

Abstract
Acute spinal cord injury initiates a complex cascade of molecular events termed ‘secondary injury’, which leads to progressive degeneration ranging from early neuronal apoptosis at the lesion site to delayed degeneration of intact white matter tracts, and, ultimately, expansion of the initial injury. These secondary injury processes include, but are not limited to, inflammation, free radical-induced cell death, glutamate excitotoxicity, phospholipase A2 activation, and induction of extrinsic and intrinsic apoptotic pathways, which are important targets in developing neuroprotective strategies for treatment of spinal cord injury. Recently, a number of studies have shown promising results on neuroprotection and recovery of function in rodent models of spinal cord injury using treatments that target secondary injury processes including inflammation, phospholipase A2 activation, and manipulation of the PTEN-Akt/mTOR signaling pathway. The present review outlines our ongoing research on the molecular mechanisms of neuroprotection in experimental spinal cord injury and briefly summarizes our earlier findings on the therapeutic potential of pharmacological treatments in spinal cord injury.
Keywords: spinal cord injury, neuroprotection, inflammation, oxidation, apoptosis, glucocorticoid receptor, phospolipase A2, microRNAs, signaling pathway
Research Highlights
The review outlines our ongoing research on the molecular mechanisms of neuroprotection in spinal cord injury and briefly summarizes our earlier findings on the therapeutic potential of pharmacological treatments in spinal cord injury.
Abbreviations
TNF-α, tumor necrosis factor-α; AP-1, activator protein-1; NF-κB, nuclear factor kappa B; ANXA1, annexin A1; iNOS, inducible nitric oxide synthase; MMP-1, matrix metalloproteinase-1; MP, methylprednisolone; GR, glucocorticoid receptor; mTOR, mammalian target of rapamycin
INTRODUCTION
Traumatic spinal cord injury leads to neurological deficits and motor and sensory dysfunctions. In the United States alone, there are approximately 265 000 people living with spinal cord injury in 2010, and an additional 12 000 new spinal cord injury cases occur every year (https://www.nscisc.uab.edu). Clinically, most traumatic spinal cord injuries are anatomically incomplete and the primary impact-induced secondary damage contributes significantly to the final extent of neural damage and ultimately to the extent of the long term disability, which represent an important target in developing neuroprotective strategies for treatment of spinal cord injury[1,2,3]. Neuroprotection aims at preventing or attenuating secondary injury.
Understanding the intimate mechanisms of secondary injury after traumatic spinal cord injury should provide us with a large number of therapeutic targets. For the last two decades, our research has focused on elucidating the molecular mechanisms that mediate secondary spinal cord injury and exploring novel neuroprotective strategies. The present review outlines our ongoing research on the molecular mechanisms of neuroprotection in experimental spinal cord injury and briefly summarizes our earlier findings on the therapeutic potential of a few promising pharmacological treatments for spinal cord injury.
INFLAMMATION
Post-traumatic inflammatory reaction has been shown to contribute to progressive tissue damage after spinal cord injury. Following spinal cord injury, inflammatory cells such as polymorphonuclear neutrophils, macrophages, and lymphocytes quickly infiltrate into the traumatized cord, and inflammatory mediators, such as eicosanoids and cytokines, are accumulated[4,5,6,7,8,9,10,11]. For example, within a few minutes post-spinal cord injury, eicosanoids such as TXA2 and PGE2 increased in the injured cord tissue[7,9]. Within the first hour post-spinal cord injury, neutrophil infiltration[4,5,12] and cytokine expression such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) at and surround the site of injury occurred[8,10,11]. The interplay between inflammatory cells and mediators likely perpetuates a progressive course of secondary injury, resulting in neuronal and glial cell death, axonal destruction and functional loss.
Activator protein-1 (AP-1) and nuclear factor kappa B (NF-κB) are two major pro-inflammatory transcription factors that are activated in inflammation[13,14]. NF-κB and AP-1 act individually or in synergy to transactivate genes that code for pro-inflammatory proteins such as cytokines and adhesion molecules, pro-inflammatory enzymes including inducible nitric oxide synthase (iNOS) and inducible cyclooxygenase II, and pro-inflammatory proteases[13,14]. We[11,15] and others[16] have shown that both AP-1 and NF-κB were activated after spinal cord injury in a rat model. Electrophoretic mobility shift assay showed that AP-1 binding activity increased after spinal cord injury, starting at 1 hour, peaking at 8 hours, and declining to basal levels by 7 days[15]. After spinal cord injury, NF-κB binding activity was also increased, peaked between 1 and 3 days post-injury[11]. Post-traumatic inflammatory reaction is a protracted process lasting for days to weeks. The time course of AP-1 and NF-kB activation in the present study is compatible for AP-1 and NF-kB to play an important role in triggering the subsequent inflammatory activities. AP-1 is a dimer composed of various Fos and Jun family proteins[17,18]. Immunostaining showed an increase in the expression of the Fos-B and c-Jun components of AP-1 in the injured cord[15]. These constituent components of AP-1 were not only localized to the cytosol but also in nuclei[15], suggesting their nuclear translocation to form the AP-1 transcription factor. The specificity of spinal cord injury -induced AP-1 activation was further confirmed by an antisense strategy directed at c-fos that blocked AP-1 activation after spinal cord injury[15]. AP-1 and NF-κB, functioning as pro-inflammatory transcription factors, transactivate a number of genes that are expressed in inflammation[14,19] including matrix metalloproteinase-1 (MMP-1)[20,21] and MMP-9[22,23]. Western blotting and immunostaining showed increased expression of MMP-1 and MMP-9 in the injured cord. Both MMP-1 and MMP-9 were expressed with high intensity in neurons and glial cells after spinal cord injury. These findings were consistent with changes of both AP-1 and NF-κB following spinal cord injury. It has been shown that excessive MMP expression leads to increased capillary permeability in numerous neurological disorders such as multiple sclerosis, infection, and ischemia[24]. Alteration of vascular permeability, a key feature of inflammation, has been shown after spinal cord injury[25]. The activation of two key pro-inflammatory transcription factors, NF-κB and AP-1, after spinal cord injury provides an underlying molecular mechanism for a post-traumatic inflammatory reaction.
TNF-α is a key inflammatory mediator, which plays a pathogenetic role in cell death in neurodegenerative diseases[26]. In both mouse and rat traumatic spinal cord injury models, an increase in TNF-α expression in the injured cord has been demonstrated at both the mRNA and protein levels as early as hours after injury[11,27,28,29]. Western blot analysis revealed that TNF-α expression began at 4 hours post-injury, peaked at 24 hours (4.5-fold increase at 1 day after spinal cord injury, P < 0.01) and declined at 3 days after spinal cord injury, which were confirmed by immunohistochemistry[11]. Immunofluorescence double labeling further revealed that within hours after spinal cord injury, increased TNF-α immunoreactivity was localized in neurons, astrocytes, oligodendrocytes, microglia, and endothelial cells in areas of the spinal cord adjacent to the lesion site, which was confirmed by immunoelectronmicroscopy[30]. Myelin breakdown was noted in oligodendrocytes that are immunopositive for TNF-α[30]. TNF-α exerts its effector actions, at least partially, through the activation of a pro-inflammatory transcription factor, NF-κB (Figure 1), which in turn upregulates such genes as iNOS, cytokines, adhesive molecules, and others[13]. Post-traumatic TNF-α expression was accompanied by an increase in NF-kB binding activity in nuclear proteins isolated from the injured cord (3.9-fold increase, P < 0.01)[11].
Figure 1.
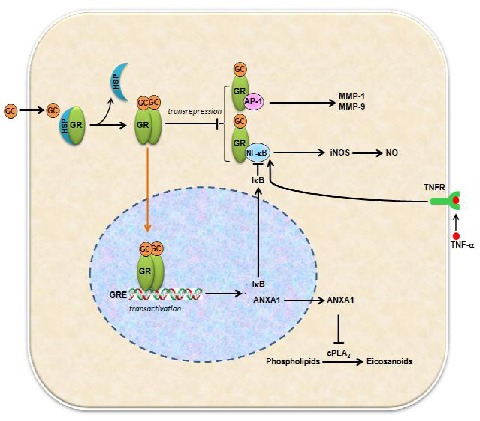
Schematic drawing illustrates possible actions of glucocorticoids (GC) and glucocorticoid receptors (GR) after acute spinal cord injury.
GC bind to GR to form activated GR (aGR) in the cytosol, which displaces heat-shock protein 90 (HSP90) and allows receptor dimerization, movement into the nucleus, and binding to the glucocorticoid response elements (GRE). This leads to transactivation of anti-inflammatory genes such as IκB and ANXA1.
IκB then sequesters NF-κB, preventing it from activating transcription of pro-inflammatory cytokines. ANXA1 inhibits cytosolic phospholipase A2 (cPLA2) activity, in turn reduces pro-inflammatory mediators such postaglandins. In addition, the aGR can interact with AP-1 and NF-κB directly to suppress pro-inflammatory mediators and cytokine production.
AP-1: Activator protein-1; NF-κB: nuclear factor kappa B; MMP-1: matrix metalloproteinase-1; MMP-9: matrix metalloproteinase-9; Inos; Inducible nitric oxide synthase; NO: nitric oxide; ANXA1: annexin A1; TNFR: tumor necrosis factor receptor; IκB: inhibitor of NF-κB.
The effect of TNF-α is mediated through its receptors TNFR1 (p55) and TNFR2 (p75). In a rat traumatic spinal cord injury model, we demonstrated that the expression of TNFR1 and TNFR2 was slightly increased at 15 minutes post-injury, reached the peak at 4 hours for TNFR2 (6.06-fold, P < 0.01) and 8 hours for TNFR1 (3.58-fold, P < 0.01), and declined markedly after 1 and 3 days[31]. Spatially, TNFR1- and TNFR2-IR were initially observed at the impact site, spread to the distant areas during the peak expression, and confined to the lesion area at later time points[31]. TNFR1 and TNFR2 were localized in neurons, oligodendrocytes, and astrocytes[31]. These results suggest that the expression of TNFR1 and TNFR2 after spinal cord injury may contribute to post-traumatic inflammatory responses of TNF-α. Our result further showed that methylprednisolone (MP), a clinically used glucocorticoid, mediated neuroprotection after spinal cord injury by inhibiting expression of TNF-α and TNFRs[11,31], suggesting TNF-α and TNFRs have deleterious effects.
However, our experiments also suggest a neuroproctive role of TNF-α in spinal cord injury[32]. Deletion of TNFR1 in knockout mice inhibited NF-κB binding activity, reduced cellular inhibitor of apoptosis protein 2 (c-IAP2) expression and increased the active form of caspase-3. After spinal cord injury, the TNFR1(-/-) mice had greater numbers of apoptotic cells, a larger lesion size, and worse functional recovery than wild-type mice. TNFR2-deficient mice had a similar, although not as pronounced, consequence as the TNFR1(-/-) mice. These findings support the argument that the TNFR-NF-κB pathway is beneficial for limiting apoptotic cell death after spinal cord injury and that a defective TNFR-NF-κB pathway results in a poorer neurological outcome. A worse functional outcome in TNFR(-/-) mice suggests that an endogenous apoptosis inhibitory mechanism mediated by TNFR activation, NF-κB, and c-IAP2 may be of pathophysiological importance. Conversely, our previous results and others suggested that neuroprotection of MP after spinal cord injury was achieved by inhibiting activation of NF-κB and its associated downstream induction of nitric oxide (NO) synthase[11,33]. These findings suggest that TNF-α may have deleterious effects, particularly early after injury, but are also critical for neural repair as the injury evolves, which is consistent with the concept of dual effects of the inflammatory response[34,35,36,37,38]. Thus, an effective therapeutic intervention must limit the acute destructive effects of the inflammatory response while also preserving its neuroprotective effects.
NEUROPROTECTION AND MECHANISMS OF MP ACTION
MP, a synthetic glucocorticoid, is the only drug used clinically to improve neurological function in patients with acute spinal cord injury. MP is also a potent anti-inflammatory agent. The mechanism of MP action is not fully understood, but our studies showed several possible mechanisms of MP. First, MP inhibits spinal cord injury -induced TNF-α expression and NF-κB activation via a glucocorticoid receptor (GR) mechanism (Figure 1)[11,15,39]. In the contusive rat spinal cord injury model, TNF-α expression and NF-κB activation have been shown to increase after spinal cord injury[11]. NF-κB activation is initiated by pro-inflammatory signaling processes such as TNF-α interaction with TNF-α receptor on the cell surface. The TNF-α-initiated signals trigger the phosphorylation of IκBα, which leads to its dissociation from NF-κB, allowing the latter to translocate into the nucleus[40]. Most significantly, MP (30 mg/kg, i.v.) given immediately after spinal cord injury reduced TNF-α expression by 55% (P < 0.01) and NF-κB binding activity[11]. MP at a dose of 30 mg/kg has also been shown to be effective in improving functional outcomes in rat spinal cord injury models[33,41]. MP also partially inhibited the injury-induced expression of TNFR1 and TNFR2, an effect which could be reversed by RU486, an antagonist of GR[31]. RU486 has also been shown to reverse MP inhibition of NF-κB activation[15]. Immunofluorescence double staining revealed a colocalization of GR and TNF-α in neurons and glial cells[39]. These findings are consistent with an anti-inflammatory role of MP in spinal cord injury. MP inhibition of NF-κB function is likely mediated by the induction of IκB, which traps NF-κB in inactive cytoplasmic complexes[42]. Second, MP inhibits post-traumatic AP-1 activation via a GR mechanism (Figure 1)[15]. Thus, for MP to inhibit NF-κB and AP-1 activation, it has to bind to GR. RU486, a GR antagonist, reversed MP inhibition of AP-1 activation after spinal cord injury[15]. Furthermore, RU486 also reversed MP inhibition of MMP-1 and MMP-9 expression after spinal cord injury[15]. These findings are again consistent with the contention that AP-1 and NF-κB are required to transactivate MMP-1 and MMP-9 and therefore are subjected to the inhibitory action of MP via a GR mechanism. Third, MP induces anti-inflammatory genes such as annexin A1 (ANXA1), interleukin 10, MAPK phosphatase I, secretory leukocyte protease inhibitor, and I-κB (Figure 1)[43,44].
POSSIBLE MECHANISMS OF GR IN MEDIATING TRAUMATIC SPINAL CORD INJURY
The GR is an intracellular receptor, which is present in the cytoplasm and nucleus of neurons and glial cells. GR can function both as a transcription factor that binds to glucocorticoid response elements (GREs) in the promoters of glucocorticoid responsive genes to activate their transcription, and as a regulator of other transcription factors (Figure 1)[43,44,45]. This receptor is typically found in the cytoplasm, but upon ligand binding, is transported into the nucleus. The glucocorticoids including MP bind to the cytosolic GR to form activated GR (aGR)[43]. The aGR has two principal mechanisms of action, transactivation of anti-inflammatory genes and transrepression of pro-inflammatory genes (Figure 1)[43,44,45]. The aGR translocates into the nucleus and binds to specific GREs in the promoter regions of anti-inflammatory genes such as ANXA1, interleukin 10, MAPK phosphatase I, secretory leukocyte protease inhibitor, and I-κB. This mechanism of action is referred to as transactivation of anti-inflammatory genes. The dominant anti-inflammatory effect of aGR is derived from aGR transrepression of pro-inflammatory genes, namely protein-protein interaction or crosstalk with other transcription factors, notably NF-κB and AP-1.
Glucocorticoids bind to intracellular GR, which in turn bind to GRE to exert ligand-activated transcription effects. Thus, glucocorticoid actions depend not only on the ligand concentration but also on the extent of GR expression. In the central nervous system (CNS) of normal adult rats, a widespread distribution of GR-IR has been demonstrated[46,47]. After spinal cord injury, an increased expression of GR was found in both the cytoplasm and nucleus of affected cells after spinal cord injury[39]. Immunohistochemistry and western blot analysis revealed an increase in GR protein expression as early as 15 minutes after injury. GR expression sharply increased at 4 hours (22-fold), peaked at 8 hours (56-fold), rapidly declined at 1 day, and returned to the baseline level at and after 3 days[39]. During its peak expression, GR was localized in neural somata and dendrites but not in axons and their terminals[39]. GR immunoreactivity was also found in oligodendrocytes and astrocytes. Interestingly, other cell types, such as endothelial cells, were GR-negative. Furthermore, the nuclear protein binding activity to GRE based on EMSA was significantly increased after injury[39]. These results suggest that, after spinal cord injury, GR expression is followed by translocation and transcription activity involving GRE. Finally, colocalization of GR and TNF-α was observed in neurons and glial cells, consistent with MP regulation of TNF-α in this model[11,39]. ANXA1 is a glucocorticoid-inducible protein, which inhibits the activity of phospholipase A2 (PLA2), and hence reduces the generation of pro-inflammatory eicosanoids. Our subsequent studies showed that expression of ANXA1 increased following spinal cord injury[48] and administration of ANXA1 reduced spinal cord injury -induced inflammatory response and tissue damage through inhibition of PLA2[49]. These findings may provide new insights into the anti-inflammatory action of MP.
REACTIVE OXYGEN SPECIES AND OXIDATIVE STRESS
Oxidative injury is a common mechanism of damage in CNS neurological disorders including spinal cord injury. Reactive oxygen species (ROS) such as hydrogen peroxide (H2 O2, an inducer of ROS)[50], superoxide (O2•-)[51], and hydroxyl radical (•OH)[52] all increased significantly in the spinal cord following injury and could occur within hours after spinal cord injury[53]. Excessive ROS production causes oxidative damage to proteins, lipids and DNA, followed by impaired cellular functions that eventually lead to severe and irreversible cell death[54]. NO is a short-lived free radical gas. The NO reacts with superoxide to generate an extremely potent ROS, peroxynitrite, that can damage cellular enzymes, membranes, and subcellular organelles through the nitration of tyrosine residues on proteins[55]. iNOS is a key enzyme that catalyzes the synthesis of NO. Nitrotyrosine (NT) is an indirect chemical indicator of toxic NO and peroxynitrite-induced cellular damage[55]. In our study[56], we observed a progressive increase in iNOS expression in the injured cord in a rat spinal cord injury model, starting at 1 day with maximal expression occurring at 7 days, as determined by western blot analysis. iNOS expression corresponded temporally to an increase in iNOS enzyme activity after spinal cord injury. In parallel with the progressive increase in iNOS activity, NT expression also increased over time after spinal cord injury. The iNOS and NT immunoreactivity was localized in neurons, astrocytes, endothelial cells and ependymal cells at the epicenter and adjacent to the region of spinal cord impact and injury. The increased expression of iNOS after spinal cord injury is consistent with spinal cord injury-induced NF-κB activation[11] which transactivates specifically the gene coding for iNOS, suggesting that NF-κB may activate iNOS gene transcription, which leads to iNOS protein expression after spinal cord injury. The inhibition of NO production with NOS antiserum reduces cell death after acute spinal cord injury[57]. The role of NOS in secondary injury is further supported by evidence of functional improvement and tissue sparing in NOS deficient mice[58]. These results suggest that the free radical formation is also an important therapeutic target for spinal cord injury.
APOPTOSIS
As early as 1997, we and others demonstrated that apoptosis is as an important mechanism of cell death after spinal cord injury[59,60]. Apoptosis of both neurons and glia contributes to spinal cord tissue damage after traumatic insults of mild to moderate severity[59]. Our determination of apoptosis relies on multiple criteria: morphology under both light and electron microscopic examination, nuclear chromatin staining with Hoechst 33342 dye and with TUNEL, DNA laddering. Within minutes after mild weight drop impact (a 10-gm weight falling 6.25 mm), neurons in the immediate impact area showed a loss of cytoplasmic Nissl substances. Over the next 7 days, this lesion area expanded and cavitated. TUNEL-positive neurons were noted primarily restricted to the gross lesion area 4-24 hours after injury, with a maximum presence at 8 hours after injury. TUNEL-positive glias were present at all stages studied between 4 hours and 14 days, with a maximum presence within the lesion area 24 hours after injury. However, at 7 days post-injury, a second wave of TUNEL-positive glial cells was noted in the white matter peripheral to the lesion and extending at least several millimeters away from the lesion center. The suggestion of apoptosis was supported by electron microscopy, as well as by nuclear staining with Hoechst 33342 dye, and by examination of DNA prepared from the lesion site. Furthermore, repeated intraperitoneal injections of cycloheximide, beginning immediately after a 12.5-mm weight drop insult, produced a substantial reduction in histological evidence of cord damage and in motor dysfunction assessed 4 weeks later. These findings support the hypothesis that apoptosis dependent on active protein synthesis contributes to the neuronal and glial cell death, as well as to the neurological dysfunction, induced by mild-to-moderate severity traumatic insults to the rat spinal cord. Thus, blocking apoptosis is a neuroprotective approach for treatment of spinal cord injury.
PLA2 AS A NOVEL TARGET FOR INTERVENTION AFTER SPINAL CORD INJURY
PLA2 is a diverse family of lipolytic enzymes that hydrolyze an ester bond at the sn-2 position of phospholipids that generates a free fatty acid such as arachidonic acid and a lysophospholipid such as lysophosphatidylcholine (Lyso-PC)[61,62,63]. Arachidonic acid can give rise to eicosanoids via cyclooxygenase (COX-1 and -2) and 5-lipoxygenase (5-LO) enzymes. Eicosanoids such as prostaglandins, thromboxanes, and leukotrienes are potent mediators of inflammation and tissue damage implicated in pathological states of numerous acute and chronic neurological disorders[61,62]. In addition, Lyso-PC is a myelinolytic agent and can act as a chemoattractant for immune cells[61,62,63]. To date, more than 19 isoforms of PLA2 have been found in the mammalian system which can be classified into three major categories: secretory PLA2 (sPLA2), cytosolic PLA2 (cPLA2) and Ca2+-independent PLA2 (iPLA2)[61,62]. All of the major PLA2s were found to be expressed in the mammalian brain and spinal cord. Our series of studies have shown that PLA2 contribute to secondary pathogenesis after spinal cord injury (Figure 2)[61,62].
Figure 2.
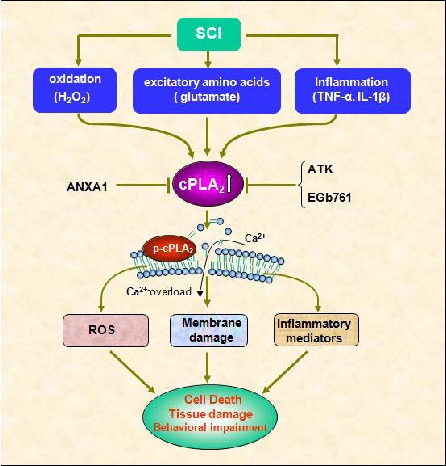
Schematic drawing shows possible role of cPLA2 in SCI.
SCI induces cPLA2 expression and activation by several toxic factors that are generated in the injured cord including inflammatory cytokines, free radicals, and excitatory amino acids.
Over-activation of cPLA2 induces cell death and tissue injury though several mechanisms.
First, cPLA2 induces membrane damage through hydrolysis of neural membrane phospholipids, resulting in alternation of membrane function such as fluidity and permeability, behavior of transporters and receptors, and calcium overload, and, eventually, leading to functional failure of excitable membranes.
Second, cPLA2 hydrolyzes membrane phospholipids to produce arachdonic acid (AA), then AA can give rise to pro-inflammatory mediators, for example eicosanoids, such as prostaglandins, thromboxanes, and leukotrienes.
Third, cPLA2-induced AA and NADPH oxidase lead to ROS production. ANXA1, ATK and EGb761 reduce neuronal death and tissue damage though inhibition of cPLA2.
cPLA2: Cytosolic phospholipase A2; AA: arachdonic acid; SCI: spinal cord injury; AA: arachdonic acid ANXA1: annexin A1; ATK: arachidonyl trifluromethyl ketone; EGb761: Ginkgo biloba extract; ROS: Reactive oxygen species; NADPH: nicotinamide adenine dinucleotide phosphate.
First and foremost, we found that PLA2 activity and expression were induced by spinal cord injury, peaking at 4 hours after injury and remaining significantly increased at 7 days[64]. Additionally, cPLA2 protein, an important isotype involved in lipid mediator generation, was also increased and peaked at 3 and 7 days after injury[64]. This isotype was localized in neurons, swollen axons, oligodendrocytes, and a subpopulation of microglia. Another important isotype, sPLA2-IIA involved in inflammation and demyelination, was increased at both mRNA and protein levels, peaking at 1 and 4 hours after the injury[65]. This isotype was localized in neurons, axons, oligodendrocytes, astrocytes, and some myelin rings.
To remove the confounding effects of injury, we directly injected PLA2 into the intact spinal cord[64,66,67]. This resulted in dose-dependent and sustained impairment in motor function[64]. Subsequent anatomical analyses revealed that injection of PLA2 induced demyelination[64,66,67] and such effect could be effectively attenuated with mepacrine, a PLA2 inhibitor[64]. Our experiments further showed that exogenous administration of PLA2 induced inflammatory and oxidative responses, as evidenced by increased expression of the inflammatory mediators TNF-α and IL-1β as well as 4-hydroxy-2(E)-nonenal, a product of lipid peroxidation and marker for oxygen free-radical-mediated membrane injury[64]. In vitro experiments revealed that exogenous PLA2 induced cultured spinal neuronal death in a dose-dependent manner, which was substantially reversed by mepacrine, the PLA2 inhibitor[64].
To further confirm the role of PLA2 in secondary pathogenesis after spinal cord injury, we investigated neuroprotective effects of ANXA1, a PLA2 inhibitor in a rat spinal cord injury model[49].
The results showed that injections of ANXA1 into the acutely injured spinal cord at 2 concentrations (5 and 20 µg) inhibited spinal cord injury -induced increases in PLA2 and myeloperoxidase activities. In addition, ANXA1 administration reduced the expression of interleukin-1β and activated caspase-3 at 24 hours, and glial fibrillary acidic protein at 4 weeks postinjury. Furthermore, ANXA1 administration significantly reversed PLA2-induced spinal cord neuronal death in vitro and reduced tissue damage and increased white matter sparing in vivo. ANXA1 administration also protected axons of long descending pathways as evidenced by increased numbers of Fluorogold-labeled neurons in selected superspinal nuclei. ANXA1 administration also significantly increased the number of animals that responded to transcranial magnetic motor-evoked potentials. These results, particularly the improvements obtained in tissue sparing and electrophysiologic measures, suggest PLA2 is a novel target for treatment of spinal cord injury, which is supported by inhibition of cPLA2 activation-mediated the protective effect of Ginkgo biloba extract (EGb761)[68].
PLA2 is induced by several toxic factors that are generated in the injured cord including inflammatory cytokines, free radicals, and EAAs[61,62]. Our in vitro experiments showed that both H2O2 and glutamate induced cPLA2 activation and neuronal death, and such effects were significantly reversed by arachidonyl trifluromethyl ketone (ATK), a cPLA2 inhibitor[68]. It is conceivable that PLA2 acts as a common pathway for multiple key mechanisms of secondary spinal cord injury, making it an attractive therapeutic target to improve tissue repair and functional recovery.
PTEN-AKT/MTOR SINGLING PATHWAY IN NEUROPROTECTION
PTEN-Akt/mTOR singling pathway is involved in cell survival (Figure 3).
Figure 3.
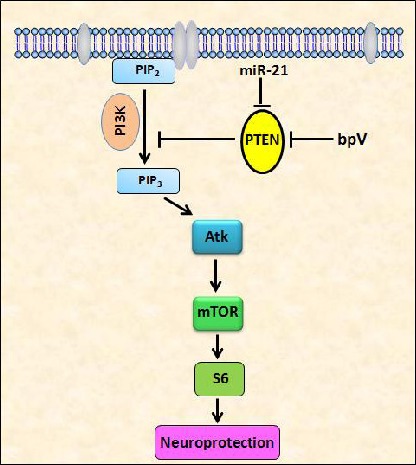
Schematic drawing shows PTEN-Akt/mTOR singling pathway in SCI.
PTEN's phosphatase converts phosphatidylinositol (3,4,5)-trisphosphate (PIP3) into phosphatidylinositol (4,5)-bisphosphate (PIP2), thus inhibiting downstream Akt and mTOR signaling.
PI3K converts PIP2 into PIP3, which can then activate Akt and mTOR, thus enhancing p-S6 expression leading to neuroprotection. Bisperoxovanadium (bpV) and miR-21 may induce Akt/mTOR activation by inhibition of PTEN, finally exert neuroprotective effects.
PTEN: phosphatase and tensin homolog; mTOR: mammalian target of rapamycin; SCI: spinal cord injury; PI3K: phosphoinositide 3-kinases; S6: ribosomal protein S6.
Phosphatase and tensin homolog (PTEN) is dual phosphatase that converts phosphatidylinositol (3,4,5)-trisphosphate (PIP3) into phosphatidylinositol (4, 5)-bisphosphate (PIP2), thus inhibiting downstream Akt and mammalian target of rapamycin (mTOR) signaling. Activation of phosphoinositide 3-kinases can phosphorylate and convert the PIP2 into PIP3, which in turn activates Akt and mTOR. PTEN inactivation has been shown to activate different downstream pathways such as Akt and mTOR, signalling and inhibit other signalling molecules such as GSK-3[69]. Recent studies demonstrated that deletion of either PTEN or tuberous sclerosis complex 1, both of which constitutively activate mTOR exerts strong neuroprotective and axon-growth-promoting effects on injured retinal ganglion cells[69,70]. Bisperoxovanadium compounds, PTEN inhibitors, have proven neuroprotective effects in several central nervous system injury/disease models[71,72,73]. More recent experiments in our laboratory showed that blocking PTEN with bisperoxovanadium produced neuroprotection and functional recovery following cervical unilateral contusive spinal cord injury[74]. In this study, two experimental groups of rats were established to (1) assess twice-daily 7 day treatment of the bisperoxovanadium, potassium bisperoxo (picolinato) vanadium, on long-term recovery of skilled forelimb activity using a novel food manipulation test, and neuroprotection 6 weeks following injury and (2) elucidate an acute mechanistic link for the action of the drug post-injury. The results showed that bisperoxovanadium promoted significant neuroprotection through reduced motorneuron death, increased tissue sparing, and minimized cavity formation in rats. Enhanced forelimb functional ability during a treat-eating assessment was also observed. Additionally, bisperoxovanadium significantly enhanced downstream Akt and mTOR signaling and reduced autophagic activity, suggesting inhibition of the phosphatase and tensin homologue deleted on chromosome 10 as a potential mechanism of bisperoxovanadium action following traumatic spinal cord injury. Overall, this study demonstrates the efficacy of a clinically applicable pharmacological therapy for rapid initiation of neuroprotection after spinal cord injury, and sheds light on the signaling involved in its action.
EFFECT OF GLIAL CELL LINE-DERIVED NEUROTROPHIC FACTOR (GDNF) ON NEUROPROTECTION
GDNF, a distant member of the transforming growth factor-β superfamily, has potent neuroprotective and neurotrophic effects on several neuronal cell types in both the central and peripheral nervous systems, which are mediated by a multisubunit receptor system consisting of a glycosyl-phosphatidylinositol-linked high-affinity ligand-binding coreceptor, GFRα1, and the transmembrane protooncogene, c-Ret[75,76,77,78]. Our experiments also show that GDNF has a neuroprotective effect on spinal cord tissues and axons of long descending pathways in adult rats after spinal cord injury[79]. Our results demonstrated that constant intrathecal infusion of the two concentrations of GDNF resulted in (1) GDNF's penetration and diffusion into the cord parenchyma surrounding the injury, (2) a 34–42% reduction of the total lesion volume measured using a stereological method, (3) a 10–13% increase in the percent of spared white matter measured at the injury epicenter, and (4) a substantial increase in the number of neurons from selective proprio- and supraspinal regions whose axons traverse the lesion and innervate the lumbar cord. These results are consistent with a recent study demonstrating that focal injections of GDNF into the spinal cord 1–2 mm rostral and caudal to a T9-10 contusive spinal cord injury significantly improved Basso Beattie Bresnahan locomotor rating scores and reduced neuronal cell death[80].
NEUROPROTECTION OF TESTOSTERONE ON MOTONEURON AND MUSCLE MORPHOLOGY
It has been shown that testosterone has neuroprotective and neurotherapeutic effects after a variety of motoneuron injuries[81,82,83,84]. Treatment with testosterone also improves motor function in spinal cord injury patients. Patients treated with testosterone had higher American Spinal Injury Association (ASIA) discharge motor scores, a result ascribed to either improved strength through the anabolic effects of testosterone on skeletal muscle or its neuroprotective/neuroregenerative effects[85]. Recently, we found that testosterone had similar beneficial effects in adult rats after spinal cord injury[86]. In this study, adult female rats received either sham or T9 spinal cord contusion injuries and were implanted with blank or testosterone-filled Silastic capsules. Four weeks later, motoneurons innervating the vastus lateralis muscle of the quadriceps were labeled with cholera toxin-conjugated horseradish peroxidase, and dendritic arbors were reconstructed in three dimensions. Contusion injury resulted in large lesions, with no significant differences in lesion volume, percent total volume of lesion, or spared white or gray matter between spinal cord injury groups. Spinal cord injury with or without testosterone treatment also had no effect on the number or soma volume of quadriceps motoneurons. However, spinal cord injury resulted in a decrease in dendritic length of quadriceps motoneurons in untreated animals, and this decrease was completely prevented by treatment with testosterone. Similarly, the vastus lateralis muscle weights and fiber cross-sectional areas of untreated spinal cord injury animals were smaller than those of sham-surgery controls, and these reductions were both prevented by testosterone treatment. No effects on motor endplate area or density were observed across treatment groups. These findings suggest that regressive changes in motoneuron and muscle morphology seen after spinal cord injury can be prevented by testosterone treatment, further supporting a role for testosterone as a neurotherapeutic agent in the injured nervous system.
MICRORNAs (miRNAs) AS NOVEL TARGETS FOR TREATMENT OF SPINAL CORD INJURY
miRNAs are a novel class of small non-coding RNAs that negatively regulate gene expression at the posttranscriptional level by binding to the 3’ untranslated region (UTR) of target mRNAs leading to their translational inhibition or degradation of the target[87]. Recent evidence suggests that expression of at least 20–30% of human protein-coding genes is modulated by miRNAs[88]. A large number of miRNAs are present in the mammalian brain and spinal cord, where they play key roles in neurodevelopment and are likely to be important mediators of plasticity[88,89,90,91]. Some miRNAs have been implicated in several neurological diseases[87,90]. Several studies also suggest the possibility of miRNA involvement in neurodegeneration[87].
In 2009, we first reported the dysregulated expression of a larger set of miRNAs in the spinal cord following traumatic injury[92], supported by subquentic studies on miRNA expression after spinal cord injury[93,94]. These altered miRNAs can be classified into 3 categories: (1) up-regulation, (2) down-regulation and (3) an early up-regulation at 4 hours followed by down-regulation at 1 and 7 days post-spinal cord injury[92]. The bioinformatics analysis indicates that the potential targets for miRNAs altered after spinal cord injury include genes encoding components that are involved in the inflammation, oxidation, and apoptosis that are known to play important roles in the pathogenesis of spinal cord injury[92]. These findings suggest that abnormal expression of miRNAs may contribute to the pathogenesis of spinal cord injury and is a potential target for therapeutic interventions following spinal cord injury.
Although the roles of miRNAs in spinal cord injury remain to be determined, increasing evidence suggests that miRNAs represent a new class of drug targets[95,96,97]. Inhibition of a particular miRNA linked to spinal cord injury with miRNA inhibitor can remove the block against expression of a therapeutic protein and conversely, administration of a miRNA mimetics can boost the endogenous miRNA population repressing a detrimental gene. For example, PTEN is a direct target of miR-21[98]. miR-21 mimics may exert neuroprotective effects on spinal cord injury by inhibition of PTEN (Figure 3). Taking advantage of their small size and the current knowledge of miRNA biogenesis, modified RNAs can be transiently delivered as a synthetic, preprocessed miRNA or anti-miRNA oligonucleotide[87]. Several studies have shown the potential of customized miRNA inhibitors to target specific pathologic miRNAs, both in vitro, and more importantly in vivo[96,99]. The anti-miRNA oligonucleotides are single stranded reverse complement oligonucleotides. The stability, binding affinity and specificity of these oligonucleotides have been improved through chemical modification[87]. The miRNA mimics are small, usually double stranded and chemically modified oligonucleotides which can be used to down-regulate a specific target protein[87]. Although there are many challenges for miRNAs as therapeutic targets such as delivery, potential off-target effects and safety, the strategy of miRNA manipulation in vivo to regulate disease-related processes is already becoming a feasible future therapeutic approach. Thus, miRNA is an attractive target for therapeutic interventions following spinal cord injury.
Acknowledgments:
We appreciate the use of the Core Facility of the Spinal Cord and Brain Injury Research Group/Stark Neurosciences Research Institute at Indiana University.
Footnotes
Funding: This work was supported by National Institutes of Health (NIH/NINDS NS059622, NS052290, NS050243, NS073636) and the Mari Hulman George Endowment Funds (XMX), and the Indiana Spinal Cord and Brain Injury Research Funds (ISDH, Grant # A70-2-079609 and A70-9-079138) (NKL & XMX).
Conflicts of interest: None declared.
(Edited by Zhao LJ/Song LP)
REFERENCES
- [1].Olivas AD, Noble-Haeusslein LJ. Phospholipase A2 and spinal cord injury: a novel target for therapeutic intervention. Ann Neurol. 2006;59:577–579. doi: 10.1002/ana.20840. [DOI] [PubMed] [Google Scholar]
- [2].Park E, Velumian AA, Fehlings MG. The role of excitotoxicity in secondary mechanisms of spinal cord injury: a review with an emphasis on the implications for white matter degeneration. J Neurotrauma. 2004;21:754–774. doi: 10.1089/0897715041269641. [DOI] [PubMed] [Google Scholar]
- [3].Hall ED, Springer JE. Neuroprotection and acute spinal cord injury: a reappraisal. NeuroRx. 2004;1:80–100. doi: 10.1602/neurorx.1.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Carlson SL, Parrish ME, Springer JE, et al. Acute inflammatory response in spinal cord following impact injury. Exp Neurol. 1998;151:77–88. doi: 10.1006/exnr.1998.6785. [DOI] [PubMed] [Google Scholar]
- [5].Mautes AE, Weinzierl MR, Donovan F, et al. Vascular events after spinal cord injury: contribution to secondary pathogenesis. Phys Ther. 2000;80:673–687. [PubMed] [Google Scholar]
- [6].Popovich PG, Wei P, Stokes BT. Cellular inflammatory response after spinal cord injury in Sprague-Dawley and Lewis rats. J Comp Neurol. 1997;377:443–464. doi: 10.1002/(sici)1096-9861(19970120)377:3<443::aid-cne10>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- [7].Resnick DK, Nguyen P, Cechvala CF. Regional and temporal changes in prostaglandin E2 and thromboxane B2 concentrations after spinal cord injury. Spine J. 2001;1:432–436. doi: 10.1016/s1529-9430(01)00130-9. [DOI] [PubMed] [Google Scholar]
- [8].Streit WJ, Semple-Rowland SL, Hurley SD, et al. Cytokine mRNA profiles in contused spinal cord and axotomized facial nucleus suggest a beneficial role for inflammation and gliosis. Exp Neurol. 1998;152:74–87. doi: 10.1006/exnr.1998.6835. [DOI] [PubMed] [Google Scholar]
- [9].Tonai T, Taketani Y, Ueda N, et al. Possible involvement of interleukin-1 in cyclooxygenase-2 induction after spinal cord injury in rats. J Neurochem. 1999;72:302–309. doi: 10.1046/j.1471-4159.1999.0720302.x. [DOI] [PubMed] [Google Scholar]
- [10].Wang CX, Olschowka JA, Wrathall JR. Increase of interleukin-1beta mRNA and protein in the spinal cord following experimental traumatic injury in the rat. Brain Res. 1997;759:190–196. doi: 10.1016/s0006-8993(97)00254-0. [DOI] [PubMed] [Google Scholar]
- [11].Xu J, Fan G, Chen S, et al. Methylprednisolone inhibition of TNF-alpha expression and NF-kB activation after spinal cord injury in rats. Brain Res Mol Brain Res. 1998;59:135–142. doi: 10.1016/s0169-328x(98)00142-9. [DOI] [PubMed] [Google Scholar]
- [12].Dusart I, Schwab ME. Secondary cell death and the inflammatory reaction after dorsal hemisection of the rat spinal cord. Eur J Neurosci. 1994;6:712–724. doi: 10.1111/j.1460-9568.1994.tb00983.x. [DOI] [PubMed] [Google Scholar]
- [13].Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- [14].Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- [15].Xu J, Kim GM, Ahmed SH, et al. Glucocorticoid receptor-mediated suppression of activator protein-1 activation and matrix metalloproteinase expression after spinal cord injury. J Neurosci. 2001;21:92–97. doi: 10.1523/JNEUROSCI.21-01-00092.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bethea JR, Castro M, Keane RW, et al. Traumatic spinal cord injury induces nuclear factor-kappaB activation. J Neurosci. 1998;18:3251–3260. doi: 10.1523/JNEUROSCI.18-09-03251.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chiu R, Boyle WJ, Meek J, et al. The c-Fos protein interacts with c-Jun/AP-1 to stimulate transcription of AP-1 responsive genes. Cell. 1988;54:541–552. doi: 10.1016/0092-8674(88)90076-1. [DOI] [PubMed] [Google Scholar]
- [18].Halazonetis TD, Georgopoulos K, Greenberg ME, et al. c-Jun dimerizes with itself and with c-Fos, forming complexes of different DNA binding affinities. Cell. 1988;55:917–924. doi: 10.1016/0092-8674(88)90147-x. [DOI] [PubMed] [Google Scholar]
- [19].Gasparini C, Feldmann M. NF-kappaB as a target for modulating inflammatory responses. Curr Pharm Des. 2012 doi: 10.2174/138161212803530763. [DOI] [PubMed] [Google Scholar]
- [20].Bond M, Baker AH, Newby AC. Nuclear factor kappa B activity is essential for matrix metalloproteinase-1 and -3 upregulation in rabbit dermal fibroblasts. Biochem Biophys Res Commun. 1999;264:561–567. doi: 10.1006/bbrc.1999.1551. [DOI] [PubMed] [Google Scholar]
- [21].Vincenti MP, Coon CI, Brinckerhoff CE. Nuclear factor kappaB/p50 activates an element in the distal matrix metalloproteinase 1 promoter in interleukin-1beta-stimulated synovial fibroblasts. Arthritis Rheum. 1998;41:1987–1994. doi: 10.1002/1529-0131(199811)41:11<1987::AID-ART14>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- [22].Yokoo T, Kitamura M. Dual regulation of IL-1 beta-mediated matrix metalloproteinase-9 expression in mesangial cells by NF-kappa B and AP-1. Am J Physiol. 1996;270:F123–130. doi: 10.1152/ajprenal.1996.270.1.F123. [DOI] [PubMed] [Google Scholar]
- [23].Bond M, Fabunmi RP, Baker AH, et al. Synergistic upregulation of metalloproteinase-9 by growth factors and inflammatory cytokines: an absolute requirement for transcription factor NF-kappa B. FEBS Lett. 1998;435:29–34. doi: 10.1016/s0014-5793(98)01034-5. [DOI] [PubMed] [Google Scholar]
- [24].Yong VW, Krekoski CA, Forsyth PA, et al. Matrix metalloproteinases and diseases of the CNS. Trends Neurosci. 1998;21:75–80. doi: 10.1016/s0166-2236(97)01169-7. [DOI] [PubMed] [Google Scholar]
- [25].Hsu CY, Hogan EL, Gadsden RH, Sr, et al. Vascular permeability in experimental spinal cord injury. J Neurol Sci. 1985;70:275–282. doi: 10.1016/0022-510x(85)90169-8. [DOI] [PubMed] [Google Scholar]
- [26].Smith JA, Das A, Ray SK, et al. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull. 2012;87:10–20. doi: 10.1016/j.brainresbull.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yakovlev AG, Faden AI. Sequential expression of c-fos protooncogene, TNF-alpha, and dynorphin genes in spinal cord following experimental traumatic injury. Mol Chem Neuropathol. 1994;23:179–190. doi: 10.1007/BF02815410. [DOI] [PubMed] [Google Scholar]
- [28].Wang CX, Nuttin B, Heremans H, et al. Production of tumor necrosis factor in spinal cord following traumatic injury in rats. J Neuroimmunol. 1996;69:151–156. doi: 10.1016/0165-5728(96)00080-x. [DOI] [PubMed] [Google Scholar]
- [29].Bartholdi D, Schwab ME. Expression of pro-inflammatory cytokine and chemokine mRNA upon experimental spinal cord injury in mouse: an in situ hybridization study. Eur J Neurosci. 1997;9:1422–1438. doi: 10.1111/j.1460-9568.1997.tb01497.x. [DOI] [PubMed] [Google Scholar]
- [30].Yan P, Li Q, Kim GM, et al. Cellular localization of tumor necrosis factor-alpha following acute spinal cord injury in adult rats. J Neurotrauma. 2001;18:563–568. doi: 10.1089/089771501300227369. [DOI] [PubMed] [Google Scholar]
- [31].Yan P, Liu N, Kim GM, et al. Expression of the type 1 and type 2 receptors for tumor necrosis factor after traumatic spinal cord injury in adult rats. Exp Neurol. 2003;183:286–297. doi: 10.1016/s0014-4886(03)00135-3. [DOI] [PubMed] [Google Scholar]
- [32].Kim GM, Xu J, Song SK, et al. Tumor necrosis factor receptor deletion reduces nuclear factor-kappaB activation, cellular inhibitor of apoptosis protein 2 expression, and functional recovery after traumatic spinal cord injury. J Neurosci. 2001;21:6617–6625. doi: 10.1523/JNEUROSCI.21-17-06617.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Constantini S, Young W. The effects of methylprednisolone and the ganglioside GM1 on acute spinal cord injury in rats. J Neurosurg. 1994;80:97–111. doi: 10.3171/jns.1994.80.1.0097. [DOI] [PubMed] [Google Scholar]
- [34].Moalem G, Leibowitz-Amit R, Yoles E, et al. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med. 1999;5:49–55. doi: 10.1038/4734. [DOI] [PubMed] [Google Scholar]
- [35].Rapalino O, Lazarov-Spiegler O, Agranov E, et al. Implantation of stimulated homologous macrophages results in partial recovery of paraplegic rats. Nat Med. 1998;4:814–821. doi: 10.1038/nm0798-814. [DOI] [PubMed] [Google Scholar]
- [36].Nawashiro H, Tasaki K, Ruetzler CA, et al. TNF-alpha pretreatment induces protective effects against focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 1997;17:483–490. doi: 10.1097/00004647-199705000-00001. [DOI] [PubMed] [Google Scholar]
- [37].Gary DS, Bruce-Keller AJ, Kindy MS, et al. Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. J Cereb Blood Flow Metab. 1998;18:1283–1287. doi: 10.1097/00004647-199812000-00001. [DOI] [PubMed] [Google Scholar]
- [38].Bruce AJ, Boling W, Kindy MS, et al. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med. 1996;2:788–794. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- [39].Yan P, Xu J, Li Q, et al. Glucocorticoid receptor expression in the spinal cord after traumatic injury in adult rats. J Neurosci. 1999;19:9355–9363. doi: 10.1523/JNEUROSCI.19-21-09355.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- [41].Chen A, Xu XM, Kleitman N, et al. Methylprednisolone administration improves axonal regeneration into Schwann cell grafts in transected adult rat thoracic spinal cord. Exp Neurol. 1996;138:261–276. doi: 10.1006/exnr.1996.0065. [DOI] [PubMed] [Google Scholar]
- [42].Auphan N, DiDonato JA, Rosette C, et al. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science. 1995;270:286–290. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- [43].Barnes PJ. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci (Lond) 1998;94:557–572. doi: 10.1042/cs0940557. [DOI] [PubMed] [Google Scholar]
- [44].Hayashi R, Wada H, Ito K, et al. Effects of glucocorticoids on gene transcription. Eur J Pharmacol. 2004;500:51–62. doi: 10.1016/j.ejphar.2004.07.011. [DOI] [PubMed] [Google Scholar]
- [45].Buckingham JC. Glucocorticoids: exemplars of multi-tasking. Br J Pharmacol. 2006;147(Suppl 1):S258–268. doi: 10.1038/sj.bjp.0706456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Fuxe K, Harfstrand A, Agnati LF, et al. Immunocytochemical studies on the localization of glucocorticoid receptor immunoreactive nerve cells in the lower brain stem and spinal cord of the male rat using a monoclonal antibody against rat liver glucocorticoid receptor. Neurosci Lett. 1985;60:1–6. doi: 10.1016/0304-3940(85)90372-6. [DOI] [PubMed] [Google Scholar]
- [47].Ahima RS, Harlan RE. Charting of type II glucocorticoid receptor-like immunoreactivity in the rat central nervous system. Neuroscience. 1990;39:579–604. doi: 10.1016/0306-4522(90)90244-x. [DOI] [PubMed] [Google Scholar]
- [48].Liu N, Han S, Lu PH, et al. Upregulation of annexins I, II, and V after traumatic spinal cord injury in adult rats. J Neurosci Res. 2004;77:391–401. doi: 10.1002/jnr.20167. [DOI] [PubMed] [Google Scholar]
- [49].Liu NK, Zhang YP, Han S, et al. Annexin A1 reduces inflammatory reaction and tissue damage through inhibition of phospholipase A2 activation in adult rats following spinal cord injury. J Neuropathol Exp Neurol. 2007;66:932–943. doi: 10.1097/nen.0b013e3181567d59. [DOI] [PubMed] [Google Scholar]
- [50].Liu D, Liu J, Wen J. Elevation of hydrogen peroxide after spinal cord injury detected by using the Fenton reaction. Free Radic Biol Med. 1999;27:478–482. doi: 10.1016/s0891-5849(99)00073-8. [DOI] [PubMed] [Google Scholar]
- [51].Liu D, Sybert TE, Qian H, et al. Superoxide production after spinal injury detected by microperfusion of cytochrome c. Free Radic Biol Med. 1998;25:298–304. doi: 10.1016/s0891-5849(98)00055-0. [DOI] [PubMed] [Google Scholar]
- [52].Liu D, Liu J, Sun D, et al. The time course of hydroxyl radical formation following spinal cord injury: the possible role of the iron-catalyzed Haber-Weiss reaction. J Neurotrauma. 2004;21:805–816. doi: 10.1089/0897715041269650. [DOI] [PubMed] [Google Scholar]
- [53].Scott GS, Cuzzocrea S, Genovese T, et al. Uric acid protects against secondary damage after spinal cord injury. Proc Natl Acad Sci U S A. 2005;102:3483–3488. doi: 10.1073/pnas.0500307102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Olanow CW. A radical hypothesis for neurodegeneration. Trends Neurosci. 1993;16:439–444. doi: 10.1016/0166-2236(93)90070-3. [DOI] [PubMed] [Google Scholar]
- [55].Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- [56].Xu J, Kim GM, Chen S, et al. iNOS and nitrotyrosine expression after spinal cord injury. J Neurotrauma. 2001;18:523–532. doi: 10.1089/089771501300227323. [DOI] [PubMed] [Google Scholar]
- [57].Sharma HS, Westman J, Olsson Y, et al. Involvement of nitric oxide in acute spinal cord injury: an immunocytochemical study using light and electron microscopy in the rat. Neurosci Res. 1996;24:373–384. doi: 10.1016/0168-0102(95)01015-7. [DOI] [PubMed] [Google Scholar]
- [58].Farooque M, Isaksson J, Olsson Y. Improved recovery after spinal cord injury in neuronal nitric oxide synthase-deficient mice but not in TNF-alpha-deficient mice. J Neurotrauma. 2001;18:105–114. doi: 10.1089/089771501750055811. [DOI] [PubMed] [Google Scholar]
- [59].Liu XZ, Xu XM, Hu R, et al. Neuronal and glial apoptosis after traumatic spinal cord injury. J Neurosci. 1997;17:5395–5406. doi: 10.1523/JNEUROSCI.17-14-05395.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Crowe MJ, Bresnahan JC, Shuman SL, et al. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med. 1997;3:73–76. doi: 10.1038/nm0197-73. [DOI] [PubMed] [Google Scholar]
- [61].Liu NK, Xu XM. Phospholipase A2 and its molecular mechanism after spinal cord injury. Mol Neurobiol. 2010;41:197–205. doi: 10.1007/s12035-010-8101-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Liu NK, Titsworth WL, Xu XM. Phospholipase A2 in CNS disorders: Implication on traumatic spinal cord and brain injuries. In: Banik N, Ray SK, editors. Handbook of Neurochemistry and Molecular Neurobiology: Brain and Spinal Cord Trauma. 3rd ed. New York: Springer; 2009. pp. 321–341. [Google Scholar]
- [63].Titsworth WL, Liu NK, Xu XM. Role of secretory phospholipase a(2) in CNS inflammation: implications in traumatic spinal cord injury. CNS Neurol Disord Drug Targets. 2008;7:254–269. doi: 10.2174/187152708784936671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Liu NK, Zhang YP, Titsworth WL, et al. A novel role of phospholipase A2 in mediating spinal cord secondary injury. Ann Neurol. 2006;59:606–619. doi: 10.1002/ana.20798. [DOI] [PubMed] [Google Scholar]
- [65].Titsworth WL, Cheng X, Ke Y, et al. Differential expression of sPLA2 following spinal cord injury and a functional role for sPLA2-IIA in mediating oligodendrocyte death. Glia. 2009;57:1521–1537. doi: 10.1002/glia.20867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Liu NK, Titsworth WL, Zhang YP, et al. Characterizing phospholipase A2-induced spinal cord injury-A comparison with contusive spinal cord injury in adult rats. Transl Stroke Res. 2011;2:608–618. doi: 10.1007/s12975-011-0089-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Titsworth WL, Onifer SM, Liu NK, et al. Focal phospholipases A2 group III injections induce cervical white matter injury and functional deficits with delayed recovery concomitant with Schwann cell remyelination. Exp Neurol. 2007;207:150–162. doi: 10.1016/j.expneurol.2007.06.010. [DOI] [PubMed] [Google Scholar]
- [68].Zhao Z, Liu N, Huang J, et al. Inhibition of cPLA2 activation by Ginkgo biloba extract protects spinal cord neurons from glutamate excitotoxicity and oxidative stress-induced cell death. J Neurochem. 2011;116:1057–1065. doi: 10.1111/j.1471-4159.2010.07160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Park KK, Liu K, Hu Y, et al. PTEN/mTOR and axon regeneration. Exp Neurol. 2010;223:45–50. doi: 10.1016/j.expneurol.2009.12.032. [DOI] [PubMed] [Google Scholar]
- [70].Park KK, Liu K, Hu Y, et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Song W, Volosin M, Cragnolini AB, et al. ProNGF induces PTEN via p75NTR to suppress Trk-mediated survival signaling in brain neurons. J Neurosci. 2010;30:15608–15615. doi: 10.1523/JNEUROSCI.2581-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Zhang QG, Wu DN, Han D, et al. Critical role of PTEN in the coupling between PI3K/Akt and JNK1/2 signaling in ischemic brain injury. FEBS Lett. 2007;581:495–505. doi: 10.1016/j.febslet.2006.12.055. [DOI] [PubMed] [Google Scholar]
- [73].Sury MD, Vorlet-Fawer L, Agarinis C, et al. Restoration of Akt activity by the bisperoxovanadium compound bpV(pic) attenuates hippocampal apoptosis in experimental neonatal pneumococcal meningitis. Neurobiol Dis. 2011;41:201–208. doi: 10.1016/j.nbd.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Walker CL, Walker MJ, Liu NK, et al. Systemic bisperoxovanadium activates Akt/mTOR, reduces autophagy, and enhances recovery following cervical spinal cord injury. PLoS One. 2012;7:e30012. doi: 10.1371/journal.pone.0030012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Jing S, Wen D, Yu Y, et al. GDNF-induced activation of the ret protein tyrosine kinase is mediated by GDNFR-alpha, a novel receptor for GDNF. Cell. 1996;85:1113–1124. doi: 10.1016/s0092-8674(00)81311-2. [DOI] [PubMed] [Google Scholar]
- [76].Treanor JJ, Goodman L, de Sauvage F, et al. Characterization of a multicomponent receptor for GDNF. Nature. 1996;382:80–83. doi: 10.1038/382080a0. [DOI] [PubMed] [Google Scholar]
- [77].Trupp M, Arenas E, Fainzilber M, et al. Functional receptor for GDNF encoded by the c-ret proto-oncogene. Nature. 1996;381:785–789. doi: 10.1038/381785a0. [DOI] [PubMed] [Google Scholar]
- [78].Trupp M, Raynoschek C, Belluardo N, et al. Multiple GPI-anchored receptors control GDNF-dependent and independent activation of the c-Ret receptor tyrosine kinase. Mol Cell Neurosci. 1998;11:47–63. doi: 10.1006/mcne.1998.0667. [DOI] [PubMed] [Google Scholar]
- [79].Iannotti C, Ping Zhang Y, Shields CB, et al. A neuroprotective role of glial cell line-derived neurotrophic factor following moderate spinal cord contusion injury. Exp Neurol. 2004;189:317–332. doi: 10.1016/j.expneurol.2004.05.033. [DOI] [PubMed] [Google Scholar]
- [80].Cheng H, Wu JP, Tzeng SF. Neuroprotection of glial cell line-derived neurotrophic factor in damaged spinal cords following contusive injury. J Neurosci Res. 2002;69:397–405. doi: 10.1002/jnr.10303. [DOI] [PubMed] [Google Scholar]
- [81].Fargo KN, Foecking EM, Jones KJ, et al. Neuroprotective actions of androgens on motoneurons. Front Neuroendocrinol. 2009;30:130–141. doi: 10.1016/j.yfrne.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Fargo KN, Sengelaub DR. Testosterone manipulation protects motoneurons from dendritic atrophy after contralateral motoneuron depletion. J Comp Neurol. 2004;469:96–106. doi: 10.1002/cne.10991. [DOI] [PubMed] [Google Scholar]
- [83].Fargo KN, Sengelaub DR. Exogenous testosterone prevents motoneuron atrophy induced by contralateral motoneuron depletion. J Neurobiol. 2004;60:348–359. doi: 10.1002/neu.20027. [DOI] [PubMed] [Google Scholar]
- [84].Jones KJ, Durica TE, Jacob SK. Gonadal steroid preservation of central synaptic input to hamster facial motoneurons following peripheral axotomy. J Neurocytol. 1997;26:257–266. doi: 10.1023/a:1018596316465. [DOI] [PubMed] [Google Scholar]
- [85].Clark MJ, Petroski GF, Mazurek MO, et al. Testosterone replacement therapy and motor function in men with spinal cord injury: a retrospective analysis. Am J Phys Med Rehabil. 2008;87:281–284. doi: 10.1097/PHM.0b013e318168bbec. [DOI] [PubMed] [Google Scholar]
- [86].Byers JS, Huguenard AL, Kuruppu D, et al. Neuroprotective effects of testosterone on motoneuron and muscle morphology following spinal cord injury. J Comp Neurol. 2012;520:2683–2696. doi: 10.1002/cne.23066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Liu NK, Xu XM. MicroRNA in central nervous system trauma and degenerative disorders. Physiol Genomics. 2011;43:571–580. doi: 10.1152/physiolgenomics.00168.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Krichevsky AM. MicroRNA profiling: from dark matter to white matter, or identifying new players in neurobiology. ScientificWorldJournal. 2007;7:155–166. doi: 10.1100/tsw.2007.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Bak M, Silahtaroglu A, Moller M, et al. MicroRNA expression in the adult mouse central nervous system. RNA. 2008;14:432–444. doi: 10.1261/rna.783108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Kosik KS. The neuronal microRNA system. Nat Rev Neurosci. 2006;7:911–920. doi: 10.1038/nrn2037. [DOI] [PubMed] [Google Scholar]
- [91].Miska EA, Alvarez-Saavedra E, Townsend M, et al. Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biol. 2004;5:R68. doi: 10.1186/gb-2004-5-9-r68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Liu NK, Wang XF, Lu QB, et al. Altered microRNA expression following traumatic spinal cord injury. Exp Neurol. 2009;219:424–429. doi: 10.1016/j.expneurol.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Strickland ER, Hook MA, Balaraman S, et al. MicroRNA dysregulation following spinal cord contusion: implications for neural plasticity and repair. Neuroscience. 2011;186:146–160. doi: 10.1016/j.neuroscience.2011.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Yunta M, Nieto-Diaz M, Esteban FJ, et al. MicroRNA dysregulation in the spinal cord following traumatic injury. PLoS One. 2012;7:e34534. doi: 10.1371/journal.pone.0034534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Esau CC, Monia BP. Therapeutic potential for microRNAs. Adv Drug Deliv Rev. 2007;59:101–114. doi: 10.1016/j.addr.2007.03.007. [DOI] [PubMed] [Google Scholar]
- [96].Mirnezami AH, Pickard K, Zhang L, et al. MicroRNAs: key players in carcinogenesis and novel therapeutic targets. Eur J Surg Oncol. 2009;35:339–347. doi: 10.1016/j.ejso.2008.06.006. [DOI] [PubMed] [Google Scholar]
- [97].Weiler J, Hunziker J, Hall J. Anti-miRNA oligonucleotides (AMOs): ammunition to target miRNAs implicated in human disease? Gene Ther. 2006;13:496–502. doi: 10.1038/sj.gt.3302654. [DOI] [PubMed] [Google Scholar]
- [98].Meng F, Henson R, Wehbe-Janek H, et al. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647–658. doi: 10.1053/j.gastro.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Medina PP, Slack FJ. Inhibiting microRNA function in vivo. Nat Methods. 2009;6:37–38. doi: 10.1038/nmeth0109-37. [DOI] [PubMed] [Google Scholar]