

Abstract
cAMP-dependent protein kinase (PKA) regulates the L-type calcium channel, the ryanodine receptor, and phospholamban (PLB) thereby increasing inotropy. Cardiac contractility is also regulated by p38 MAPK, which is a negative regulator of cardiac contractile function. The aim of this study was to identify the mechanism mediating the positive inotropic effect of p38 inhibition. Isolated adult and neonatal cardiomyocytes and perfused rat hearts were utilized to investigate the molecular mechanisms regulated by p38. PLB phosphorylation was enhanced in cardiomyocytes by chemical p38 inhibition, by overexpression of dominant negative p38α and by p38α RNAi, but not with dominant negative p38β. Treatment of cardiomyocytes with dominant negative p38α significantly decreased Ca2+-transient decay time indicating enhanced sarco/endoplasmic reticulum Ca2+-ATPase function and increased cardiomyocyte contractility. Analysis of signaling mechanisms involved showed that inhibition of p38 decreased the activity of protein phosphatase 2A, which renders protein phosphatase inhibitor-1 phosphorylated and thereby inhibits PP1. In conclusion, inhibition of p38α enhances PLB phosphorylation and diastolic Ca2+ uptake. Our findings provide evidence for novel mechanism regulating cardiac contractility upon p38 inhibition.
Keywords: Phospholamban, SERCA2a, p38, Cardiac contractility
1. Introduction
Cardiac contractile performance is mainly regulated by Frank–Starling mechanism and by local and circulating catecholamines, which bind to cardiomyocyte β-adrenergic receptors (β-ARs) on cardiomyocyte cell membrane. β-ARs coupling to Gs induces the cAMP-dependent protein kinase (PKA) activity of leading to phosphorylation of a set of proteins involved in regulation of cardiac excitation–contraction coupling, including L-Type Ca2+ channels (LTCC), ryanodine receptors (RyR), troponin I, the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA2a), and phospholamban (PLB). PLB is a phosphoprotein, which tightly regulates SERCA2a activity and thus indirectly controls Ca2+ reuptake to sarcoplasmic reticulum (SR) during diastole [1,2]. In its non-phosphorylated form PLB is a negative regulator of SERCA2a. Upon phosphorylation (mainly by PKA and calcium/calmodulin dependent protein kinase (CaMKII)), PLB releases SERCA2a from inhibition. In opposite, PLB is dephosphorylated by protein phosphatase-1 (PP1), which suppresses SERCA2a function, thus reducing Ca2+ reuptake to SR and cardiac contractility [3]. PP1 is under tight regulation of inhibitor-1 (I-1), which in its active state depresses PP1 activity thereby maintaining PLB phosphorylation and SERCA2a activity [4].
p38 MAPK is phosphorylated and activated by two upstream MAP kinase kinases (MAPKKs, or MKKs), namely MKK3 and MKK6, whereas p38 MAPK is dephosphorylated mainly by dual-specificity protein phosphatases (DUSPs), also known as MAPK phosphatases. The p38 MAPK is activated by various pro-inflammatory stimuli, and p38 MAPK pathway is considered a therapeutic target in inflammatory disorders. In the heart, p38 MAPK is involved in regulation of numerous processes including cardiomyocyte apoptosis, cardiac hypertrophy, fibrosis and cardiac contractile function [5–7].
The first evidence of a role for p38 MAPK in regulating cardiac contractility was provided by Zheng et al. who showed that a chemical inhibitor of p38, SB203580 markedly enhances the β-AR-mediated contractile response and by Liao et al. who additionally showed that forced activation of p38 via activated MKK3 depresses the cardiomyocyte contractility [8,9]. Thereafter, several studies using both genetic and pharmacological approaches have indicated that p38 inhibition enhances cardiomyocyte contractile function [10–14]. However, the mechanisms by which p38 regulates intrinsic cardiac contractility are not fully understood. Moreover, the role of major cardiac p38 isoforms, p38α and p38β, in the regulation of inotropic response is not known. It has been shown that inhibition of p38 increases Ca2+-sensitivity of myofilaments [8], whereas forced activation of p38 enhances dephosphorylation of α-tropomyosin and decreases ATPase activity [13].
In a previous study, we showed that pharmacological inhibition of p38 further enhances endothelin-1 (ET-1) induced cardiac contractile function [14]. Importantly, we provided evidence that p38 inhibition may regulate SERCA2a function. In addition, in a recent study, mice deficient for DUSP1 and DUSP4 showed increased activation of p38 in the heart and defective Ca2+ uptake into SR, while the prolonged [Ca2+]i decay could be restored by treatment with p38 inhibitor SB731445 [11]. Moreover, when mice deficient for DUSP1/DUSP4 were crossed with mice lacking PLB, the LV contractile function was restored. These data thus suggest that p38 regulates mechanisms controlling Ca2+ homeostasis. Herein, we identify p38α as the key p38 isoform regulating intrinsic cardiac contractile function. We use chemical inhibitors, recombinant adenoviruses and RNAi to show that inhibition of p38α enhances PLB phosphorylation and SERCA2a function and provide evidence for the underlying molecular mechanisms.
2. Methods
Detailed Materials and Methods section is found in the Online Data Supplement.
All animal protocols were approved by the Animal Use and Care Committee of the University of Oulu. Neonatal cardiomyocytes (NRVMs) were used for molecular biological analyses. Adult cardiomyocytes and ex vivo heart samples were used for molecular biological and functional analyses.
2.1. Ca2+ transient measurements
Isolated adult rodent cardiomyocytes were plated onto 35 mm glass-bottom laminin-coated dishes. Serum free media was removed and cells were kept in BDM-free Tyrode’s solution containing 1.25 mM CaCl2. When applicable, adenoviral infection was done 18 h prior to measurement. Cells were loaded either with 2 μM Fluo-4-AM (#14201, Invitrogen) or 5 μM Rhod2-AM (#1245MP Invitrogen) for 20 min at 37 °C. After that, cells were rinsed and pre-incubated 15 min at 37 °C to ensure de-esterification of Fluo-4-AM before measurements, Rhod2-labeled cells were measured immediately after rinsing. SB203580 (10 μM) was added 1 h before loading the cells with Fluo-4 AM, and DMSO was added to control cells in the same volume. Cells were paced at 0.5 Hz 40–50 V (pulse duration 1 ms). The fluorescence signals were recorded with Zeiss Cell Observer SD (Carl Zeiss, Germany) spinning disc confocal microscope (Fluo-4 excitation: 488 nm, emission: 500–550 nm; Rhod2 excitation: 561 nm, emission: 598–660 nm; 40×/0.75 objective) with a sampling rate of 100 frames/s. Analyses of Ca2+-transients were performed with Carl Zeiss Axiovision and OriginPro. Ca2+-transients are expressed as a background subtracted F/F0−ratio, where F is the background subtracted fluorescence intensity and F0 is the background subtracted minimum fluorescence value measured from each cell at rest.
2.2. Statistical analysis
Results are presented as mean ± S.E. or S.D. Data were analyzed with Student’s t test and for multiple experimental groups one way analysis of variance followed by Bonferroni post hoc test was used. Differences were considered statistically significant at the level of p < 0.05.
3. Results
3.1. Inhibition of p38α enhances PLB phosphorylation in cardiomyocytes
To better understand the role of p38 MAPKs in controlling cardiac calcium regulatory proteins we first studied if inhibition of p38 in isolated cardiomyocytes is sufficient to induce PLB phosphorylation. Treatment of neonatal rat ventricular cardiomyocytes (NRVM) by p38 inhibitor SB203580 (10 μM) increased phosphorylation of PLB at Ser16 (Fig. 1A). Phosphorylation of PLB Ser16 was also modestly enhanced when p38 inhibitor-treated cells were stimulated with the Gq agonist ET-1 (100 nM, Fig. 1A). Inhibition of p38, however, had no effect on PLB Thr17 phosphorylation (Fig. 1A). Phosphorylation status of heat-shock protein 27 (Hsp27), a p38 target, confirms the inhibition of p38 (Fig. 1A). To determine if there is a difference between the cardiac p38 isoforms in regulating the response, NRVMs were infected with adenoviruses encoding either for dominant negative p38α (dn-p38α) or dominant negative p38β (dn-p38β). Immunoblot analysis showed that overexpression of dn-p38α, but not dn-p38β, is sufficient to enhance PLB phosphorylation at Ser16 (Fig. 1B). dn-p38 adenoviruses had no significant effect on total PLB protein. Similar to overexpression of dn-p38α, p38α depletion by RNAi enhanced Ser16 PLB phosphorylation (Fig. 1C). p38α siRNA efficiently downregulated p38α mRNA levels, but had no effect on p38β expression (Suppl. Fig. 1A). We then asked if forced activation of p38α or p38β is sufficient to modulate PLB phosphorylation. As shown in Fig. 1D, forced activation of p38α markedly reduced Ser16 PLB phosphorylation. Analysis of PLB protein levels, however, showed that forced activation of either of the cardiac p38 isoforms caused significant reduction in total PLB protein (Fig. 1D). In summary, inhibition of p38α, but not p38β, enhances PLB phosphorylation.
Fig. 1.
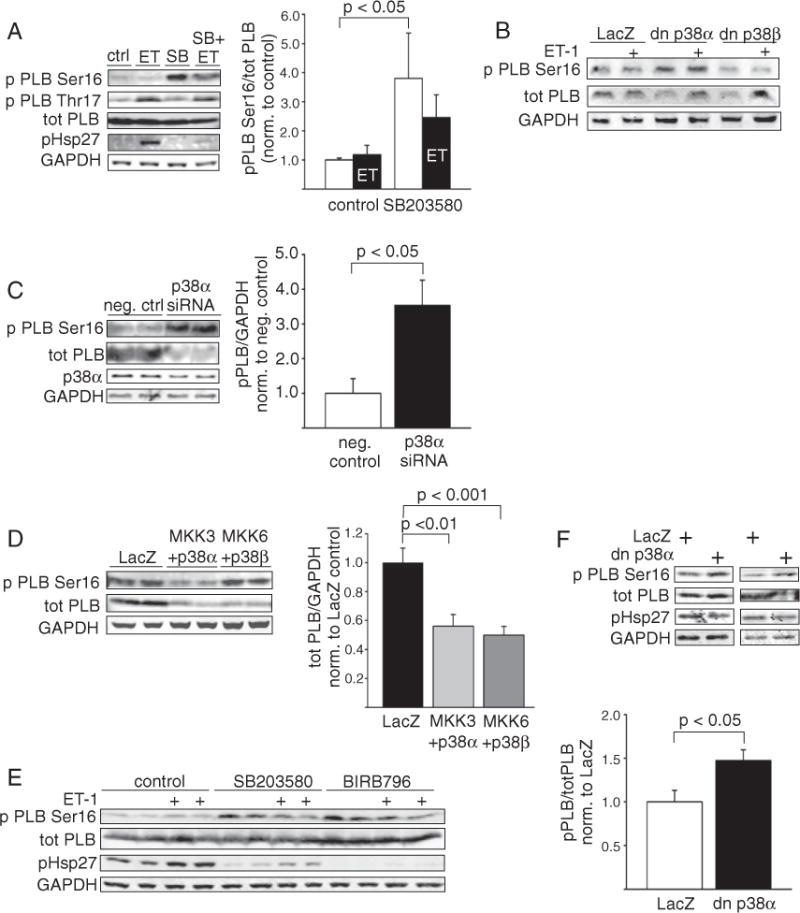
Inhibition ofp38 enhances PLB phosphorylation at Ser16. A, Immunoblot analysis and quantification of Ser16 phosphorylated and total PLB, and immunoblot analysis of PLB phosphorylation at Thr17 in NRVMs treated either with vehicle (DMSO),ET-1 (100 nM, 1 h), SB203580 (10 μM) orET-1 + SB203580. Phosphorylated Hsp27 was used as p38 inhibition control and GAPDH as loading control (mean ± SEM, n = 8–12). B, Immunoblot analysis of Ser16 phosphorylated and total PLB in dn-p38α (2 MOI), dn-p38β (2 MOI) and LacZ (2 MOI) infected NRVMs. C, Immunoblot analysis and quantification of Ser16 phosphorylated PLB and p38α in NRVMs transfected with negative control or p38α siRNA (150 nM, mean ± SEM, n = 5–8). D, Immunoblot analysis of Ser16 phosphorylated and total PLB and quantification of PLB protein levels in NRVMs overexpressing LacZ, MKK3 + p38α or MKK6 + p38β, virus dose 2 + 2 MOI in each well (mean ± SEM, n = 8–12). E, Immunoblot analysis of Ser16 phosphorylated and total PLB in ARVMs treated either with vehicle (DMSO), SB203580 (10 μM) or BIRB796 (1 μM) alone or in combination with ET-1 (100 nM). F, Representative western blot and quantification of PLB in ARVMs infected with dn-p38α (100 MOI) or LacZ (control) (mean ± SEM, n = 6). PLB, phospholamban; SERCA2a, sarcoplasmic reticulum Ca2+ATPase; NRVM, neonatal rat ventricular myocyte; ET-1, endothelin-1; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; dn-p38α/β, dominant negative p38α/β; ARVMs, and adult rat ventricular cardiomyocytes.
Similar to data on protein level, activation of either of the p38 isoforms resulted in almost complete abolishment of PLB mRNA (Suppl. Fig. 1B). Forced p38α and p38β activation also significantly suppressed SERCA2a and calsequestrin (CASQ2) mRNA levels, whereas sodium-calcium exchanger (NCX) mRNA levels were not affected (Suppl. Fig. 1B). There is actually data from previous studies showing that activation of p38 in vitro or in vivo suppresses SERCA2a expression [15,16] thereby worsening diastolic Ca2+ uptake and cardiomyocyte contractility. On the other hand, overexpression of dn-p38α resulted in significant decrease in SERCA2a mRNA levels, and overexpression of dn-p38β resulted in a decrease in PLB, SERCA2a, CASQ2 and NCX mRNA levels (Suppl. Fig. 1C). Depletion of p38α by siRNA modestly decreased PLB mRNA levels, but had no effect on SERCA2a mRNA levels (Suppl. Fig. 1D). Pharmacological inhibition of p38 with SB203580 (10 μM), however, had no effect on PLB, SERCA2a, CASQ2 or NCX mRNA levels (Suppl. Fig. 1E). In summary, overexpression of either wild type or dominant negative p38, but not chemical p38 inhibition or p38α depletion by RNAi, results in decreased SERCA2a expression. These findings suggest a possible non-kinase role for p38 in regulating its downstream targets.
For possible further analysis of functional effects of p38 inhibition on Ca2+ handling/SERCA2a function, we then assessed the effects of p38 inhibition on PLB phosphorylation in adult cardiomyocytes. Treatment of adult rat ventricular myocytes (ARVMs) with SB203580 (10 μM) caused a marked increase in PLB phosphorylation at Ser16 (Fig. 1E). Similar to findings with SB203580, treatment of ARVMs with BIRB796 (1 μM), a p38 inhibitor structurally different from SB203580, markedly enhanced PLB phosphorylation at Ser16. Similar to findings with NRVMs, co-treatment of cells with ET-1 and either of the p38 inhibitors also appeared to increase PLB phosphorylation compared to treatment with ET-1 alone. To assess the relative role of the p38α isoform, adult rat cardiomyocytes were infected with adenovirus encoding for dominant negative p38α. Similar to findings in NRVMs, p38α inhibition enhanced PLB Ser16 phosphorylation (Fig. 1F). Thus, our earlier beneficial results of p38 inhibition on PLB phosphorylation in neonatal cardiomyocytes could be duplicated in adult cardiomyocytes. Furthermore, these p38α-dependent effects on PLB Ser16 validate adult cardiomyocytes as a paradigm in which to further explore the function of other critical regulatory components of contractile response.
3.2. Inhibition of p38α enhances SERCA2a function and cardiomyocyte contractility
To determine if inhibition of p38 and enhanced PLB phosphorylation were sufficient to affect SERCA2a function, we assessed the effect of p38 inhibition on Ca2+ cycling in adult cardiomyocytes upon p38 inhibition. As shown in Fig. 2A, treatment of isolated adult mouse cardiomyocytes with SB203580 (10 μM) resulted in significant decrease in Ca2+-transient decay time (τ = 270 ± 29 ms vs. 203 ± 50 ms, p < 0.01). On the other hand, Ca2+-transient amplitude was not affected by p38 inhibition (Fig. 2A), indicating that p38 inhibition had no effect on ryanodine receptors (RyR) or L-type Ca2+ channels (LTCC). Analysis of Ca2+ transients upon inhibition of p38α isoform alone revealed a marked decrease in Ca2+-transient decay time in dn-p38α infected cells compared to LacZ infected cells (τ = 336 ± 98 ms vs. 149 ± 55 ms, p < 0.001, Fig. 2B). Similar to findings with chemical inhibitor, dn-p38α had no effect on Ca2+-transient amplitude (Fig. 2B).
Fig. 2.
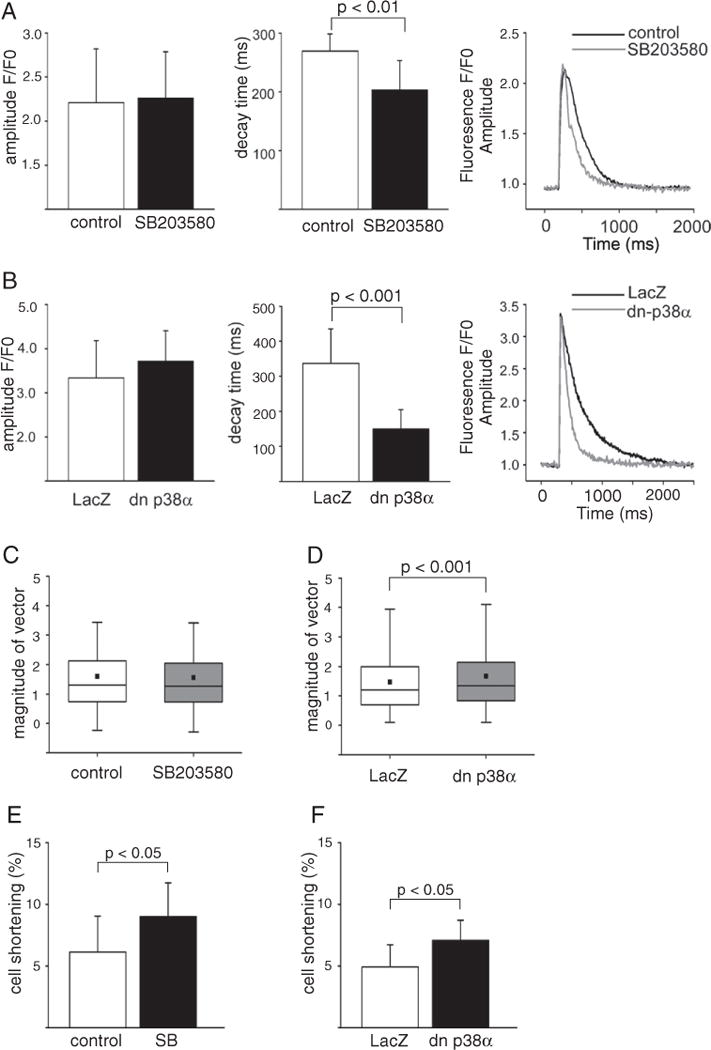
Inhibition of p38 induces shortening in Ca2+ decay time. A–B, Ca2+-transient average amplitudes, decay times (ms) and representative fluorescence signals measured from Fluo-4 loaded cells at 0.5 Hz pacing frequency in AMVMs treated either with vehicle (DMSO) or SB203580 (10 μM) alone (A) or LacZ or dn-p38α (100 MOI) adenoviruses (B) under basal conditions. A–B, data is shown as mean ± SD; Ca2+-transient of 11–29 cardiomyocytes were measured for each group. C–D, Box plot graphs of contractility measurements by particle image velocimetry analysis. NRVMs were either treated with SB203580 (10 μM,C) or infected with dn p38α (2 MOI, D), DMSO and LacZ as controls, respectively (data was obtained from three independent experiments). E–F, Analysis of cell shortening in ARVMs. E, Freshly isolated cardiomyocytes were treated with either SB203580 (10 μM) or DMSO (control). F, ARVMs were infected with dn p38α (100 MOI) or LacZ (100 MOI, control). Total of 7–11 myocytes were measured for each group from two different hearts. E–F, data is shown as mean ± SD. AMVMs, adult mouse ventricular cardiomyocytes; ARVMs, adult rat ventricular cardiomyocytes; ET-1, endothelin-1; dn-p38α, dominant negative p38α; NRVM, neonatal rat ventricular cardiomyocytes.
Analysis of cardiomyocyte contractility in NRVMs by particle image velocimetry showed that treatment of cardiomyocytes with SB203580 had no significant effect on contractility (Fig. 2C, Suppl. Fig. 2A). Inhibition of p38α by dn-p38α resulted in significant increase in contractility compared with LacZ infected when measured by particle image velocimetry analysis (Fig. 2D, Suppl. Fig. 2B, Suppl. Videos 1–2). On the other hand, analysis of cardiomyocyte contractility in ARVMs showed that treatment with SB203580 (10 μM) significantly enhanced cell shortening (6.1 ± 2.7% vs. 9.0 ±2.9%, p < 0.05, Fig. 2E). Analysis of ARVMs upon inhibition of p38α isoform alone also revealed a marked increase in cell shortening in dn-p38α infected cells compared to LacZ infected cells (4.9 ± 1.8% vs. 7.1 ± 1.6%, p < 0.05, Fig. 2F).
To determine if p38 inhibition was sufficient to modulate Ca2+ cycling upon activation of Gq-dependent signaling, ET-1 stimulus was added to the medium. Consistent with the observed effects on PLB phosphorylation, there was a significant decrease in Ca2+ decay time also in cells cotreated with ET-1 and SB203580 compared to cells treated with ET-1 alone (τ = 332 ± 29 ms vs. 187 ± 38 ms, p < 0.001, Suppl. Fig. 2C). Notably, the decrease in Ca2+ decay time upon p38α inhibition also persisted in cells treated with ET-1 (τ = 220 ± 78 ms vs. 139 ± 33 ms, p < 0.001, Suppl. Fig. 2D). Thus, consistent with increased PLB phosphorylation, inhibition of p38α enhances SERCA2a function as witnessed by a decrease in Ca2+-transient decay time.
3.3. The response to p38 inhibition requires intrinsic PKA activity
An increase in Ca2+-transient amplitude due to activation of RyR or LTCC is a hallmark of PKA activation. Our data thus indicate that p38 inhibition does not induce holistic PKA activation. We then asked if modulation of PKA activity would affect the response to p38 inhibition. PKA is activated by Gs-induced adenylyl cyclase, which is regulated by ubiquitously expressed G protein-coupled receptor kinases (GRKs) that phosphorylate GPCRs triggering their desensitization. GRK2 is the major cardiac GRK and elevated in patients with heart failure [17]. We considered that modulating GRK2 would provide a physiological paradigm to both enhance and inhibit the PKA activity (see Fig. 6 further below). Overexpression of βARKct (carboxyl terminus of GRK2), whichis similar to GRK2 depletion stimulates cAMP formation, resulted in a marked increase in phosphorylation of PLB Ser16 (Fig. 3A). However, βARKct did not further augment PLB Ser16 phosphorylation afforded by p38 inhibition. To inhibit PKA activity, ARVMs were then infected with an adenovirus encoding for wild type GRK2 (wtGRK2). As shown in Fig. 3B, inhibition of p38 by SB203580 enhanced PLB phosphorylation in LacZ infected control cells, and the response was modestly attenuated in cells overexpressing wtGRK2. Similarly, overexpression of dn-p38α increased PLB phosphorylation in LacZ infected cells, but response was weaker in cells overexpressing GRK2 (Fig. 3C).
Fig. 6.
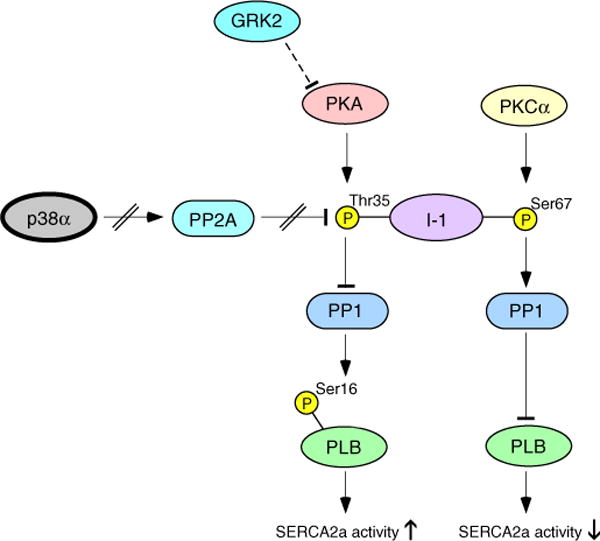
Regulation of PLB by inhibition of p38α. A schematic view of effects of p38α inhibition on signaling elements regulating PLB phosphorylation. PKA, protein kinase A; PKCα, protein kinase C alpha; I-1, inhibitor 1; PP1, protein phosphatase 1; PP2A, protein phosphatase 2A; ERK, extracellular signal-regulated kinase; PLB, phospholamban; GRK2, G-protein-coupled-kinase-2.
Fig. 3.
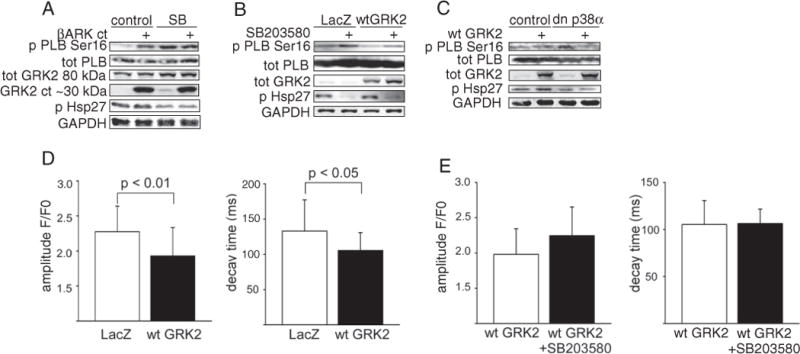
The response to p38 inhibition requires intrinsic PKA activity. A–C, Representative immunoblots of Ser16 phosphorylation and total PLB, GRK2, phosphorylated Hsp27 (p38 inhibition control) and GAPDH (loading control) in ARVMs. Cardiomyocytes were infected with βARKct (A) or wtGRK2 (B, C). p38 was inhibited either pharmacologically by SB203580 (10 μM, A–B) or by dn-p38α adenovirus (C). D–E, Averaged amplitude and decay times (ms) of intracellular Ca2+-transients measured with Rhod2. ARVMs were infected either with LacZ or wtGRK2, and treated either with vehicle (DMSO) or SB203580 (10 μM, 1 h prior measurement, E). A total of 13–57 cardiomyocytes from minimum two different hearts were measured in each group (mean ± SD). A–E, Total amount of virus was 100 MOI in all experiments, LacZ as control virus. PLB, phospholamban; I–1, inhibitor-1 ; Hsp27, heat shock protein 27; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; βARKct, carboxy-terminus of GRK2; GRK, G-protein-coupled receptor kinase; NRVMs, neonatal rat ventricular cardiomyocytes; ARVMs, adult rat ventricular cardiomyocytes; wtGRK2, wild type GRK2; dn-p38α, dominant negative p38α.
To investigate the effect of p38 inhibition on SERCA2a function upon PKA inhibition, we analyzed Ca2+ transients in adult rat cardiomyocytes overexpressing wtGRK2. Overexpression of wtGRK2 attenuated Ca2+ transient amplitude compared to LacZ infected cells (2.27 ± 0.37 vs. 1.93 ± 0.4, p < 0.01) and shortened decay time of Ca2+ transient (τ = 133 ± 43 ms vs. 105 ± 25 ms, p < 0.05, Fig. 3D). As suggested by attenuated response in PLB Ser16 phosphorylation upon p38 inhibition, inhibition of p38 in cardiomyocytes overexpressing GRK2 had no effect on Ca2+-transient decay time (Fig. 3E). Taken together, these data indicate that intrinsic PKA activity is required for shortening in [Ca2+]i decay time afforded by p38 inhibition.
3.4. Inhibition of p38 enhances GRK2 phosphorylation
We then considered the option that inhibition of p38 could modulate the activity of cardiac GRKs. Analysis of GRK2 phosphorylation showed that treatment of adult rat cardiomyocytes with SB203580 enhanced phosphorylation of GRK2 Ser670 (Suppl. Fig. 3A). It has been previously shown that phosphorylation of GRK2 Ser670 is catalyzed by extracellular signal-regulated kinase (ERK) [18]. Increased GRK2 Ser670 phosphorylation was also observed in cells treated with ET-1 alone or in combination with SB203580. As shown in Suppl. Fig. 3B, overexpression of dn-p38α also resulted in increased phosphorylation of GRK2 Ser670, and increased phosphorylation persisted in cells overexpressing wtGRK2. To address the functional significance of the Ser670 site of GRK2, we infected adult cardiomyocytes with adenoviruses encoding for wtGRK2 or GRK2 harboring a phosphorylation resistant mutation at Ser670 residue (GRK2S670A). As shown in Suppl. Fig. 3C, increased PLB phosphorylation afforded by p38 inhibition was attenuated in cardiomyocytes overexpressing wtGRK2 compared to cardiomyocytes infected with LacZ. However, the p38 inhibition-induced PLB phosphorylation persisted in cells overexpressing GRK2S670A. These data thus indicate that phosphorylation of GRK2 in response to p38 inhibition does not mediate the enhanced PLB phosphorylation, but GRK2 phosphorylation may rather serve as a negative feedback mechanism.
3.5. Inhibition of p38α enhances phosphorylation of I-1 at Thr35
The Ser16 PLB is phosphorylated by PKA and dephosphorylated by PP1 [3]. The activity of PP1 is controlled by I-1, which suppresses the PP1 activity when phosphorylated at Thr35 by PKA. This PKA-dependent activation of I-1 is antagonized by phosphorylation of I-1 at the proline-directed Ser67 [19]. To explore a possible role for the I-1–PP1 cascades in p38-dependent regulation of PLB function, we assessed the effect of p38 inhibition on I-1 Thr35 and Ser67 phosphorylation. Overexpression of dn-p38α had no clear effect on phosphorylation of I-1 at Ser67, whereas overexpression of dn-p38β modestly enhanced phosphorylation of I-1 at Ser67 (Fig. 4A). However, overexpression of dn-p38α, but not dn-p38β, markedly enhanced phosphorylation of I-1 at Thr35 (Fig. 4B). Similarly, p38α depletion by RNAi potentiated Thr35 I-1 phosphorylation (Fig. 4C). On the other hand, overexpression of GRK2 resulted in reduced phosphorylation of I-1 Thr35 and blunted the response to p38 inhibition (Fig. 4D). As a more physiological paradigm, we also infused isolated rat hearts with p38 inhibitor SB203580 with and without ET-1. In the previous study, we showed that p38 inhibition in these conditions enhances cardiac contractility and results in increased phosphorylation of PLB [14]. Similar to findings in isolated cardiomyocytes, analysis of protein samples extracted from hearts ex vivo showed increased phosphorylation of I-1 at Thr35 but no effect on I-1 Ser67 following administration of p38 inhibitor (Fig. 4E). These data suggest that p38 inhibition enhances I-1 activity leading to increased PLB phosphorylation and enhanced cardiac function also in intact heart.
Fig. 4.
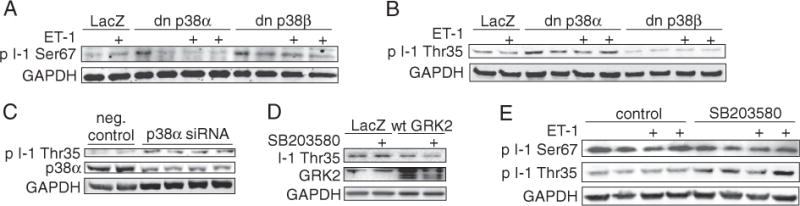
Inhibition of p38α enhances phosphorylation of inhibitor-1 at Thr35. Immunoblot analyses of Ser67 (A) and Thr35 (B) I-1 phosphorylation in NRVMs, infected either with dn-p38α or dn-p38β, LacZ as control. Virus dose was 2 MOI in each well. C, Immunoblot of Thr35 I-1 phosphorylation in NRVMs, silenced either with negative control or p38α siRNA (150 nM). D, Immunoblot analysis of I-1 Thr35 phosphorylation in NRVMs infected either with LacZ (control) or wtGRK2 (2 MOI), treated with SB203580 (10 μM). E, Immunoblot analyses of Ser67 and Thr35 I-1 phosphorylation in Langendorff perfused hearts, treated either with vehicle, SB203580 (3 μM), ET-1 (3 nM) or SB203580 + ET-1 (3 μM + 3 nM) for 10 min. At the end of the experiment, total protein was extracted from left ventricular tissue samples. GAPDH as loading control for all western blots. I-1, inhibitor-1; NRVM, neonatal rat ventricular myocyte; ET-1, endothelin-1 ; dn-p38α/β, dominant negative p38α/β; PLB, phospholamban; GRK2, G-protein coupled kinase-2; wtGRK2, wild type GRK2; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
3.6. p38 inhibition reduces PP2A activity
We then asked if the increased Thr35 phosphorylation of I-1 could be due to modulation of phosphatase activity by p38. The Thr35 residue of I-1 is a known target for protein phosphatase 2A (PP2A) [20]. To assess the effect of PP2A inhibition on PLB and I-1 phosphorylation, neonatal cardiomyocytes were treated with PP2A inhibitor okadaic acid (OA). As shown in Fig. 5A, treatment of cells with OA (1 nM) increased phosphorylation of both PLB Ser16 and I-1 Thr35. The magnitude of the response was similar to that achieved by SB203580. However, phosphorylation of eIF2α Ser51, a known target of PP1 [21], was also modestly enhanced by SB203580 and OA, further indicating that p38 inhibition results in inhibition of PP1 activity (Fig. 5A). Direct analysis of PP2A activity showed that treatment of cells with SB203580 and OA resulted in a comparable decrease in PP2A activity (Fig. 5B). To further investigate the role of PP2A, cardiomyocytes were treated with fostriecin, a specific inhibitor of PP2A [22]. As shown in Fig. 5C, fostriecin treatment resulted in increased phosphorylation of PLB Ser16 and I-1 Thr35. p38 inhibition thus reduces the activity of PP2A, which renders I-1 Thr35 phosphorylated and results in inhibition of PP1. In addition to I-1 Thr35, cardiac troponin I (cTnI) is a known target for PP2A in cardiomyocytes [23]. Analysis of phosphorylation of troponin I revealed an increase in phosphorylation of Ser22/23 (Ser 23/24 in humans) in response to p38 inhibition with SB203580 (Fig. 5D). There is also previous data suggesting that p38 regulates ERK via a PP2A dependent mechanism [24]. In agreement, p38 inhibition in cardiomyocytes resulted in enhanced ERK phosphorylation (Fig. 5E). The enhanced ERK phosphorylation was independent of PKA, since the response was also present when PKA was inhibited by overexpression of GRK2. Taken together, these data indicate that inhibition of p38 reduces PP2A activity and results in enhanced phosphorylation PP2A target I-1 Thr35.
Fig. 5.
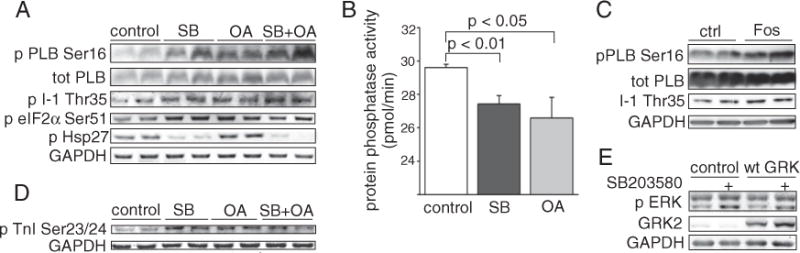
p38 inhibition reduces PP2A activity. A, Immunoblot analysis of Ser16 phosphorylated PLB, total PLB, Thr35 phosphorylated I-1, Ser51 phosphorylated eIF2α and phosphorylated Hsp27 in NRVMs treated either with SB203580 (10 μM) and okadaic acid (1 nM) or their combination. B, Protein phosphatase-2A assay, NRVMs were treated either with SB203580 (10 μM) or okadaic acid (1 nM) and phosphate transfer was determined. C, Immunoblot analysis of Ser16 phosphorylated PLB, total PLB and Thr35 phosphorylated I-1 in NRVMs treated with fostriecin (1 μM) for 1 h. D, Immunoblot analysis of phosphorylated TnI at Ser 23/24 in NRVMs treated either with DMSO (control), SB203580 (10 μM), okadaic acid (1 nM) or their combination. E, Immunoblot analysis of ERK1/2 phosphorylation, ARVMs infected either with dn-p38α or wtGRK2. GAPDH was usedas a loading control. OA, okadaic acid; PLB, phospholamban; I-1, inhibitor-1; eIF2α, eukaryotic initiation factor 2α; Hsp27, heat shock protein 27; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; NRVM, neonatal rat ventricular myocyte; TnI, troponin I; ERK, extracellular signal-regulated kinase; wtGRK2, wild type GRK2.
4. Discussion
It is well established that p38 is a negative regulator of cardiac contractile function. The underlying molecular mechanism, however, is not fully understood, and studies thus far have not addressed the isoform-specific effects of p38. In this study, we show that inhibition of p38α enhances PLB Ser16 phosphorylation and diastolic Ca2+ uptake in cardiomyocytes. Notably, the decrease in Ca2+-transient decay time persists even when dn-p38α infected cells are treated with Gq agonist ET-1 (which activates both p38α and p38β) thus strengthening the concept of key role for p38α regulating the lusitropic response. In agreement, phosphorylation of PLB is nearly abolished by activation of p38α, but not by activation of p38β. Analysis of cardiomyocyte contractile amplitude further showed that inhibition of p38α is sufficient to increase the contractility. These findings reveal a novel mechanism for regulation of cardiac contractility by p38 MAPK.
During a single contraction cycle the main part (~92% in adult mouse) of the released Ca2+ is pumped back into the SR by SERCA2a [25]. Impaired myocardial diastolic function in failing hearts is at least partly attributed to decreased SERCA2a levels and increasing SERCA2a levels in the heart by viral gene transfer has shown promise for treatment of heart failure [26]. PLB is an essential phosphoprotein regulating function of SERCA2a in the SR and changes in its phosphorylation associate with functional alterations in cardiac contractility [2]. PLB is regulated by phosphorylation on residues Ser16 and Thr17 by PKA and by CaMKII, respectively [27]. PP1 is the predominating phosphatase dephosphorylating PLB. Ubiquitously expressed I-1 is the first recognized regulator of PP1, and inhibits PP1 when phosphorylated at Thr35 by PKA resulting in increased phosphorylation of PLB at Ser16 [28]. Ablation of I-1 enhances PP1 activity and results in a mild decrease in cardiac contractility and in a significantly blunted β-adrenergic contractile response [29]. I-1 overexpression, on the other hand, decreases PP1 activity, enhances PLB phosphorylation and enhances cardiac contractile function [30]. I-1 is also regulated by phosphorylation at Ser67 by protein kinase C-α (PKCα), and increased phosphorylation of I-1 at Ser67 attenuates I-1 activity depressing cardiac contractile function [31]. In agreement, inducible overexpression of either constitutively active mutant of I-1 or PKCα phosphorylation-deficient mutant of I-1 is sufficient to enhance cardiac contractility [32].
Previous data on effect of p38 on I-1 do not exist. Our data demonstrates that Ser67 I-1 phosphorylation is not affected by overexpression of dn-p38α, but overexpression of dn-p38β modestly increases it. To the contrary, phosphorylation of I-1 at Thr35 is enhanced by dn-p38α, but not by dn-p38β. In agreement, depletion of p38α by siRNA results in increased phosphorylation of Thr35 I-1. These data are thus consistent with the observed increase in PLB Ser16 phosphorylation upon inhibition of p38α, but not upon inhibition of p38β. Since p38 inhibition had no effect on Ca2+-transient amplitude, we considered that enhanced I-1 Thr35 phosphorylation could be due to decreased phosphatase activity rather than enhanced PKA activity. Previous data shows that Thr35 of I-1 is dephosphorylated by PP2A [20]. Our current data shows that treatment of cardiomyocytes with PP2A inhibitor OA enhances I-1 Thr35 and PLB Ser16 phosphorylation. Notably, a similar response is achieved by treatment of cells with p38 inhibitor SB203580. Direct assessment of PP2A activity further shows that treatment of cardiomyocytes with OA or SB203580 results in a comparable decrease in PP2A activity. In addition, inhibition of p38 also results in enhanced phosphorylation of cTnI and ERK, known targets of PP2A. PKA-induced cTnI phosphorylation is known to promote relaxation and it may also contribute to positive inotropy [33]. The enhanced phosphorylation of cTnI upon p38 inhibition may thus contribute to enhanced inotropic response to p38 inhibition in vivo. Taken together, these data demonstrate that p38 inhibition decreases PP2A activity, which renders I-1 Thr35 phosphorylated resulting in inhibition of PP1 and in an increase in PLB Ser16 phosphorylation (Fig. 6). However, our data does not exclude the possibility that additional mechanisms might also contribute to response to p38 inhibition.
Inhibition of p38 has been shown to enhance PKA activity and cardiac contractility in response to β-adrenergic stimuli [9]. Our data, however, indicates that p38 inhibition in nonstimulated cardiomyocytes does not enhance PKA activity. PKA activity, however, is requisite for the response to p38 inhibition, since overexpression of wtGRK2 (which blunts cAMP formation and PKA activity) was sufficient to prevent the shortening in Ca2+ decay time afforded by p38 inhibition. p38 inhibition-induced enhancement of SERCA2a function thus does not require extracellular stimuli, but is dependent on intrinsic PKA activity.
Enhancement of myofilament Ca2+ sensitivity by inhibition of p38 facilitates improvement in cardiac contractility without excess energy or oxygen consumption. Our current data, however, indicate that p38 also regulates SERCA2a function. Enhancing SERCA2a function comes at high energy cost, though, since transfer of 2 mol of Ca2+ into SR requires hydrolysis of 1 mol of ATP [34]. This could be of importance since p38 is considered a valid target for the treatment of ischemic heart disease. Our current findings would suggest that it might not be desirable to inhibit p38α under conditions of decreased oxygen supply to the heart, when limiting cardiac energy consumption is the major therapeutic goal. On the other hand, SERCA2a gene therapy has shown promise in the treatment of heart failure and diastolic dysfunction. Enhancing SERCA2a function by inhibiting p38α could thus be a useful strategy to improve cardiac diastolic function.
5. Conclusions
In summary, we demonstrate here that inhibition of p38α induces phosphorylation of PLB, enhances diastolic Ca2+ uptake in cardiomyocytes and enhances cardiomyocyte contractility. We show that p38 inhibition does not induce holistic PKA activation in cardiomyocytes, whereas intrinsic PKA activity is required for the response to p38 inhibition. Our data indicates that inhibition of p38α reduces the activity of PP2A and thereby enhances phosphorylation of PKA target I-1 Thr35 and inhibits PP1. These data reveal a novel mechanism for p38 inhibition-induced increase in intrinsic cardiac contractility. Our findings suggest that p38α inhibition affords improved cardiac relaxation, which could be beneficial in the treatment of diastolic heart failure.
Supplementary Material
Acknowledgments
We thank Marja Arbelius and Kirsi Salo for their technical assistance.
Funding
This work was supported by the Academy of Finland [Center of Excellence to H.R. and grants 127730, 131020 and 218044 to R.K.]; Biocenter Oulu [to L.K.]; Finnish Foundation for Cardiovascular Research [to L.K., H.R., R.K.]; Aarne Koskelo Foundation [to L.K.]; Emil Aaltonen Foundation [to L.K.]; the National Institutes of Health [to J.A.B.]; and the Sigrid Juselius Foundation [to H.R., R.K.].
Non-standard abbreviations and acronyms
- AMVM
adult mouse ventricular myocyte
- ARVM
adult rat ventricular myocyte
- dn
dominant negative
- ET-1
endothelin-1
- GRK
G protein coupled receptor kinase
- I-1
inhibitor-1
- MAPK
mitogen-activated protein kinase
- NRVM
neonatal rat ventricular myocyte
- OA
okadaic acid
- PLB
phospholamban
- PP1
protein phosphatase-1
- PP2A
protein phosphatase-2A
- SERCA2a
sarco/endoplasmic reticulum Ca2+ ATPase
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.yjmcc.2013.12.005.
Footnotes
Conflict of interest
None.
References
- 1.Eisner D, Bode E, Venetucci L, Trafford A. Calcium flux balance in the heart. J Mol Cell Cardiol. 2013;58:110–7. doi: 10.1016/j.yjmcc.2012.11.017. [DOI] [PubMed] [Google Scholar]
- 2.Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 3.Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res. 2012;110:1646–60. doi: 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wittkopper K, Dobrev D, Eschenhagen T, El-Armouche A. Phosphatase-1 inhibitor-1 in physiological and pathological beta-adrenoceptor signalling. Cardiovasc Res. 2011;91:392–401. doi: 10.1093/cvr/cvr058. [DOI] [PubMed] [Google Scholar]
- 5.Baines CP, Molkentin JD. STRESS signaling pathways that modulate cardiac myocyte apoptosis. J Mol Cell Cardiol. 2005;38:47–62. doi: 10.1016/j.yjmcc.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 6.Rose BA, Force T, Wang Y. Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol Rev. 2010;90:1507–46. doi: 10.1152/physrev.00054.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kerkela R, Force T. p38 mitogen-activated protein kinase: a future target for heart failure therapy? J Am Coll Cardiol. 2006;48:556–8. doi: 10.1016/j.jacc.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 8.Liao P, Wang SQ, Wang S, Zheng M, Zheng M, Zhang SJ, et al. p38 mitogen-activated protein kinase mediates a negative inotropic effect in cardiac myocytes. Circ Res. 2002;90:190–6. doi: 10.1161/hh0202.104220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng M, Zhang SJ, Zhu WZ, Ziman B, Kobilka BK, Xiao RP. Beta 2-adrenergic receptor-induced p38 MAPK activation is mediated by protein kinase A rather than by Gi or gbeta gamma in adult mouse cardiomyocytes. J Biol Chem. 2000;275:40635–40. doi: 10.1074/jbc.M006325200. [DOI] [PubMed] [Google Scholar]
- 10.Bellahcene M, Jacquet S, Cao XB, Tanno M, Haworth RS, Layland J, et al. Activation of p38 mitogen-activated protein kinase contributes to the early cardiodepressant action of tumor necrosis factor. J Am Coll Cardiol. 2006;48:545–55. doi: 10.1016/j.jacc.2006.02.072. [DOI] [PubMed] [Google Scholar]
- 11.Auger-Messier M, Accornero F, Goonasekera SA, Bueno OF, Lorenz JN, van Berlo JH, et al. Unrestrained p38 MAPK activation in Dusp1/4 double-null mice induces cardiomyopathy. Circ Res. 2013;112:48–56. doi: 10.1161/CIRCRESAHA.112.272963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kerkela R, Ilves M, Pikkarainen S, Tokola H, Ronkainen VP, Majalahti T, et al. Key roles of endothelin-1 and p38 MAPK in the regulation of atrial stretch response. Am J Physiol Regul Integr Comp Physiol. 2011;300:R140–9. doi: 10.1152/ajpregu.00853.2009. [DOI] [PubMed] [Google Scholar]
- 13.Vahebi S, Ota A, Li M, Warren CM, de Tombe PP, Wang Y, et al. p38-MAPK induced dephosphorylation of alpha-tropomyosin is associated with depression of myocardial sarcomeric tension and ATPase activity. Circ Res. 2007;100:408–15. doi: 10.1161/01.RES.0000258116.60404.ad. [DOI] [PubMed] [Google Scholar]
- 14.Szokodi I, Kerkela R, Kubin AM, Sarman B, Pikkarainen S, Konyi A, et al. Functionally opposing roles of extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase in the regulation of cardiac contractility. Circulation. 2008;118:1651–8. doi: 10.1161/CIRCULATIONAHA.107.758623. [DOI] [PubMed] [Google Scholar]
- 15.Andrews C, Ho PD, Dillmann WH, Glembotski CC, McDonough PM. The MKK6-p38 MAPK pathway prolongs the cardiac contractile calcium transient, downregulates SERCA2, and activates NF-AT. Cardiovasc Res. 2003;59:46–56. doi: 10.1016/s0008-6363(03)00329-8. [DOI] [PubMed] [Google Scholar]
- 16.Liao P, Georgakopoulos D, Kovacs A, Zheng M, Lerner D, Pu H, et al. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci U S A. 2001;98:12283–8. doi: 10.1073/pnas.211086598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ungerer M, Bohm M, Elce JS, Erdmann E, Lohse MJ. Altered expression of beta-adrenergic receptor kinase and beta 1-adrenergic receptors in the failing human heart. Circulation. 1993;87:454–63. doi: 10.1161/01.cir.87.2.454. [DOI] [PubMed] [Google Scholar]
- 18.Pitcher JA, Tesmer JJ, Freeman JL, Capel WD, Stone WC, Lefkowitz RJ. Feedback inhibition of G protein-coupled receptor kinase 2 (GRK2) activity by extracellular signalregulated kinases. J Biol Chem. 1999;274:34531–4. doi: 10.1074/jbc.274.49.34531. [DOI] [PubMed] [Google Scholar]
- 19.Bibb JA, Nishi A, O’Callaghan JP, Ule J, Lan M, Snyder GL, et al. Phosphorylation of protein phosphatase inhibitor-1 by Cdk5. J Biol Chem. 2001;276:14490–7. doi: 10.1074/jbc.M007197200. [DOI] [PubMed] [Google Scholar]
- 20.El-Armouche A, Bednorz A, Pamminger T, Ditz D, Didie M, Dobrev D, et al. Role of calcineurin and protein phosphatase-2A in the regulation of phosphatase inhibitor-1 in cardiac myocytes. Biochem Biophys Res Commun. 2006;346:700–6. doi: 10.1016/j.bbrc.2006.05.182. [DOI] [PubMed] [Google Scholar]
- 21.Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–9. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- 22.Weinbrenner C, Baines CP, Liu GS, Armstrong SC, Ganote CE, Walsh AH, et al. Fostriecin, an inhibitor of protein phosphatase 2A, limits myocardial infarct size even when administered after onset of ischemia. Circulation. 1998;98:899–905. doi: 10.1161/01.cir.98.9.899. [DOI] [PubMed] [Google Scholar]
- 23.Deshmukh PA, Blunt BC, Hofmann PA. Acute modulation of PP2a and troponin I phosphorylation in ventricular myocytes: studies with a novel PP2a peptide inhibitor. Am J Physiol Heart Circ Physiol. 2007;292:H792–9. doi: 10.1152/ajpheart.00225.2006. [DOI] [PubMed] [Google Scholar]
- 24.Junttila MR, Li SP, Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 2008;22:954–65. doi: 10.1096/fj.06-7859rev. [DOI] [PubMed] [Google Scholar]
- 25.Bers DM. Cardiac excitation–contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 26.Kho C, Lee A, Hajjar RJ. Altered sarcoplasmic reticulum calcium cycling-targets for heart failure therapy. Nat Rev Cardiol. 2012;9:717–33. doi: 10.1038/nrcardio.2012.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simmerman HK, Collins JH, Theibert JL, Wegener AD, Jones LR. Sequence analysis of phospholamban. Identification of phosphorylation sites and two major structural domains. J Biol Chem. 1986;261:13333–41. [PubMed] [Google Scholar]
- 28.Nicolaou P, Hajjar RJ, Kranias EG. Role of protein phosphatase-1 inhibitor-1 in cardiac physiology and pathophysiology. J Mol Cell Cardiol. 2009;47:365–71. doi: 10.1016/j.yjmcc.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carr AN, Schmidt AG, Suzuki Y, del MF Sato Y, Lanner C, et al. Type 1 phosphatase, a negative regulator of cardiac function. Mol Cell Biol. 2002;22:4124–35. doi: 10.1128/MCB.22.12.4124-4135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pathak A, del Monte F, Zhao W, Schultz JE, Lorenz JN, Bodi I, et al. Enhancement of cardiac function and suppression of heart failure progression by inhibition of protein phosphatase 1. Circ Res. 2005;96:756–66. doi: 10.1161/01.RES.0000161256.85833.fa. [DOI] [PubMed] [Google Scholar]
- 31.Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, et al. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med. 2004;10:248–54. doi: 10.1038/nm1000. [DOI] [PubMed] [Google Scholar]
- 32.Wittkopper K, Fabritz L, Neef S, Ort KR, Grefe C, Unsold B, et al. Constitutively active phosphatase inhibitor-1 improves cardiac contractility in young mice but is deleterious after catecholaminergic stress and with aging. J Clin Invest. 2010;120:617–26. doi: 10.1172/JCI40545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Layland J, Grieve DJ, Cave AC, Sparks E, Solaro RJ, Shah AM. Essential role of troponin I in the positive inotropic response to isoprenaline in mouse hearts contracting auxotonically. J Physiol. 2004;556:835–47. doi: 10.1113/jphysiol.2004.061176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inesi G, Kurzmack M, Lewis D. Kinetic and equilibrium characterization of an energy-transducing enzyme and its partial reactions. Methods Enzymol. 1988;157:154–90. doi: 10.1016/0076-6879(88)57074-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.