

Abstract
Innate immune system forms the first line of defense against microbial infections, as it exerts an immediate response. Innate immunity works through Toll-like receptors (TLRs) which functions as primary sensors of pathogens. TLR activates multiple signaling cascades leading to the induction of genes responsible for the release of inflammatory cytokines and type I interferon. Thus, they induce antimicrobial responses and also have an instructive role in adaptive immunity. However, TLR-mediated inflammation is said to be responsible for many of the destructive host responses in inflammatory diseases like periodontitis. Hence, therapeutics targeting TLRs are being used to treat disease such as HIV, Hepatitis B, asthma etc. Recently, synthetic TLR agonists are tried as novel vaccine adjuvant in treating periodontal diseases. This paper reviews the scope of TLR-based therapeutics in treating periodontitis.
Keywords: Periodontitis, toll like receptors, toll like receptor agonists
INTRODUCTION
Invasion of a host by a pathogenic agent initiates a cascade of immune responses through interactions between the pathogen-borne virulence factors and immune mechanisms of the host. Host–pathogen interaction occurs through the recognition of conserved molecular patterns called pathogen-associated molecular patterns (PAMPs). Receptors that recognize these PAMPs are germline encoded and are called as pattern recognition receptors (PRRs).[1,2] Toll-like receptors (TLRs) belong to the class of signaling PRRs.[3]
Periodontitis is an infection-driven chronic inflammatory disease, and TLRs play a pivotal role in its pathogenesis. Recent research has suggested that TLRs are essential in maintaining the periodontium in a healthy state. However, when excessively activated or inadequately controlled, they may contribute to chronic inflammatory diseases and autoimmune diseases.[4]
Hence, development of drugs that inhibit adverse host reactions and promote beneficial responses would be of great use to treat periodontitis. This article reviews about recent developments of therapeutic agents targeting TLRs and the issues involved in it.
Toll-like receptors
TLRs are evolutionarily conserved from the worm Caenorhabditis elegans to mammals.[5] Toll gene products were first discovered by Anderson et al., in 1985, in the fruit fly, Drosophila melanogaster. Charles Janeway and Ruslov Medzhitov identified the first human homolog of Drosophilia toll in 1997, initially termed as human Toll and subsequently as TLR 4.[6]
TLRs function as key PRRs of the innate immune system. TLRs are type I transmembrane glycoproteins comprising of leucine-rich repeat (LRR) motifs in the pathogen-binding ectodomains and an intracellular signaling domain that is homologous to interleukin-1 receptor (IL-1R), known as Toll/IL-1 receptor (TIR) domain.[7] The extracellular domains of TLR family proteins contain 16-28 LRRs. The LRR family comprises approximately 6000 proteins. The intracellular cytoplasmic domains have a common fold containing a five-stranded β-sheet surrounded by five α-helices. These are involved in a wide variety of functions including immune responses, signal transduction, cell-cycle regulation, enzyme regulation, etc.[8]
TLR family receptors interact with variety of ligands, ranging from hydrophilic nucleic acids to hydrophobic LPS or lipoproteins. They exist as homomultimers or heteromultimers even without their ligands.[5]
TLR expression has been implicated in diverse cell types which includes immune cells such as mast cells, NK cells, B cells, T cells, dendritic cells, neutrophils, macrophages and non-immune cells such as fibroblasts, airway and gut epithelial cells, etc., Till date 13 TLRs have been recognized in mice and 11 in humans.[9,10] TLRs 1,2,4,5 and 6 recognize extracellular microbial structures and are expressed on the host cell surface. For example, TLR 2 and 4 recognize bacterial lipoproteins and LPS, respectively. TLR 3, 7, 8, and 9 detect viral or bacterial nucleic acids and are located on endosomes, lysosomes, etc.[9]
TLR activation
TLRs serve as important signal-transducing elements which alert the host through pattern recognition of diverse microbial products. Moreover, development of adaptive immunity is controlled through activation of TLRs present on innate immune cells. This activation results in production of cytokines and chemokines critical for T-cell priming and differentiation.
TLRs bind to pathogen-derived factors and also products of inflamed tissue. This causes activation of transcription factors such as nuclear factor kappa of activated B cell (NF-κB) and the interferon regulatory factors (IRF) which lead to the induction of immune and inflammatory genes and thereby release of inflammatory cytokines and type I interferons.[11]
TLR signaling occurs via two different pathways: myeloid differentiation88 (MyD88)-dependent pathway and MyD88-independent pathway [Figure 1].[9] TLR 1, 2, 4, 5, 6, 7, 8 and 9 utilize the MyD88-dependent pathway. TLR 3 and 4 utilize the MyD88-independent pathway or Toll IL-1 receptor domain containing adaptor-inducing interferon β (TRIF) pathway. TIR domain containing adaptor proteins such as MyD88, TRIF, TIRAP (Toll/interleukin-1 receptor containing adaptor protein), TRAM (TRIF related adaptor molecule) play a major role in downstream signaling to elicit specific immune responses.[5,11]
Figure 1.

Flowchart describing TLR signaling pathways
MyD88-dependent pathway
The MyD88-dependent pathway is analogous to signaling pathways through IL-1 receptors. MyD88 recruits IL-1 receptor-activated kinase 4 (IRAK 4) which facilitates IRAK 4-mediated phosphorylation of IRAK 1. Activated IRAK 4 associates with tumor necrosis factor receptor-associated family 6 (TRAF-6) leading to two distinct signaling pathways. One pathway leads to activation of activated protein 1 (AP-1) through mitogenactivating protein kinases (MAPKs). The other pathway activates the IKK (Inhibitor of nuclear factor-κB (IκB) kinase) complex which causes degradation of IκB leading to nuclear translocation of transcription factor NF-κB. Both AP-1 and NF-κB induce the production of pro-inflammatory cytokines.[11,12]
MyD88 independent/TRIF dependent pathway
TLR 4 downstream signaling occurs with the association of adaptor proteins TRAM and TRIF. This in turn activates IRF 3 which leads to the production of IFN-β. TLR 3 signaling occurs via TRIF leading to activation of IRF 3 and production of type I interferons.[12]
Damage associated molecular patterns
In addition to recognizing ligands derived from pathogens, numerous endogenous ligands termed damage-associated molecular patterns (DAMP) released from sites with tissue injury and cell death are also detected by TLRs. DAMPs associated with necrotic cells include high mobility group box 1 (HMGB1), heat shock proteins (HSP), purine metabolites, etc., Extracellular sources of DAMPs are those generated from dying cells and at sites of tissue repair and remodeling, e.g., hyaluronan, heparin sulfate etc., TLR 2 and 4 are commonly involved in the detection of these DAMPs [Table 1].[13]
Table 1.
DAMPs and their associated TLRs

This implies that TLR-mediated immune response may be activated in the absence of foreign microbes. This idiopathic activation of TLRs may be responsible for the occurrence of various autoimmune diseases (systemic lupus erythematosus), chronic inflammatory diseases and infectious diseases.[14]
Toll-like receptors in the pathogenesis of periodontal disease
Periodontitis is a chronic inflammatory disease which implies that TLRs play a key role in chronic inflammation and autoimmunity by inducing the production of high levels of pro inflammatory cytokines.[15,16] Periodontal health represents a dynamic state, hence the pro-inflammatory and anti-inflammatory activities have to be optimally balanced. When this homeostasis is disturbed by the pathogens, TLRs become activated.
Gingival epithelial cell express TLR 2, 3, 4, 5, 6 and 9 that are also the first to detect microbial invasion. Once these TLRs bind to their respective PAMPs, antimicrobial peptides and pro-inflammatory cytokines are released immediately. If the microbial attack is uncontrolled, excessive stimulation of TLRs and increased production of pro-inflammatory cytokines occur leading to tissue destruction. Once the epithelial barrier is breached by the pathogens and their cytotoxic products, TLRs present on the non-immune cells such as the fibroblasts, osteoblasts and osteoclasts also get activated. As a result, pooling of cytokines occurs further contributing to the destruction of periodontal tissues. Thus, uncontrolled activation of TLRs results in destructive rather than protective response.[9]
TLR 2 is readily detectable in gingival epithelia, being denser in the spinous epithelial layer than in the basal layer.[15] There is upregulation of TLR 2, 4, 7 and 9 (but not TLR 5) in periodontitis lesions.[16]
Effects of TLRs on osteoclasts are quite complex. TLR 2 and 4 induce receptor activator of NF-kappaB ligand (RANKL) expression and osteoclast differentiation[16] but TLR 9 inhibits RANKL-induced osteoclast differentiation.[17] In early osteoclast precursors, TLR 2,4,9 inhibit their further differentiation, whereas in cells which are already under osteoclastic differentiation TLR enhances the survival of mature osteoclasts.[18,19]
TLR 4 plays a pivotal role in the pathogenesis of periodontitis due to several reasons. Lipopolysaccharides (LPS) are the major virulence factor of gram-negative bacteria and putative periodontal pathogen P. gingivalis. TLR 4 binds to LPS with the help of LPS-binding protein, MD2 and CD14 and plays an important role in various immune and inflammatory diseases such as sepsis, rheumatoid arthritis, ischemia/reperfusion injury, allergy and periodontitis. TLR 4 utilizes both MyD88-dependent and TRIF-dependent pathways. It also undergoes dimerization with TLR 2 which extends additional activation of TLR 4.[20]
Thus, TLRs are constitutively expressed in healthy periodontium and their upregulation by periodontal pathogens suggests that TLRs form the major role in pathogenesis of periodontitis.
Prevention of aberrant activation of TLRs
Negative regulation of TLR signaling
-
Synthetic substitutes
- TLR agonists
- TLR antagonist.
Negative regulation of TLR signaling
Negative controlling of TLR occurs by different mechanisms [Table 2].
Table 2.

Soluble decoy TLRs
It is well established that soluble decoy receptors induced by various stimuli provide important negative regulatory mechanisms for cytokines and chemokines and for their respective receptor interactions.[21,22] Similarly, there are also soluble decoy TLRs (sTLRs) that could be effective in blocking TLR signaling. Although there exist only a single copy of the TLR4 gene, several mRNA products have been detected indicating the presence of TLR4 isoforms.[23]
Dissociation of adaptor complexes:[24,25] TRAM adaptor with GOLD (TAG) domain, identified as a variant of TRAM, competes with TRAM for TRIF binding and inhibits the TRIF-dependent pathway
Sterile alpha and armadillo motif-containing protein (SARM) blocks TRIF complex formation by directly binding to TRIF
IRF5 directly interacts with MyD88 resulting in shutdown of IRF5-dependent gene induction.[24,25]
Degradation of signal proteins
Dendritic cells with tyrosine-protein kinase receptor 3 [Tyro3, Axl and Mer (TAM)] ligands suppress TLR-induced cytokine production. TAM signaling upregulates SOCS 1 (suppressors of cytokine signaling 1) and SOCS 3 expression through activation of STAT1 (signal tranducers and activator of transcription 1).[24,25]
Transcriptional regulation
MicroRNAs (miRNAs) are short double-stranded RNA molecules involved in gene silencing mechanisms. They are essential regulators in key immune pathways. miR-155, miR-21, miR-146a are of main concern due to their expression levels following TLR activation.[24,25]
miR-146a targets TRAF-6 and IRAK 1 implicating it as a negative regulator fine-tuning the immune response.
miR-155 or onco miR targets TAB2, RIP1 (receptor interacting protein 1) and IKKε (Inhibitor of nuclear factor κB kinase activator) causing negative regulation of immune signaling particularly in Kaposi's sarcoma and Hodgkins lymphoma.[26]
Pathogen recognition
Pathogens have developed strategies to evade TLR signaling. For example, Porphyromonas gingivalis alters the proportion of lipid A moieties and increases its virulence through manipulation of innate immune response. It causes degradation of essential TLR co-receptor (CD14) or immunostimulatory cytokines.[24,25]
Other mechanisms of negative regulation are through reduction of TLR expression and regulation of TLR effect by apoptosis.
Drawbacks of negative regulators
Though numerous negative regulators have been identified, these could not tightly control the activation due to various reasons.
Loss of individual negative regulators leads to hyperactivation of TLR signaling
Requires combination or synergistic effects of negative regulators
Multiple regulators targeting the same molecule
Pathogens acquire skillful strategies to suppress host response.[25]
Synthetic analogs of TLRs have been studied either as single agents or in combination with tumor antigens. These synthetic substitutes act directly on dendritic cells and also trigger NK cells to kill tumor cells. They can induce potent immunity in humans in addition to clinical responses.[27]
TLRs as vaccine adjuvants
Adjuvants are compounds that are generally added to killed whole organism or subunit vaccine to enhance the immune response against co-inoculated antigens. They improve the efficacy of vaccine in newborns, elderly or in immunocompromised individuals, e.g., of adjuvants alum, oil etc.
Adjuvants exert their actions by recruiting professional antigen-presenting cells (APCs) to the vaccination site, increasing the delivery of antigens to APCs, or by activating APCs to produce cytokines and by triggering T-cell responses.[28,29]
Mechanism of action of TLR adjuvants
Various mechanisms have been proposed to study the action of TLR adjuvants.[4] Dendritic cells communicate between the peripheral and lymphoid tissues and bridge the innate and adaptive immunities.[28,29] Dendritic cells mature when they interact with the pathogens or PAMPs. These matured dendritic cells express major histocompatibility (MHC) complexes and co-stimulatory molecules (e.g., CD40, CD80). On reaching the lymph nodes, activated dendritic cells produce cytokines and activate naive T lymphocytes. Activated T lymphocytes produce a Th1 or Th2 immune response.[30]
TLR agonists which are similar but less toxic when compared to these PAMPs can also cause dendritic cell maturation.[31] TLR signaling increases the expression of costimulatory molecules CD80/86 needed for clonal T-cell expansion.
For this reason, agonists of TLRs and other PRRs are attractive targets as vaccine adjuvants to maximize beneficial immunologic effects.
TLR agonists
TLR agonists are potential agents to be used as vaccine adjuvant. These are small molecular mimics of natural ligands which are less toxic but have improved pharmacodynamic and pharmacokinetic properties.[32]
Commonly used TLR agonist:
Monophosphoryl lipid A (MPLA): 3-O-deacylated monophosphoryl lipid A (MPLA) is generated from its parent compound, LPS. It is only 0.1% toxic when compared to its parent moiety. It induces cytokine cascades of both Th1 and Th2 type. Uses: Prophylactic and therapeutic vaccine against infectious disease and cancers.[32]
Guanosine-containing compounds (loxoribine) and imidazoquinolines (imiquimod, resiquimod and S-27609): These are synthetic analogs of DNA or RNA oligonucleotides. Imiquimod activates TLR 7 whereas resiquimod acts through TLR 8.[33]
Uses: Imiquimod and resiquimod are used for treatment of cutaneous disorders caused by papilloma virus.
Short Oligodeoxynucleotides (CpG ODN): These are analogues of short oligonucleotides (CpG motifs) which are recognized by TLR 9. They are mainly used in the treatment of human lupus. Based on the nucleotide sequence and length, CpGs are classified into class A, B and C.[32]
Classes A and C–activate B cells (lysosomal portion) and plasmacytoid DCs and induce IFN-α production
Class B–activates B cells (endosomal portion) and causes maturation of DCs.
Clinical uses of TLR agonists
Mainly used as vaccine adjuvants for allergic diseases, cancers (melanoma, lung cancer), infectious diseases (HBV, HIV, influenza, anthrax etc.). Clinical trials have been successfully conducted for most of the vaccines [Table 3].[34]
Table 3.
Clinical development: Vaccines and vaccine adjuvants
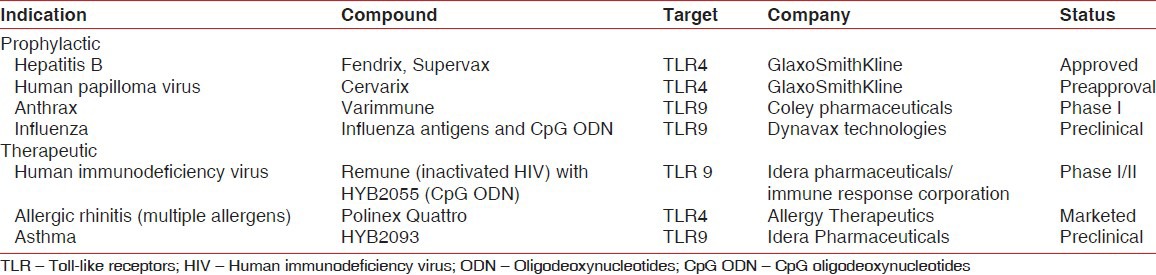
Disadvantages of TLR agonists
TLR agonist can cause chronic inflammation if improperly linked to TLR-containing organelles
Non-specific activation of immune cells as an anti-tumor or anti-infectious agent might result in autoimmune diseases
CpG ODN can cause systemic lupus erythematosus (SLE).
Aluminum hydroxide (Alum) is an adjuvant that is the component of many FDA-approved vaccines. TLR agonists are more effective when given in combination with these adjuvants.[34]
TLR Antagonists
These are developed as structural analogs of agonists which bind to TLR but fail to induce signal transduction. Consequently, the agonisitic action of TLR ligands for the induction of the inflammatory cascade is prevented.
Eritoran and resatorid: Eritoran is synthetic analog that inhibits LPS from activating TLR 4. It suppresses TNF-α production. Resatorid acts as a selective inhibitor of signaling from the intracellular domain of TLR 4.
Uses: Septic shock.[32]
DV1079 and CPG52364: DV1079 is a bifunctional inhibitor of TLR 7 and 9. It blocks IFN-α production. CPG52364, is an antagonists of TLR 7, 8, 9.
Uses: Autoimmune disorders such as SLE, psoriasis, rheumatoid arthritis.[32]
Drawback of TLR antagonists
Risk of increasing the susceptibility to infectious agents and tumors due to blocking of multiple TLRs.[32]
Scope of TLR as an adjuvant for periodontal vaccine
Periodontitis is basically microbe-induced inflammatory destruction of the periodontium. The pathogenesis is mediated through the host immune response against the pathogen leading to subsequent tissue destruction. It is still not clear as to which type of T-cell response is protective and which is destructive in periodontal disease. There is supportive, but not conclusive, evidence that T-helper 1 cells and their cytokines characterize early ⁄ stable periodontal lesions.
On the contrary, few studies have ascribed destructive effects to T-helper 1 cells.[35,36] If the T-helper 1 response cannot be sustained, this may lead to T-helper 2-driven disease progression characterized by increased infiltration of plasma cells secreting low-affinity, nonprotective antibodies. Hence, it is important to clarify which constitutes protective versus destructive host response in periodontitis.
The choice of adjuvant is a critical parameter, not only for enhancing the magnitude of specific immune responses, but also for modulating the T-cell response to the desired outcome. Lack of such knowledge in periodontal vaccine development may result in vaccine-induced host responses that could potentially do more harm than good. With such inadequacies in periodontal vaccines the scope for TLR-based therapeutics can complicate the situation further.
CONCLUSION
The importance of TLR in immune homeostasis is well recognized in the past decade. Now it is clear that TLR function must be regulated and controlled at the sites of inflammation. This has led to the development of various TLR-based therapeutic drugs.
TLRs are better targets when compared to cytokines or other downstream processes. Their role starts early both in inflammatory and infectious conditions so inhibiting them might be very potent. In infection-driven pathologic conditions (such as periodontitis), TLRs play the lead role in the initiation of inflammation resulting in the production of endogenous ligands (DAMPs). These endogenous ligands cause further stimulation of TLRs and release of inflammatory cytokines. Thus, targeting TLRs might lead to remission from chronic inflammatory conditions.[20]
Although a number of negative regulators for TLRs have been identified, single negative regulator could not bring about the intended effect resulting in the requirement of synergistic combinations of negative regulators. Adding to this are the polymorphic variants of negative regulators which have been reported in diseases such as inflammatory bowel disease, rheumatoid arthritis, SLE etc.[32]
Considering these issues associated with negative regulators, artificial or synthetic substitutes have been developed to manipulate at different levels in the TLR signaling cascade. These are expected to restore inflammatory diseases, overcome uncontrolled inflammation and counteract against infections. But these TLR agonists and antagonists when used for a prophylactic purpose have the chance of stimulating innate immune response at the subthreshold level leading to development of autoimmune disease and cancers.
Targeting TLR receptors with synthetic substitutes (TLR agonists and antagonists) for the prevention of various diseases and malignancies holds great potential in the medical field despite the drawbacks and pitfalls. These drugs show convincing evidence that they induce appropriate microenvironment to create effective immune responses in eradicating underlying diseases. But strategies to target the sites of disease or application of the drug can improve the outcome by focusing the alteration of the microenvironment only at the intended site of action, thus preventing the indiscriminate activation of immune cells at non-diseased sites.
In addition to delivery problems, TLR agonists have a short half life and poor bioavailability. TLR agonist-based therapy can be amended by selecting specific delivery vehicles, targeted delivery and combinations with other therapeutic modalities.[37]
Apart from the problems related to use of synthetic substitutes, periodontitis is a multifactorial disease and does not possess a single causative organism. The next major issue is that TLR recognizes both microbial and non-microbial ligands (DAMPs). Hence, precise targeting of a pathogen or a TLR becomes extremely difficult. Therefore, the emerging TLR-based therapeutics warrant extensive research starting from the selection of delivery vehicles to targeted TLR therapy.
TLR-based therapeutics may hold promise for treating periodontal diseases only if the issues related to the disease and synthetic substitutes have been solved. Further, long-term clinical studies are necessary to demonstrate the beneficial effect of TLR-based therapeutics.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Janeway CA, Medzhitov R. Innate Immune recognition. Annu Rev Immunol. 2002;20:197–16. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 2.Kumar H, Kawai T, Akira S. Pathogen Recognition by the innate immune system. Int Rev Immunol. 2011;30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- 3.Medzhitov R. The Innate Immune system. In: Paul WE, editor. Fundamental of Immunology. Philadelphia: Lippincott Williams and Wilkins Publishers; 2008. pp. 427–50. [Google Scholar]
- 4.Hajishengallis G. Toll gates to periodontal host modulation and vaccine therapy. Periodontol 2000. 2009;51:181–207. doi: 10.1111/j.1600-0757.2009.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akira S, Takeda K. Toll-like receptor signaling. Nat Rev Immunol. 2004;4:499–11. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 6.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophilia Toll protein signals activation of adaptive immunity. Nature. 1997;188:394–7. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 7.Pandey S, Agrawal DK. Immunobiology of Toll like receptors: Emerging trends. Immunol Cell Biol. 2006;84:333–41. doi: 10.1111/j.1440-1711.2006.01444.x. [DOI] [PubMed] [Google Scholar]
- 8.Jin MS, Lee JO. Structure of the Toll-like receptor family and its ligand complexes. Immunity. 2008;29:182–91. doi: 10.1016/j.immuni.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 9.Hans M, Hans VM. Toll – like receptors and their dual role in periodontitis. J Oral Sci. 2011;53:263–71. doi: 10.2334/josnusd.53.263. [DOI] [PubMed] [Google Scholar]
- 10.Kumar H, Kawal T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- 11.Takeda K, Akira S. Toll like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 12.Anderson KV. Toll signaling pathways in the innate immune response. Curr Opin Immunol. 2000;12:13–9. doi: 10.1016/s0952-7915(99)00045-x. [DOI] [PubMed] [Google Scholar]
- 13.Grace Y. Chen Gabriel Nunez. Sterile inflammation: Sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–37. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gearing AJ. Targeting toll like receptors for drug development: A summary of commercial approaches. Immunol Cell Biol. 2007;85:490–4. doi: 10.1038/sj.icb.7100102. [DOI] [PubMed] [Google Scholar]
- 15.Kusumoto Y, Hirano H, Saitoh K, Yamada S, Takedachi M, Nozaki T, et al. Human gingival epithelial cells produce chemotactic factors interleukin-8 and monocyte chemo attractant protein-1 after stimulation with Porphyromonas gingivalis via toll-like receptor 2. J Periodontol. 2004;75:370–9. doi: 10.1902/jop.2004.75.3.370. [DOI] [PubMed] [Google Scholar]
- 16.Mori Y, Yoshimura A, Ukai T, Lien E, Espevik T, Hara Y. Immunohistochemical localization of Toll-like receptors 2 and 4 in gingival tissue from patients with periodontitis. Oral Microbiol Immunol. 2003;18:54–8. doi: 10.1034/j.1399-302x.2003.180109.x. [DOI] [PubMed] [Google Scholar]
- 17.Kajita K, Honda T, Amanuma R, Domon H, Okui T, Ito H, et al. Quantitative messenger RNA expression of Toll-like receptors and interferon-α1 in gingivitis and periodontitis. Oral Microbiol Immunol. 2007;22:398–402. doi: 10.1111/j.1399-302X.2007.00377.x. [DOI] [PubMed] [Google Scholar]
- 18.Kim KW, Cho ML, Lee SH, Oh HJ, Kang CM, Ju JH, et al. Human rheumatoid synovial fibroblasts promote osteoclastogenic activity by activating RANKL via TLR-2 and TLR-4 activation. Immunol Lett. 2007;110:54–64. doi: 10.1016/j.imlet.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 19.Amcheslavsky A, Bar-Shavit Z. Interleukin (IL)-12 mediates the anti-osteoclastogenic activity of CpG-oligodeoxynucleotides. J Cell Physiol. 2006;207:244–50. doi: 10.1002/jcp.20563. [DOI] [PubMed] [Google Scholar]
- 20.O’Neill, Bryant CE, Doyle SL. Therapeutic targeting of Toll-like receptors for infectious and inflammatory diseases and cancer. Pharmacol Rev. 2009;61:177–97. doi: 10.1124/pr.109.001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takami M, Kim N, Rho J, Choi Y. Stimulation by toll-like receptors inhibits osteoclast differentiation. J Immunol. 2002;169:1516–23. doi: 10.4049/jimmunol.169.3.1516. [DOI] [PubMed] [Google Scholar]
- 22.Colotta F, Dower SK, Sims JE, Mantovani A. The type II ‘decoy’ receptor: A novel regulatory pathway for interleukin 1. Immunol Today. 1994;15:562–6. doi: 10.1016/0167-5699(94)90217-8. [DOI] [PubMed] [Google Scholar]
- 23.Qureshi ST, Larivière L, Leveque G, Clermont S, Moore KJ, Gros P, et al. Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4) J Exp Med. 1999;18:615–25. doi: 10.1084/jem.189.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liew FY, Xu D, Brint EK, O’Neill LA. Negative regulation of Toll Like receptor mediated immune responses. Nat Rev Immunol. 2005;5:446–58. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 25.Kondo T, Kawai T, Akira S. Dissecting negative regulation of Toll like receptor signaling. Trends Immunol. 2012;33:449–57. doi: 10.1016/j.it.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 26.Quinn SR, O’Neill LA. A trio of microRNAs that control toll like receptor signaling. Int Immunol. 2011;23:421–5. doi: 10.1093/intimm/dxr034. [DOI] [PubMed] [Google Scholar]
- 27.Bharadwaj N. TLR agonists: Are they good adjuvants? Cancer J. 2010;16:382–91. doi: 10.1097/PPO.0b013e3181eaca65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steinman RM. The dendritic cell system and its role in immunogenicity. Ann Rev Immunol. 1991;9:271–96. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 29.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 30.Gemmell E, Yamazaki K, Seymour GJ. The role of T cells in periodontal disease: Homeostasis and autoimmunity. Periodontol 2000. 2007;43:14–40. doi: 10.1111/j.1600-0757.2006.00173.x. [DOI] [PubMed] [Google Scholar]
- 31.Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002;20:709–60. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- 32.Makkouk A, Abdelnoor AM. The potential use of toll like receptor (TLR) agonists and antagonists as prophylactic and/or therapeutic agents. Immunopharmacol Immunotoxicol. 2009;31:331–8. doi: 10.1080/08923970902802926. [DOI] [PubMed] [Google Scholar]
- 33.Gorden KB, Gorsk KS, Gibson SJ, Kedl RM, Kieper WC, Qiu X, et al. Synthetic TLR agonists reveal functional differences between human TLR7 and TLR8. J Immunol. 2005;174:1259–68. doi: 10.4049/jimmunol.174.3.1259. [DOI] [PubMed] [Google Scholar]
- 34.Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll like receptor agonists and antagonists. Nat Med. 2007;13:552–9. doi: 10.1038/nm1589. [DOI] [PubMed] [Google Scholar]
- 35.Takeichi O, Haber J, Kawai T, Smith DJ, Moro I, Taubman MA. Cytokine profiles of T-lymphocytes from gingival tissues with pathological pocketing. J Dent Res. 2000;79:1548–5. doi: 10.1177/00220345000790080401. [DOI] [PubMed] [Google Scholar]
- 36.Ukai T, Mori Y, Onoyama M, Hara Y. Immunohistological study of interferon-gamma- and interleukin-4-bearing cells in human periodontitis gingiva. Arch Oral Biol. 2001;46:901–8. doi: 10.1016/s0003-9969(01)00057-7. [DOI] [PubMed] [Google Scholar]
- 37.Engel AL, Holt GE, Lu H. The pharmacokinetics of Toll-like receptor agonists and the impact on immune system. Expert Rev Clin Pharmacol. 2011;4:275–89. doi: 10.1586/ecp.11.5. [DOI] [PMC free article] [PubMed] [Google Scholar]