

Abstract
Objective
A severe but treatable form of immune-mediated encephalitis is associated with antibodies in serum and cerebrospinal fluid (CSF) against the GluN1 subunit of the N-methyl-D-aspartate receptor (NMDAR). Prolonged exposure of hippocampal neurons to antibodies from patients with anti-NMDAR encephalitis caused a reversible decrease in the synaptic localization and function of NMDARs. However, acute effects of the antibodies, fate of the internalized receptors, type of neurons affected, and whether neurons develop compensatory homeostatic mechanisms were unknown and are the focus of this study.
Methods
Dissociated hippocampal neuron cultures and rodent brain sections were used for immunocytochemical, physiological, and molecular studies.
Results
Patient antibodies bind to NMDARs throughout the rodent brain, and decrease NMDAR cluster density in both excitatory and inhibitory hippocampal neurons. They rapidly increase the internalization rate of surface NMDAR clusters, independent of receptor activity. This internalization likely accounts for the observed decrease in NMDAR-mediated currents, as no evidence of direct blockade was detected. Once internalized, antibody-bound NMDARs traffic through both recycling endosomes and lysosomes, similar to pharmacologically induced NMDAR endocytosis. The antibodies are responsible for receptor internalization, as their depletion from CSF abrogates these effects in hippocampal neurons. We find that although anti-NMDAR antibodies do not induce compensatory changes in glutamate receptor gene expression, they cause a decrease in inhibitory synapse density onto excitatory hippocampal neurons.
Interpretation
Our data support an antibody-mediated mechanism of disease pathogenesis driven by immunoglobulin-induced receptor internalization. Antibody-mediated downregulation of surface NMDARs engages homeostatic synaptic plasticity mechanisms, which may inadvertently contribute to disease progression. Ann Neurol 2014;76:108–119
Glutamatergic transmission is central to many functions thought to depend on synaptic plasticity, including learning and memory, cognition, and behavior.1,2 Several newly described immune-mediated encephalitides that target synaptic antigens have offered novel insights into the link between synapse function and human cognition and behavior.3,4 One form of autoimmune encephalitis is associated with antibodies against the N-methyl-D-aspartate receptor (NMDAR).5,6 Consistent with the prominent role of NMDARs in glutamatergic transmission as well as activity-dependent plasticity, symptoms of anti-NMDAR encephalitis include sudden behavioral, memory, and personality changes that progress to seizures, autonomic instability, and coma. If left untreated, irreversible deficits and death can occur. Immunotherapy treatment leads to a substantial to full recovery for about 80% of patients.7
NMDARs, along with α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) and kainate receptors, mediate glutamatergic synaptic transmission and have a prominent role in synaptic plasticity, learning, and behavior. Pharmacological blockade or genetic reduction of NMDARs alters learning and memory,8–10 excitatory–inhibitory balance,11,12 and behavior.13–15 Defects in glutamate signaling have been linked to neuropsychiatric disorders, and NMDAR hypofunction has been proposed to be part of the pathophysiological mechanisms underlying schizophrenia.16 Subanesthetic doses of NMDAR blockers such as phencyclidine and ketamine are psychotomimetic in humans and rodents, and cause the stereotypic movements, autonomic instability, and seizures that are characteristic of anti-NMDAR encephalitis.17,18
The striking parallels between patient symptoms and the consequences of NMDAR hypofunction described above underscore the importance of determining the mechanisms of antibody-mediated dysfunction in this disease. Patient antibodies cause a selective, reversible decrease of NMDAR surface density, synaptic localization, and currents in vitro.6,19,20 Here, we explored mechanisms of disease pathogenesis, investigating whether patient antibodies preferentially bind to NMDARs on specific types of neurons or brain regions, the time course of receptor internalization, whether antibodies directly antagonize the receptor, whether components besides immunoglobulins within patient cerebrospinal fluid (CSF) can contribute to downregulation of NMDARs, and whether neurons engage homeostatic mechanisms in response to the decrease in glutamatergic transmission. Understanding the acute mechanisms of antibody-mediated dysfunction sets the stage for future studies in in vivo models of anti-NMDAR encephalitis.
Materials and Methods
Cell Culture and Treatment
Hippocampal neurons were prepared and maintained from embryonic day 18 rat pups as previously described.19 Neurons were treated on in vitro day 14 (DIV14; unless otherwise noted) with CSF from patients or controls at a dilution of 1:20, and drugs at the following concentrations: amino-phosphonovaleric acid (APV), 50μM; picrotoxin, 10μM; NMDA, 1mM; glycine, 10μM. Cerebrospinal fluid and serum were obtained from randomly selected patients with well-characterized clinical manifestations of anti-NMDAR encephalitis. Antibodies to the NMDAR were demonstrated as previously reported.6 Control samples were obtained from patients undergoing CSF screening for various disorders not associated with antibodies against the NMDAR.
Immunostaining
Immunostaining protocols for cultured neurons and rodent brain sections have been described in detail elsewhere.19 Neurons were treated as specified in the text and incubated with the following primary antibodies: to label NMDARs, anti-GluN1 (Millipore, Billerica, MA; AB9864R, 1:100) and anti-GluN1 (Sigma, St Louis, MO; G8913,1:100); inhibitory neurons, anti–glutamic acid decarboxylase 6 (GAD6; Developmental Studies Hybridoma Bank, Iowa City, IA; 1:20; the monoclonal antibody was developed by Dr David I. Gottlieb at Washington University School of Medicine and is maintained at the University of Iowa); presynaptic terminals, anti-bassoon (Stressgen Bioreagents, Ann Arbor, MI; VAM-PS003, 1:400); recycling endosomes, anti-Rab11 (Zymed Laboratories, San Francisco, CA; 71–5300, 1:100); lysosomes, anti-Lamp1 (Enzo Life Sciences, Plymouth Meeting, PA; ADI-VAM-EN001, 1:100); γ-aminobutyric acid receptors (GABAARs), anti-GABAAβ2/3 (Millipore, 05–474, 1:500); and presynaptic inhibitory terminals, anti-vesicular GABA transporter (vGAT; Synaptic Systems, Göttingen, Germany; 131-004, 1:1,000). Manufacturer's websites provide controls for specificity of all primary antibodies used. Omission of primary antibodies was used as a control for each of the secondary antibodies, which were raised in goat against rabbit, mouse, or guinea pig immunoglobulin G (IgG) and conjugated to various Alexa Fluor dyes. All secondary antibodies were obtained from Invitrogen (Carlsbad, CA). Coverslips were mounted and imaged on a confocal microscope (Leica, Wetzlar, Germany; TCS SP5) and analyzed using interactive software (custom-written ImageJ macros).19
To selectively label internalized NMDARs, surface NMDARs were bound by incubation with patient CSF (1:20). After treatment, coverslips were incubated with unconjugated goat anti-human secondary antibody at 1:10. Then, neurons were fixed, permeabilized, and stained with fluorescently conjugated goat anti-human secondary antibody, which only labeled internalized receptors due to the saturation of surface receptors by unlabeled antibody.
To label surface and internal receptors, after incubation with patient antibodies, the remaining surface antibody-bound receptors were labeled with a fluorescent secondary antibody. Neurons were fixed, permeabilized, and stained with a different secondary antibody against human IgG to label the internalized antibody-bound receptors, as well as anti-GluN1 to label the total population of NMDARs. To induce NMDAR internalization pharmacologically, we used 2 different treatment protocols. The first was a 24-hour incubation with picrotoxin, which blocks inhibitory GABAAR-mediated synaptic transmission, causing a homeostatic decrease of NMDARs.21 The second was a 15-minute exposure to NMDA plus glycine, which activates the NMDAR and causes rapid internalization.22
Electrophysiology
Electrophysiology protocols have been described in detail elsewhere.19 Whole cell voltage clamp recordings were made from DIV17–21 neurons that had been treated for 30 minutes with patient or control CSF, or 24 hours with patient F(ab) fragments. Extracellular physiological solution was (in millimolars): 119 NaCl, 5 KCl, 2 CaCl2, 30 glucose, 10 HEPES, pH = 7.4. For excitatory currents, intracellular pipet solution was (in millimolars): 100 cesium gluconate, 0.2 ethyleneglycoltetraacetic acid (EGTA), 5 MgCl2, 2 adenosine triphosphate, 0.3 guanosine triphosphate, 40 HEPES, pH = 7.2. For inhibitory currents, intracellular solution was (in millimolars): 140 KCl, 2 MgCl2, 11 EGTA, 10 HEPES, 7 glucose, pH = 7.3. Tetrodotoxin (TTX) (1μM) was used to block action potentials, picrotoxin (10μM) was used to block GABAAR-mediated miniature inhibitory postsynaptic currents (mIPSCs), cyano-nitroquinoxaline-dione (CNQX) (10μM) was used to block AMPAR-mediated miniature excitatory postsynaptic currents (mEPSCs), and APV (50μM) was used to block NMDAR-mediated mEPSCs. Spontaneous miniature postsynaptic currents were detected and analyzed using MiniAnalysis (Synaptosoft, Leonia, NY). NMDAR and AMPAR components of mEPSCs were separated temporally by their distinct kinetics. The amplitude of the NMDAR-mediated current was determined in a window between 15 and 25 milliseconds after the peak of the averaged AMPAR-mediated component, which has a fast, <1-millisecond rise time.19
F(ab) Fragment Preparation
F(ab) fragments were prepared from patients' serum whole IgG using a kit according to the manufacturer's directions (Pierce Fab Micro Preparation Kit; Thermo Scientific, Waltham, MA).
Immunodepletion
Patient CSF was incubated on 50:50 Protein A and rProtein G agarose column (Invitrogen) equilibrated in binding buffer (0.01M sodium phosphate, pH 7.0, 0.15M sodium chloride). The column was rocked for 1 hour at room temperature, and CSF was collected and stored at −20°C until use.
Statistical Analysis
Data sets were analyzed using a Mann-Whitney U test or 1-way analysis of variance test.
Results
Patient and Commercial GluN1 Antibodies Were Similarly Distributed after Immunostaining
Patient antibodies stained rat hippocampus in sections more intensely than other brain regions, such as cortex, striatum, and hindbrain.6 To determine whether this pattern was due to a modification of the epitope specific to the hippocampus, or due to a higher expression of NMDARs within the hippocampus, we asked whether the staining pattern of patient antibodies was the same as that of 2 commercially available antibodies that recognize epitopes within the intracellular, C-terminal domain of GluN1.
Staining with commercial antibodies produced the same enrichment in hippocampal immunoreactivity as patient CSF (Fig 1A). Patient and commercial antibodies both stained cortex, striatum, and cerebellum as well as hippocampus, although less intensely. The difference in overall staining intensity between the sample on the left (Patient 1) and on the right (Patient 2) is due to a difference in the patients' antibody titers. Double labeling with patient and commercial antibodies shows their considerable colocalization in the dendritic layers of the dentate gyrus (see Fig 1B). These results suggest that the observed preferential hippocampal staining of patient antibodies was due to a higher density of NMDARs within the hippocampus compared with other regions.
FIGURE 1.
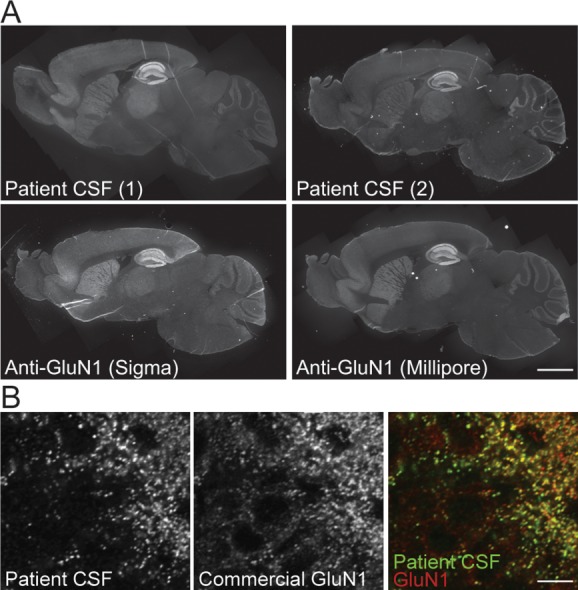
Patient and commercial GluN1 antibodies were similarly distributed after immunostaining. (A) Sagittal mouse brain sections immunostained for GluN1 with 2 patient cerebrospinal fluid (CSF) samples (top) or 2 commercial anti-GluN1 antibodies against C-terminal epitopes (bottom). The pattern of N-methyl-D-aspartate receptor (NMDAR) localization is similar, with the greatest intensity of immunoreactivity in the hippocampus and less in cortex, striatum, and cerebellum. Scale bar = 1mm. (B) Cellular localization in hippocampal dentate gyrus neurons stained with patient CSF (left, green in overlay) or commercial anti-GluN1 (middle, red in overlay), demonstrating colabeling of NMDAR clusters throughout the neuropil. Scale bar = 20μm.
Patient Antibodies Decreased Surface NMDARs on Excitatory and Inhibitory Hippocampal Neurons
We asked whether the effects of patient antibody treatment differed between excitatory and inhibitory neurons, potentially disrupting the excitatory–inhibitory balance, as occurs following administration of some NMDAR antagonists that cause symptoms similar to anti-NMDAR encephalitis.11,23 Cultured hippocampal neurons were treated with control or patient CSF, then stained for surface NMDARs, total GluN1, and GAD6 to label inhibitory neurons. Inhibitory neurons were identified by strong immunoreactivity of GAD6 in cell bodies.
In excitatory, GAD-negative neurons, patient CSF treatment caused a significant decrease in surface NMDAR cluster density compared with control CSF (Fig 2). A similar result was seen in inhibitory, GAD-positive neurons. Patient antibodies also caused a decrease in the intensity of surface NMDAR clusters. There was no difference in the density of surface NMDAR clusters recognized by patient antibodies in these 2 populations of neurons in control conditions. Thus, there is a lack of regional or neuronal subtype specificity in the binding and effects of patient antibodies, suggesting broad actions of anti-NMDAR antibodies on cell types across brain regions.
FIGURE 2.
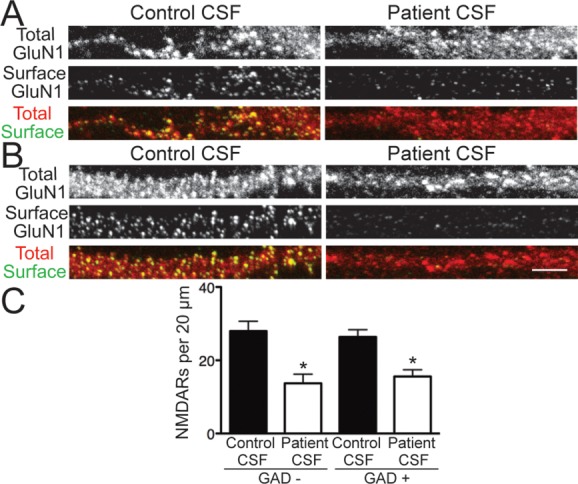
Patient antibodies decreased surface N-methyl-D-aspartate receptors (NMDARs) on excitatory and inhibitory hippocampal neurons. (A, B) Hippocampal neurons immunostained for surface GluN1, total GluN1, and glutamic acid decarboxylase 6 (GAD6) after treatment for 24 hours with control or patient cerebrospinal fluid (CSF) and imaged with confocal microscopy. (A) GAD−, excitatory neurons; (B) GAD+, inhibitory neurons. Surface NMDARs were defined as the colocalization of nonpermeabilized patient antibody staining, which recognized an extracellular epitope, and permeabilized commercial GluN1 staining, which recognized an intracellular epitope. Scale bar = 5μm. (C) Quantification of surface NMDAR density on excitatory, GAD− neurons, and inhibitory, GAD+ neurons (n = 12–28 cells per condition, 3 independent experiments). Treatment with patient CSF caused a similar, significant reduction in surface NMDAR clusters on both excitatory and inhibitory neurons compared to control CSF treatment (excitatory, 27.97 ± 2.67 vs 13.71 ± 2.51; inhibitory, 26.38 ± 1.96 vs 15.6 ± 1.83). *p < 0.05, 1-way analysis of variance.
Patient Antibodies Increased the Rate of NMDAR Internalization in a Time-Dependent and Activity-Independent Manner
We next wanted to study the time course and dynamics of NMDAR internalization. Hippocampal neurons were treated with patient antibodies or patient F(ab) fragments (previously shown not to decrease surface NMDAR clusters19) for 15 minutes to 48 hours.
Surface NMDAR cluster density was significantly decreased compared with F(ab) treatment after 12 hours of exposure to patient antibodies (Fig 3A, B). There was no further reduction with longer treatment. The accumulation of internalized NMDAR clusters followed the same time course, also reaching a maximum following 12 hours of treatment.
FIGURE 3.

Patient antibodies increased the rate of N-methyl-D-aspartate receptor (NMDAR) internalization in a time-dependent and activity-independent manner. (A) Hippocampal neurons were treated for various lengths of time with patient cerebrospinal fluid (CSF) or F(ab) fragments and then immunostained for surface antibody-bound GluN1, internalized antibody-bound GluN1, and total GluN1. F(ab) fragments were used to monitor the constitutive, antibody-independent turnover of NMDARs. Coverslips were imaged, and NMDAR cluster density was analyzed. Scale bar = 5μm. (B) Quantification of surface and internalized NMDARs following treatment (n = 17–55 cells per condition). Surface NMDAR density was significantly decreased after 12 hours of patient CSF treatment compared with patient antibody-derived F(ab) fragments (86.02 ± 7.46% vs 140.2 ± 8.68%), after which surface levels reached a plateau. This was paralleled by an increase over time in the density of internalized NMDARs (by 12 hours, 72.01 ± 9.67% vs 17.83 ± 4.26%). *p < 0.05, 1-way analysis of variance (ANOVA). (C) Hippocampal neurons were treated for 24 hours with control or patient CSF with or without amino-phosphonovaleric acid (APV), then immunostained for surface GluN1, total GluN1, and presynaptic terminal marker bassoon. Scale bar = 5μm. (D) Quantification of synaptic NMDAR density (n = 12–18 cells per condition, 3 independent experiments). Patient CSF caused a significant reduction in NMDAR density in both the presence (46.86 ± 4.17% of control CSF) and absence (52.18 ± 6.19% of control CSF) of APV. *p < 0.05, 1-way ANOVA.
We next examined whether internalization was stimulated by any mechanism other than antibody-mediated crosslinking. For example, activation of glutamate receptors can prime them for internalization.22 Agonism by patient antibodies could decrease surface NMDAR levels by similar mechanisms. This could change our understanding of the effects of patient antibodies on NMDARs, and point to new options for patient care. To test this possibility, hippocampal neurons were treated with control or patient CSF with or without APV, an NMDAR blocker, then stained for surface NMDARs, bassoon (for presynaptic terminals), and total GluN1.
As expected, patient CSF caused a significant decrease in synaptic NMDAR clusters (see Fig 3C, D). This effect was not mitigated by the presence of APV, demonstrating that antibody-mediated internalization was independent of NMDAR activity and did not occur as a compensatory response to agonism of the receptor. Therefore, the mechanism of internalization was likely due primarily to the effects of NMDAR crosslinking by patient antibodies.
Patient Antibody-Bound Receptors Trafficked through Recycling Endosomes and Lysosomes
In anti-NMDAR encephalitis, NMDARs are bound by immunoglobulins during endocytosis, a scenario that would not occur under physiological conditions of receptor internalization. To examine whether intracellular NMDAR trafficking was influenced by this unique situation, we compared patient antibody treatment with 2 pharmacological manipulations that caused NMDAR internalization, picrotoxin (a GABAAR blocker) and NMDA/glycine.
Hippocampal neurons were treated with 1 of the following: patient CSF for 24 hours, patient F(ab) fragments for 24 hours, F(ab) fragments plus picrotoxin for 24 hours, or F(ab) fragments for 24 hours plus NMDA/glycine for 15 minutes (see Materials and Methods). F(ab) fragments allowed us to track NMDAR internalization induced by pharmacological treatment, without the complication of full patient IgG further stimulating endocytosis (see Fig 3A, B). Following treatment, neurons were stained for internalized NMDARs, and Rab11 and Lamp1, markers for recycling endosomes and lysosomes, respectively.
Patient CSF, picrotoxin, and NMDA/glycine all caused a significant increase in NMDAR internalization compared with F(ab) fragments alone (Fig 4). Between the 3 conditions that promoted internalization, there was no significant change in the proportion of internalized NMDARs that localized to recycling endosomes or lysosomes. Additionally, for all 3 conditions, a greater percentage of internalized receptors colocalized with recycling endosomes than lysosomes. These data suggest that the postendocytic trafficking of NMDARs is not affected by IgG binding the receptor.
FIGURE 4.
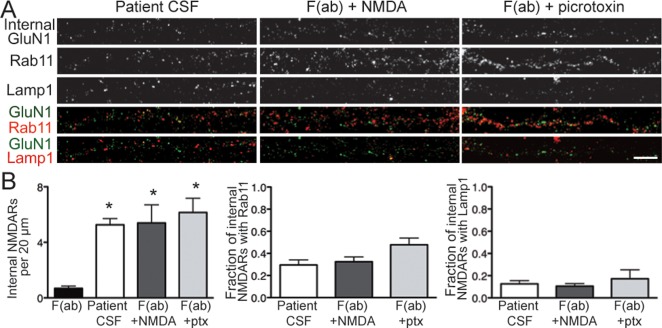
Patient antibody-bound N-methyl-D-aspartate receptors (NMDARs) trafficked through recycling endosomes and lysosomes. (A) Hippocampal neurons were treated with 1 of the following: F(ab) fragments generated from patient antibodies, patient cerebrospinal fluid (CSF; full immunoglobulin G), F(ab) plus NMDA and glycine, or F(ab) plus picrotoxin. Neurons were then immunostained for internalized antibody-bound GluN1, Rab11 (to mark recycling endosomes), and Lamp1 (to mark lysosomes). Scale bar = 5μm. (B) Quantification of intracellular trafficking of NMDARs following treatment (n = 15–19 cells per condition, 3 independent experiments). Left panel shows quantification of internalized NMDAR clusters following treatment; middle panel shows quantification of internalized NMDAR clusters colocalized with recycling endosome marker Rab11; right panel shows quantification of internalized NMDAR clusters colocalized with lysosome marker Lamp1. After 24 hours, there was a significant increase in internalized NMDAR cluster density following treatment with patient CSF, F(ab) plus NMDA, and F(ab) plus picrotoxin (ptx) compared with F(ab) fragments alone (5.26 ± 0.45, 5.40 ± 1.31, 6.15 ± 1.03 vs 0.68 ± 0.17). *p < 0.05, 1-way analysis of variance (ANOVA). There were no significant differences in the intracellular localization of NMDARs between the 3 treatments used to induce internalization (recycling endosomes: 29.56 ± 4.54%, 32.46 ± 4.37%, 47.83 ± 6.01%; lysosomes: 12.03 ± 2.80%, 10.16 ± 2.25%, 17.21 ± 8.12%); 1-way ANOVA, p > 0.05.
Patient CSF Caused NMDAR Hypofunction through Immunoglobulin-Induced Receptor Internalization
Patient antibodies could potentially modulate receptor function independently of their ability to internalize NMDARs because the epitope is within the N-terminus of GluN1,24 a region that also contains the ligand-binding domain. To test this possibility, we first performed whole cell patch clamp recordings of mEPSCs from hippocampal neurons on DIV17–21 that had been treated with control or patient CSF for 30 minutes, a time point before significant internalization of NMDARs has occurred (see Fig 3A, B). NMDAR-mediated current amplitudes from neurons treated for only 30 minutes with patient CSF were not significantly different from current amplitudes in neurons treated with control CSF (Fig 5; p > 0.05). Consistent with this finding, we also observed that exposure for 24 hours to F(ab) fragments, which were incapable of triggering receptor internalization (see Fig 3A, B), did not result in a significant decrease in NMDAR-mediated current amplitude. Both of these findings were in contrast to the large reduction in NMDAR-mediated mEPSC amplitude that resulted from 24 hours of treatment with patient CSF,19 and provide physiological evidence to support the notion that antibody-mediated NMDAR hypofunction results from receptor internationalization and not an acute antagonism of receptor function.
FIGURE 5.
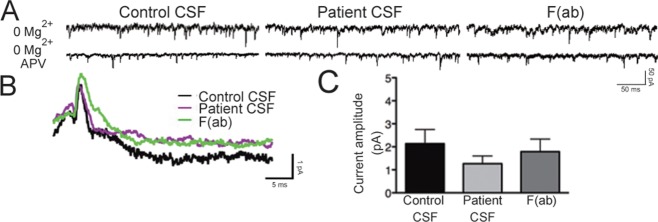
Patient antibodies do not acutely antagonize the N-methyl-D-aspartate receptor (NMDAR). (A) Representative traces of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor (AMPAR)-mediated and NMDAR-mediated miniature excitatory postsynaptic currents (mEPSCs) from whole cell patch clamp recordings of hippocampal neurons following treatment with control or patient cerebrospinal fluid (CSF) for 30 minutes or F(ab) fragments for 24 hours. Recordings were made in the presence of tetrodotoxin, picrotoxin, and 0mM Mg2+ to isolate dual glutamatergic currents. In the lower traces, amino-phosphonovaleric acid (APV) was added to block the NMDAR-mediated portion of the mEPSC. (B) Representative averaged NMDAR-mediated mEPSCs recorded from neurons in different treatment conditions. The difference between the 0mM Mg2+ condition and the 0mM Mg2+ plus APV condition is plotted, showing the NMDAR current. (C) Quantification of NMDAR current amplitude following treatment (n = 5–10 cells per condition). Amplitude was not significantly different: 2.14 ± 0.62pA, control CSF; 1.26 ± 0.34pA, patient CSF; 1.79 ± 0.55pA, F(ab) fragments; 1-way analysis of variance, p > 0.05.
We next asked whether other factors within patient CSF, such as cytokines or complement components, were necessary for NMDAR internalization. We previously demonstrated that IgGs from patient serum and CSF were sufficient to decrease NMDAR cluster density,19 but whether they were also necessary to do so remained unknown. Hippocampal neurons were stained with IgG-depleted patient CSF to confirm complete depletion, then treated with control CSF, patient CSF, or IgG-depleted CSF, and stained for surface and total NMDARs and bassoon.
Depleted CSF did not retain immunoreactivity on hippocampal neurons, indicating that IgGs were successfully depleted from the samples (Fig 6). Unlike patient CSF, treatment with depleted CSF did not reduce NMDAR cluster density, demonstrating that in the absence of IgG, patient CSF no longer caused NMDAR internalization.
FIGURE 6.

Patient antibodies are directly pathogenic. (A) Immunoglobulin G (IgG) was removed from patient cerebrospinal fluid (CSF) with protein A and protein G agarose beads. Hippocampal neurons were immunostained with control CSF, patient CSF, or IgG-depleted CSF. Similar to control CSF, depleted CSF did not stain neurons. Scale bar = 20μm. (B) Hippocampal neurons were treated for 24 hours with control CSF, patient CSF, or depleted CSF, then immunostained for surface GluN1, total GluN1, and bassoon. Scale bar = 5μm. (C) Quantification of synaptic N-methyl-D-aspartate receptor (NMDAR) density following treatment (n = 19–21 cells per condition, 3 independent experiments). Depletion of IgG from patient CSF abrogated the reduction in synaptic NMDAR cluster density (depleted CSF, 99.15 ± 6.47% of control CSF; patient CSF, 57.70 ± 7.10% of control CSF). *p < 0.05, 1-way analysis of variance.
These data showed that the removal of patient antibodies from CSF abrogated both neuronal staining and the effects of patient CSF on surface NMDAR density. Together with previous experiments using purified IgG from patient CSF,19 these results demonstrated that immunoglobulins within patient CSF were both necessary and sufficient to cause the loss of surface NMDARs.
Patient Antibodies Caused Homeostatic Plasticity of Inhibitory Synapse Density but Not Gene Expression
We next assessed whether homeostatic plasticity mechanisms were engaged after patient antibody-induced NMDAR internalization. We verified that we were able to detect homeostatic plasticity in our hippocampal cultures using a pharmacological blocker of NMDARs, APV, which had previously been shown to elicit an increase in surface NMDAR levels.25 Hippocampal neurons were treated for 24 hours with APV, then stained for surface and total NMDARs and bassoon.
In contrast to patient CSF, which caused a significant decrease in synaptic NMDAR cluster density (Fig 7A, C), APV treatment resulted in a significant increase. This confirmed that homeostatic plasticity was detectable in our experimental system.
FIGURE 7.

Patient antibodies cause a homeostatic decrease of inhibitory synapse density. (A) Hippocampal neurons were treated for 24 hours with control cerebrospinal fluid (CSF), patient CSF, or amino-phosphonovaleric acid (APV), then immunostained for surface GluN1, total GluN1, and bassoon. Scale bar = 5μm. (B) Quantification of synaptic N-methyl-D-aspartate receptor (NMDAR) density following treatment (n = 18–20 cell per condition, 3 independent experiments). Patient CSF caused a significant decrease in cluster density (43.27 ± 4.48% of control CSF), whereas APV caused a significant increase (160.4 ± 12.69% of control CSF). *p < 0.05, 1-way analysis of variance. (C) Representative traces of γ-aminobutyric acid receptor (GABAAR)-mediated miniature inhibitory postsynaptic currents (mIPSCs) from whole cell patch clamp recordings of hippocampal neurons following treatment with control or patient CSF for 24 hours. Recordings were made in the presence of tetrodotoxin, cyano-nitroquinoxaline-dione (CNQX), and APV. (D) Quantification of mIPSC amplitude and interevent interval following treatment (n = 5–6 cells per condition). Amplitude (45.49 ± 6.97pA, control CSF; 40.74 ± 3.43, patient CSF) and interevent interval (1,734 ± 474.1 milliseconds, control CSF; 1,139 ± 154.3 milliseconds, patient CSF) were not significantly different. Mann–Whitney test, p > 0.05. (E) Hippocampal neurons were treated with control or patient CSF for 24 hours, then immunostained for GABAAR and vesicular γ-aminobutyric acid transporter (vGAT). Scale bar = 5μm. (F) Quantification of inhibitory synapse density onto excitatory neurons following treatment (n = 30 cells per condition, 3 independent experiments). Patient CSF caused a significant decrease in inhibitory synapse density (87.52 ± 3.00% of control treatment). *p < 0.05, Mann–Whitney test.
Similar to APV treatment, neurons may attempt to increase NMDAR insertion in response to patient antibodies. This would be undetectable due to antibody-mediated internalization of any newly inserted receptors. Alternatively, neurons may upregulate NMDAR transcription after internalization. Many genes are transcriptionally regulated by activity,26 and some forms of homeostatic plasticity are transcription-dependent.27 To evaluate this possibility, quantitative polymerase chain reaction was performed to determine the levels of NMDAR subunit mRNA following patient antibody or APV treatment for 24 hours, but no changes were detected in GluN1, GluN2A, or GluN2B, or in any of the GluN1 C-terminal splice variants (data not shown). We also measured transcriptional changes in these splice variants after 7 days of treatment, but mRNA levels were still unchanged (data not shown).
We next assayed whether inhibitory synapses displayed homeostatic plasticity in response to the NMDAR hypofunction caused by patient antibodies. Evidence suggests a link between NMDAR dysregulation and impairments in inhibition,28,29 for example, the frequent occurrence of seizures in encephalitis patients that is indicative of defects in inhibitory tone. Hippocampal neurons were treated on DIV17–20 with control or patient CSF for 24 hours, then whole cell patch clamp recordings of mIPSCs were made. Neither the amplitude nor the interevent interval of mIPSCs was changed by patient CSF treatment (see Fig 7C, D; p > 0.05). Gene expression of GAD1 and GAD2, enzymes responsible for GABA production in inhibitory neurons, was also unchanged (data not shown).
Finally, we treated hippocampal neurons on DIV14 with control or patient CSF for 24 hours, then fixed and stained for the inhibitory GABAAR and inhibitory presynaptic marker vGAT. We found that patient antibody treatment caused a significant decrease in inhibitory synapse density (see Fig 7B, D) onto excitatory neurons, which were identified by a lack of vGAT staining within the cell body. Together, our data showed that although loss of NMDARs did not stimulate transcriptional changes of NMDAR subunits or splice variants, neurons were able to partly adjust their inhibitory tone in a compensatory direction.
Discussion
This study revealed several novel findings related to the effects of antibodies from patients with anti-NMDAR encephalitis, including the lack of neuron subtype specificity, time course of the effects, fate of internalized receptors, and homeostatic responses to NMDAR loss. In addition, the results confirmed that the major mechanism of dysfunction was loss of NMDARs due to IgG-mediated internalization, which likely occurs throughout the brain. The process of internalization plateaued after 12 hours, reaching a steady state that persisted throughout the duration of treatment. This may reflect the presence of a population of NMDARs unaffected by antibodies, or represent a state of equilibrium between the rate of internalization and the rate of receptor insertion into the plasma membrane.
Internalization of NMDARs can be promoted by receptor activity.22,30 We were unable to block patient antibody-mediated NMDAR internalization with APV, suggesting that patient antibodies did not exert activity-dependent effects. Electrophysiological analyses also excluded direct blockade as a prominent mechanism of antibody-mediated NMDAR hypofunction. These results, along with the finding that patient F(ab) fragments did not cause internalization and that patient IgG was necessary and sufficient for internalization, suggested that antibody-mediated NMDAR internalization was solely due to crosslinking of the receptors. A similar mechanism had been demonstrated for myasthenia gravis and Lambert–Eaton syndrome.31 Moreover, human autopsy studies of anti-NMDAR encephalitis patients showed deposits of IgG and decreased NMDARs, without evidence of cytotoxic T-cell mechanisms, all supporting an antibody-mediated pathogenesis.5,32
NMDARs dynamically traffic into and out of the synapse during physiological processes, such as synapse maturation and synaptic plasticity.1 This cycling is largely governed by regulatory signals within the C-terminal portions of the receptor complexes that mediate clathrin-dependent endocytosis.33–35 We showed that internalized, antibody-bound receptors entered a recycling and a degradation pathway, although the proportion of internalized receptors in recycling compartments consistently exceeded that within lysosomes. Even with different manipulations to induce NMDAR internalization, the trajectory of postendocytic trafficking was unchanged, excluding an immunoglobulin-specific effect.
Homeostatic plasticity is important for maintaining the stability of neuronal network activity in the face of potentially destabilizing changes in the strengths of individual synapses.36 The NMDAR hypofunction in anti-NMDAR encephalitis led us to the question of whether patient antibody treatment can induce homeostatic changes in cultured neurons. We did not detect gene expression changes in any NMDAR subunits with either APV or patient antibody treatment, concluding that transcriptional regulation was not a major locus of homeostatic plasticity in this system.
We noted the similarity in some symptoms between anti-NMDAR encephalitis and schizophrenia. Several lines of evidence in animal models and humans had led to the hypothesis that NMDAR hypofunction in inhibitory interneurons led to disinhibition in corticolimbic regions, underpinning core symptom domains in schizophrenia.16,37 We examined whether patient antibodies may have had disparate effects on excitatory and inhibitory neurons, but these populations of neurons were indistinguishable by our analyses.
Inhibitory neurons exert complex control over excitatory cell firing, both because of their exuberant connectivity (in cortex, a single interneuron can contact more than half of the local pyramidal neurons) and because of the diverse effects they have on neuronal spiking, depending on the location of the synaptic connections (axon initial segment, distal dendrites, soma, etc).38 Therefore, even a small change in the magnitude of connectivity could have profound effects on network excitability and precision. We found that although neither expression of the GABA synthesizing enzymes, GAD1 and GAD2, nor amplitude and interevent interval of mIPSCs was changed by patient CSF treatment, GABAergic synapse density onto excitatory neurons was decreased. We suspect this difference in results between the electrophysiology and staining experiments was due to the increased power of immunostaining experiments, which sampled ∼30 neurons per condition, as opposed to the physiology experiments, which measured mIPSCs from 6 neurons per condition.
This was consistent with literature showing that genetic or pharmacological NMDAR dysfunction can lead to a loss of inhibitory tone, but indicated a distinct mechanism that may contribute in this disease. Although a loss of glutamatergic drive to inhibitory interneurons is often considered the pathogenic event leading to disinhibition in NMDAR hypofunction models, our data suggested that an additional phenomenon could be the homeostatic downregulation of inhibitory synapses onto excitatory neurons.
It will be important to extend these findings to an in vivo context to establish the pathophysiological mechanisms leading to dysfunction in patients. However, it is interesting to consider the implications of engaging homeostatic plasticity mechanisms in the presence of anti-NMDAR antibodies. Examples of homeostatic responses to altered levels of activity have been found or hypothesized to occur in several neurological disorders, including epilepsy, myasthenia gravis, Alzheimer disease, and schizophrenia.39 In schizophrenia, and NMDA hypofunction models, loss of inhibition may contribute to symptom profile and disease progression. Understanding the mechanisms that drive circuit-level changes underlying the behavioral and neurological symptoms of the disorder will be important to link the pathophysiologic events of anti-NMDAR encephalitis with those of other disorders with similar neuropsychiatric manifestations.
Acknowledgments
This work was supported by a grant from the NIH National Institute of Neurologic Disorders and Stroke (RO1NS077851; R.J.B.-G., J.D.); a McKnight Neuroscience of Brain Disorders Award (R.J.B.-G., J.D.); Fundació la Marató de TV3, Spain (101530; R.J.B.-G., J.D.); Instituto Carlos III FIS (PI11/01780; J.D.); and Fondo de Investigaciones Sanitarias de la Seguridad Socia, Spain (PI11/01789; J.D.).
We thank M. O. Scott and M. Maronski for technical assistance and L. McCracken for maintaining patient clinical and sample databases.
Authorship
E.H.M., X.P., and A.J. designed and executed the experiments. E.H.M. wrote the first draft of the manuscript. R.J.B.-G. and J.D. advised on experiment execution, data analysis, and presentation and edited the manuscript. T.D.P. advised on the electrophysiological experiment execution and analyses.
Potential Conflicts of Interest
J.D.: patents licensed to Euroimmun, Mayo Clinic, Athena. R.J.B.-G.: personal fees, Pfizer.
References
- 1.Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- 2.Kessels HW, Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenfeld MR, Dalmau J. Anti-NMDA-receptor encephalitis and other synaptic autoimmune disorders. Curr Treat Options Neurol. 2011;13:324–332. doi: 10.1007/s11940-011-0116-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lai M, Hughes EG, Peng X, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol. 2009;65:424–434. doi: 10.1002/ana.21589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dalmau J, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61:25–36. doi: 10.1002/ana.21050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7:1091–1098. doi: 10.1016/S1474-4422(08)70224-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013;12:157–165. doi: 10.1016/S1474-4422(12)70310-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- 9.Cui Z, Wang H, Tan Y, et al. Inducible and reversible NR1 knockout reveals crucial role of the NMDA receptor in preserving remote memories in the brain. Neuron. 2004;41:781–793. doi: 10.1016/s0896-6273(04)00072-8. [DOI] [PubMed] [Google Scholar]
- 10.Davis S, Butcher SP, Morris RG. The NMDA receptor antagonist D-2-amino-5-phosphonopentanoate (D-AP5) impairs spatial learning and LTP in vivo at intracerebral concentrations comparable to those that block LTP in vitro. J Neurosci. 1992;12:21–34. doi: 10.1523/JNEUROSCI.12-01-00021.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kehrer C, Maziashvili N, Dugladze T, Gloveli T. Altered excitatory-inhibitory balance in the NMDA-hypofunction model of schizophrenia. Front Mol Neurosci. 2008;1:6. doi: 10.3389/neuro.02.006.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grunze HC, Rainnie DG, Hasselmo ME, et al. NMDA-dependent modulation of CA1 local circuit inhibition. J Neurosci. 1996;16:2034–2043. doi: 10.1523/JNEUROSCI.16-06-02034.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98:427–436. doi: 10.1016/s0092-8674(00)81972-8. [DOI] [PubMed] [Google Scholar]
- 14.Newcomer JW, Krystal JH. NMDA receptor regulation of memory and behavior in humans. Hippocampus. 2001;11:529–542. doi: 10.1002/hipo.1069. [DOI] [PubMed] [Google Scholar]
- 15.Belforte JE, Zsiros V, Sklar ER, et al. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci. 2010;13:76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olney JW, Newcomer JW, Farber NB. NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res. 1999;33:523–533. doi: 10.1016/s0022-3956(99)00029-1. [DOI] [PubMed] [Google Scholar]
- 17.Krystal JH, Karper LP, Seibyl JP, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans: psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- 18.Lahti AC, Weiler MA, Tamara M, et al. Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology. 2001;25:455–467. doi: 10.1016/S0893-133X(01)00243-3. [DOI] [PubMed] [Google Scholar]
- 19.Hughes EG, Peng X, Gleichman AJ, et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci. 2010;30:5866–5875. doi: 10.1523/JNEUROSCI.0167-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mikasova L, De Rossi P, Bouchet D, et al. Disrupted surface cross-talk between NMDA and ephrin-B2 receptors in anti-NMDA encephalitis. Brain. 2012;135:1606–1621. doi: 10.1093/brain/aws092. [DOI] [PubMed] [Google Scholar]
- 21.Watt AJ, van Rossum MC, MacLeod KM, et al. Activity coregulates quantal AMPA and NMDA currents at neocortical synapses. Neuron. 2000;26:659–670. doi: 10.1016/s0896-6273(00)81202-7. [DOI] [PubMed] [Google Scholar]
- 22.Nong Y, Huang YQ, Ju W, et al. Glycine binding primes NMDA receptor internalization. Nature. 2003;422:302–307. doi: 10.1038/nature01497. [DOI] [PubMed] [Google Scholar]
- 23.Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gleichman AJ, Spruce LA, Dalmau J, et al. Anti-NMDA receptor encephalitis antibody binding is dependent on amino acid identity of a small region within the GluN1 amino terminal domain. J Neurosci. 2012;32:11082–11094. doi: 10.1523/JNEUROSCI.0064-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rao A, Craig AM. Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron. 1997;19:801–812. doi: 10.1016/s0896-6273(00)80962-9. [DOI] [PubMed] [Google Scholar]
- 26.West AE, Greenberg ME. Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb Perspect Biol. 2011;3(6) doi: 10.1101/cshperspect.a005744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57:819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- 28.Lisman JE, Coyle JT, Green RW, et al. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008;31:234–242. doi: 10.1016/j.tins.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27:11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vissel B, Krupp JJ, Heinemann SF, Westbrook GL. A use-dependent tyrosine dephosphorylation of NMDA receptors is independent of ion flux. Nat Neurosci. 2001;4:587–596. doi: 10.1038/88404. [DOI] [PubMed] [Google Scholar]
- 31.Drachman DB, Angus CW, Adams RN, et al. Myasthenic antibodies cross-link acetylcholine receptors to accelerate degradation. N Engl J Med. 1978;298:1116–1122. doi: 10.1056/NEJM197805182982004. [DOI] [PubMed] [Google Scholar]
- 32.Martinez-Hernandez E, Horvath J, Shiloh-Malawsky Y, et al. Analysis of complement and plasma cells in the brain of patients with anti-NMDAR encephalitis. Neurology. 2011;77:589–593. doi: 10.1212/WNL.0b013e318228c136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roche KW, Standley S, McCallum J, et al. Molecular determinants of NMDA receptor internalization. Nat Neurosci. 2001;4:794–802. doi: 10.1038/90498. [DOI] [PubMed] [Google Scholar]
- 34.Scott DB, Michailidis I, Mu Y, et al. Endocytosis and degradative sorting of NMDA receptors by conserved membrane-proximal signals. J Neurosci. 2004;24:7096–7109. doi: 10.1523/JNEUROSCI.0780-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lavezzari G, McCallum J, Dewey CM, Roche KW. Subunit-specific regulation of NMDA receptor endocytosis. J Neurosci. 2004;24:6383–6391. doi: 10.1523/JNEUROSCI.1890-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turrigiano G. Homeostatic signaling: the positive side of negative feedback. Curr Opin Neurobiol. 2007;17:318–324. doi: 10.1016/j.conb.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 37.Coyle JT. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell Mol Neurobiol. 2006;26:365–384. doi: 10.1007/s10571-006-9062-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Isaacson JS, Scanziani M. How inhibition shapes cortical activity. Neuron. 2011;72:231–243. doi: 10.1016/j.neuron.2011.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wondolowski J, Dickman D. Emerging links between homeostatic synaptic plasticity and neurological disease. Front Cell Neurosci. 2013;7:223. doi: 10.3389/fncel.2013.00223. [DOI] [PMC free article] [PubMed] [Google Scholar]