

Abstract
Diabetic nephropathy is the major cause of end-stage renal failure throughout the world in both developed and developing countries. Diabetes affects all cell types of the kidney, including endothelial cells, tubulointerstitial cells, podocytes and mesangial cells. During the past decade, the importance of podocyte injury in the formation and progression of diabetic nephropathy has been established and emphasized. However, recent findings provide additional perspectives on pathogenesis of diabetic nephropathy. Glomerular endothelial damage is already present in the normoalbuminuric stage of the disease when podocyte injury starts. Genetic targeting of mice that cause endothelial injury leads to accelerated diabetic nephropathy. Tubulointerstitial damage, previously considered to be a secondary effect of glomerular protein leakage, was shown to have a primary significance in the progression of diabetic nephropathy. Emerging evidence suggests that the glomerular filtration barrier and tubulointerstitial compartment is a composite, dynamic entity where any injury of one cell type spreads to other cell types, and leads to the dysfunction of the whole apparatus. Accumulation of novel knowledge would provide a better understanding of the pathogenesis of diabetic nephropathy, and might lead to a development of a new therapeutic strategy for the disease.
Keywords: Cell type specific, Conditional targeting, Diabetic nephropathy
Introduction
Diabetic nephropathy is a potentially fatal diabetic vascular complication characterized by slowly increasing proteinuria and a gradual decrease in renal function. Diabetic nephropathy is the leading cause of end-stage renal failure in Japan and Western countries, affecting alarmingly large numbers of people worldwide1. In Japan, diabetic nephropathy accounts for 44% of newly-induced hemodialysis, and a governmental survey estimated that there are approximately 100,000 hemodialysis patients. The number of patients is increasing rapidly, making diabetic nephropathy a critical social and economical problem.
The pathogenesis of diabetic nephropathy has been intensely investigated, and the roles of various mechanisms has been established, and include the effect of high glucose, polyol pathway activation, renin–angiotensin system activation, reactive oxygen species (ROS), activation of the protein kinase C pathway, increase of advanced glycation end-product (AGE) and glomerular hyperfiltration2,3. These changes lead to various cellular responses, expression of secretory factors and extracellular matrices that ultimately result in disruption of the glomerular filtration barrier, and histological changes including mesangial expansion, nodular glomerular sclerosis and tubulointerstitial fibrosis (Figure1)4.
Figure 1.

Schematic diagram on pathogenesis of diabetic nephropathy. In diabetic conditions, high glucose, activation of the polyol pathway, glomerular hyperfiltration, activation of the renin–angiotensin–aldosterone system (RAA), increase of advanced glycation end-product (AGE), increased reactive oxygen species (ROS), activation of diacylglycerol (DAG)/protein kinase C (PKC) pathway and an increase in Tgfb lead to abnormal cellular responses, such as overproduction of extracellular matrices and inflammation in the kidney. Diabetes also affects the production of nitric oxide (NO), Angpt2 and glycocalyx in the glomerular endothelial cells, and leads to endothelial injury. In podocytes, reduced vascular endothelial growth factor (VEGF) A expression increased mechanistic target of rapamycin (mTOR) signaling, and insulin resistance leads to podocyte dysfunction resulting in podocyte detachment and apoptosis. Furthermore, recent findings show that tubulointerstitial fibrosis also plays significant roles. These cell types are tightly connected together, and dysfunctions in one compartment can spread to other cell types.
During the past decade, ‘podocentric’ experiments have accumulated a huge amount of knowledge on the roles of podocytes in diabetic nephropathy. In contrast, recent findings have provided an additional perspective that other cell types are also affected at very early stages of diabetic nephropathy and contribute to the progression of the disease. To emphasize this concept, the current review will highlight the emerging roles of glomerular endothelial cells and tubulointerstitial cells in the pathogenesis of diabetic nephropathy, and juxtapose them to the roles of podocytes.
Glomerular Endothelial Cells
Glomerular endothelial cells are highly fenestrated, with 50- to 80-nm pores that go through the cytoplasm5. The luminal surface of endothelial cells is covered by a thick layer of glycocalyx that includes proteoglycans (PGs), such as syndecan, glypican, perlecan and versican, as well as their glycosaminoglycan (GAG) side chains, heparan sulfate and chondroitin sulfate6. Endothelial GAGs maintain the negative charge of the endothelial glycocalyx, and are believed to be a significant component of the glomerular charge barrier6.
Endothelial Cells are a Significant Part of the Glomerular Filtration Barrier
Because of the huge size of fenestration compared with that of circulating proteins, such as albumin, it has been believed that glomerular endothelial cells do not contribute to the filtration of macromolecules. However, emerging studies show its importance in the glomerular filtration barrier. Digestion of GAGs with heparanase, chondroitinase and hyaluronidase decrease the thickness of the glycocalyx layer, and the negative charge density of the glomerular filtration barrier, resulting in the increase of the fractional clearance of albumin7,8. In addition, elusion of non-covalently bound components of the endothelial glycocalyx by infusion of hypersonic sodium chloride into the renal artery leads to a 12-fold increase in the fractional clearance of albumin without detectable change of the ultrastructure9. In disease models, chronic infusion of hyaluronidase causes proteinuria in apolipoprotein E (ApoE) knockout mice10, and Adriamycin injection largely decreases the thickness of the glycocalyx and proteoglycan expression in the glomeruli11. These experiments provide strong evidence that the endothelial glycocalyx forms a significant part of the glomerular filtration barrier.
Role of Endothelial Cells in Diabetic Nephropathy
Several diabetic animal models show reduced endothelial glycocalyx. In non-obese diabetic (NOD) mice, long-term diabetes causes a threefold increase in the fractional clearance for negatively charged albumin compared with controls. In contrast, the fractional clearance for neutral Ficoll that has a similar size to albumin is not increased. This change is accompanied by a decrease in glycocalyx proteins including versican and decorin12. Therefore, the authors concluded that the charge barrier, rather than the size barrier, is affected in NOD mice glomeruli. Disruption of the charge barrier before that of the size barrier might show that endothelial damage occurs before podocyte/glomerular basement membrane (GBM) damage in NOD mice. In addition, staining for lectin that binds to hyaluronic acid and heparin sulfates of the endothelial glycocalyx is attenuated in streptozotocin (STZ)-induced diabetic rats13 and Zucker fatty rats14.
How about human beings? It has long been known that endothelial injury, assessed by elevation of plasma von Willebrand factor, is seen in both type 1 and type 2 diabetic patients15,16. An extensive study using state-of-the-art technology showed that the volume of endothelial glycocalyx in the total body and the thickness of the glycocalyx layer in retinal and sublingual arteries are reduced in type 1 diabetic patients. In that study, the decrease of endothelial glycocalyx was more severe in the group with albuminuria17. A similar reduction of the endothelial glycocalyx was also observed in type 2 diabetic patients18. In addition, very important studies have shown morphological abnormalities of renal glomerular endothelium in diabetic patients. Toyoda et al.19 showed that the fraction of fenestrated endothelium is reduced from 41% in controls to 32% in normo- and microalbuminuric patients, and further to 25% in macroalbuminuric patients with type 1 diabetes. Recently, Weil et al.20 reported a similar decrease of fenestrated area in type 2 diabetes patients (Figure2). Interestingly, podocyte detachment also starts in the normoalbuminuric stage and correlates to albuminuria20. In that study, the endothelial fenestration fraction correlated with the urinary albumin creatinine ratio more closely compared with podocyte detachment. Although podocyte detachment has been considered to be a critical event in various types of glomerular diseases, including diabetic nephropathy21, these studies clearly showed that endothelial damage simultaneously occurs when podocytes are damaged. This notion could raise a question against the concept that podocyte injury is the primary event and endothelial damage occurs as a secondary reaction22,23. These findings rather support the concept that the glomerular filtration barrier is a composite multilayered structure, and injury in one layer might spread to any other layers and affect the whole function of the glomerular filtration barrier.
Figure 2.
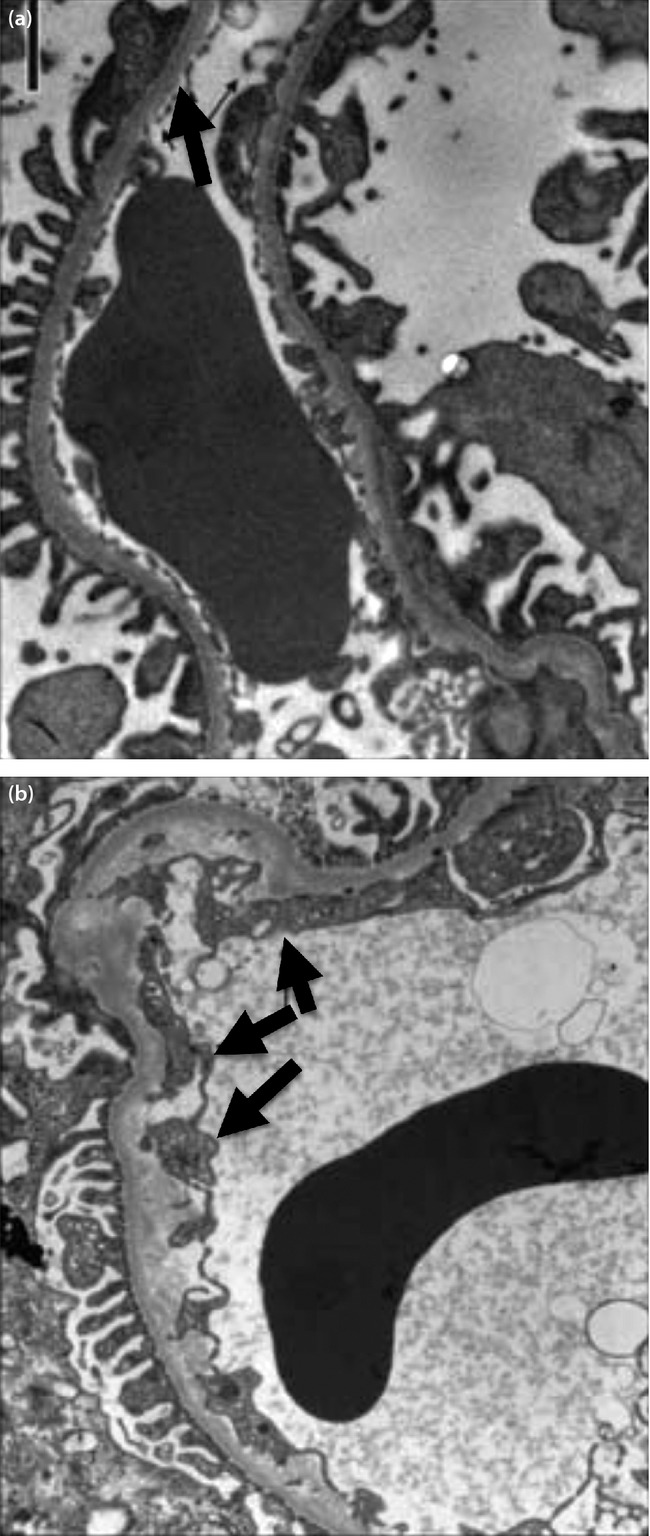
Endothelial injury is already present in the normoalbuminuric stage. (a) The ultrastructure of glomerular filtration barrier from a normal subject, note intact endothelial fenestration. (b) Sample from a normoalbuminuric Pima Indian patient with type 2 diabetes showing intact podocyte foot processes, but normal endothelial fenestrae are absent (arrows; adapted from Weil et al.20 with permission).
Endothelial Dysfunction and Diabetic Nephropathy: Lessons from Genetically Targeted Mice
Various cytokines and factors are secreted by/have effects on endothelial cells to maintain the glomerular filtration barrier. Emerging evidence using genetically targeted mice prove their importance in diabetic nephropathy.
Nitric Oxide
In diabetic animals and humans, bioavailability of the nitric oxide (NO) is reduced24. In cultured endothelial cells, high glucose inhibits endothelial NO synthase (eNOS) activity25. However, in type 2 diabetic patients, eNOS expression is increased in kidney glomeruli by immunohistochemistry, and the increase was correlated with more severe vascular complications26. Similarly, protein expression of eNOS in STZ-induced diabetic rats is increased in afferent arterioles and glomeruli27. In contrast, eNOS required cofactors including tetrahydrobiopterin (BH4) to produce NO. If eNOS is ‘uncoupled’, peroxynitrite (ONOO–), a putative reactive oxygen species, is produced instead of NO. In endothelial cells, high glucose results in a uncoupling of eNOS, reduction of NO production and increased superoxide production28. This uncoupling might explain the discrepancy between elevated expression of eNOS and reduced NO production in the diabetic condition.
The requirement of eNOS in the glomerular endothelium in diabetes was examined using two diabetic animal models combined with genetic deletion of eNOS. Both db/db and STZ mice bred with eNOS knockout mice showed dramatic albuminuria, increased glomerular basement membrane thickness, mesangial expansion, and focal segmental and nodular sclerosis29,30. The potential mechanism of the enhanced nephropathy was an uncoupling of the vascular endothelial growth factor A (VEGFA)–eNOS axis, with enhanced VEGF expression and impaired NO production, which led to excessive endothelial proliferation. These phenotypes were at least partially mediated by intraglomerular hypertension, because lowering blood pressure rescues the glomerular lesion in the diabetic eNOS deficient mice31. These results provide robust evidence that endothelial dysfunction results in enhancement of diabetic nephropathy and suggest NO as a potential therapeutic target.
VEGFA
A large amount of VEGFA is produced by podocytes. The secreted VEGFA go across the GBM and bind to kinase insert domain protein receptor (Kdr; also known as VEGF receptor 2) expressed on the endothelial cells. The VEGFA–Kdr axis is essential for the formation and maintenance of the glomerular filtration barrier32,33. Podocyte-specific deletion of VEGFA leads to impaired recruitment of endothelial cells into glomeruli, failure in formation of glomerular filtration barrier and congenital nephrotic syndrome33. Mice that carry haploinsufficient VEGFA allele in podocytes show an endothelial swelling and loss of fenestration in glomeruli known as endotheliosis – a feature seen in thrombotic microangiopathies (TMA)33. Overexpression of VEGFA in podocytes leads to a collapse of the glomerular tuft33. In addition, patients on anti-VEGF therapy sometimes develop proteinuria as a result of TMA of the glomeruli34. Indeed, deletion of VEGFA alleles from adult mice podocytes results in TMA34.
The role of VEGFA in diabetic nephropathy has been a controversy. An increase of VEGFA expression was shown in the glomeruli and tubulointerstitium in STZ diabetic rats and in type 2 diabetic mice35–37. As VEGFA is a potent stimulator that destabilizes endothelial cells and induces vascular permeability, some investigators attempted to block VEGFA signaling to treat diabetic nephropathy in mice38,39. In db/db mice, an inhibitor for tyrosine kinase of Kdr reduced urinary albumin excretion (UAE)40,41. In Zucker diabetic rats, the neutralizing antibody for VEGF reduced glomerular hypertrophy, but did not improve UAE40. Therefore, these reports appear to support the notion that VEGF worsens diabetic nephropathy.
However, reports on VEGFA expression in human diabetic nephropathy are inconsistent. Hohenstein et al.42 reported enhanced VEGFA expression in glomeruli of type 2 diabetes patients by immunostaining. In contrast, Baelde et al.43 showed reduction of Vegfa messenger ribonucleic acid expression in human type 2 diabetic glomeruli by Affymetrix microarray43. Several additional reports showed that Vegfa expression was decreased in both the glomeruli and tubulointerstitium, and was correlated with reduced renal microvascular density, tubular epithelial atrophy, mesangial expansion and proteinuria44,45. These results rather support the notion that VEGF is protective.
Recently, two genetic mice models shed a new light onto this controversy. Veron et al.46 produced a mouse model that carries podocyte-specific inducible overexpression of Vegf164, and rendered the mice diabetic by STZ injection. The results showed accelerated nephropathy with Kimmelstiel–Wilson like nodular glomerulosclerosis and massive proteinuria46. In contrast, Sivaskandarajah et al.47 showed that inducible deletion of VEGFA in adult podocytes results in severe enhancement of diabetic nephropathy using a STZ-induced mice model. In that report, the diabetic mutant showed severe glomerulosclerosis, enhanced proteinuria and increased apoptosis in the kidneys47. These results clearly show that the levels of VEGFA in the glomeruli require ‘fine tuning’, and either overdose or suppression could result in exacerbation of diabetic nephropathy. This tight and subtle regulation is similar to that of VEGFA in glomerular development.
Angiopoietins
Another family of angiogenic factors required for maintenance of glomerular endothelial cells is angiopoietin–Tek signaling. Angiopoietin 1 (Angpt1) and angiopoietin 2 (Angpt2) are both ligands to Tek tyrosine kinase (Tek/Tie-2)48,49. Angiopoietin 1 binds to the Tek receptor expressed on endothelial cells, and cause its tyrosine phosphorylation, but Angpt2 works as an antagonist while not activating any intracellular signaling. However, some data suggest that Angpt2 activates phosphorylation of Tek in certain conditions50. Angpt1 is believed to stabilize the blood vessel, reduce vascular permeability and support the survival of endothelial cells. Angpt1 conventional knockout mice die at embryonic day 12.549 and Angpt2 knockout is also perinatal lethal51.
Recent analysis using conditional alleles showed critical roles of Angpt1 in glomeruli. Jeansson et al. showed that deletion of Angpt1 at embryonic day 10.5 resulted in abnormal glomerular development with a single capillary loop without mesangial migration that is reminiscent to the phenotype of Pdgfb/Pdgfrb mutants52,53. In contrast, loss of the Angpt1 allele at e13.5 does not cause any phenotype, showing that Angpt1 is only required when the vasculature is undergoing dynamic remodeling.
The role of angiopoietins in diabetes has been shown by several reports. In diabetic patients, Angpt2 expression is increased54. On the other hand, diabetic animal models show a decrease of Angpt1 and increase of Angpt2. Furthermore, STZ-induced diabetic mice with whole-body or glomerular-specific Angpt1 deletion develop increased urinary albumin excretion, severe mesangial expansion, glomerular sclerosis and early mortality53. A study using db/db mice showed that treatment with recombinant adenovirus-expressing cartilage oligomeric matrix protein (COMP)-Ang-1, a potent Angpt-1 variant, resulted in improvement in diabetic renal damage55. Recently, STZ diabetic mice with podocyte-specific overexpression of Angpt1 also showed a similar protective effect on diabetic nephropathy56. Taken together, the Angpt1–Tek axis plays an important protective role in glomerular endothelial cell function in the diabetic condition. Drugs that target Angpt1 and its downstream molecules might provide potentially useful therapeutic strategies.
Tubulointerstitial Cells
Although it has been widely accepted that glomerular injury is the main component of diabetic nephropathy, plenty of evidence has shown that tubulointerstitial changes are present and are involved in its progression57. Tubulointerstitium includes the tubular system, interstitial cells and vascular system, and accounts for as much as 90% of kidney volume58.
Histological Abnormality of Tubulointerstitium Correlates with Progression of Diabetic Nephropathy
Histologically, early diabetic kidneys show tubular hypertrophy, thickening of the tubular basement membrane and interstitial inflammation with mononuclear cell infiltration59. When it progresses, they show tubular atrophy and tubulointerstitial fibrosis. Interestingly, tubulointerstitial expansion closely correlates with elevation of serum creatinine in both type 1 and type 2 diabetes60–62. Taft et al.63 followed 47 patients with diabetes for 4 years, and carried out renal biopsies at the beginning and the end of the study. The results showed that cortical tubulointerstitial fibrosis more closely correlated to the decrease of creatinine clearance than glomerular changes63. This association seems to be weaker in the microalbuminuric stage64, suggesting that tubulointerstitial fibrosis is a good determinant of moderate-to-severe renal injury rather than early diabetic nephropathy59.
In addition, abnormalities in glomerulotubular junctions have been reported in diabetic kidneys. A study on renal biopsy samples of type 1 diabetes patients showed that 4% of glomeruli from microalbuminuric patients show some glomerulotubular junctional abnormalities, including connections of glomeruli to atrophic tubules. Furthermore, in the macroalbuminuric stage, 71% of glomeruli showed glomerulotubular junction abnormalities, including 8% glomeruli without any tubule connected (atubular glomeruli)65,66. Type 2 diabetic glomeruli also show atubular glomeruli and connection to atrophic tubules from the stage of microalbuminuria67. These data suggest that, even though tubulointerstitial changes are dominantly seen in the advanced stage, subtle abnormality is already present in the microalbuminuric time-point.
Increase of Tubular Markers in Urine of Early Diabetic Patients
Data of urinary biomarker also show that tubular damage starts at an early stage of diabetic nephropathy. Studies on type 1 diabetic children showed that urinary N-acetyl-b-D-glucosaminidase (NAG) is increased in diabetes patients compared with normal controls, and is correlated with urinary albumin excretion and glycemic control. This increase was already seen in the microalbuminuric stage68,69. Intriguingly, within microalbuminuric type 1 diabetic patients, a group that shows low levels of urinary NAG and kidney injury molecule 1 (KIM-1) tends to show regression of albuminuria after 2 years, suggesting that tubular dysfunction is a critical component of the early course of diabetic nephropathy69.
Does Tubulointerstitial Change Affect Glomerular Structure and Function–Tubuloglomerular Feedback?
It has been considered that the glomeruli is the primary site of injury, and the tubulointerstitial change is a secondary reaction to elevated intratubular protein as a result of glomerular leakage. Recent studies suggested that tubular changes could lead to alteration in glomerular structure and function.
Using genetically modified mice, VEGFA was overexpressed in renal tubular cells on administration of doxycyline. The mutant mice showed an interstitial fibrosis and tubular cysts with dense network of peritubular capillaries. The mice did not show significant proteinuria; however, they developed glomerular changes including marked mesangial expansion similar to that seen in diabetic nephropathy. The authors observed downregulation of VEGFA expression in podocytes, and concluded that tubular overproduction of VEGFA resulted in suppression of VEGFA in podocytes that led to interference of cross-talk between podocytes and the endocapillary compartment (Figure3)70.
Figure 3.

Genetic overexpression of vascular endothelial growth factor A (VEGFA) in tubules results in glomerular disease. (a) A glomerulus from a control mouse. (b) A glomerulus from a tubular-specific VEGFA overexpression mouse that shows dilated capillaries at the periphery and nodule-like mesangial expansion, showing that tubular dysfunction results in glomerular disease (reproduced from Hakroush et al.70 with permission).
Another study also provides evidence for tubuloglomerular feedback. Deletion of sirtuin 1 (Sirt1), a NAD+ regulated deacetylase, specifically in tubules leads to increased albuminuria and effacement of podocyte foot processes. Additionally, STZ-induced diabetes aggravated the nephropathy of tubule-specific Sirt1 knockout mice. Furthermore, overexpression of Sirt1 in tubules protected the mice from the glomerular ultrastructural changes and albuminuria in STZ mice. In human diabetic nephropathy with proteinuria, Sirt1 was downregulated and Claudin1 was upregulated. These results suggest that Sirt1 in proximal tubules protects against diabetic nephropathy and influence podocyte function71. Taken together, these experiments clearly show that tubulointerstitial change can affect glomerular structure and function.
Epithelial to Mesenchymal Transformation in Diabetic Nephropathy
Epithelial to mesenchymal transformation (EMT) is a cellular process through which epithelial cells undergo ‘de-differentiation’; lose epithelial characteristics; express mesenchymal markers, such as alpha-smooth muscle actin, desmin and vimentin; and acquire a matrix-producing fibroblast-like phenotype. A study using genetically tagged proximal tubules showed that up to 36% of the interstitial fibroblasts were derived from proximal tubules in a unilateral ureter obstruction mouse model72. A recent analysis reported that 35% of the myofibroblasts were derived from bone marrow, 10% from endothelial cells and 5% from tubular cells through an EMT program in a unilateral ureter obstruction model73,74. In diabetic nephropathy, EMT-like changes of the tubules, such as upregulation of vimentin and decrease of E-cadherin, have been observed both in vitro and in vivo75,76. In addition, tubular expressions of mesenchymal markers are also shown in renal biopsy samples from human diabetic nephropathy77. In contrast, however, overexpression of transforming growth factor beta (Tgfb) in tubular cells, a multifunctional cytokine that is believed to be the key mediator of EMT, resulted in total decomposition of tubular cells and fibrosis, but not mesenchymal transformation of tubular cells78. Therefore, there is no solid evidence that tubular cells transform into matrix-producing interstitial fibroblasts in diabetic nephropathy. In terms of endothelial to mesenchymal transformation, Li et al.79 carried out genetic tag experiments using Tie2-Cre mice, and showed that infusion of AGE to mice resulted in fibroblasts derived from endothelial cells79.
Podocytes
Morphology of Podocytes
Podocytes are terminally differentiated cells that wrap the glomerular capillary loops from outside, providing physical support and secretary cytokines. They develop microtubule-based thick primary foot processes and fine actin-based secondary foot processes that interdigitate each other. The foot processes from the neighboring podocytes are connected with each other by a specialized intercellular junction called a slit diaphragm (SD). SD consists of proteins including nephrin, podocin, Cd2-ap and actin-binding protein alpha-actinin 4. The mutations of these proteins cause focal and segmental glomerulosclerosis in mice and humans, thus SD is considered to be essential in macromolecular filtration80.
Diabetic Nephropathy and Podocytes
The early key events in diabetic nephropathy include loss of podocytes in glomeruli. A landmark study was carried out by Pagtalunan et al.81, and showed that the number of podocytes per glomeruli were reduced in renal biopsies from Pima Indian type 2 diabetes patients with macroproteinuria. The loss of podocytes was correlated with the flattening of podocyte foot processes81. Subsequent analysis showed that the decrease of podocyte number is a good predictor of progression of albuminuria82. Similar findings are also reported on the nephropathy of type 1 diabetic patients19,83.
The mechanism of podocyte loss is considered to be detachment and apoptosis. Podocyte apoptosis is observed in many animal diabetic models including Akita mice, db/db84 mice and STZ induced rats85, and could at least partially be mediated by an increase of Tgfb, Smad786, AGE87, angiotensin II88 and reactive oxygen species84. In human type 2 diabetic kidneys, apoptosis was seen in both tubular and glomerular compartments, some was obviously in podocytes89. In contrast, under diabetic conditions, some podocytes detach from the GBM and fall into the urine. The detached ‘urinary’ podocytes are viable and can propagate ex vivo90. Indeed, patients of type 2 diabetes show urinary excretion of podocytes, and it worsens as the disease progresses from normoalbiminuria, microalbuminuria and to macroalbuminuria91. Interestingly, the urinary podocyte detachment can be reduced by administration of hydroxymethylglutaryl-CoA (HMG-CoA) reductase inhibitors92.
Factors Involved in Podocyte Function in Diabetic Nephropathy
Proteins of Slit Diaphragm
Nephrin is a 180-kDa transmembrane protein that belongs to the immunoglobulin superfamily. In 1998, Karl Triggvasson et al.93 group identified the nephrin (NPHS1) gene, and showed that its mutations caused the congenital nephrotic syndrome of the Finnish type. The nephrin cytoplasmic tail includes three tyrosine-aspartic acid-x-valine (YDxV) residues. These residues are phosphorylated by Src family kinases, recruit SH2/SH3 containing Nck adaptor proteins resulting in reorganization of actin cytoskelton. Deletion of Nck1/2 in podocytes leads to massive congenital proteinuria94,95. Many reports have shown downregulation of nephrin in diabetic nephropathy in rodent models and human patients96–98. Some reports suggest that the decrease of nephrin is partially mediated by the renin–angiotensin system, AGE and protein kinase C pathway98–101. In contrast, there are also several reports showing upregulation of nephrin expression in mice diabetic kidneys41,102. The cause of this diversity is still unknown, but could be a difference of disease stages. In addition, other slit diaphragm proteins, such as podocin and synaptpodin, are also significantly reduced in human diabetic nephropathy103.
Insulin Signaling
Podocytes express insulin receptor and increase their glucose uptake on insulin stimulation104. In addition, knockdown of nephrin in podocytes attenuates insulin-dependent glucose uptake in podocytes, showing that nephrin is required for insulin function in podocytes105. Recently, a study on a genetic mouse model that lacks insulin receptor in podcytes was reported. The mice showed massive proteinuria and histological abnormality similar to diabetic nephropathy. In addition, in vitro studies using an immortalized human podocyte cell line suggest that insulin sends signals through mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K), and regulates actin cytoskeleton remodeling in podocytes106. These studies show that local insulin resistance affects podocyte function and leads to progression of diabetic nephropathy, thus demonstrating the importance of the therapy that increases insulin sensitivity in podocytes.
mTor Signaling
The mechanistic target of rapamycin (mTor) is a ubiquitously expressed serine-threonine kinase. mTor binds to regulatory-associated protein of mTOR (Rptor), or Rptor independent companion of mTor (Rictor), and forms mTORC1 and mTORC2, respectively107. mTORC1 is activated by amino acids, stress, oxygen and growth factors, and regulates protein translation, ribosomal biogenesis, cell growth and autophagy. mTORC2 responds to growth factors, and regulates cell survival, growth, metabolism and cytoskeletal rearrangement107. Rapamycin acutely suppresses mTORC1 function108; however, in certain contexts, it also inhibits mTORC2109. Interestingly, organ transplant patients using rapamycin sometimes develop proteinuria, suggesting the importance of mTor signaling in glomeruli108. Indeed, mice studies showed that the mTor pathway is essential for podocyte function. Deletion of mTor or Rptor specifically from podocytes leads to massive proteinuria and renal failure110,111.
In contrast, mTor activity was increased in renal biopsy samples of diabetic patients and in diabetic db/db mice111,112. Activation of mTORC1 by deletion of its suppressor, Tsc1, in podocytes results in proteinuria, podocyte loss, mesangial expansion and thickening of glomerular basement membrane112. Furthermore, haploinsufficiency of Rptor protected mice from diabetic nephropathy in STZ mice and db/db mice, showing a decrease in urinary albumin excretion and mesangial expansion111,112. These results show the critical roles of mTor signaling in diabetic nephropathy, and that either overactivation or elimination of the mTor pathway leads to podocyte dysfunction and glomerular disease. Recent analysis showed that podocyte-specific ablation of Rictor or Akt2, a potent target of mTORC2, results in accelerated nephropathy induced by heminephrectomy, showing that mTORC2 is also involved in progression of chronic kidney disease (CKD)113.
Notch Signaling
The Notch pathway regulates cell proliferation, differentiation, cell fate specification and organ development, and is conserved in all metazoans114. Notch is a transmembrane receptor that binds to cell surface ligands, such as Delta-like or Jagged, on neighboring cells. On interaction of these ligands, Notch intracellular domain (NICD) is cleaved by gamma-secretase, and then NICD translocates to the nucleus where it forms transcriptional complexes with deoxyribonucleic acid (DNA) binding proteins including RBPj, CBF1 and Su114.
Although the importance of the Notch pathway has been well documented in renal development, it is mostly silenced in adult normal kidneys115. However, NICD is increased in podocytes of human diabetic nephropathy and focal segmental glomerulosclerosis, and in db/db mice. Conditional overexpression of NICD in podocytes resulted in proteinuria and glomerulosclerosis. In addition, deletion of RBPj, an essential transcriptional partner of NICD, or administration of gamma-secretase inhibitor, protected rats from proteinuric renal models including diabetic nephropathy115. The Notch pathway might mediate high glucose-induced apoptosis in podocytes116,117.
Wnt/beta-Catenin Signaling
Wnts are a family of highly-conserved secreted glycoproteins that regulate cell proliferation, cell fate determination, organogenesis and tumorgenesis. Beta-catenin is the central component of the canonical Wnt pathway. On binding of Wnts to their receptor, Frizzled, a series of downstream signalings including Dishvelled, Axin, adenomatous polyposis coli (APC) and glycogen synthase-3beta (GSK-3beta) are activated, and eventually lead to dephosphorylation of beta-catenin. Dephosphorylated beta-catenin escapes the ubiquitin-mediated degradation, then the stabilized beta-catenin translocates to the nucleus where it binds to DNA and regulates transcription of various target genes.
Wnt beta-catenin signaling is activated in podocytes of diabetic nephropathy in humans and STZ-induced diabetic mice118,119. Pharmacological activation118, as well as podocyte-specific stabilization of beta-catenin, leads to albuminuria119. Interestingly, deletion of beta-catenin in podocytes and expression of Wnt inhibitor Dickkopf-related protein 1 (Dkk1) result in enhancement of diabetic nephropathy induced by STZ119, suggesting that both hypo- and hyperactivation of the beta-catenin pathway promotes proteinuric diseases. In vitro studies showed that Wnt/beta-catenin signaling regulates podocyte motility, adhesion, apoptosis and expression of podocyte differentiation markers including nephrin119. Interestingly, beta-catenin is a key molecule that regulates the cell fate determination when podocytes differentiate from renal vesicles120.
Tgfb
Many reports show that Tgfb is a central factor that contributes to the progression of glomerulosclerosis. Increased expression of Tgfb1 is observed in human diabetic kidneys at early and late stages of nephropathy, and the expression of Tgfb is correlated with glycemic control121,122. In db/db mice, elevation of Tgfb, its receptor Tgf beta receptor 2 (Tgfbr2) and nuclear translocation of Smad3, a main signaling transducer of Tgfb signaling, were observed123. Tgfb level was elevated in the glomeruli of STZ-induced diabetic rats by micropuncture124. In addition, overexpression of Tgfb in hepatocytes results in multiple organ phenotype including glomerulonephritis and renal fibrosis125. Recently, overexpression of Tgfb receptor 1 specifically in podocytes resulted in massive glomerulosclerosis126. Interestingly, this phenotype was mediated by Endothelin1 release by podocytes, which leads to mitochondrial oxidative stress in adjacent endothelial cells in a paracrine manner, thus emphasizing the importance of cross-talk between podocytes and endothelial cells.
As mentioned earlier, Tgfb is reported to induce podocyte apoptosis through the P38-MAPK pathway86. Smad7 is one of the downstream targets of Tgfb signaling, and is also known as a potent inhibitor of the classical Smad signaling pathway, forming a negative feedback. Smad7 knockout mice show exacerbated diabetic nephropathy with increased albuminuria, enhanced renal fibrosis and increased expressions of inflammatory factors. In the diabetic Smad7 knockout kidneys, both the Tgfb–Smad pathway and nuclear factor-kappa B pathway are activated. Overexpression of Smad7 by gene transfer specifically to kidneys attenuated microalbuminuria127. Although Smad7 is reported to induce podocyte apoptosis86, these data suggest that Smad7 plays a protective role in diabetic nephropathy, highly likely through suppression of Tgfb signaling pathways.
In the immortalized podocyte cell line, Tgfb decreased podocyte markers, such as nephrin and Zo-1, and increased mesenchymal markers, including alpha smooth muscle actin and vimentin, showing that Tgfb induces de-differentiation in podocytes similar to epithelial mesenchymal transformation128. In addition, it has been shown that Tgfb increases the expression of extracellular matrices including fibronectin and type IV collagen in podocytes, and contributes to the thickening of GBM129,130.
Soluble Flt1
Soluble fms-related tyrosine kinase 1 (sFlt1) is an alternatively spliced soluble form of VEGF receptor 1 (VEGFR-1)/Flt-1, and works as a decoy ligand to Kdr. An increase of sFlt1 using adenovirus results in a marked decrease of endothelial fenestrae in mice glomeruli131. sFlt blood levels are higher in pre-eclampsia patients, and injection of sFlt1 to pregnant rats causes hypertension and proteinuria132. A recent report showed a surprising additional function of sFlt1 on non-endothelial cells through Kdr-independent mechanisms. Deletion of Flt1 in podocytes leads to reorganization of their cytoskeleton, massive proteinuria and renal failure. The allele that lacks the intracellular kinase domain of Flt1 rescues this proteinuric phenotype, suggesting that full-length protein is not critically required. The authors showed that sFlt binds to the lipid microdomain on the podocyte surface, and promote cellular adhesion and actin reorganization133. Interestingly, mice with overexpression of sFlt1 in podcytes were protected from STZ-induced changes in glomeruli. Transgenic diabetic mice show a decrease in urinary albumin excretion, mesangial expansion, podocyte foot processes fusion and glomerular basement membrane thickening134. These results show an interesting autocrine function of sFlt1 that protects podocytes in addition to its role as a decoy on endothelial VEGFA signaling. However, systemic overexpression of sFlt1 will not be useful, because intramuscular injection of adeno-associated virus 1 encoding human soluble Flt1 resulted in improvement in podocyte injury, but exacerbated tubulointerstitial damage, showing a conflicting effect of sFlt1 in podocytes and interstitial compartment135.
Rho GTPases
Rho guanosine triphosphatases (GTPases) are known as master regulators of actin cytoskeleton. The classical members of this family are Rho, Rac1 and Cdc42. Activation of Cdc42 leads to fillipodia formation, Rac1 promotes lamellipodia formation and RhoA mainly regulates formation of stress fibers136. Many reports suggested its importance in podocyte biology. Podocyte-specific deletion of Cdc42 leads to a congenital nephrotic syndrome and renal failure with defect in actin polymerization at intracellular domain of nephrin137, whereas the other two Rho guanosine triphosphatases did not show any proteinuria when deleted in podocytes. Activation of RhoA in podocytes results in focal segmental glomerulosclerosis138. It has been shown that RhoA is activated in the renal cortex of diabetic db/db mice139. Several trials have been carried out to suppress RhoA in diabetic rodent models. Administration of Fasudil, a Rho-kinase inhibitor, resulted in attenuation of urinary albumin excretion, ultrastructural chances of glomerular filtration barrier and mesangial expansion in db/db mice139. A similar protective effect was also observed in STZ-induced diabetic mice and rats140–142. These effects included suppression of Tgfb1 and connective tissue growth factor, and hypoxia-inducible factor 1 alpha signaling141,142.
Conclusion
Diabetic nephropathy affects many cellular functions, some of them are common across cell types and some are cell-type specific. Recent studies showed distinct roles of each compartment, and factors that are required for cross-talk between cell types. An accumulation of a better understanding on the pathogenesis of diabetic nephropathy would provide a novel opportunity for the development of a new therapeutic strategy.
Acknowledgments
We deeply thank all the members of the diabetes/metabolism group at Chiba University Graduate School of Medicine, Clinical Cell Biology for fruitful discussion. The authors declare no conflict of interest.
References
- Atkins RC, Zimmet P. Diabetic kidney disease: Act now or pay later. Nephrol Dial Transplant. 2010;25:331–333. doi: 10.1093/ndt/gfp757. [DOI] [PubMed] [Google Scholar]
- Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev. 2013;93:137–188. doi: 10.1152/physrev.00045.2011. [DOI] [PubMed] [Google Scholar]
- Rask-Madsen C, King GL. Vascular complications of diabetes: Mechanisms of injury and protective factors. Cell Metab. 2013;17:20–33. doi: 10.1016/j.cmet.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolset SO, Reinholt FP, Jenssen T. Diabetic nephropathy and extracellular matrix. J Histochem Cytochem. 2012;60:976–986. doi: 10.1369/0022155412465073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa Y, Cina D, Quaggin SE. Chapter 22 Glomerular Cell Biology. Seldin and Giebisch's the Kidney: Physiology & Pathophysiology. Amsterdam, Boston: Elsevier Inc., Academic Press; 2012. [Google Scholar]
- Haraldsson B, Nystrom J, Deen WM. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev. 2008;88:451–487. doi: 10.1152/physrev.00055.2006. [DOI] [PubMed] [Google Scholar]
- Jeansson M, Haraldsson B. Morphological and functional evidence for an important role of the endothelial cell glycocalyx in the glomerular barrier. Am J Physiol Renal Physiol. 2006;290:F111–F116. doi: 10.1152/ajprenal.00173.2005. [DOI] [PubMed] [Google Scholar]
- Jeansson M, Haraldsson B. Glomerular size and charge selectivity in the mouse after exposure to glucosaminoglycan-degrading enzymes. J Am Soc Nephrol. 2003;14:1756–1765. doi: 10.1097/01.asn.0000072742.02714.6e. [DOI] [PubMed] [Google Scholar]
- Friden V, Oveland E, Tenstad O. The glomerular endothelial cell coat is essential for glomerular filtration. Kidney Int. 2011;79:1322–1330. doi: 10.1038/ki.2011.58. [DOI] [PubMed] [Google Scholar]
- Meuwese MC, Broekhuizen LN, Kuikhoven M. Endothelial surface layer degradation by chronic hyaluronidase infusion induces proteinuria in apolipoprotein E-deficient mice. PLoS ONE. 2010;5:e14262. doi: 10.1371/journal.pone.0014262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeansson M, Bjorck K, Tenstad O. Adriamycin alters glomerular endothelium to induce proteinuria. J Am Soc Nephrol. 2009;20:114–122. doi: 10.1681/ASN.2007111205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeansson M, Granqvist AB, Nystrom JS. Functional and molecular alterations of the glomerular barrier in long-term diabetes in mice. Diabetologia. 2006;49:2200–2209. doi: 10.1007/s00125-006-0319-z. [DOI] [PubMed] [Google Scholar]
- Satoh M, Kobayashi S, Kuwabara A. In vivo visualization of glomerular microcirculation and hyperfiltration in streptozotocin-induced diabetic rats. Microcirculation. 2010;17:103–112. doi: 10.1111/j.1549-8719.2009.00010.x. [DOI] [PubMed] [Google Scholar]
- Kuwabara A, Satoh M, Tomita N. Deterioration of glomerular endothelial surface layer induced by oxidative stress is implicated in altered permeability of macromolecules in Zucker fatty rats. Diabetologia. 2010;53:2056–2065. doi: 10.1007/s00125-010-1810-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberton RP, Goodman AD, Kassoff A. Von Willebrand factor (VIII R:Ag), fibronectin, and insulin-like growth factors I and II in diabetic retinopathy and nephropathy. Diabetes. 1984;33:125–129. doi: 10.2337/diab.33.2.125. [DOI] [PubMed] [Google Scholar]
- Vukovich TC, Schernthaner G, Knobi PN. The effect of near-normoglycemic control on plasma factor VIII/von Willebrand factor and fibrin degradation products in insulin-dependent diabetic patients. J Clin Endocrinol Metab. 1989;69:84–89. doi: 10.1210/jcem-69-1-84. [DOI] [PubMed] [Google Scholar]
- Nieuwdorp M, Mooij HL, Kroon J. Endothelial glycocalyx damage coincides with microalbuminuria in type 1 diabetes. Diabetes. 2006;55:1127–1132. doi: 10.2337/diabetes.55.04.06.db05-1619. [DOI] [PubMed] [Google Scholar]
- Broekhuizen LN, Lemkes BA, Mooij HL. Effect of sulodexide on endothelial glycocalyx and vascular permeability in patients with type 2 diabetes mellitus. Diabetologia. 2010;53:2646–2655. doi: 10.1007/s00125-010-1910-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda M, Najafian B, Kim Y. Podocyte detachment and reduced glomerular capillary endothelial fenestration in human type 1 diabetic nephropathy. Diabetes. 2007;56:2155–2160. doi: 10.2337/db07-0019. [DOI] [PubMed] [Google Scholar]
- Weil EJ, Lemley KV, Mason CC. Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney Int. 2012;82:1010–1017. doi: 10.1038/ki.2012.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanamura K, Tojo A, Fujita T. Urinary and glomerular podocytes in patients with chronic kidney diseases. Clin Exp Nephrol. 2014;18:95–103. doi: 10.1007/s10157-013-0814-8. [DOI] [PubMed] [Google Scholar]
- Satchell SC. The glomerular endothelium emerges as a key player in diabetic nephropathy. Kidney Int. 2012;82:949–951. doi: 10.1038/ki.2012.258. [DOI] [PubMed] [Google Scholar]
- Salmon AH, Satchell SC. Endothelial glycocalyx dysfunction in disease: Albuminuria and increased microvascular permeability. J Pathol. 2012;226:562–574. doi: 10.1002/path.3964. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Tanabe K, Croker BP. Endothelial dysfunction as a potential contributor in diabetic nephropathy. Nat Rev Nephrol. 2011;7:36–44. doi: 10.1038/nrneph.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du XL, Edelstein D, Dimmeler S. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J Clin Invest. 2001;108:1341–1348. doi: 10.1172/JCI11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenstein B, Hugo CP, Hausknecht B. Analysis of NO-synthase expression and clinical risk factors in human diabetic nephropathy. Nephrol Dial Transplant. 2008;23:1346–1354. doi: 10.1093/ndt/gfm797. [DOI] [PubMed] [Google Scholar]
- Sugimoto H, Shikata K, Matsuda M. Increased expression of endothelial cell nitric oxide synthase (ecNOS) in afferent and glomerular endothelial cells is involved in glomerular hyperfiltration of diabetic nephropathy. Diabetologia. 1998;41:1426–1434. doi: 10.1007/s001250051088. [DOI] [PubMed] [Google Scholar]
- Brodsky SV, Gao S, Li H. Hyperglycemic switch from mitochondrial nitric oxide to superoxide production in endothelial cells. Am J Physiol Heart Circ Physiol. 2002;283:H2130–H2139. doi: 10.1152/ajpheart.00196.2002. [DOI] [PubMed] [Google Scholar]
- Zhao HJ, Wang S, Cheng H. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J Am Soc Nephrol. 2006;17:2664–2669. doi: 10.1681/ASN.2006070798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Sato W, Glushakova O. Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. J Am Soc Nephrol. 2007;18:539–550. doi: 10.1681/ASN.2006050459. [DOI] [PubMed] [Google Scholar]
- Kosugi T, Heinig M, Nakayama T. Lowering blood pressure blocks mesangiolysis and mesangial nodules, but not tubulointerstitial injury, in diabetic eNOS knockout mice. Am J Pathol. 2009;174:1221–1229. doi: 10.2353/ajpath.2009.080605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eremina V, Baelde HJ, Quaggin SE. Role of the VEGF–a signaling pathway in the glomerulus: Evidence for crosstalk between components of the glomerular filtration barrier. Nephron Physiol. 2007;106:32–37. doi: 10.1159/000101798. [DOI] [PubMed] [Google Scholar]
- Eremina V, Sood M, Haigh J. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111:707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequeira-Lopez ML, Weatherford ET, Borges GR. The microRNA-processing enzyme dicer maintains juxtaglomerular cells. J Am Soc Nephrol. 2010;21:460–467. doi: 10.1681/ASN.2009090964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper ME, Vranes D, Youssef S. Increased renal expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR-2 in experimental diabetes. Diabetes. 1999;48:2229–2239. doi: 10.2337/diabetes.48.11.2229. [DOI] [PubMed] [Google Scholar]
- Tsuchida K, Makita Z, Yamagishi S. Suppression of transforming growth factor beta and vascular endothelial growth factor in diabetic nephropathy in rats by a novel advanced glycation end product inhibitor, OPB-9195. Diabetologia. 1999;42:579–588. doi: 10.1007/s001250051198. [DOI] [PubMed] [Google Scholar]
- Cohen MP, Chen S, Ziyadeh FN. Evidence linking glycated albumin to altered glomerular nephrin and VEGF expression, proteinuria, and diabetic nephropathy. Kidney Int. 2005;68:1554–1561. doi: 10.1111/j.1523-1755.2005.00567.x. [DOI] [PubMed] [Google Scholar]
- de Vriese AS, Tilton RG, Elger M. Antibodies against vascular endothelial growth factor improve early renal dysfunction in experimental diabetes. J Am Soc Nephrol. 2001;12:993–1000. doi: 10.1681/ASN.V125993. [DOI] [PubMed] [Google Scholar]
- Flyvbjerg A, Dagnaes-Hansen F, De Vriese AS. Amelioration of long-term renal changes in obese type 2 diabetic mice by a neutralizing vascular endothelial growth factor antibody. Diabetes. 2002;51:3090–3094. doi: 10.2337/diabetes.51.10.3090. [DOI] [PubMed] [Google Scholar]
- Schrijvers BF, Flyvbjerg A, Tilton RG. A neutralizing VEGF antibody prevents glomerular hypertrophy in a model of obese type 2 diabetes, the Zucker diabetic fatty rat. Nephrol Dial Transplant. 2006;21:324–329. doi: 10.1093/ndt/gfi217. [DOI] [PubMed] [Google Scholar]
- Sung SH, Ziyadeh FN, Wang A. Blockade of vascular endothelial growth factor signaling ameliorates diabetic albuminuria in mice. J Am Soc Nephrol. 2006;17:3093–3104. doi: 10.1681/ASN.2006010064. [DOI] [PubMed] [Google Scholar]
- Hohenstein B, Hausknecht B, Boehmer K. Local VEGF activity but not VEGF expression is tightly regulated during diabetic nephropathy in man. Kidney Int. 2006;69:1654–1661. doi: 10.1038/sj.ki.5000294. [DOI] [PubMed] [Google Scholar]
- Baelde HJ, Eikmans M, Doran PP. Gene expression profiling in glomeruli from human kidneys with diabetic nephropathy. Am J Kidney Dis. 2004;43:636–650. doi: 10.1053/j.ajkd.2003.12.028. [DOI] [PubMed] [Google Scholar]
- Bortoloso E, Del Prete D, Dalla Vestra M. Quantitave and qualitative changes in vascular endothelial growth factor gene expression in glomeruli of patients with type 2 diabetes. Eur J Endocrinol. 2004;150:799–807. doi: 10.1530/eje.0.1500799. [DOI] [PubMed] [Google Scholar]
- Lindenmeyer MT, Kretzler M, Boucherot A. Interstitial vascular rarefaction and reduced VEGF-A expression in human diabetic nephropathy. J Am Soc Nephrol. 2007;18:1765–1776. doi: 10.1681/ASN.2006121304. [DOI] [PubMed] [Google Scholar]
- Veron D, Bertuccio CA, Marlier A. Podocyte vascular endothelial growth factor (Vegf(1)(6)(4)) overexpression causes severe nodular glomerulosclerosis in a mouse model of type 1 diabetes. Diabetologia. 2011;54:1227–1241. doi: 10.1007/s00125-010-2034-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaskandarajah GA, Jeansson M, Maezawa Y. Vegfa protects the glomerular microvasculature in diabetes. Diabetes. 2012;61:2958–2966. doi: 10.2337/db11-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisonpierre PC, Suri C, Jones PF. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis [see comments] Science. 1997;277:55–60. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- Suri C, Jones PF, Patan S. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis [see comments] Cell. 1996;87:1171–1180. doi: 10.1016/s0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- Yuan HT, Khankin EV, Karumanchi SA. Angiopoietin 2 is a partial agonist/antagonist of Tie2 signaling in the endothelium. Mol Cell Biol. 2009;29:2011–2022. doi: 10.1128/MCB.01472-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale NW, Thurston G, Hackett SF. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by Angiopoietin-1. Dev Cell. 2002;3:411–423. doi: 10.1016/s1534-5807(02)00217-4. [DOI] [PubMed] [Google Scholar]
- Betsholtz C, Lindblom P, Bjarnegard M. Role of platelet-derived growth factor in mesangium development and vasculopathies: Lessons from platelet-derived growth factor and platelet-derived growth factor receptor mutations in mice. Curr Opin Nephrol Hypertens. 2004;13:45–52. doi: 10.1097/00041552-200401000-00007. [DOI] [PubMed] [Google Scholar]
- Jeansson M, Gawlik A, Anderson G. Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J Clin Invest. 2011;121:2278–2289. doi: 10.1172/JCI46322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HS, Blann AD, Chong AY. Plasma vascular endothelial growth factor, angiopoietin-1, and angiopoietin-2 in diabetes: Implications for cardiovascular risk and effects of multifactorial intervention. Diabetes Care. 2004;27:2918–2924. doi: 10.2337/diacare.27.12.2918. [DOI] [PubMed] [Google Scholar]
- Lee S, Kim W, Moon SO. Renoprotective effect of COMP-angiopoietin-1 in db/db mice with type 2 diabetes. Nephrol Dial Transplant. 2007;22:396–408. doi: 10.1093/ndt/gfl598. [DOI] [PubMed] [Google Scholar]
- Dessapt-Baradez C, Woolf AS, White KE. Targeted glomerular angiopoietin-1 therapy for early diabetic kidney disease. J Am Soc Nephrol. 2014;25:33–42. doi: 10.1681/ASN.2012121218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonventre JV. Can we target tubular damage to prevent renal function decline in diabetes? Semin Nephrol. 2012;32:452–462. doi: 10.1016/j.semnephrol.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert RE, Cooper ME. The tubulointerstitium in progressive diabetic kidney disease: More than an aftermath of glomerular injury? Kidney Int. 1999;56:1627–1637. doi: 10.1046/j.1523-1755.1999.00721.x. [DOI] [PubMed] [Google Scholar]
- Najafian B, Alpers CE, Fogo AB. Pathology of human diabetic nephropathy. Contrib Nephrol. 2011;170:36–47. doi: 10.1159/000324942. [DOI] [PubMed] [Google Scholar]
- Bader R, Bader H, Grund KE. Structure and function of the kidney in diabetic glomerulosclerosis. Correlations between morphological and functional parameters. Pathol Res Pract. 1980;167:204–216. doi: 10.1016/S0344-0338(80)80051-3. [DOI] [PubMed] [Google Scholar]
- Lane PH, Steffes MW, Fioretto P. Renal interstitial expansion in insulin-dependent diabetes mellitus. Kidney Int. 1993;43:661–667. doi: 10.1038/ki.1993.95. [DOI] [PubMed] [Google Scholar]
- Ueno M, Kawashima S, Nishi S. Tubulointerstitial lesions in non-insulin dependent diabetes mellitus. Kidney Int Suppl. 1997;63:S191–S194. [PubMed] [Google Scholar]
- Taft JL, Nolan CJ, Yeung SP. Clinical and histological correlations of decline in renal function in diabetic patients with proteinuria. Diabetes. 1994;43:1046–1051. doi: 10.2337/diab.43.8.1046. [DOI] [PubMed] [Google Scholar]
- Fioretto P, Steffes MW, Sutherland DE. Sequential renal biopsies in insulin-dependent diabetic patients: Structural factors associated with clinical progression. Kidney Int. 1995;48:1929–1935. doi: 10.1038/ki.1995.493. [DOI] [PubMed] [Google Scholar]
- Najafian B, Crosson JT, Kim Y. Glomerulotubular junction abnormalities are associated with proteinuria in type 1 diabetes. J Am Soc Nephrol. 2006;17:S53–S60. doi: 10.1681/ASN.2005121342. [DOI] [PubMed] [Google Scholar]
- Najafian B, Kim Y, Crosson JT. Atubular glomeruli and glomerulotubular junction abnormalities in diabetic nephropathy. J Am Soc Nephrol. 2003;14:908–917. doi: 10.1097/01.asn.0000057854.32413.81. [DOI] [PubMed] [Google Scholar]
- White KE, Marshall SM, Bilous RW. Prevalence of atubular glomeruli in type 2 diabetic patients with nephropathy. Nephrol Dial Transplant. 2008;23:3539–3545. doi: 10.1093/ndt/gfn351. [DOI] [PubMed] [Google Scholar]
- Watts GF, Vlitos MA, Morris RW. Urinary N-acetyl-beta-D-glucosaminidase excretion in insulin-dependent diabetes mellitus: Relation to microalbuminuria, retinopathy and glycaemic control. Diabete Metab. 1988;14:653–658. [PubMed] [Google Scholar]
- Gibb DM, Tomlinson PA, Dalton NR. Renal tubular proteinuria and microalbuminuria in diabetic patients. Arch Dis Child. 1989;64:129–134. doi: 10.1136/adc.64.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakroush S, Moeller MJ, Theilig F. Effects of increased renal tubular vascular endothelial growth factor (VEGF) on fibrosis, cyst formation, and glomerular disease. Am J Pathol. 2009;175:1883–1895. doi: 10.2353/ajpath.2009.080792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa K, Wakino S, Simic P. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat Med. 2013;19:1496–1504. doi: 10.1038/nm.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwano M, Plieth D, Danoff TM. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg EM, Potenta SE, Sugimoto H. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol. 2008;19:2282–2287. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBleu VS, Taduri G, O'Connell J. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19:1047–1053. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns WC, Twigg SM, Forbes JM. Connective tissue growth factor plays an important role in advanced glycation end product-induced tubular epithelial-to-mesenchymal transition: Implications for diabetic renal disease. J Am Soc Nephrol. 2006;17:2484–2494. doi: 10.1681/ASN.2006050525. [DOI] [PubMed] [Google Scholar]
- Holian J, Qi W, Kelly DJ. Role of Kruppel-like factor 6 in transforming growth factor-beta1-induced epithelial-mesenchymal transition of proximal tubule cells. Am J Physiol Renal Physiol. 2008;295:F1388–F1396. doi: 10.1152/ajprenal.00055.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastaldi MP, Ferrario F, Giardino L. Epithelial-mesenchymal transition of tubular epithelial cells in human renal biopsies. Kidney Int. 2002;62:137–146. doi: 10.1046/j.1523-1755.2002.00430.x. [DOI] [PubMed] [Google Scholar]
- Koesters R, Kaissling B, Lehir M. Tubular overexpression of transforming growth factor-beta1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am J Pathol. 2010;177:632–643. doi: 10.2353/ajpath.2010.091012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Qu X, Yao J. Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes. 2010;59:2612–2624. doi: 10.2337/db09-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greka A, Mundel P. Cell biology and pathology of podocytes. Annu Rev Physiol. 2012;74:299–323. doi: 10.1146/annurev-physiol-020911-153238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagtalunan ME, Miller PL, Jumping-Eagle S. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest. 1997;99:342–348. doi: 10.1172/JCI119163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer TW, Bennett PH, Nelson RG. Podocyte number predicts long-term urinary albumin excretion in Pima Indians with Type II diabetes and microalbuminuria. Diabetologia. 1999;42:1341–1344. doi: 10.1007/s001250051447. [DOI] [PubMed] [Google Scholar]
- White KE, Bilous RW, Marshall SM. Podocyte number in normotensive type 1 diabetic patients with albuminuria. Diabetes. 2002;51:3083–3089. doi: 10.2337/diabetes.51.10.3083. [DOI] [PubMed] [Google Scholar]
- Susztak K, Raff AC, Schiffer M. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–233. [PubMed] [Google Scholar]
- Menini S, Iacobini C, Oddi G. Increased glomerular cell (podocyte) apoptosis in rats with streptozotocin-induced diabetes mellitus: Role in the development of diabetic glomerular disease. Diabetologia. 2007;50:2591–2599. doi: 10.1007/s00125-007-0821-y. [DOI] [PubMed] [Google Scholar]
- Schiffer M, Bitzer M, Roberts IS. Apoptosis in podocytes induced by TGF-beta and Smad7. J Clin Invest. 2001;108:807–816. doi: 10.1172/JCI12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang PY, Yu Q, Fang W. Advanced glycation endproducts induce podocyte apoptosis by activation of the FOXO4 transcription factor. Kidney Int. 2007;72:965–976. doi: 10.1038/sj.ki.5002456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Ding G, Zhu J. Angiotensin II infusion induces nephrin expression changes and podocyte apoptosis. Am J Nephrol. 2008;28:500–507. doi: 10.1159/000113538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzola D, Gandolfo MT, Ferrario F. Apoptosis in the kidneys of patients with type II diabetic nephropathy. Kidney Int. 2007;72:1262–1272. doi: 10.1038/sj.ki.5002531. [DOI] [PubMed] [Google Scholar]
- Petermann AT, Pippin J, Krofft R. Viable podocytes detach in experimental diabetic nephropathy: Potential mechanism underlying glomerulosclerosis. Nephron Exp Nephrol. 2004;98:e114–e123. doi: 10.1159/000081555. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Ushiyama C, Suzuki S. Urinary excretion of podocytes in patients with diabetic nephropathy. Nephrol Dial Transplant. 2000;15:1379–1383. doi: 10.1093/ndt/15.9.1379. [DOI] [PubMed] [Google Scholar]
- Takemoto M, Ishikawa T, Onishi S. Atorvastatin ameliorates podocyte injury in patients with type 2 diabetes complicated with dyslipidemia. Diabetes Res Clin Pract. 2013;100:e26–e29. doi: 10.1016/j.diabres.2012.12.018. [DOI] [PubMed] [Google Scholar]
- Kestila M, Lenkkeri U, Mannikko M. Positionally cloned gene for a novel glomerular protein–nephrin–is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1:575–582. doi: 10.1016/s1097-2765(00)80057-x. [DOI] [PubMed] [Google Scholar]
- Blasutig IM, New LA, Thanabalasuriar A. Phosphorylated YDXV motifs and Nck SH2/SH3 adaptors act cooperatively to induce actin reorganization. Mol Cell Biol. 2008;28:2035–2046. doi: 10.1128/MCB.01770-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones N, Blasutig IM, Eremina V. Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature. 2006;440:818–823. doi: 10.1038/nature04662. [DOI] [PubMed] [Google Scholar]
- Bonnet F, Cooper ME, Kawachi H. Irbesartan normalises the deficiency in glomerular nephrin expression in a model of diabetes and hypertension. Diabetologia. 2001;44:874–877. doi: 10.1007/s001250100546. [DOI] [PubMed] [Google Scholar]
- Kelly DJ, Aaltonen P, Cox AJ. Expression of the slit-diaphragm protein, nephrin, in experimental diabetic nephropathy: Differing effects of anti-proteinuric therapies. Nephrol Dial Transplant. 2002;17:1327–1332. doi: 10.1093/ndt/17.7.1327. [DOI] [PubMed] [Google Scholar]
- Langham RG, Kelly DJ, Cox AJ. Proteinuria and the expression of the podocyte slit diaphragm protein, nephrin, in diabetic nephropathy: Effects of angiotensin converting enzyme inhibition. Diabetologia. 2002;45:1572–1576. doi: 10.1007/s00125-002-0946-y. [DOI] [PubMed] [Google Scholar]
- Doublier S, Salvidio G, Lupia E. Nephrin expression is reduced in human diabetic nephropathy: Evidence for a distinct role for glycated albumin and angiotensin II. Diabetes. 2003;52:1023–1030. doi: 10.2337/diabetes.52.4.1023. [DOI] [PubMed] [Google Scholar]
- Davis BJ, Forbes JM, Thomas MC. Superior renoprotective effects of combination therapy with ACE and AGE inhibition in the diabetic spontaneously hypertensive rat. Diabetologia. 2004;47:89–97. doi: 10.1007/s00125-003-1256-8. [DOI] [PubMed] [Google Scholar]
- Menne J, Meier M, Park JK. Nephrin loss in experimental diabetic nephropathy is prevented by deletion of protein kinase C alpha signaling in-vivo. Kidney Int. 2006;70:1456–1462. doi: 10.1038/sj.ki.5001830. [DOI] [PubMed] [Google Scholar]
- Aaltonen P, Luimula P, Astrom E. Changes in the expression of nephrin gene and protein in experimental diabetic nephropathy. Lab Invest. 2001;81:1185–1190. doi: 10.1038/labinvest.3780332. [DOI] [PubMed] [Google Scholar]
- Koop K, Eikmans M, Baelde HJ. Expression of podocyte-associated molecules in acquired human kidney diseases. J Am Soc Nephrol. 2003;14:2063–2071. doi: 10.1097/01.asn.0000078803.53165.c9. [DOI] [PubMed] [Google Scholar]
- Coward RJ, Welsh GI, Yang J. The human glomerular podocyte is a novel target for insulin action. Diabetes. 2005;54:3095–3102. doi: 10.2337/diabetes.54.11.3095. [DOI] [PubMed] [Google Scholar]
- Coward RJ, Welsh GI, Koziell A. Nephrin is critical for the action of insulin on human glomerular podocytes. Diabetes. 2007;56:1127–1135. doi: 10.2337/db06-0693. [DOI] [PubMed] [Google Scholar]
- Welsh GI, Hale LJ, Eremina V. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab. 2010;12:329–340. doi: 10.1016/j.cmet.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. The pharmacology of mTOR inhibition. Sci Signal. 2009;2:pe24. doi: 10.1126/scisignal.267pe24. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sengupta S. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Cina DP, Onay T, Paltoo A. Inhibition of MTOR disrupts autophagic flux in podocytes. J Am Soc Nephrol. 2012;23:412–420. doi: 10.1681/ASN.2011070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godel M, Hartleben B, Herbach N. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest. 2011;121:2197–2209. doi: 10.1172/JCI44774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Mori H, Wang J. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest. 2011;121:2181–2196. doi: 10.1172/JCI44771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canaud G, Bienaime F, Viau A. AKT2 is essential to maintain podocyte viability and function during chronic kidney disease. Nat Med. 2013;19:1288–1296. doi: 10.1038/nm.3313. [DOI] [PubMed] [Google Scholar]
- Kopan R. Notch signaling. Cold Spring Harb Perspect Biol. 2012;4:a011213. doi: 10.1101/cshperspect.a011213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niranjan T, Bielesz B, Gruenwald A. The Notch pathway in podocytes plays a role in the development of glomerular disease. Nat Med. 2008;14:290–298. doi: 10.1038/nm1731. [DOI] [PubMed] [Google Scholar]
- Wang XM, Yao M, Liu SX. Interplay between the Notch and PI3K/Akt pathways in high glucose-induced podocyte apoptosis. Am J Physiol Renal Physiol. 2014;306:F205–F213. doi: 10.1152/ajprenal.90005.2013. [DOI] [PubMed] [Google Scholar]
- Gao F, Yao M, Shi Y. Notch pathway is involved in high glucose-induced apoptosis in podocytes via Bcl-2 and p53 pathways. J Cell Biochem. 2013;114:1029–1038. doi: 10.1002/jcb.24442. [DOI] [PubMed] [Google Scholar]
- Dai C, Stolz DB, Kiss LP. Wnt/beta-catenin signaling promotes podocyte dysfunction and albuminuria. J Am Soc Nephrol. 2009;20:1997–2008. doi: 10.1681/ASN.2009010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Gruenwald A, Suh JH. Wnt/beta-catenin pathway in podocytes integrates cell adhesion, differentiation, and survival. J Mol Biochem. 2011;286:26003–26015. doi: 10.1074/jbc.M111.223164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewko B, Endlich N, Kriz W. C-type natriuretic peptide as a podocyte hormone and modulation of its cGMP production by glucose and mechanical stress. Kidney Int. 2004;66:1001–1008. doi: 10.1111/j.1523-1755.2004.00848.x. [DOI] [PubMed] [Google Scholar]
- Sharma K, McGowan TA. TGF-beta in diabetic kidney disease: Role of novel signaling pathways. Cytokine Growth Factor Rev. 2000;11:115–123. doi: 10.1016/s1359-6101(99)00035-0. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Nakamura T, Noble NA. Expression of transforming growth factor beta is elevated in human and experimental diabetic nephropathy. Proc Natl Acad Sci USA. 1993;90:1814–1818. doi: 10.1073/pnas.90.5.1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SW, Isono M, Chen S. Increased glomerular and tubular expression of transforming growth factor-beta1, its type II receptor, and activation of the Smad signaling pathway in the db/db mouse. Am J Pathol. 2001;158:1653–1663. doi: 10.1016/s0002-9440(10)64121-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SN, Hirschberg R. Growth factor ultrafiltration in experimental diabetic nephropathy contributes to interstitial fibrosis. Am J Physiol Renal Physiol. 2000;278:F554–F560. doi: 10.1152/ajprenal.2000.278.4.F554. [DOI] [PubMed] [Google Scholar]
- Sanderson N, Factor V, Nagy P. Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc Natl Acad Sci USA. 1995;92:2572–2576. doi: 10.1073/pnas.92.7.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daehn I, Casalena G, Zhang T. Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J Clin Invest. 2014;124:1608–1621. doi: 10.1172/JCI71195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HY, Huang XR, Wang W. The protective role of Smad7 in diabetic kidney disease: Mechanism and therapeutic potential. Diabetes. 2011;60:590–601. doi: 10.2337/db10-0403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman-Edelstein M, Thomas MC, Thallas-Bonke V. Dedifferentiation of immortalized human podocytes in response to transforming growth factor-beta: A model for diabetic podocytopathy. Diabetes. 2011;60:1779–1788. doi: 10.2337/db10-1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M, Maezawa Y, Yokote K. Mice lacking Smad3 are protected against streptozotocin-induced diabetic glomerulopathy. Biochem Biophys Res Commun. 2003;305:1002–1007. doi: 10.1016/s0006-291x(03)00885-4. [DOI] [PubMed] [Google Scholar]
- Maezawa Y, Yokote K, Sonezaki K. Influence of C-peptide on early glomerular changes in diabetic mice. Diabetes Metab Res Rev. 2006;22:313–322. doi: 10.1002/dmrr.612. [DOI] [PubMed] [Google Scholar]
- Kamba T, Tam BY, Hashizume H. VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am J Physiol Heart Circ Physio. 2006;290:H560–H576. doi: 10.1152/ajpheart.00133.2005. [DOI] [PubMed] [Google Scholar]
- Maynard SE, Min JY, Merchan J. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Sison K, Li C. Soluble FLT1 binds lipid microdomains in podocytes to control cell morphology and glomerular barrier function. Cell. 2012;151:384–399. doi: 10.1016/j.cell.2012.08.037. [DOI] [PubMed] [Google Scholar]
- Ku CH, White KE, Dei Cas A. Inducible overexpression of sFlt-1 in podocytes ameliorates glomerulopathy in diabetic mice. Diabetes. 2008;57:2824–2833. doi: 10.2337/db08-0647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosugi T, Nakayama T, Li Q. Soluble Flt-1 gene therapy ameliorates albuminuria but accelerates tubulointerstitial injury in diabetic mice. Am J Physiol Renal Physiol. 2010;298:F609–F616. doi: 10.1152/ajprenal.00377.2009. [DOI] [PubMed] [Google Scholar]
- Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol. 2004;265:23–32. doi: 10.1016/j.ydbio.2003.06.003. [DOI] [PubMed] [Google Scholar]
- Scott RP, Hawley SP, Ruston J. Podocyte-specific loss of Cdc42 leads to congenital nephropathy. J Am Soc Nephrol. 2012;23:1149–1154. doi: 10.1681/ASN.2011121206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Jiang R, Aoudjit L. Activation of RhoA in podocytes induces focal segmental glomerulosclerosis. J Am Soc Nephrol. 2011;22:1621–1630. doi: 10.1681/ASN.2010111146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolavennu V, Zeng L, Peng H. Targeting of RhoA/ROCK signaling ameliorates progression of diabetic nephropathy independent of glucose control. Diabetes. 2008;57:714–723. doi: 10.2337/db07-1241. [DOI] [PubMed] [Google Scholar]
- Komers R, Oyama TT, Beard DR. Rho kinase inhibition protects kidneys from diabetic nephropathy without reducing blood pressure. Kidney Int. 2011;79:432–442. doi: 10.1038/ki.2010.428. [DOI] [PubMed] [Google Scholar]
- Matoba K, Kawanami D, Okada R. Rho-kinase inhibition prevents the progression of diabetic nephropathy by downregulating hypoxia-inducible factor 1alpha. Kidney Int. 2013;84:545–554. doi: 10.1038/ki.2013.130. [DOI] [PubMed] [Google Scholar]
- Gojo A, Utsunomiya K, Taniguchi K. The Rho-kinase inhibitor, fasudil, attenuates diabetic nephropathy in streptozotocin-induced diabetic rats. Eur J Pharmacol. 2007;568:242–247. doi: 10.1016/j.ejphar.2007.04.011. [DOI] [PubMed] [Google Scholar]