

Abstract
Compounds acting via the GPCR neurotensin receptor type 2 (NTS2) display analgesic effects in relevant animal models. Using a pharmacophore model based on known NT receptor nonpeptide compounds, we screened commercial databases to identify compounds that might possess activity at NTS2 receptor sites. Modification of our screening hit to include structural features known to be recognized by NTS1 and NTS2, led to the identification of the novel NTS2 selective nonpeptide, N-{[6-chloro-4-(2,6-dimethoxyphenyl)quinazolin-2-yl]carbonyl}-Lleucine (9). This compound is a potent partial agonist in the FLIPR assay with a profile of activity similar to that of the reference NTS2 analgesic nonpeptide levocabastine (5).
Keywords: Neurotensin, NTS2 receptor, Levocabastine, SR142948a, SR48692, FLIPR assay, pain
The identification of novel analgesics remains a key goal of medicinal chemistry. Despite years of effort, the opioids remain the treatment of choice for severe acute pain even with their deleterious adverse effect profile that includes constipation, respiratory depression as well as development of tolerance and addiction. Also, patients experiencing chronic pain, a persistent pain that can follow from peripheral nerve injury, often fail to find relief with opioids. Although antidepressant and antiepileptic drugs are currently the treatment of choice for this type of pain, it is estimated that more than half of these patients are not treated adequately. Thus, the identification of nonopioid analgesics that are also effective for management of chronic pain would represent a significant advancement of the field.
The tridecapeptide neurotensin (NT, Glu-Leu-Tyr-Glu-Asn-Lys-Pro-Arg-Arg-Pro-Tyr-Ile-Leu), identified forty years ago from bovine hypothalamus, operates via interaction with two G-protein coupled receptors named NTS1 and NTS2 (NTR1, NTR2.) and the multi-ligand type-I transmembrane receptor sortilin (NTS3).1-3 NT acts as both a neuromodulator and neurotransmitter in the CNS and periphery and oversees a host of biological functions including regulation of dopamine pathways, 1hypothermia, hypotension and importantly, nonopioid analgesia 4-6. Although the latter behavior highlighted the potential for NT-based analgesics, the lions’ share of early research efforts were aimed at development of NT-based antipsychotics acting at the NTS1 receptor site. Interestingly, this work failed to produce nonpeptide compounds despite intense discovery efforts. Undeterred, researchers focused on the active fragment of the NT peptide (NT(8-13), 1, Chart 1), to create a host of peptide-based compounds that, to this day, remain at the forefront of NT research.7-14
Chart 1.

Structures of neurotensin reference peptides (1, 2), reference nonpeptides (3-5), and recently described NTS2 selective nonpeptide compounds (6,7) and title compound (9).
Studies with NTS1 and NTS2 have shown that NT, and NT-based compounds, modulate analgesia via both of these receptor subtypes.15,16 These studies also revealed that NT compounds are active against both acute and chronic pain and that there exists a synergy between NT and opioid-mediated analgesia17-20. Together, these findings highlight the NT system as a potential source of novel analgesics that could act alone or in concert with opioid receptor-based drugs.18,21
Many of these compounds produce analgesia along with hypothermia and hypotension, behaviors attributed to signaling via the NTS1 receptor. 22,23 In vivo evidence in support of these findings has been provided using the NTS2-selective peptide NT79 (2) as it was found to be active in models of acute pain but without effect on temperature or blood pressure.12 These results were recently confirmed by the development of the compound ANG2002, a conjugate of NT and the brain-penetrant peptide Angiopep-2, which is effective in reversing pain behaviors induced by the development of neuropathic and bone cancer pain.24 Taken together, the promise of activity against both acute and chronic pain as well as a more balanced ratio of desired versus adverse effect profile directed our discovery efforts towards NTS2-selective analgesics.
The work to identify NT-based antipsychotics was directed at the NTS1 receptor as little was known about the NTS2 receptor at that time. This suggested to us that the failure to find nonpeptide compounds might be a phenomenon peculiar to NTS1 and that this barrier would not exist for NTS2. Three nonpeptide compounds in total were known to bind NTS1 and/or NTS2 and these included two pyrazole analogs SR48692 (3) and SR142948a (4) and levocabastine (5). While compounds 3 and 4 were found to antagonize the analgesic and neuroleptic activities of NT in a variety of animal models, 5 showed selectivity for NTS2 versus NTS1 and analgesic properties in animal models of acute and chronic pain16,25-28 thus demonstrating that nonpeptide NTS2-selective analgesic compounds could be identified.
To find novel nonpeptide compounds, we developed a medium throughput FLIPR assay in a CHO cell line stably expressing rNTS2 based on reports that compound 3 mediated calcium release at the NTS2 receptor in this cell line. We planned to follow up this assay with a binding assay using [125I]NT to confirm interaction with NTS2.29,30 Profiling compounds 3, 4, 5 and NT in our FLIPR assay revealed that 3 and 4 were full agonists whereas levocabastine (5) behaves as a potent partial agonist and NT was an antagonist of the calcium release mediated by 4. This evidence guided our discovery efforts toward identification of compounds displaying either potent partial agonist or antagonist activity in the FLIPR assay.
We reported recently that this effort resulted in the identification of several nonpeptide compounds that met these criteria. The first of these was the NTS2 selective potent partial agonist 6 (NTRC-739) that was obtained from an SAR study starting with the nonselective full agonist 3.30 More recently, we identified an NT-like antagonist 7 starting from the screening of a library of substituted indole compounds based upon the NTS1 partial agonist 8. 31,32 In this article, we report the discovery of N-{[6-chloro-4-(2,6-dimethoxyphenyl)quinazolin-2-yl]carbonyl}-L-leucine (NTRC-808, 9) a novel nonpeptide chemotype that shows potent partial agonist activity in the FLIPR functional assay and is selective for NTS2 versus NTS1. The discovery of this compound was accomplished in a different manner from the two described above as we used a pharmacophore model and database mining strategy to obtain our initial hit compound rather than adjusting the activity of a scaffold previously shown to bind NT receptors. The details of the effort that provided 9 are described below.
The target compounds described herein were synthesized as shown in Scheme 1 using methods similar to those reported by Castellano et al.33 Thus, benzoxazinone 1134 was available starting from 2-amino-5-chlorobenzoic acid (10) by treatment with acetic anhydride at reflux. This was then coupled with lithiated veratrole at 0 °C followed by hydrolysis with HCl at reflux to give intermediate 12.35 Synthesis of the key phenylquinazoline carboxylic acid intermediate 13 was then accomplished by treating 12 first with glyoxylic acid and ammonium acetate, followed by stirring under air in dimethylformamide (DMF) for three days. The target compounds 14b, 9 and 16b were then available by first coupling 13 to the appropriate amino acid ester using O-benzotriazol-1-yl-N,N,N’,N’-tetramethyluronium hexafluorophosphate (HBTU) and triethylamine to give ester intermediates 14a, 15 and 16a. Target compounds 14b and 9 were prepared by treating 14a and 15 with trifluoroacetic acid (TFA) in CH2Cl2 while 16b was available via hydrolysis of 16a using LiOH and dioxane.
Scheme 1.

Synthetic route to target compounds 9, 14b and 16b. Reagents and conditions: (i) Ac2O, Et2O, reflux; (ii) veratrole, n-BuLi, 0°C; HCl, EtOH, H2O; (iii) aqueous glyoxalic acid, NH4OAc; (iv) DMF, RT, air, 3 days; (v) HBTU, Et3N, CH2Cl2, L-cyclohexylglycine tert-butyl ester•HCl; (vi) HBTU, Et3N, CH2Cl2, L-leucine tert-butyl ester•HCl; (vii) HBTU, Et3N, CH2Cl2, 1-aminocyclohexylcarboxylic acid methyl ester•HCl; (viii) TFA, CH2Cl2; (ix) LiOH, dioxane.
Our initial work to identify nonpeptide NTS2 receptor based analgesics began with SAR studies of known NTS1 and NTS2 active nonpeptide chemotypes (such as 3, 5 and 8) as we did not have access to vast chemical libraries that could be screened for starting points. We thus relied on small, targeted libraries of compounds based on the chemotypes defined above that could be screened using our FLIPR assays.
As an alternative method of identifying starting points, we also explored a scaffold-hopping method using molecular modeling and chemical library database mining to obtain novel chemotypes. To accomplish this, we developed a 3D-pharmacophore hypothesis based on commonalties of the training set that included compounds 3, 4, 5 and 6. Conformational libraries of each were developed using an MMFF94 force field and 20 genetic algorithm runs based on independent random number seeds and a radial dielectric screening model of 80. Libraries were then filtered for unique conformations using both a 30-degree torsion angle uniqueness criteria and symmetry. A 3D-pharmacophore was then developed using MOLMOD36,37 employing the conformational libraries, a distance tolerance of 1.5 angstroms and 3-10 kcal energy windows to obtain the distance matrix (Figure 1).
Figure 1.

3D-Pharmacophore derived from compounds 3, 4, 5, and 6.
This consisted of a central set of hydrogen bond acceptors and donors (1, 2 and 3), a terminal hydrophobic tail (4), 2-centroids of aromatic rings (5 and 6) and a carboxyl group (7) near the terminus of the ligands. Because of the training set used, this model necessarily embodies many of the same elements delineated by Quéré in the pharmacophore of NTS1 antagonists.38 We then conducted virtual screening of the Life Chemicals Stock Collection and the Chembridge Express Hit Collection and obtained a set of compounds that were compliant with the 3D-pharmacophore.
We purchased 36 compounds identified in the search described above and screened for agonist and antagonist activity in parallel at a single concentration (10 μM) at NTS1 and NTS2. Only one of the 36 compounds purchased was active, compound 17 in Chart 2. This compound demonstrated very weak antagonist activity (~20% inhibition) at NTS2 but was devoid of activity at NTS1. Attempts to obtain dose response curves for 17 proved problematic due to the combination of low activity and low solubility. However, this endeavor was considered an overall success as the strategy identified a previously unidentified nonpeptide scaffold that could be used to develop novel NTS2 ligands.
Chart 2.
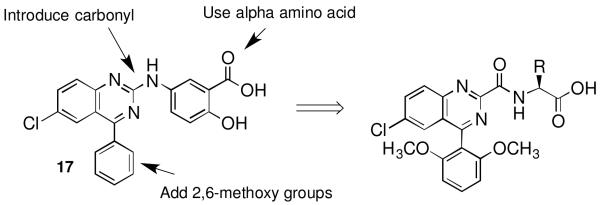
Targeted modifications of latent hit 17 to improve activity at NT receptors
To improve upon this, we devised analogues of 17 that incorporated molecular features previously shown to promote activity at the NT receptors. The highest priority was given to those features that were embodied in the training set but absent in 17. The highlights of this plan are illustrated in Chart 2 and included the addition of a carbonyl group to provide an H-bond acceptor/donor in the central region of future analogues. This had an added benefit for analogue generation as the carboxylate precursor would enable parallel synthesis efforts. In addition to this, we planned to add 2,6-dimethoxy groups to the phenyl ring in 17 as these are known to be important to NT receptor recognition, calcium mobilization and selectivity.30,38,39 Finally, we believed that replacement of the aminobenzoic acid in 17 for alpha amino acids known to be active at NT receptors would improve overall activity and receptor recognition. From previous experience, we chose the amino acids illustrated in Scheme 1 as the first set to be investigated.30,38,39
The data from the FLIPR assay (NTS2) and binding assays (NTS1 and NTS2) for 9, 14b and 16b are provided in Table 1 along with the data obtained for our reference compounds NT, 4 and 5. The target compounds 9, 14b and 16b, as a group, failed to show activity in the calcium mobilization assay at NTS1 (data not shown). Taken together, their failure to compete with [125I]NT at NTS1 supports the notion that their lack of calcium mobilization in the NTS1 FLIPR assay follows directly from a lack of interaction with NTS1.
Table 1.
Data for reference compounds NT, 4, 5, 6 and test compounds 9, 14b and 16b at the rNTSl and rNTS2 receptors
In the NTS2 assay, however, these same compounds revealed a greatly improved activity relative to 17. Compound 9, with the isobutyl side chain, proved to be the most active of the three target compounds tested. Unlike 17, it displayed potent partial agonist activity with an EC50 of 14 nM and Emax of 18% relative to 4, Figure 2. This activity profile is very similar to that previously described for the NTS2 selective compounds levocabastine (5) and 6.30 In the binding assay, its affinity fell between that of 5 and 6, with a Ki of 88 nM.
Figure 2.

Compound 9 shows potent partial agonist activity versus the agonist standard compound 4 in the rNTS2 FLIPR assay.
Target compound 14b, bearing the L-cyclohexylglycine side chain, did not show partial agonist activity in the functional assay at NTS2 but instead antagonized the activity of 4 with an IC50 of 1067 nM. This was an improvement over the screening hit 17 but still 25-fold less active than NT in the functional assay as indicated in Table 1. It was also less active in binding with a Ki 5-fold greater than 9 at 474 nM.
Target compound 16b, with the symmetrically disposed aminocyclohexyl side chain, showed partial agonist activity in the FLIPR assay like 9, but with much lower activity, roughly 10-fold less (EC50 values of 145 and 14 nM respectively) though both shared similar efficacies (Emax of 26 versus 18% respectively). Comparison of the binding assay data revealed that while 16b was selective for NTS2 it was also 11-fold less effective than 9 in competing with NT for binding at NTS2. Overall, the modifications made to compound 17, demonstrated significantly improved activity for NTS2 and provided a novel nonpeptide chemotype selective for the NTS2 receptor.
A comparison of the NTS2 FLIPR data collected for the benzoquinazoline-based compounds 9, 14b and 16b to that obtained from other scaffolds using an identical set of amino acids, provides insight into the SAR of calcium signaling at NTS2. This is readily appreciated by inspection of the data provided in Table 2. Here the data from test compounds 9, 14b and 16b bearing the benzoquinazoline scaffold (A) are compared to that obtained from two different pyrazole-based scaffolds (B and C) using the set of amino acids that included L-leucine, L-cyclohexylglycine and 2-aminocyclohexane carboxylic acid (19-23).30,39 The most readily apparent trend in the data shows that scaffolds A and C are more alike functionally compared with A and B as the former pair exhibited only partial agonist or antagonist behavior whereas B displays near full agonist behavior in two examples (19 and 20). These similarities in data appear to align with the fact that scaffolds A and C are more alike in overall size compared with B.
Table 2.
NTS2 calcium signaling as a function of amino acid and scaffold combination.
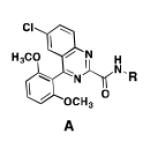
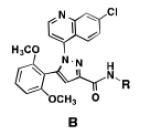
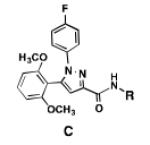
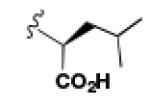
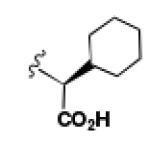
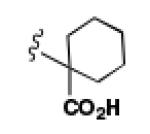
However, the counter trend must also be appreciated. Scaffold A is most active when substituted with the smallest side chain (isobutyl), whereas both of the pyrazole-based scaffolds are most active with the larger, more bulky side chains (compare compounds 9 and 23). This dichotomy in behavior appears to be related to the difference in scaffold structure. However, at this juncture, given the limited data set, we cannot ascertain whether it is the difference in the rigidity of the backbone or the different heterocycles that make up the backbone that is responsible for this phenomenon. Neither can we determine if both conditions must exist. This will only be revealed through testing of additional compounds.
Altogether, this study demonstrates that novel NTS2 nonpeptide scaffolds can be obtained using a virtual screening approach even with a pharmacophore model based on a limited training set. The discovery of compound 9 provides a novel nonpeptide potent partial agonist in the FLIPR assay that is selective for NTS2 versus NTS1 and is significant since compounds known to show antipsychotic and analgesic activity in vivo possess either partial agonist (5) or antagonist (NT) activity in the NTS2 FLIPR assay. The novel scaffold provides another dimension of data from which to draw and will contribute to our understating of the SAR of calcium signaling at NTS2. We are continuing our SAR studies with this scaffold to improve activity as well as building a larger data set that will be useful for comparison with other nonpeptide scaffolds. We are also working to establish an in vitro to in vivo correlation for compound 9 in relevant animal models to determine if it is an analgesic or an antagonist of NTS2 mediated analgesia.
Supplementary Material
Acknowledgements
This research was supported by the National Institute on Drug Abuse, Grant DA029961.We gratefully acknowledge the NIMH Chemical Synthesis and Drug Supply Program for providing us with the samples of SR142948a (4), and SR48692 (3).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Supplementary material including experimental details for compound synthesis and biological testing as well as catalog numbers of the compounds identified for testing in our database mining, can be obtained from the online version at …
References and Notes
- 1.Caraway R, Leeman SE. J Biol Chem. 1973;248:6854. [PubMed] [Google Scholar]
- 2.Kitabgi P. Curr Opin Drug Discov Devel. 2002;5:764. [PubMed] [Google Scholar]
- 3.Sarret P, Beaudet A. In: Handbook of Chemical Neuroanatomy. Bjorkland A, Hokfelt T, Quinton P, editors. Vol. 20. Elsivier; Amsterdam: 2003. p. 323. [Google Scholar]
- 4.Clineschmidt BV, McGuffin JC. Eur J Pharmacol. 1977;46:395. doi: 10.1016/0014-2999(77)90236-9. [DOI] [PubMed] [Google Scholar]
- 5.Nemeroff CB, Osbahr AJ, 3rd, Manberg PJ, Ervin GN, Prange AJ., Jr. Proc Natl Acad Sci. 1979;76:5368. doi: 10.1073/pnas.76.10.5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nemeroff CB, Hernandez DE, Luttinger D, Kalivas PW, Prange AJ., Jr. Ann N Y Acad Sci. 1982;400:330. doi: 10.1111/j.1749-6632.1982.tb31579.x. [DOI] [PubMed] [Google Scholar]
- 7.Folkers K, Chang D, Humphries J, Caraway R, Leeman SE, Bowers CY. Proc Natl Acad Sci. 1976;73:3833. doi: 10.1073/pnas.73.11.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wustrow DJ, Davis MD, Akunne HC, Corbin AE, Wiley JN, Wise LD, Heffner TG. Biorg Med Chem Lett. 1995;5:997. [Google Scholar]
- 9.Briody S, Boules M, Oliveros A, Fauq I, Richelson E. Behav Brain Res. 207:118. doi: 10.1016/j.bbr.2009.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akunne HC, Demattos SB, Whetzel SZ, Wustrow DJ, Davis DM, Wise LD, Cody WL, Pugsley TA, Heffner TG. Biochem Pharmacol. 1995;49:1147. doi: 10.1016/0006-2952(95)98512-8. [DOI] [PubMed] [Google Scholar]
- 11.Hughes FM, Shaner BE, May LA, Zotian L, Brower JO, Woods RJ, Cash M, Morrow D, Massa F, Mazella J, Dix TA. J Med Chem. 2010;53:4623. doi: 10.1021/jm100092s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boules M, Liang Y, Briody S, Miura T, Fauq I, Oliveros A, Wilson M, Khaniyev S, Williams K, Li Z, Qi Y, Katovich M, Richelson E. Brain Res. 2010;1308:35. doi: 10.1016/j.brainres.2009.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Held C, Plomer M, Hubner H, Meltretter J, Pischetsrieder M, Gmeiner P. ChemMedChem. 2013;8:75. doi: 10.1002/cmdc.201200376. [DOI] [PubMed] [Google Scholar]
- 14.Bredeloux P, Cavelier F, Dubuc I, Vivet B, Costentin J, Martinez J. J Med Chem. 2008;51:1610. doi: 10.1021/jm700925k. [DOI] [PubMed] [Google Scholar]
- 15.Smith KE, Boules M, Williams K, Richelson E. Behav Brain Res. 2012;232:93. doi: 10.1016/j.bbr.2012.03.044. [DOI] [PubMed] [Google Scholar]
- 16.Sarret P, Esdaile MJ, Perron A, Martinez J, Stroh T, Beaudet A. J Neurosci. 2005;25:8188. doi: 10.1523/JNEUROSCI.0810-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roussy G, Dansereau MA, Baudisson S, Ezzoubaa F, Belleville K, Beaudet N, Martinez J, Richelson E, Sarret P. Mol Pain. 2009;5:38. doi: 10.1186/1744-8069-5-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tetreault P, Beaudet N, Perron A, Belleville K, Rene A, Cavelier F, Martinez J, Stroh T, Jacobi AM, Rose SD, Behlke MA, Sarret P. FASEB J. 2013;27 doi: 10.1096/fj.12-225540. [DOI] [PubMed] [Google Scholar]
- 19.Boules M, Shaw A, Liang Y, Barbut D, Richelson E. Brain Res. 2009;1294:22. doi: 10.1016/j.brainres.2009.07.086. [DOI] [PubMed] [Google Scholar]
- 20.Boules M, Johnston H, Tozy J, Smith K, Li Z, Richelson E. Behav Pharmacol. 2011;22:573. doi: 10.1097/FBP.0b013e3283474a3a. [DOI] [PubMed] [Google Scholar]
- 21.Dobner PR. Peptides. 2006;27:2405. doi: 10.1016/j.peptides.2006.04.025. [DOI] [PubMed] [Google Scholar]
- 22.Tyler-McMahon BM, Stewart JA, Farinas F, McCormick DJ, Richelson E. Eur J Pharmacol. 2000;390:107. doi: 10.1016/s0014-2999(99)00877-8. [DOI] [PubMed] [Google Scholar]
- 23.Fantegrossi WE, Ko MC, Woods JH, Richelson E. Pharmacol Biochem Behav. 2005;80:341. doi: 10.1016/j.pbb.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 24.Demeule M, Beaudet N, Regina A, Besserer-Offroy E, Murza A, Tetreault P, Belleville K, Che C, Larocque A, Thiot C, Beliveau R, Longpre JM, Marsault E, Leduc R, Lachowicz JE, Gonias SL, Castaigne JP, Sarret P. J Clin Invest. 2014;124:1199. doi: 10.1172/JCI70647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gully D, Canton M, Boigegrain R, Jeanjean F, Molimard JC, Poncelet M, Gueudet C, Heaulme M, Leyris R, Brouard A, et al. Proc Natl Acad Sci. 1993;90:65. doi: 10.1073/pnas.90.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gully D, Labeeuw B, Boigegrain R, Oury-Donat F, Bachy A, Poncelet M, Steinberg R, Suaud-Chagny MF, Santucci V, Vita N, Pecceu F, Labbe-Jullie C, Kitabgi P, Soubrie P, Le Fur G, Maffrand JP. J Pharmacol Exp Ther. 1997;280:802. [PubMed] [Google Scholar]
- 27.Awouters F, Niemegeers CJ, Jansen T, Megens AA, Janssen PA. Agents and actions. 1992;35:12. doi: 10.1007/BF01990945. [DOI] [PubMed] [Google Scholar]
- 28.Tasaka K, Kamei C, Tsujimoto S, Yoshida T, Aoki I. Arzneimittel-Forschung. 1990;40:1295. [PubMed] [Google Scholar]
- 29.Gendron L, Perron A, Payet MD, Gallo-Payet N, Sarret P, Beaudet A. Mol Pharmacol. 2004;66:1421. doi: 10.1124/mol.104.002303. [DOI] [PubMed] [Google Scholar]
- 30.Thomas JB, Giddings AM, Wiethe RW, Olepu S, Warner KR, Sarret P, Gendron L, Longpre JM, Zhang Y, Runyon SP, Gilmour BP. J Med Chem. 2014;57:5318. doi: 10.1021/jm5003843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomas JB, Giddings AM, Wiethe RW, Olepu S, Warner KR, Sarret P, Gendron L, Longpre JM, Zhang Y, Runyon SP, Gilmour BP. J Med Chem. 2014;57:7472. doi: 10.1021/jm500857r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fan Y, Lai MH, Sullivan K, Popiolek M, Andree TH, Dollings P, Pausch MH. Biorg Med Chem Lett. 2008;18:5789. doi: 10.1016/j.bmcl.2008.09.075. [DOI] [PubMed] [Google Scholar]
- 33.Castellano S, Taliani S, Milite C, Pugliesi I, Da Pozzo E, Rizzetto E, Bendinelli S, Costa B, Cosconati S, Greco G, Novellino E, Sbardella G, Stefancich G, Martini C, Da Settimo F. J Med Chem. 2012;55:4506. doi: 10.1021/jm201703k. [DOI] [PubMed] [Google Scholar]
- 34.Miki T, Kori M, Mabuchi H, Banno H, Tozawa R, Nakamura M, Itokawa S, Sugiyama Y, Yukimasa H. Bioorg Med Chem. 2002;10:401. doi: 10.1016/s0968-0896(01)00290-5. [DOI] [PubMed] [Google Scholar]
- 35.Tomisek A, Christensen BE. J Am Chem Soc. 1948;70:1701. doi: 10.1021/ja01185a008. [DOI] [PubMed] [Google Scholar]
- 36.Harris D, Clayton T, Cook J, Sahbaie P, Halliwell RF, Furtmüller R, Huck S, Sieghart W, DeLorey TM. J Med Chem. 2008;51:3788. doi: 10.1021/jm701433b. [DOI] [PubMed] [Google Scholar]
- 37.Harris DL, Delorey TM, He X, Cook JM, Loew GH. Eur J Pharmacol. 2000;401:271. doi: 10.1016/s0014-2999(00)00462-3. [DOI] [PubMed] [Google Scholar]
- 38.Quéré L, Boigegrain R, Jeanjean F, Gully D, Evrard G, Durant F. J Chem Soc, Perkin Trans. 1996;2:2639. [Google Scholar]
- 39.Thomas JB, Navarro H, Warner KR, Gilmour B. Biorg Med Chem Lett. 2009;19:1438. doi: 10.1016/j.bmcl.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.