

Abstract
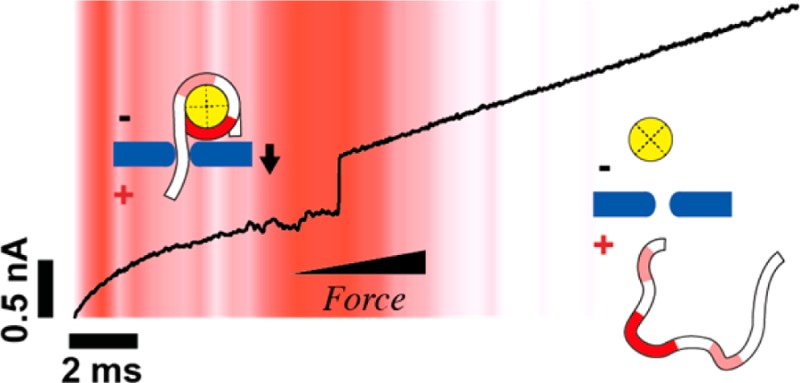
Nucleosomes are the fundamental repeating units of chromatin, and dynamic regulation of their positioning along DNA governs gene accessibility in eukaryotes. Although epigenetic factors have been shown to influence nucleosome structure and dynamics, the impact of DNA methylation on nucleosome packaging remains controversial. Further, all measurements to date have been carried out under zero-force conditions. In this paper, we present the first automated force measurements that probe the impact of CpG DNA methylation on nucleosome stability. In solid-state nanopore force spectroscopy, a nucleosomal DNA tail is captured into a pore and pulled on with a time-varying electrophoretic force until unraveling is detected. This is automatically repeated for hundreds of nucleosomes, yielding statistics of nucleosome lifetime vs electrophoretic force. The force geometry, which is similar to displacement forces exerted by DNA polymerases and helicases, reveals that nucleosome stability is sensitive to DNA sequence yet insensitive to CpG methylation. Our label-free method provides high-throughput data that favorably compares with other force spectroscopy experiments and is suitable for studying a variety of DNA–protein complexes.
Keywords: nucleosome, methylation, nanopore, force, spectroscopy, solid-state
The nucleosome, which comprises 147 base pairs of DNA wound 1.7 times around a histone octamer,1 is the fundamental organizational unit of eukaryotic chromatin. Apart from keeping genomic DNA condensed in the nucleus, nucleosomes sterically hinder the accessibility of specific genomic regions along DNA to proteins that effect transcription regulation and DNA repair.2−4 Although their positioning along the genome is to some extent sequence dependent,5 accessibility of nucleosomal regions is dynamically altered via ATP-dependent remodelers6,7 and epigenetic modifications to DNA and histone proteins. However, despite evidence that some epigenetic modifications modulate the intrinsic nucleosome stability,8 our knowledge of the impact of various histone and DNA modifications on nucleosome stability is largely limited by difficulties in existing techniques for assessing these interactions at high-throughput.
Nucleosomal interactions have been studied using a variety of bulk and single-molecule techniques, the latter of which can provide information on both stability and dynamics. Single-molecule techniques include equilibrium measurements such as Förster resonance energy transfer (FRET), which probes time-dependent structural fluctuations using a distance-dependent fluorescence pair,9 as well as optical tweezers10−13 and atomic force microscopy,14,15 which apply force to end-tethered DNA molecules. The incessant need to chemically modify the DNA or histone proteins for all of these experiments can sometimes lead to experiment-specific variability that complicates data interpretation. For example, although CpG DNA methylation is a well-recognized epigenetic mark involved in gene expression, its effect on the stability and dynamics of nucleosomes is highly controversial: some FRET and other fluorescence-based studies have reported increases in the rigidity and compaction of nucleosomes upon DNA methylation,16,17 whereas other studies have reported a looser, more open conformation for the methylated state.18,19 To our knowledge, however, no measurement of the impact of DNA methylation on the stability of unlabeled nucleosomes under applied force has been carried out to date.
Single-molecule force spectroscopy experiments are nonequilibrium techniques in which piconewton-range loads applied against biomolecular complexes are used to monitor their dynamics and stability. The particular configuration in which force is applied impacts the property that is measured and the interpretation of the result. In conventional single-molecule stretching experiments, for example, changing the direction of the applied force with respect to the nucleosome spool can influence the force signatures.20 Geometry can also present a limitation on the type of interactions that are probed. For example, DNA–histone interactions at the axis of symmetry, the so-called dyad axis, are difficult to study in stretching experiments because the histone octamer may not completely unbind upon DNA stretching.21 This problem was recently tackled by pulling apart the nucleosomal DNA from the same side (i.e., 5′ and 3′ strands are pulled apart).13,22 However, an unraveling pathway that involves unzipping of the double-stranded DNA (dsDNA) helix wound around the core octamer is not necessarily comparable to a pathway where the DNA helix remains intact. In addition, arduous chemical modification is required for these experiments. Presumably due to these complexities, single-molecule force spectroscopy studies that probe the influence of DNA or histone modifications have yet to be reported.
A different single-molecule approach that can be well suited for studying DNA–protein complexes is nanopore-based resistive sensing.23 In this technique, a dilute solution of charged biomolecules is contacted with a nanopore-containing membrane and an electrochemical transmembrane bias is applied. The steady-state ion current that results from the applied bias creates a nanoscale-localized electric field that electrophoretically captures biomolecules and draws them through the pore. Stochastic transport of one biomolecule at a time is detected by measurements of discrete fluctuations in the transmembrane ionic current. Nanopore force spectroscopy experiments are based on capturing a portion of a biomolecular complex within a pore and applying electrophoretic force while monitoring the time of complex rupture. Because electrophoretic force is applied directly to the trapped/confined molecule, no chemical modification is required, which greatly simplifies sample preparation for experiments. Using lipid-embedded protein channels such as α-hemolysin, force spectroscopy has been used for studying the stability of DNA secondary structures like hairpins24−28 or aptamers,29 as well as an exonuclease I–ssDNA complex.30 However, the reliance on protein channels for spectroscopy studies presents limitations on the size of biomolecules that can be studied, also setting upper limits on the bias that can be applied without rupturing the supporting lipid membrane. Solid-state nanopores, on the other hand, show great potential for studying nucleic acid–protein interactions.31−34 The reduced membrane fragility allows a wider range of forces to be applied, and the pore geometry can be fine-tuned using advanced nanofabrication techniques to suit the application. Using solid-state nanopores with diameters greater than 2.5 nm, several dsDNA–protein complexes35−37 have been studied. In contrast, biological channels that allow dsDNA transport are rarely reported.38,39
Our group recently reported nanopore-based unraveling of nucleosomes at constant electrophoretic force.40 In this study, we have found that nucleosomes are only detected at moderately high voltages, which presumably is due to the large barrier associated with threading of the nucleosomal dsDNA tail into the pore. This limitation of constant voltage experiments to the large-force regime has prompted us to develop the first electrophoretic force spectroscopy for probing unlabeled nucleosomes, used here for assessing the influence of sequence and CpG DNA methylation on nucleosome stability.
Results
Our measurement setup is depicted schematically in Figure 1a. Initially, a capturing voltage of 350–550 mV is applied until threading of a ∼ 20–50 bp-long nucleosomal dsDNA tail into a 2.6–2.8 nm diameter pore is detected (I). The current drop that marks DNA threading signals a hardware trigger to reduce the voltage (see Materials and Methods section) and then ramp it upward at a constant loading rate (2.5 V/s–40 V/s) (II). Because the nucleosome dimensions (∼11 nm) are larger than the pore dimensions, the electrophoretically pulled dsDNA tail gradually strains the complex until the nucleosome is ruptured, after which DNA is rapidly (10–50 μs) released from the pore (III). The release event, which signals nucleosome rupture, is detected as a fast opening transition in the ionic current. In all of our experiments we have used a narrow range of pore dimensions (diameters of 2.6–2.8 nm, effective pore lengths of 5–8 nm), as determined via the ionic current traces.41,42
Figure 1.
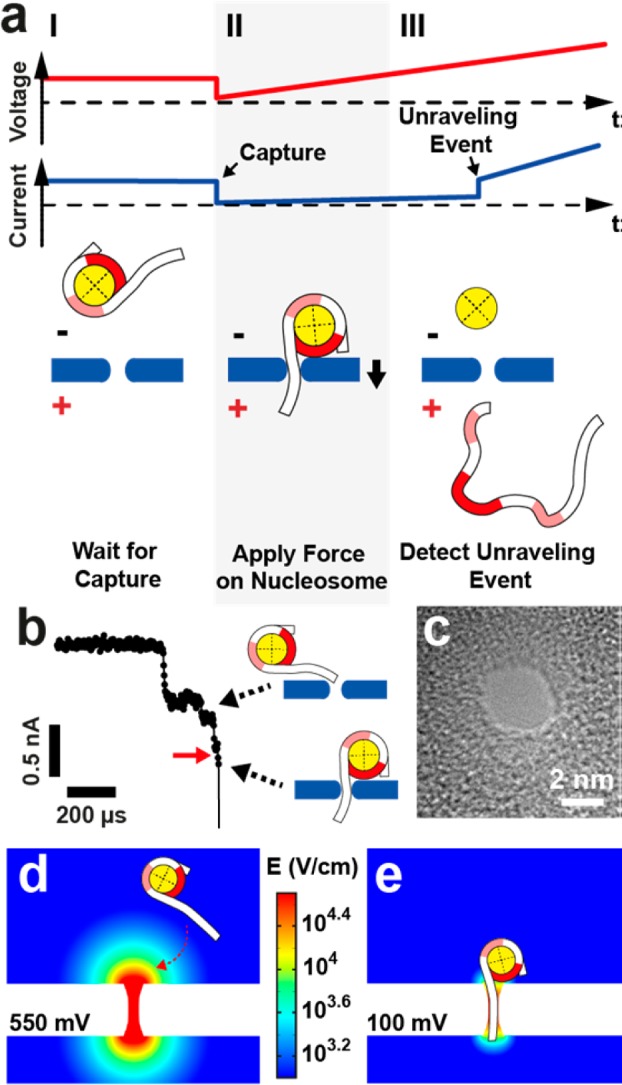
Electrophoretic Force Spectroscopy. (a) Schematic depiction of the measurement principle. I: A constant voltage is applied to capture a mononucleosome. II: A potential ramp is triggered, applying increasing force on the nucleosome. III: When the nucleosome unravels, the DNA molecule escapes from the pore and a stepwise increase in the current signal at a specific transition voltage is observed. Light and bold red shaded areas on the nucleosomal DNA represent off-dyad and on-dyad sites of interaction, respectively. (b) Representative current trace of a capture event at 400 mV voltage; red arrow indicates trigger level. (c) Transmission electron microscope (TEM) image of one of the ∼2.8 nm silicon nitride nanopores used in our experiments. (d) Finite-element simulations (COMSOL) of the electric field of a pore with dimensions as in (c) at a capture voltage of 550 mV (log scale, V/cm). (e) Same as in (d), except the voltage is reduced to 100 mV after nucleosome capture.
Nucleosome Capture
As observed in constant voltage nanopore experiments,40 nucleosome-associated current blockades feature a multilevel structure with one or more shallow current blockade levels that precede a deep blockade level. We attribute the shallow levels to nucleosome interactions with the pore before threading of the DNA tail into the pore lumen has occurred and the deep blockade level to capture of a DNA tail. Therefore, in our acquisition protocol, we set the trigger level for nucleosome capture to a value that suggests complete DNA threading, as shown by the red arrow in Figure 1b (for more details see Supporting Information Figure S1).
A representative transmission-electron microscopy (TEM) image of one of the pores used in our experiments is shown in Figure 1c. Our used pore diameters (∼2.8 nm) are only slightly larger than the cross-sectional diameter of a DNA molecule (∼2.2 nm), which efficiently excludes nucleosome entry into the pore. However, for this measurement a DNA tail must be properly oriented in order for DNA threading to occur, and consequently, we have found that nucleosome capture requires much higher voltages as compared with free DNA capture.40 To illustrate the impact of applied voltage on nucleosome capture, we performed finite-element simulations. In Figure 1d, the color map shows the electric field distribution when a capturing voltage of 550 mV is applied (log scale). At this voltage, the electric field above the pore is large enough to electrophoretically trap the nucleosome at the pore mouth until its DNA tail is threaded. Upon hardware-based detection of tail threading the voltage is reduced to 100 mV (Figure 1e), which produces a finer, more localized electrophoretic force to the dsDNA tail at the onset of the force spectroscopy experiment. In addition to high-throughput, this active control approach allows us to access a lower range of voltages, in which we expect an increased sensitivity to alterations in biomolecular interactions.
Nucleosome Unraveling
We first performed force spectroscopy studies on nucleosomes assembled from sea urchin 5S rDNA (total DNA length = 208 bp; see Supporting Information Figure S2). This positioning sequence is known to form nucleosomes with a relative energy gain of 0.5 kcal/mol compared to a pool of random DNA sequences.43 For each detected capture event, we recorded the ionic current before and during the triggered voltage ramp. A representative set of 50 overlaid current traces obtained in a 5S nucleosome unraveling experiment is shown in Figure 2a. For the sake of discussion, we highlight a model trace in white. At the beginning of the voltage ramp the current was in a low state that corresponds to an occupied pore. At some critical voltage (∼235 mV for the model trace) an opening transition is observed, signaling nucleosome rupture. The red background in the image reflects the relative frequency of unraveling at each particular voltage range during the experiment (n = 314). In rare cases, the pore remained in its blocked state throughout the voltage ramp; such events were not considered in our analysis. Apart from these long-lived events, the traces show two distinct regions in which events are frequently seen. Below the traces in Figure 2a, we show the distribution of transition voltages shown, which reveals a dominant population centered at ∼230 mV that we attribute to nucleosome unraveling. In addition to this well-defined population, we observe the tail of a second minor population that vanishes as the voltage reaches 150 mV. By performing a control measurement using a 250 bp dsDNA fragment, we were able to assign the minor population to free dsDNA, which is also observed in our experiments, although infrequently and at unmeasurably low peak voltage values. Because our sample contains both free DNA and nucleosomes, we tuned our experimental conditions so that the relative capture frequency of nucleosomes to DNA is maximized. By performing this experiment using various loading rates (i.e., voltage ramp speeds), we found that the peak voltage (Vpeak) of nucleosome unraveling scales with the logarithm of the loading rate in the range of 2.5–40 V/s (see Figure 2b). In Figure 2c, we calculated the nucleosome lifetime as a function of bias using eq 1 (see Materials and Methods section), where only events assigned to the nucleosome population in the transition voltage distribution were taken into account. All data obtained from three different pores at various loading rates collapse onto a single master curve, confirming the applicability of the transformation defined by eq 1.44 We also evaluated the possible influence of capture voltage on the unraveling statistics by performing measurements at two different capture voltages (400 mV and 550 mV) for identical ramp parameters (20 V/s, 50 mV starting voltage). Although we found the rate of nucleosome capture to increase at the higher voltage, in Figure 2b, we show that the most probable nucleosome unraveling voltage for the 20 V/s ramps are independent of the capture voltage, implying that our capture conditions are not nucleosome-destructive. Finally, when comparing the obtained lifetimes with our previous constant voltage data,40 we find a systematic trend in which constant voltage lifetimes are always lower than our ramp data (Figure 2c, black squares). These consistently higher values obtained in constant voltage experiments arise from the way in which we performed our analysis: for the constant voltage data, we measured the full duration of the event including the shallow current level at the beginning, whereas in voltage ramp experiments, any prethreading time is not taken into account.
Figure 2.
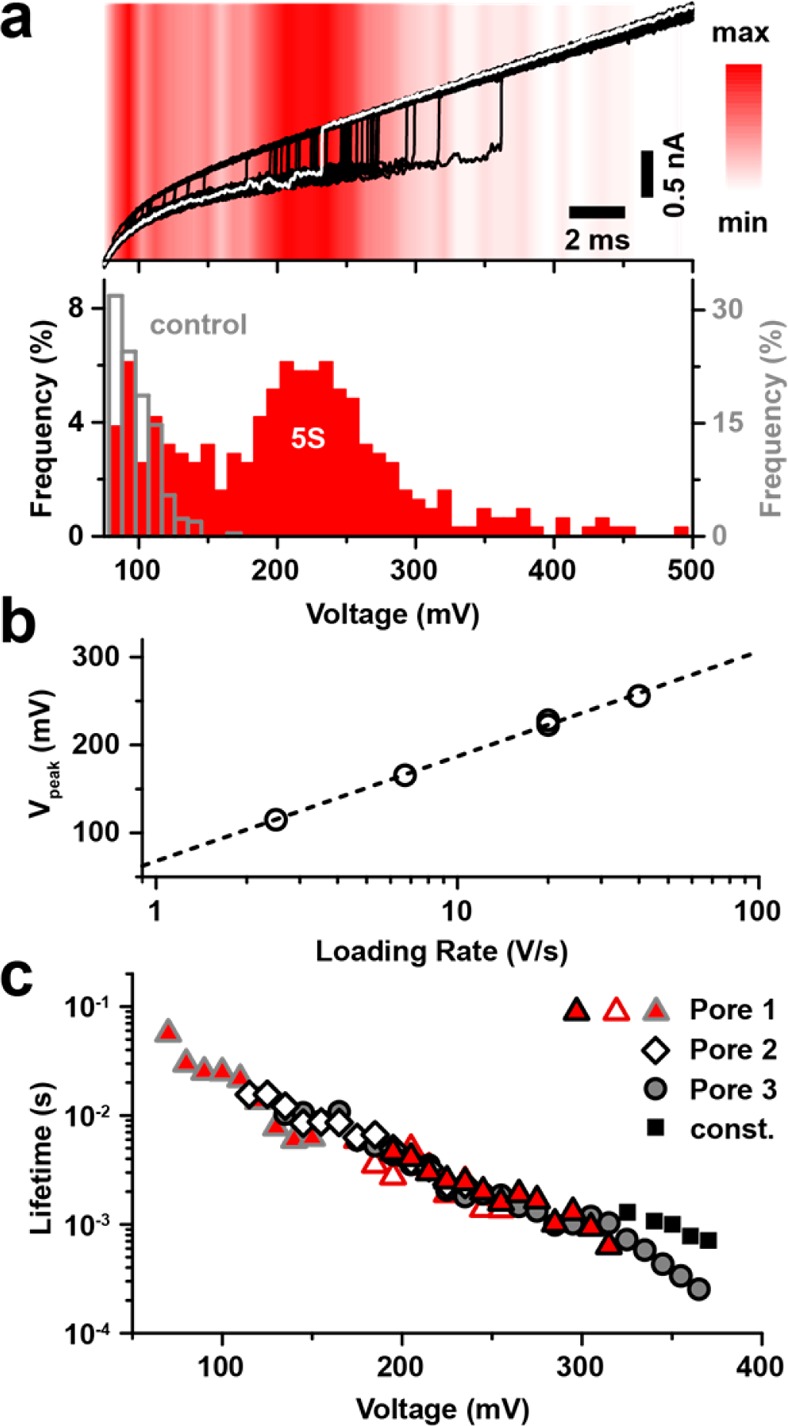
Nucleosome unraveling experiments. (a) Top: Current traces acquired during triggered potential ramps in the presence of nucleosomes assembled from the sea urchin 5S sequence (subset of 50 representative traces with an opening transition, 20 V/s loading rate, typical trace highlighted in white). Red color shading: Distribution of opening transitions for the complete data set (314 detected transitions). Bottom: Corresponding transition voltage histograms for the data set (red) and for a 250 bp DNA control (gray). (b) Peak position of the nucleosome-attributed population as a function of loading rate. Dashed line represents a logarithmic fit. For a loading rate of 20 V/s, the peak position is shown for two experiments with different capture voltages (400 mV and 550 mV). (c) Voltage-dependent lifetime of 5S nucleosome structures determined from measurement data using eq 1. Data from three different pores (triangles, diamonds, and circles) at loading rates of 2.5 V/s (red triangles with gray border), 6.7 V/s (diamonds), 20 V/s (red triangles with black border and open triangles), and 40 V/s (gray spheres) are shown. Data from constant voltage nanopore experiments40 are shown for comparison (black squares). Pores with diameters d = 2.6–2.8 nm and effective length heff = 5–8 nm were used.
Our findings give rise to a rather straightforward interpretation: logarithmic regimes of the most probable transition voltages across a wide range of loading rates are associated with an irreversible crossing of a single energy barrier at a fixed location along the unbinding pathway.45,46 Likewise, the validity of the transformation used for the voltage-dependent lifetime representation of our data (eq 1) is coupled to the assumption of an escape over a single barrier. As nanopore force spectroscopy experiments do not provide information on the unraveling coordinates, escape over individual energy barriers within a multibarrier landscape cannot be resolved. Thus, even for a multibarrier energy landscape, our findings imply that one distinct energy barrier limits the transition across our experimental bias range. Should multiple barriers with comparable energy govern the transition, the lifetime transformation shown in eq 1 would not reflect lifetime values at constant voltage. To explain our observation of a single barrier, we turn to the prior literature: A recent study by Hall et al.13 in which nucleosomal DNA was unzipped using optical tweezers has identified three major regions of interaction that can be categorized as on-dyad and off-dyad interactions. Whereas on-dyad interactions that are localized at the H3–H4 tetramer were found to be the strongest, off-dyad interactions are significantly weaker. Further, single-molecule FRET studies9 have revealed a salt-induced decrease in off-dyad stability. This salt dependence is presumably due to a shift in the balance of DNA bending and electrostatic interaction with the histone core, which is screened more efficiently at elevated salt concentrations. Given that our experiments were conducted at an ionic strength that is similar to that inside of a eukaryotic nucleus,47 we conclude that the nucleosome dyad is the most energy-costly barrier of the unraveling process in our experiments.
Sequence Dependence
Next, we studied the sensitivity of electrophoretic force spectroscopy to positioning sequence by comparing the 5S nucleosome to a similar nucleosome construct assembled from the Widom 601 clone43 (total DNA length = 189 bp; see Supporting Information Figure S2). Compared to 5S rDNA, a relative energy gain of 2.8 kcal/mol was determined for this SELEX-generated positioning sequence, which is known to form the most stable and most accurately positioned nucleosome assembly. As its selection was mainly driven by the interaction of its central ∼70 bp with the H3–H4 tetramer,48 dyad interactions are particularly strong for this sequence. In Figure 3, we display raw ramp traces for 5S (red) and Widom 601 (blue) nucleosomes under identical conditions (loading rate = 40 V/s). In agreement with its higher stability, we observe for 601 nucleosomes considerably higher transition voltages than for 5S nucleosomes: Vpeak is 260 mV for the 5S sample, whereas it is 330 mV for the 601 sample. We note that full DNA tail threading was achieved in all experiments, as indicated by the similar changes in conductance levels between open and occupied pore states in all experiments (ΔG = 2.6 ± 0.1 nS).
Figure 3.
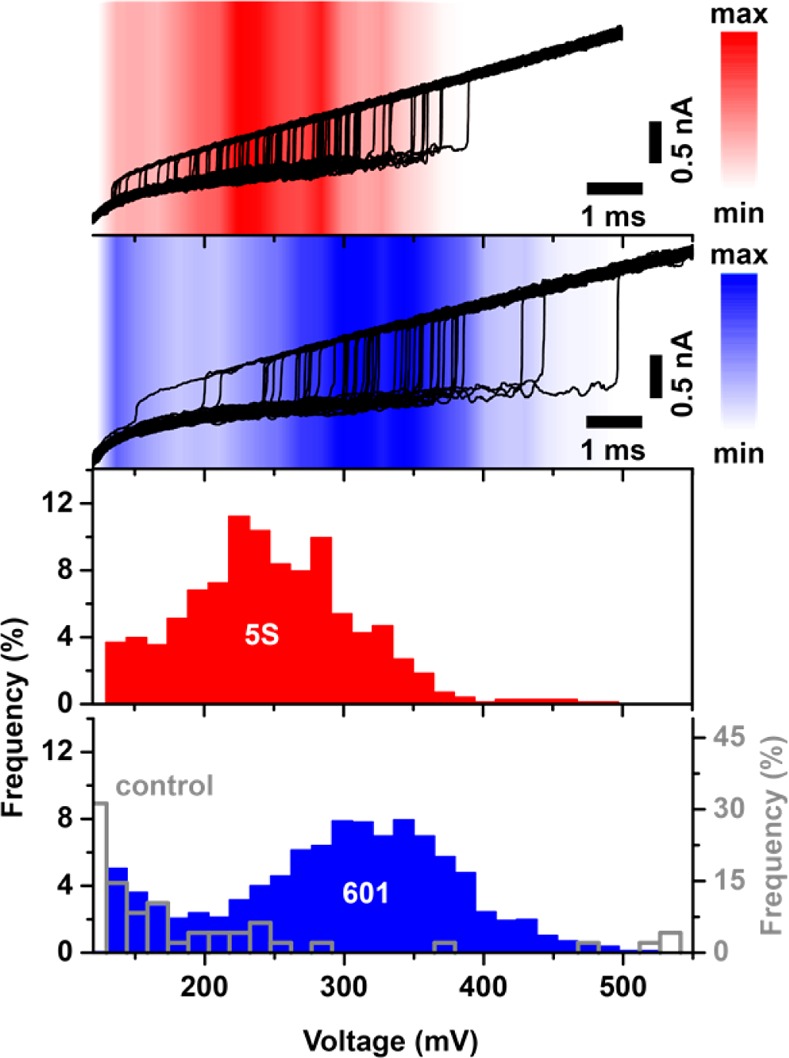
Impact of the positioning sequence on unraveling characteristics. Top: Current traces acquired during triggered potential ramps in the presence of nucleosomes assembled from the sea urchin 5S (red) and the Widom 601 sequence (blue) (subsets of 50 representative traces with an opening transition, 40 V/s loading rate). Color shading: Distribution of opening transitions for the complete data sets (703 and 1547 detected transitions for 5S and 601, respectively). Bottom: Corresponding transition voltage histograms for 5S nucleosomes (red), 601 nucleosomes (blue), and a 250 bp DNA only control (open gray bars).
In Figure 4a, voltage-dependent lifetime data are analyzed for both positioning sequences by compiling data from experiments using multiple pores and different loading rates. Similarly to the 5S nucleosomes, unraveling lifetimes for the Widom nucleosomes at different loading rates (10 V/s and 40 V/s) fall on the same master curve. Furthermore, triggered constant voltage experiments (see Figure 4a gray symbols and Supporting Information Figure S3) reveal a good agreement of lifetimes with transformed ramp data. Despite various potential pitfalls from pore-to-pore variability and possible ionic strength fluctuations, we are able to resolve both positioning sequences, and we find that Widom 601 lifetimes are universally ∼5-fold longer than 5S lifetimes.
Figure 4.
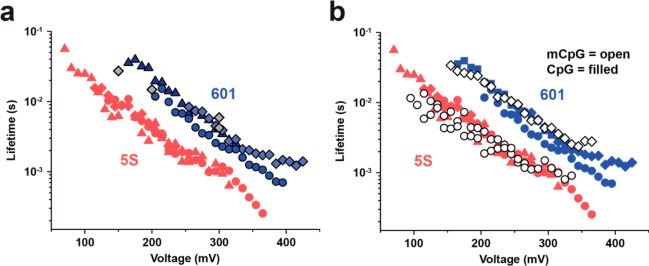
Nucleosome lifetime comparison. (a) Voltage-dependent lifetime of nucleosomes assembled from the Widom 601 (blue symbols) and the 5S positioning sequence (red symbols, shown for comparison). The 601 data was obtained from three different pores at loading rates of 10 V/s (triangles) and 40 V/s (diamonds and circles). Gray diamonds: Lifetime for 601 nucleosomes obtained in triggered constant voltage experiments. (b) Voltage-dependent lifetime of nucleosomes assembled from methylated DNA (mCpG) using the Widom 601 (open diamonds) and the 5S positioning sequence (open circles). Data from (a) are shown for comparison. Scatters in lifetime data for the same nucleosome reflects pore-to-pore variability.
Influence of DNA Methylation
Finally, we studied the impact of DNA methylation using our method. Both DNA sequences we study here contain >10 CpG sites in their positioning sequences (see Supporting Information Figure S2). First, we used a methyltransferase to methylate CpG sites on the unassembled DNA of both positioning sequences (5S rDNA and Widom 601, for details on the methylation protocol, see Materials and Methods section). After confirming DNA methylation via methylation-specific digestion, we reconstituted nucleosomes from the methylated DNA and histones and performed force spectroscopy on the assembled nucleosomes. In Figure 4b, we present lifetime vs voltage data for CpG methylated (mCpG, open markers) and unmethylated (CpG, solid markers) nucleosomes. For the Widom 601 data, we clearly see that methylation does not impact the overall nucleosome stability, as indicated by the similar lifetime vs voltage trajectory. For the 5S sequence, we find a slight decrease in stability that is marked by a less than 2-fold reduction in mean lifetimes in the 100–150 mV voltage range. However, these experiments on both sequences show that the difference in stability upon methylation in both nucleosomes is far smaller than differences we observed for the 5S and Widom 601 samples, for example. Though a more exhaustive study to ascertain the generality of this result, our data for these sequences suggests a minor impact of methylation on nucleosome packaging and stability.
Discussion/Conclusion
In this work, we investigated the influence of sequence and CpG methylation on the stability of unlabeled mononucleosomes. Nanopore-based electrophoretic force spectroscopy was used here to capture a nucleosomal DNA tail and gradually unravel it under increasing force. In contrast with experiments that are conducted at constant voltage, the use of dynamic force spectroscopy was helpful for our nucleosome study because a large bias is typically required for DNA tail capture, whereas the smaller bias regime allows our technique to probe behavior under a weaker electrophoretic force, in which longer nucleosome lifetimes are obtained. The automated approach in our experiment offers high throughput without requiring any chemical labeling or long tail DNA handles (20–30 bp tails are sufficient for our study). Results for the sea urchin 5S rDNA positioning sequence align reasonably well with prior constant force nanopore experiments from our group.40 We have observed a single logarithmic loading rate dependence on the peak unraveling voltage, which supports a mechanism that involves crossing of a single energy barrier, which we interpret to reflect the most dominant interaction, that is, of nucleosomal DNA with the H3–H4 tetramer in the dyad region. For the high-affinity Widom 601 positioning sequence, we obtain lifetimes that are half an order of magnitude higher, and despite minor pore-to-pore scatter, our method demonstrates the required sensitivity to discern these two positioning sequences. We demonstrated its use by showing for the first time nucleosome unraveling experiments on CpG methylated DNA sequences. Surprisingly, for the sequences we tested, CpG methylation did not affect the nucleosome assembly nor unraveling trajectories, which suggests that DNA methylation plays alternative role in nucleosomal maintenance, for example, transcription factor binding modulation. Whereas methylation did show an effect on off-dyad nucleosome equilibrium dynamics in recent fluorescence-based studies16−19 and for some periodic CpG patterns in a MNase digestion assay,8 our method implies that the central H3–H4 tetramer interactions on the dyad region are hardly affected by DNA methylation. Because in the dyad region the DNA is less bent,49,50 we expect that changes in DNA mechanical properties will not greatly influence the nucleosome stability,51 and thus, our observations appear consistent with previous findings. This confirmation of methylation-independent nucleosome stability also points to other possible mechanisms by which DNA methylation alters gene expression, for example, modulating the binding of transcription activators/repressors.
Due to its high throughput and simplicity, our technique is well suited to screen different epigenetic markers and assess their influence on nucleosome stability under external force.
Nanopore-based force spectroscopy can probe biomolecular interactions with high throughput and a unique geometry, as compared with optical/magnetic tweezers and atomic force microscopy. Much like the action of processivity-enhancing motors in prokaryotes and eukaryotes, our method applies either a constant or a time-varying force to a nucleosomal dsDNA tail in order to displace histones that interact with the pulled DNA. We have found little to no impact of DNA methylation on nucleosome stability, which suggests that histone modifications play a greater role on these systems. Fortuitously, the label-free nature of our method is ideally suited for studying the influence of histone core modifications such as methylation, acetylation or phosphorylation on nucleosome structure, which is highly challenging for other single-molecule experiments due to many chemical modifications that are required.
Materials and Methods
Nanopore Fabrication
A detailed description of the nanopore fabrication process can be found elsewhere.42 The substrate membrane devices were prepared from 500-μm-thick ⟨100⟩ silicon wafers passivated with a 2.5 μm SiO2 thermal layer and coated with a 40 nm-thick silicon nitride layer (LPCVD-grown). Freestanding silicon nitride membranes were released in each device using optical lithography and wet chemical etching, and membranes were thinned using e-beam lithography and reactive ion etching. Finally, nanopores were drilled by exposing the membrane to the highly focused beam of a transmission electron microscope (JEOL 2010FEG). Nanopore devices were cleaned by treatment with a hot piranha solution and a copious water rinse prior to each experiment.
Nucleosome Assembly and DNA Methylation
Monoucleosomes were prepared as previously described, with some slight changes.40 Purified histones from the Epimark Nucleosome Assembly Kit (NEB E5350) and either 5s rDNA (NEB) or biotinylated Nucleosome Assembly 601 Sequence DNA (Widom 601) (Epicypher 18-0001) were combined following the dilution assembly protocol for 25 pmol. Briefly, 5 M NaCl was mixed with either unmethylated or methylated DNA (methylation of Widom 601 DNA described below) and dimer and tetramer histone proteins for each reaction. Additional amounts of 10 mM Tris were added to dilute NaCl as per the protocol. Incubations were 30 min at 24 °C. Once reactions were complete, nucleosome assembly was determined by gel shift analysis. Gel shift was completed by running 10 μL of the assembly reaction on a 6% polyacrylamide gel, staining the DNA with SYBR-safe (Life Technologies S33102), and imaging with a Chemi-doc imager (Biorad) (Supporting Information Figure S4).
DNA was methylated using CpG Methyltransferase (M.SssI) (NEB M0226M). Specifically, 15 μg of DNA was incubated with 640 μM S-adenosylmethionine (SAM) and M. SssI enzyme (5 μL of 20 U/μL) in the provided buffer for 4 h at 37 °C. Another 2.5 μL of 32 mM SAM was added and the incubation extended for an addition 2 h at 37 °C. The enzyme was then inactivated at 65 °C for 20 min. DNA was purified with the GeneJet PCR Extraction Kit (Thermo Scientific K0701) following the provided protocol except the DNA was eluted with water. DNA was quantified, lyophilized, and resuspended at the appropriate concentration (10 μM). Methylation of DNA was determined through restriction digestion with the MspI (NEB R0106M) and HpaII (NEB R0171M) isoschizomer restriction enzymes as per the provided protocols. Both of these enzymes target CCGG sequences for digestion, but only HpaII is blocked when the inner cytosine is methylated. The Nucleosome Assembly 601 DNA sequence contains one CCGG site near the center of the sequence. Digested samples were run on a 6% polyacrylamide gel to detect the digested DNA fragments (Supporting Information Figure S4).
Measurement Setup
All experiments were performed at room temperature (23 ± 1 °C). A cleaned nanopore chip was mounted between two aqueous compartments using a fast-curing silicone elastomer (Ecoflex 5, Smooth-On Inc.). Each compartment was filled with buffer (265 mM KCl, 83 mM NaCl, 1 mM EDTA, 10 mM Tris, pH 8) and an Ag/AgCl electrode. Ionic current was driven through the nanopore and recorded using an Axopatch 200B (Molecular Devices) Patch Clamp Amplifier, interfaced to a computer using synchronized NI PCI-6230 (potential ramp output) and NI PCIe-6351 (current acquisition) DAQ cards (National Instruments). Using custom LabVIEW code, the card’s built-in FPGA chip was programmed to trigger voltage ramps on falling edges in the current signal with selected threshold levels and with a response time in the microsecond range (without the measurement cell attached). The true response of our system (including the measurement cell) is given by its RC time constant, typically ∼300 μs. At the end of each ramp, a reverse bias pulse was applied (−400 mV for 20–40 ms) to prevent long-lived pore blocks. Current recordings were filtered at 100 kHz using the amplifier’s built-in Bessel filter and digitized at sampling rates of at least 250 kHz.
Data Analysis
All data were analyzed using custom MATLAB scripts. The raw current traces were median-filtered using a 40–80 μs time window, and a derivative-based step detection algorithm was used to identify the transition voltages. Traces corresponding to clogged states of the pore, as indicated by a reduced open pore current prior to capture were removed automatically beforehand. As demonstrated previously, the lifetime as a function of bias voltage can be obtained from unfolding/unraveling voltage distributions using the expression44
| 1 |
where p(V|V̇) is the unraveling probability as a function of voltage V and loading rate V̇. This expression is not based on a specific functional form of τ(V) and provides model-independent estimates of τ(V) from the measurement data. However, it is based on the assumption that the underlying transition is irreversible and can be described as the escape over a single energy barrier under quasi-adiabatic conditions, that is, intrinsic thermal relaxation processes are assumed much faster than the time scale of the loading rate. If the transition is governed by multiple energy barriers, this transformation will not reflect constant voltage behavior and data obtained at different loading rates are not expected to collapse on a master curve.52
Acknowledgments
We thank Vera Arnaut and Korbinian Kapsner for assistance with data analysis. This work was funded by grants from the National Institutes of Health (R21-HG6873 and R01-HG6321, M.W.) and by the Deutsche Forschungsgemeinschaft (SFB 863—TP A8, Nanosystems Initiative Munich).
Supporting Information Available
Capture traces, constant voltage experiments, DNA sequences, and gel of methylation. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
M.L. and A.I. conceived and designed the experiments. S.C. fabricated the nanopore devices. S.R.M.K. reconstituted nucleosomes and performed methylation. M.L. and A.I. performed the nanopore experiments. M.L., A.I., and M.W. analyzed the data. M.L., M.W., and F.C.S. wrote the manuscript, and all other authors commented on it.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Luger K.; Mader A. W.; Richmond R. K.; Sargent D. F.; Richmond T. J. Crystal Structure of the Nucleosome Core Particle at 2.8 Angstrom Resolution. Nature 1997, 389, 251–260. [DOI] [PubMed] [Google Scholar]
- Workman J. L.; Kingston R. E. Alteration of Nucleosome Structure as a Mechanism of Transcriptional Regulation. Annu. Rev. Biochem. 1998, 67, 545–579. [DOI] [PubMed] [Google Scholar]
- Jin J.; Bai L.; Johnson D. S.; Fulbright R. M.; Kireeva M. L.; Kashlev M.; Wang M. D. Synergistic Action of RNA Polymerases in Overcoming the Nucleosomal Barrier. Nat. Struct. Mol. Biol. 2010, 17, 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studitsky V. M.; Kassavetis G. A.; Geiduschek E. P.; Felsenfeld G. Mechanism of Transcription through the Nucleosome by Eukaryotic RNA Polymerase. Science 1997, 278, 1960–1963. [DOI] [PubMed] [Google Scholar]
- Segal E.; Fondufe-Mittendorf Y.; Chen L.; Thastrom A.; Field Y.; Moore I. K.; Wang J.-P. Z.; Widom J. A Genomic Code for Nucleosome Positioning. Nature 2006, 442, 772–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narlikar G. J.; Sundaramoorthy R.; Owen-Hughes T. Mechanisms and Functions of ATP-Dependent Chromatin-Remodeling Enzymes. Cell 2013, 154, 490–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman G. D. Mechanisms of Atp-Dependent Nucleosome Sliding. Curr. Opin Str Biol. 2010, 20, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collings C.; Waddell P.; Anderson J. Effects of DNA Methylation on Nucleosome Stability. Nucleic Acids Res. 2013, 41, 2918–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gansen A.; Valeri A.; Hauger F.; Felekyan S.; Kalinin S.; Toth K.; Langowski J.; Seidel C. A. M. Nucleosome Disassembly Intermediates Characterized by Single-Molecule Fret. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 15308–15313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brower-Toland B. D.; Smith C. L.; Yeh R. C.; Lis J. T.; Peterson C. L.; Wang M. D. Mechanical Disruption of Individual Nucleosomes Reveals a Reversible Multistage Release of DNA. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 1960–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y.; Bustamante C. Pulling a Single Chromatin Fiber Reveals the Forces That Maintain Its Higher-Order Structure. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemmen G. J.; Sim R.; Haushalter K. A.; Ke P. C.; Kadonaga J. T.; Smith D. E. Forced Unraveling of Nucleosomes Assembled on Heterogeneous DNA Using Core, Histones, Nap-1, and Acf. J. Mol. Bio 2005, 351, 89–99. [DOI] [PubMed] [Google Scholar]
- Hall M. A.; Shundrovsky A.; Bai L.; Fulbright R. M.; Lis J. T.; Wang M. D. High-Resolution Dynamic Mapping of Histone-DNA Interactions in a Nucleosome. Nat. Struct. Mol. Biol. 2009, 16, 124–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuba S. H.; Karymov M. A.; Liu Y.; Lindsay S. M.; Zlatanova J. Mechanically Stretching Single Chromatin Fibers. Gene Ther. Mol. Biol. 1999, 4, 209–301. [Google Scholar]
- Wang H.; Bash R.; Yodh J. G.; Hager G.; Lindsay S. M.; Lohr D. Using Atomic Force Microscopy to Study Nucleosome Remodeling on Individual Nucleosomal Arrays in Situ. Biophys. J. 2004, 87, 1964–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy J. S.; Wei S.; Lee J. Y.; Tan S.; Chu S.; Lee T.-H. DNA Methylation Increases Nucleosome Compaction and Rigidity. J. Am. Chem. Soc. 2010, 132, 1782–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. Y.; Lee T.-H. Effects of DNA Methylation on the Structure of Nucleosomes. J. Am. Chem. Soc. 2011, 134, 173–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Useche I.; Ke J.; Tian Y.; Shim D.; Howell S. C.; Qiu X.; Yuan C. DNA Methylation Regulated Nucleosome Dynamics. Sci. Rep. 2013, 3, 2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Useche I.; Yuan C. The Effect of DNA Cpg Methylation on the Dynamic Conformation of a Nucleosome. Biophys. J. 2012, 103, 2502–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihardja S.; Spakowitz A. J.; Zhang Y.; Bustamante C. Effect of Force on Mononucleosomal Dynamics. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 15871–15876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrovolskaia I. V.; Arya G. Dynamics of Forced Nucleosome Unraveling and Role of Nonuniform Histone-DNA Interactions. Biophys. J. 2012, 103, 989–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shundrovsky A.; Smith C. L.; Lis J. T.; Peterson C. L.; Wang M. D. Probing Swi/Snf Remodeling of the Nucleosome by Unzipping Single DNA Molecules. Nat. Struct. Mol. Biol. 2006, 13, 549–554. [DOI] [PubMed] [Google Scholar]
- Bayley H.; Martin C. R. Resistive-Pulse Sensing - from Microbes to Molecules. Chem. Rev. 2000, 100, 2575–2594. [DOI] [PubMed] [Google Scholar]
- Mathé J.; Visram H.; Viasnoff V.; Rabin Y.; Meller A. Nanopore Unzipping of Individual DNA Hairpin Molecules. Biophys. J. 2004, 87, 3205–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakane J.; Wiggin M.; Marziali A. A Nanosensor for Transmembrane Capture and Identification of Single Nucleic Acid Molecules. Biophys. J. 2004, 87, 615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropini C.; Marziali A. Multi-Nanopore Force Spectroscopy for DNA Analysis. Biophys. J. 2007, 92, 1632–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner S.; Bessonov A.; Gerland U.; Simmel F. C. Sequence-Dependent Unfolding Kinetics of DNA Hairpins Studied by Nanopore Force Spectroscopy. J. Phys.: Condens. Matter 2010, 22, 454119. [DOI] [PubMed] [Google Scholar]
- Renner S.; Geltinger S.; Simmel F. C. Nanopore Translocation and Force Spectroscopy Experiments in Microemulsion Droplets. Small 2010, 6, 190–194. [DOI] [PubMed] [Google Scholar]
- Arnaut V.; Langecker M.; Simmel F. C. Nanopore Force Spectroscopy of Aptamer–Ligand Complexes. Biophys. J. 2013, 105, 1199–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornblower B.; Coombs A.; Whitaker R. D.; Kolomeisky A.; Picone S. J.; Meller A.; Akeson M. Single-Molecule Analysis of DNA-Protein Complexes Using Nanopores. Nat. Methods 2007, 1–3. [DOI] [PubMed] [Google Scholar]
- Carlsen A. T.; Zahid O. K.; Ruzicka J. A.; Taylor E. W.; Hall A. R. Selective Detection and Quantification of Modified DNA with Solid-State Nanopores. Nano Lett. 2014, 5488–5492. [DOI] [PubMed] [Google Scholar]
- Raillon C.; Granjon P.; Graf M.; Radenovic A. In Detection of Rnap-DNA Complexes Using Solid State Nanopores; IEEE Engineering in Medicine and Biology Society (EMBC): Piscataway, NJ, 2013; pp 4106–4109. [DOI] [PubMed] [Google Scholar]
- Zhao Q.; Sigalov G.; Dimitrov V.; Dorvel B.; Mirsaidov U.; Sligar S.; Aksimentiev A.; Timp G. Detecting Snps Using a Synthetic Nanopore. Nano Lett. 2007, 7, 1680–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorvel B.; Sigalov G.; Zhao Q.; Comer J.; Dimitrov V.; Mirsaidov U.; Aksimentiev A.; Timp G. Analyzing the Forces Binding a Restriction Endonuclease to DNA Using a Synthetic Nanopore. Nucleic Acids Res. 2009, 37, 4170–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabard-Cossa V.; Wiggin M.; Trivedi D.; Jetha N. N.; Dwyer J. R.; Marziali A. Single-Molecule Bonds Characterized by Solid-State Nanopore Force Spectroscopy. ACS Nano 2009, 3, 3009–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeets R. M. M.; Kowalczyk S. W.; Hall A. R.; Dekker N. H.; Dekker C. Translocation of Reca-Coated Double-Stranded DNA through Solid-State Nanopores. Nano Lett. 2009, 9, 3089–3096. [DOI] [PubMed] [Google Scholar]
- Soni G. V.; Dekker C. Detection of Nucleosomal Substructures Using Solid-State Nanopores. Nano Lett. 2012, 12, 3180–3186. [DOI] [PubMed] [Google Scholar]
- Wendell D.; Jing P.; Geng J.; Subramaniam V.; Lee T. J.; Montemagno C.; Guo P. Translocation of Double-Stranded DNA through Membrane-Adapted Phi29 Motor Protein Nanopores. Nat. Nanotechnol. 2009, 4, 765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschini L.; Soskine M.; Biesemans A.; Maglia G., A Nanopore Machine Promotes the Vectorial Transport of DNA across Membranes, Nat. Commun. 2013, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivankin A.; Carson S.; Kinney S. R. M.; Wanunu M. Fast, Label-Free Force Spectroscopy of Histone-DNA Interactions in Individual Nucleosomes Using Nanopores. J. Am. Chem. Soc. 2013, 135, 15350–15352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk S. W.; Grosberg A. Y.; Rabin Y.; Dekker C. Modeling the Conductance and DNA Blockade of Solid-State Nanopores. Nanotechnology 2011, 22, 315101. [DOI] [PubMed] [Google Scholar]
- Wanunu M.; Dadosh T.; Ray V.; Jin J.; McReynolds L.; Drndic M. Rapid Electronic Detection of Probe-Specific MicroRNAs Using Thin Nanopore Sensors. Nat. Nanotechnol. 2010, 5, 807–814. [DOI] [PubMed] [Google Scholar]
- Lowary P. T.; Widom J. New DNA Sequence Rules for High Affinity Binding to Histone Octamer and Sequence-Directed Nucleosome Positioning. J. Mol. Biol. 1998, 276, 19–42. [DOI] [PubMed] [Google Scholar]
- Dudko O. K.; Hummer G.; Szabo A. Theory, Analysis, and Interpretation of Single-Molecule Force Spectroscopy Experiments. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 15755–15760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans E.; Ritchie K. Dynamic Strength of Molecular Adhesion Bonds. Biophys. J. 1997, 72, 1541–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkel R.; Nassoy P.; Leung A.; Ritchie K.; Evans E. Energy Landscapes of Receptor-Ligand Bonds Explored with Dynamic Force Spectroscopy. Nature 1999, 397, 50–53. [DOI] [PubMed] [Google Scholar]
- Dick D. A. The Distribution of Sodium, Potassium and Chloride in the Nucleus and Cytoplasm of Bufo Bufo Oocytes Measured by Electron Microprobe Analysis. J. Physiol. 1978, 284, 37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thåström A.; Bingham L. M.; Widom J. Nucleosomal Locations of Dominant DNA Sequence Motifs for Histone–DNA Interactions and Nucleosome Positioning. J. Mol. Biol. 2004, 338, 695–709. [DOI] [PubMed] [Google Scholar]
- Ioshikhes I.; Bolshoy A.; Derenshteyn K.; Borodovsky M.; Trifonov E. N. Nucleosome DNA Sequence Pattern Revealed by Multiple Alignment of Experimentally Mapped Sequences. J. Mol. Biol. 1996, 262, 129–139. [DOI] [PubMed] [Google Scholar]
- Sivolob A. V.; Khrapunov S. N. Translational Positioning of Nucleosomes on DNA: The Role of Sequence-Dependent Isotropic DNA Bending Stiffness. J. Mol. Biol. 1995, 247, 918–931. [DOI] [PubMed] [Google Scholar]
- Pérez A.; Castellazzi C. L.; Battistini F.; Collinet K.; Flores O.; Deniz O.; Ruiz M.; Torrents D.; Eritja R.; Soler-López M.; et al. Impact of Methylation on the Physical Properties of DNA. Biophys. J. 2012, 102, 2140–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Dudko O. K. A Transformation for the Mechanical Fingerprints of Complex Biomolecular Interactions. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 16432–16437. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.