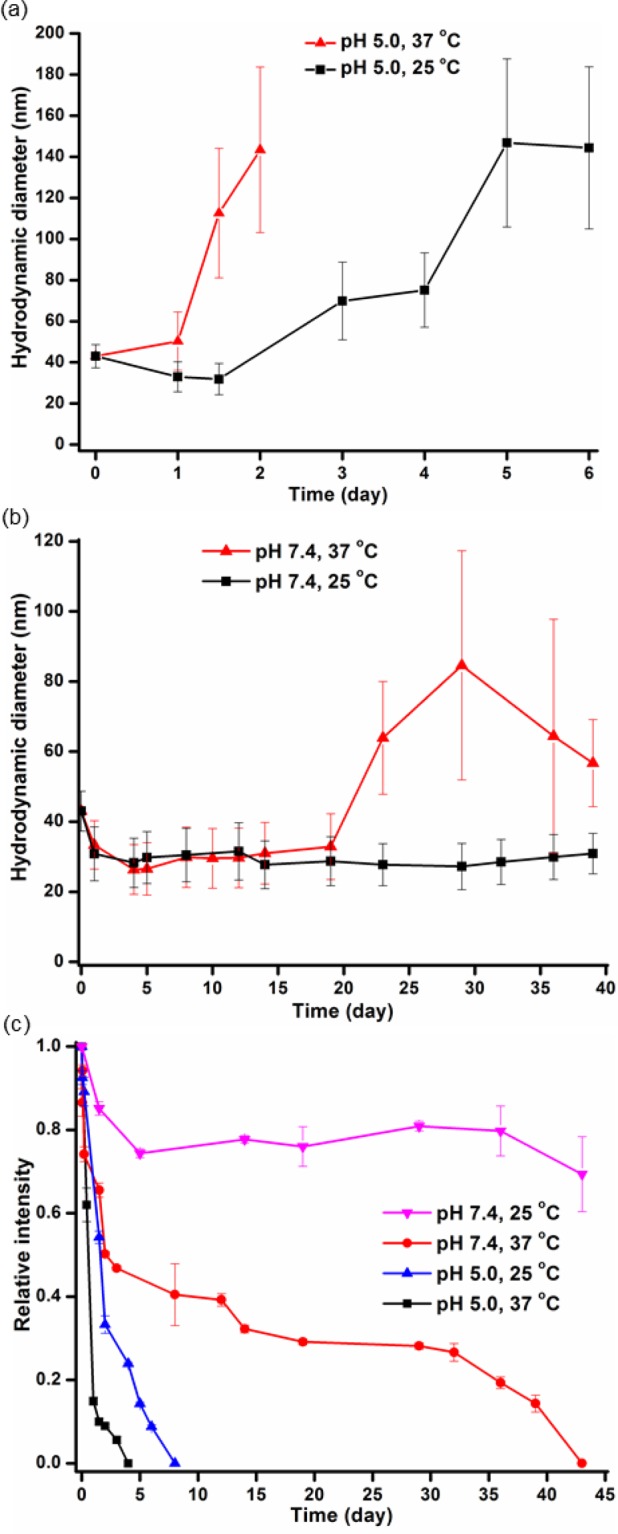
Study of the micelle stability by using DLS. Changes in the hydrodynamic diameter of micelles at pH 5.0 (a) or pH 7.4 (b) and at a temperature of 25 °C (black line) or 37 °C (red line) over a period of time. (c) Changes in the relative intensity of micelles in different environments, pH and temperature, over a period of time. The average values and their standard deviations, from three measurements, are shown.
Identification of the Degradation Products by ESI, GC, and MALDI-TOF Mass Spectrometry
There have been several reports where the hydrolytic or enzymatic degradation behavior of PPE-containing micelles were studied by using titration,44 NMR spectroscopy,44,49 GPC,45−48 and/or DLS41 methods; however, to the best of our knowledge, the identification of the actual degradation products of PPE has not been performed directly. In this study, we successfully identified the degradation products of PPE qualitatively using electrospray ionization (ESI), gas chromatography (GC), and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry. For this study, nanoparticles in aqueous solution at pH 5.0 were incubated at 37 °C until they were not detectable by DLS, and the complete disappearance of 31P resonance signal from the intact PEVEP backbone was confirmed by 31P NMR spectroscopy. The mixture solution containing the degradation products was analyzed by using ESI, GC, and MALDI-TOF MS.
The presence of oligomers and phosphoric acids was confirmed by ESI MS (Figure 7), demonstrating the hydrolytic degradability of phosphoester linkages of PPE. Tandem mass spectrometry was performed on most of the precursor ions as a way of verifying the chemical structures of these ions. The MS/MS product ions are listed along with the structures in Figure 7.
Figure 7.

ESI MS analysis of the degradation products of 8. Mass spectra in negative ion mode; m/z range of 100–2000 (a) and 50–180 (b). See Figures S5 and S6 for MS/MS spectra of f1 and g1.
Two series of oligomers (f and g series) that differ by 44 Da were observed (Figure 7). In order to verify that the f series were not the fragments of the g series, i.e., by losing vinyl alcohol (m/z 44), MS/MS of the lower mass precursor ions m/z 309 and 353 were performed (Figures S5 and S6). The predominant fragment ion for m/z 309 was 141 while that for m/z 353 was 185, thus both losing a neutral repeating unit, 168. In addition, a loss of ethylene glycol was observed for both ions. However, unlike the precursor ion m/z 353, m/z 309 lost vinyl dihydrogen phosphate to produce ion e. This major difference implied that the end group of the f series oligomers is 2-hydroxyethyl dihydrogen phosphate, while that of g series oligomers is bis(2-hydroxyethyl) hydrogen phosphate. Therefore, the f series were not the fragment ions from the g series.
Because of its unionizable nature by electrospray ionization, the presence of ethylene glycol, as one of the degradation products, was confirmed by GC MS with electron ionization (EI) (Figure S7). Neither vinyl- nor vinyl ether-containing compounds were detected using GC MS, which agreed with our observation by 1H NMR spectroscopy, shown in Figure 5. Again, this absence of vinyl or vinyl ether functionalities within the degradation products substantiated our hypothesis of a simultaneous hydrolysis of the vinyl ether moieties during the degradation process of the PPE backbone. Finally, MALDI-TOF MS analysis of the mixture of the degradation products verified the presence of the intact PEG block segment with a single distribution having a spacing of 44 Da, corresponding to a PEG repeat unit (Figure S8).
Cytotoxicity of Micelles of mPEG44-b-PEVEP337 and Their Degradation Products 8
The cytotoxicities of the parent micelles, 7, and their degradation products, 8, were evaluated toward two cell lines, RAW 264.7 mouse macrophages and OVCAR-3 human ovarian adenocarcinoma cells, at a concentration range from 3 to 3000 μg/mL for 24 h (Figure 8). Both 7 and 8 maintained high cell viability over the range of the tested concentrations in both cell lines. We have previously observed low cytotoxicity and immunotoxicity of PPE-based micelles with different surface charges, their shell cross-linked analogues, and their degradation products even though we were not able to identify the degradation products at that time.50 These PPE-based nanoparticles are expected to have broad implications in clinical nanomedicine as alternative vehicles to those involved in several of the currently available medications, with precise control over their molecular structures and overall architectures.
Figure 8.
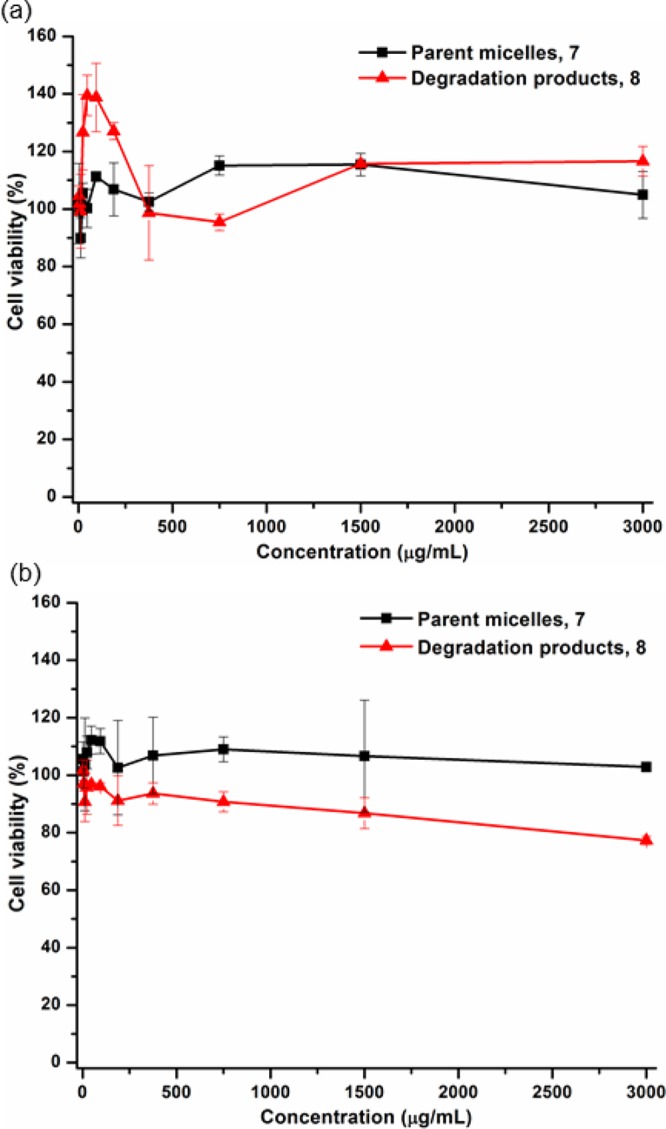
Cytotoxicity of the parent micelles of mPEG44-b-PEVEP337 (black line) and their degradation products 8 (red line) at a concentration range of 3–3000 μg/mL for 24 h in RAW 264.7 mouse macrophages (a) and OVCAR-3 human ovarian adenocarcinoma cells (b).
Conclusions
In conclusion, a novel polyphosphoester with ethylene glycol vinyl ether side chain functionality was developed as a versatile template for postpolymerization modifications, and its degradability and biocompatibility were investigated. A well-defined (PDI ≤1.05) homopolymer with vinyl ether side chain functionality was prepared by conducting ROP using an organocatalyst, DBU. The kinetic study of this homopolymerization revealed an excellent controllability during ROP with predetermined molecular weights and narrow molecular weight distributions. Subsequently, the vinyl ether side chain moieties displayed chemical availability and reactivity upon conjugation with hydroxyl- or thiol-containing model small molecules via three different types of conjugation chemistries—thiol–ene “click” reaction, acetalization, or thio–acetalization reaction—resulting in modified polymers that contained either stable thio–ether or hydrolytically labile acetal or thio–acetal linkages. Despite the relatively low conversion percentages observed during acetalization and thio–acetalization, ca. 18 and 8%, respectively, we anticipate that these degrees of conjugation efficiency would be adequate to achieve a sufficient loading of diagnostic and/or therapeutic molecules into this nanoparticle system. Meanwhile, amphiphilic diblock copolymers, mPEG44-b-PEVEP33, were also prepared by ROP, and they afforded well-defined micelles with a narrow and monomodal size distribution in water. The degradation study of the prepared micelles demonstrated a full acid-catalyzed hydrolytic degradation behavior of both the side chain functionalities and the backbone linkages. Finally, the parent micelles and their degradation products, as identified qualitatively by mass spectrometry, were found to be nontoxic toward RAW 264.7 mouse macrophages and OVCAR-3 human ovarian adenocarcinoma cells. The fundamental understanding of selective hydrolysis of the vinyl ether and/or acetal/thio–acetal moieties for the introduction of hydroxyl groups to the PPE system, which was conventionally limited to a cyclic PPE monomer, as a potential protecting group strategy is currently under investigation. Moreover, incorporation of biologically active molecules into these PEVEP-based functional, degradable polymers via the presented conjugation chemistries is underway.
Acknowledgments
We gratefully acknowledge financial support from the National Heart Lung and Blood Institute as a Program of Excellence in Nanotechnology (HHSN268201000046C) and from the National Institute of Diabetes and Digestive and Kidney Diseases (R01-DK082546). The Welch Foundation is gratefully acknowledged for support through the W. T. Doherty-Welch Chair in Chemistry, Grant No. A-0001.
Supporting Information Available
Experimental section and additional data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Kamber N. E.; Jeong W.; Waymouth R. M.; Pratt R. C.; Lohmeijer B. G. G.; Hedrick J. L. Chem. Rev. 2007, 107, 5813–5840. [DOI] [PubMed] [Google Scholar]
- Tempelaar S.; Mespouille L.; Coulembier O.; Dubois P.; Dove A. P. Chem. Soc. Rev. 2013, 42, 1312–1336. [DOI] [PubMed] [Google Scholar]
- Clément B.; Grignard B.; Koole L.; Jérôme C.; Lecomte P. Macromolecules 2012, 45, 4476–4486. [Google Scholar]
- Sanders D. P.; Coady D. J.; Yasumoto M.; Fujiwara M.; Sardon H.; Hedrick J. L. Poly. Chem. 2014, 5, 327–329. [Google Scholar]
- Sanders D. P.; Fukushima K.; Coady D. J.; Nelson A.; Fujiwara M.; Yasumoto M.; Hedrick J. L. J. Am. Chem. Soc. 2010, 132, 14724–14726. [DOI] [PubMed] [Google Scholar]
- Meldal M.; Tornøe C. W. Chem. Rev. 2008, 108, 2952–3015. [DOI] [PubMed] [Google Scholar]
- Tasdelen M. A. Polym. Chem. 2011, 2, 2133–2145. [Google Scholar]
- Lowe A. B. Polym. Chem. 2010, 1, 17–36. [Google Scholar]
- Hoyle C. E.; Bowman C. N. Angew. Chem., Int. Ed. 2010, 49, 1540–1573. [DOI] [PubMed] [Google Scholar]
- Iha R. K.; Wooley K. L.; Nyström A. M.; Burked D. J.; Kade M. J.; Hawker C. J. Chem. Rev. 2009, 109, 5620–5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder W. H.; Sachsenhofer R. Macromol. Rapid Commun. 2007, 28, 15–54. [Google Scholar]
- Fournier D.; Hoogenboom R.; Schubert U. S. Chem. Soc. Rev. 2007, 36, 1369–1380. [DOI] [PubMed] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. [DOI] [PubMed] [Google Scholar]
- Thirumurugan P.; Matosiuk D.; Jozwiak K. Chem. Rev. 2013, 113, 4905–4979. [DOI] [PubMed] [Google Scholar]
- Gillies E. R.; Jonsson T. B.; Fréchet J. M. J. J. Am. Chem. Soc. 2004, 126, 11936–11943. [DOI] [PubMed] [Google Scholar]
- Gillies E. R.; Frechet J. M. J. Chem. Commun. 2003, 1640–1641. [DOI] [PubMed] [Google Scholar]
- Murthy N.; Thng Y. X.; Schuck S.; Xu M. C.; Fréchet J. M. J. J. Am. Chem. Soc. 2002, 124, 12398–12399. [DOI] [PubMed] [Google Scholar]
- Tang R.; Ji W.; Panus D.; Palumbo R. N.; Wang C. J. Controlled Release 2011, 151, 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.; Du F.; Wang Y.; Ji S.; Liang D.; Yu L.; Li Z. Biomacromolecules 2007, 9, 109–115. [DOI] [PubMed] [Google Scholar]
- Bae Y.; Fukushima S.; Harada A.; Kataoka K. Angew. Chem., Int. Ed. 2003, 42, 4640–4643. [DOI] [PubMed] [Google Scholar]
- Filippov S. K.; Franklin J. M.; Konarev P. V.; Chytil P.; Etrych T.; Bogomolova A.; Dyakonova M.; Papadakis C. M.; Radulescu A.; Ulbrich K.; Stepanek P.; Svergun D. I. Biomacromolecules 2013, 14, 4061–4070. [DOI] [PubMed] [Google Scholar]
- Delplace V.; Couvreur P.; Nicolas J. Polym. Chem. 2014, 5, 1529–1544. [Google Scholar]
- Steen K. H.; Steen A. E.; Reeh P. W. J. Neurosci. 1995, 15, 3982–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannock I. F.; Rotin D. Cancer Res. 1989, 49, 4373–4384. [PubMed] [Google Scholar]
- Helmlinger G.; Sckell A.; Dellian M.; Forbes N. S.; Jain R. K. Clin. Cancer Res. 2002, 8, 1284–1291. [PubMed] [Google Scholar]
- Gillies E. R.; Goodwin A. P.; Fréchet J. M. J. Bioconjugate Chem. 2004, 15, 1254–1263. [DOI] [PubMed] [Google Scholar]
- Heffernan M. J.; Murthy N. Bioconjugate Chem. 2005, 16, 1340–1342. [DOI] [PubMed] [Google Scholar]
- Bachelder E. M.; Beaudette T. T.; Broaders K. E.; Dashe J.; Fréchet J. M. J. J. Am. Chem. Soc. 2008, 130, 10494–10495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natalello A.; Tonhauser C.; Frey H. ACS Macro Lett. 2013, 2, 409–413. [DOI] [PubMed] [Google Scholar]
- Satoh K.; Poelma J. E.; Campos L. M.; Stahl B.; Hawker C. J. Polym. Chem. 2012, 3, 1890–1898. [Google Scholar]
- Tonhauser C.; Schüll C.; Dingels C.; Frey H. ACS Macro Lett. 2012, 1, 1094–1097. [DOI] [PubMed] [Google Scholar]
- Li Y.; Du W.; Sun G.; Wooley K. L. Macromolecules 2008, 41, 6605–6607. [Google Scholar]
- Mangold C.; Dingels C.; Obermeier B.; Frey H.; Wurm F. Macromolecules 2011, 44, 6326–6334. [Google Scholar]
- Rickerby J.; Prabhakar R.; Ali M.; Knowles J.; Brocchini S. J. Mater. Chem. 2005, 15, 1849–1856. [Google Scholar]
- Wang H.; He J.; Zhang M.; Tao Y.; Li F.; Tam K. C.; Ni P. J. Mater. Chem. B 2013, 1, 6596–6607. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Wang H.; Liu J.; Deng L.; Liu J.; Dong A.; Zhang J. Biomacromolecules 2013, 14, 3973–3984. [DOI] [PubMed] [Google Scholar]
- Gu Y.; Zhong Y.; Meng F.; Cheng R.; Deng C.; Zhong Z. Biomacromolecules 2013, 14, 2772–2780. [DOI] [PubMed] [Google Scholar]
- Lim Y. H.; Heo G. S.; Cho S.; Wooley K. L. ACS Macro Lett. 2013, 2, 785–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson T. P.; Lonnecker A. T.; Heo G. S.; Zhang S.; Dove A. P.; Wooley K. L. Biomacromolecules 2013, 14, 3346–3353. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Zou J.; Zhang F.; Elsabahy M.; Felder S. E.; Zhu J.; Pochan D. J.; Wooley K. L. J. Am. Chem. Soc. 2012, 134, 18467–18474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; Zhang S.; Zhang F.; Loftis A.; Pavía-Sanders A.; Zou J.; Fan J.; Taylor J.-S. A.; Wooley K. L. Adv. Mater. 2013, 25, 5609–5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson T. P.; Lim Y. H.; Flores J. A.; Heo G. S.; Zhang F.; Zhang S.; Samarajeewa S.; Raymond J. E.; Wooley K. L. Langmuir 2014, 30, 631–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.-C.; Yuan Y.-Y.; Du J.-Z.; Yang X.-Z.; Wang J. Macromol. Biosci. 2009, 9, 1154–1164. [DOI] [PubMed] [Google Scholar]
- Baran J.; Penczek S. Macromolecules 1995, 28, 5167–5176. [Google Scholar]
- Wang Y.-C.; Tang L.-Y.; Li Y.; Wang J. Biomacromolecules 2008, 10, 66–73. [DOI] [PubMed] [Google Scholar]
- Iwasaki Y.; Nakagawa C.; Ohtomi M.; Ishihara K.; Akiyoshi K. Biomacromolecules 2004, 5, 1110–1115. [DOI] [PubMed] [Google Scholar]
- Wang Y.-C.; Tang L.-Y.; Sun T.-M.; Li C.-H.; Xiong M.-H.; Wang J. Biomacromolecules 2007, 9, 388–395. [DOI] [PubMed] [Google Scholar]
- Du J.-Z.; Chen D.-P.; Wang Y.-C.; Xiao C.-S.; Lu Y.-J.; Wang J.; Zhang G.-Z. Biomacromolecules 2006, 7, 1898–1903. [DOI] [PubMed] [Google Scholar]
- Liu J.; Huang W.; Pang Y.; Zhu X.; Zhou Y.; Yan D. Biomacromolecules 2010, 11, 1564–1570. [DOI] [PubMed] [Google Scholar]
- Elsabahy M.; Zhang S.; Zhang F.; Deng Z. J.; Lim Y. H.; Wang H.; Parsamian P.; Hammond P. T.; Wooley K. L. Sci. Rep. 2013, 3:3313, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.