

Abstract
In the United States, few Food and Drug Administration (FDA)–approved options exist for the treatment of focal cartilage and osteochondral lesions. Developers of products for cartilage repair face many challenges to obtain marketing approval from the FDA. The objective of this review is to discuss the necessary steps for FDA application and approval for a new cartilage repair product. FDA Guidance Documents, FDA Panel Meetings, scientific organization recommendations, and clinicaltrials.gov were reviewed to demonstrate the current thinking of FDA and the scientific community on the regulatory process for cartilage repair therapies. Cartilage repair therapies can receive market approval from FDA as medical devices, drugs, or biologics, and the specific classification of product can affect the nonclinical, clinical, and regulatory strategy to bring the product to market. Recent FDA guidance gives an outline of the required elements to bring a cartilage repair product to market, although these standards are often very general. As a result, companies have to carefully craft their study patient population, comparator group, and clinical endpoint to best showcase their product’s attributes. In addition, regulatory strategy and manufacturing process validation need to be considered early in the clinical study process to allow for timely product approval following the completion of clinical study. Although the path to regulatory approval for a cartilage repair therapy is challenging and time-consuming, proper clinical trial planning and attention to the details can eventually save companies time and money by bringing a product to the market in the most expeditious process possible.
Keywords: articular cartilage, regulatory affairs, clinical trials, FDA
Introduction
Articular cartilage damage can occur through a variety of traumatic and degenerative diseases and can lead to pain and lack of mobility of the knee. There are 2 distinct categories of cartilage damage: focal lesions and degenerative lesions. Focal lesions are well-defined defects, often caused by trauma, osteochondritis dissecans, or osteonecrosis. Degenerative lesions are typically poorly demarcated and usually caused as a result of ligament instability, meniscal injuries, malalignment, or osteoarthritis.1 For centuries, the limited capacity for articular cartilage repair has been noted,2 because of the cartilage tissue’s low quantity of cells and avascular structure, leading to a lack of a typical wound healing response when injured. As such, once cartilage is damaged, there is little intrinsic ability for it to heal, thus resulting in further degeneration and pain.
Although many Food and Drug Administration (FDA)–approved therapies treat symptomatic effects of cartilage damage, such as COX inhibitors and hyaluronic acid injections, there has been no evidence of structural improvement with these conservative treatment modalities.3 For more aggressive late-stage cartilage damage and osteoarthritis, total knee arthroplasty is indicated. However, for earlier stage degeneration or focal cartilage damage, several surgical techniques are used to elucidate a repair response or replace damaged cartilage tissue. Microfracture and subchondral drilling are commonly used to stimulate a repair response in the joint but often lead to the formation of fibrocartilage instead of smooth hyaline cartilage. In addition, the positive effects of these treatments may be short-lived and effective in only younger patients.4 Autologous mosaicplasty and allograft treatments are also used for cartilage repair, but mosaicplasty can result in donor site morbidity and viable allograft tissue may be difficult to obtain because of lack of substantial quantity of donor tissue.5
The process of autologous chondrocyte implantation has been developed to repair focal cartilage defects, using the patient’s own cells expanded in a commercial laboratory setting and re-implanted in the patient. The only product commercially available in the United States for autologous chondrocyte implantation is Genzyme’s Carticel®, having its FDA approval process documented in the literature.6 Carticel initially obtained approval as an unregulated cell therapy in 1995. Following subsequent guidance from the FDA,7 Genzyme obtained an accelerated approval for Carticel in 1997, submitting clinical data from Europe, preregulation US registry data, and committed to a postmarketing approval study. Current cartilage repair technologies face a more stringent review process in their path to market, as the FDA has developed a greater body of knowledge on cartilage repair therapies since the approval of Carticel. The objective of this review is to discuss the various regulatory pathways and nonclinical and clinical data required for bringing a new cartilage repair product to market in the United States.
Methods
A review of FDA materials was performed to determine the current thinking of the FDA on the regulatory pathways for cartilage repair products. This review encompassed regulatory statutes, FDA guidance documents, and transcripts from FDA Advisory Committee meetings. Additional recommendations on nonclinical and clinical studies were obtained from publications by the International Cartilage Repair Society (ICRS). In addition, a search on clinicaltrials.gov with the search term “articular cartilage” was performed in March 2012 to demonstrate the current state of clinical trials for cartilage repair therapies.
Results
Regulatory Pathways for Cartilage Repair Products
The FDA regulatory pathway required for a cartilage repair product can significantly affect the timing and strategy required to bring the product to market, specifically because of the quality and quantity of nonclinical and clinical data required for FDA approval. Cartilage repair therapies can receive market approval from the FDA as medical devices, drugs, or biologics. An overview of the centers of the FDA responsible for different types of products is located in Table 1. In some circumstances, the cartilage repair technology is composed of multiple components that require FDA review, such as a device containing a biological agent, or a construct composed of cells and a scaffold. The FDA Office of Combination Products determines which FDA Center has jurisdiction over a particular technology seeking review based on the primary mechanism of action for the technology. It is possible for a technology to require approval through more than one Center as a combination product, with the Office of Combination Products assigning primary review oversight to one center. An overview of the classification of these types of products is located in Table 2.
Table 1.
Food and Drug Administration Approval Pathways for Cartilage Repair Products
| Classification | Center responsible for review | Clinical study name | Submission required for approval |
|---|---|---|---|
| Drug | Center for Drug Evaluation and Research (CDER) | Investigational New Drug (IND) | New Drug Application (NDA) |
| Device | Center for Devices and Radiological Health (CDRH) | Investigational Device Exemption (IDE) | Pre-Market Application (PMA) |
| Biologic | Center for Biologics Evaluation and Research (CBER) | IND | Biologics Licensing Application (BLA) |
Table 2.
Food and Drug Administration Classification of Cartilage Repair Products
| Classification | Primary mechanism of action | Examples |
|---|---|---|
| Drug | Compound metabolically induces repair | Injectable compounds to stimulate growth or prevent tissue loss |
| Device | Provides structural support and/or matrix for intrinsic tissue growth, not dependent on being metabolized for primary intended purpose | Collagenous matrices, biphasic synthetic osteochondral implants, hyaluronic acid injection |
| Biologic | Cell-based product or therapeutic protein | Autologous or allogeneic cellular products, recombinant proteins |
To demonstrate the types of clinical studies in cartilage repair currently underway, a March 2012 search on clinicaltrials.gov with keywords “articular cartilage” was performed that uncovered 179 clinical studies. Eighty studies were removed as dietary supplements or not related to regulated cartilage repair products (nonproduct procedures such as microfracture or meniscectomy), and 15 studies were removed as they were treatments for systemic inflammatory diseases, such as rheumatoid arthritis, rather than localized joint repair. Figure 1 shows a breakdown of the remaining 84 studies based on technology type, ranging from autologous or allogeneic cellular products, devices, and injectable drugs. Each of these product classifications has a different regulatory pathway and presents different challenges to obtain FDA approval for product sales and marketing.
Figure 1.
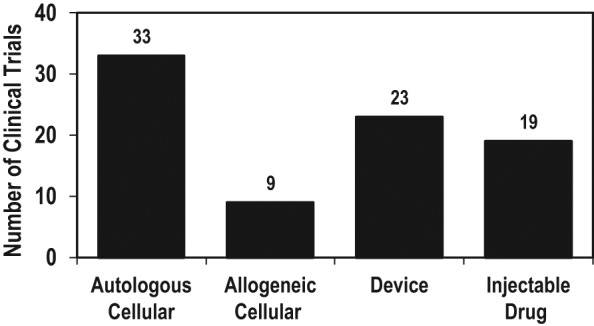
Articular cartilage clinical trials, reported on clinicaltrials.gov, March 2012.
Autologous cellular products
Autologous cellular products use a patient’s own cells to repopulate and repair defects in the joint space and usually involve an initial procedure to remove tissue or blood. The patient’s cartilage or mesenchymal stem cells are proliferated in a laboratory setting before implantation surgery. Some therapies create fully formed cartilage constructs through months of ex vivo cell and tissue culture. Autologous cellular products constitute the largest percentage of clinical studies for cartilage repair, as Genzyme has paved the way with this class of technology with its Carticel product.
Autologous cellular products are regulated as biologics or combination products, and the Center for Biologics Evaluation and Research (CBER) is designated as the lead center for FDA review of these products.8 Approval of these biologics requires a phased approach with dosing (phase I), smaller scale safety/probable effectiveness (phase II), and larger scale randomized safety and effectiveness studies (phase III). Each phase requires Investigational New Drug (IND) approval for commencement of these studies in the United States. Following the completion of phase III clinical trials, a Biologics Licensing Application (BLA) is submitted to CBER for review to determine marketing approval for the product.
Allogeneic cellular products
Although not as commonplace as the use of autologous cells, a number of allogeneic cellular products are currently under clinical study. These products use donor cells or tissue from human donors to repair damaged cartilage and range from morselized tissue to transgenic cells engineered to secrete growth factors. Allogeneic cell sources limit the need for initial tissue biopsy, as is required with autologous therapies, but may cause an immune response. Allogeneic cellular products are usually regulated as biologics or combination products and, as such, require approval under a BLA in the same process as an autologous cellular product.
However, certain allogeneic cellular and tissue products fall beyond the scope of marketing application through the FDA. Cells, tissues, and tissue-based products that are minimally manipulated, used in a homologous fashion, have no systemic effects, and are not used in combination with other products are considered donor tissue and do not require a marketing application with clinical safety and efficacy data.9 For example, morselized cartilage tissue is considered “minimally manipulated” and is regulated as graft tissue and not a biologic.
Injectable drugs
Drugs currently under development for the repair of cartilage are compounds developed to stimulate a repair response by a patient’s own joint tissue. Injectable compounds for cartilage repair are classified as drugs and regulated by the Center for Drug Evaluation and Research (CDER).8 One exception is the case of viscosupplements, such as hyaluronic acid, that are not intended for cartilage repair but for viscosupplementation and symptom management; these products are regulated as devices through the Center for Devices and Radiological Health (CDRH). Approval of drugs requires a phased approach with separate dosing (phase I), smaller scale safety/probable effectiveness (phase II), and larger scale randomized safety and effectiveness studies (phase III). Each phase requires IND approval for commencement of these studies in the United States. Following the completion of phase III clinical trials, a NDA is submitted to CDER for review to determine product approval.
Devices
Devices for cartilage repair have been developed to fill cartilage defects and/or create a basis for self-repair, ranging from injectable substances that fill defects to scaffolds that support cartilage repair, with potential other procedures such as microfracture involved to create a cellular response for repair. As no class II devices are available for the treatment of articular cartilage defects, the 510(k) process is not applicable for these technologies. The general classification for devices intended to repair cartilage defects is class III (product code NCO), and class III devices must be approved through the Pre-Market Application (PMA) process.
The PMA process requires demonstration of safety and effectiveness through clinical studies, usually with a comparison to a previously approved product or standard of care. The FDA will request that these clinical studies be performed in the United States under an Investigational Device Exemption (IDE), likely as a prospective randomized clinical trial with predefined endpoints. Following the completion of a pivotal IDE trial, a PMA is submitted to CDRH for review to determine product approval.
Nonclinical Data and Testing
Manufacturers of cartilage repair therapies should provide the FDA with nonclinical data sufficient to establish a scientific rationale for clinical investigation of a product and demonstrate an acceptable safety profile of the product prior to initiating a human clinical study (21 CFR 312.23(a)(8)). In vitro testing of cartilage repair therapies is useful in demonstrating proof of concept in a laboratory environment prior to initiation of animal studies. Mechanical testing may also be appropriate for repair products that carry a mechanical load and/or experience mechanical loading, specifically compression or compression shear testing. Animal testing should be performed to determine the biological response to the product (proof of concept), durability of the response, toxicology, and dose response. Certain animal testing should be performed in an animal model sufficiently large enough to determine proof of concept, such as goats, sheep, or horses, although there is no perfect animal model of articular cartilage injury.10,11 Recent recommendations from the ICRS support the use of in vitro, small animal, and large animal studies, and provide specific recommendations on the design and execution of nonclinical testing to support proof of concept and give the necessary information to initiate clinical study.12 Ongoing non-clinical testing is advised to address any safety or effectiveness questions or issues that arise over the course of clinical study, and FDA may request additional testing be performed prior to initiation of clinical study, during clinical study, and prior to regulatory approval.
Clinical Trials, Patient Population, and Clinical Endpoints
The Food and Drug Administration has recently published a guidance document on the IND/IDE studies for cartilage repair therapies13 that provides a general outline for the design of IND and IDE studies for cartilage repair. However, due to the variety of potential cartilage repair or replacement technologies, a great amount of flexibility is possible in the design of a clinical trial. In addition, ICRS has issued recommendations for clinical studies of cartilage repair,14 although the recommendations are intended to cover a broad range of cartilage repair technologies and are somewhat general. Since a clear-cut standard is not present for these studies, sponsor companies should carefully craft their study patient population, comparator group, and clinical endpoint to best showcase their product’s attributes.
Patient population
A variety of different indications can be treated utilizing cartilage repair therapies, with treatments based on the severity of symptoms and cartilage damage and ranging from small focal defects in cartilage to osteochondral defects to damage along a large portion of the joint’s surface. Study sponsors should determine a patient population appropriate for their treatment and develop inclusion and exclusion criteria to encompass this population. In specifying the patient population, it is important to include what concomitant diseases and disorders are allowable for clinical trial inclusion and to incorporate that information in the inclusion and exclusion criteria. The ultimate goal in crafting inclusion and exclusion criteria for the clinical study is to create a homogeneous patient cohort that best represents the indications for use of the product. However, inclusion and exclusion criteria should not be so limited that patient enrollment would be hindered by heightened specificity of clinical trial subjects, and subsequently lead to extremely limited indications for use in the labeling of the approved product.
Choice of comparator group
Comparator groups possible for cartilage repair therapies include placebo, sham, standard of care, and active control groups. In general, FDA requires statistical superiority in primary endpoint for the treatment over placebo, sham, and standard of care controls. However, active comparator groups are commonly used for cartilage repair studies, since both blinding and ethical issues are present in using a placebo, sham, or standard of care control group. FDA Guidance notes that potential comparator groups include microfracture, debridement, osteochondral autograft transplantation (mosiacplasty), autologous chondrocyte implantation (ACI), autogenous perichondral or periosteal grafts, and osteochondral allografts.13 The comparator group chosen for the study should be reflective of the standard of care associated with treating the proposed indication for the investigational treatment. Microfracture is commonly used as the comparator group, as it is the standard of care for treatment of most focal chondral lesions. However, a FDA Advisory Panel has recommended that, when comparing to microfracture, investigational therapies should demonstrate superiority, since no consistent treatment effect size has been determined for microfracture, and microfracture outcomes are highly dependent on the age of the patient.15 ACI can also be used as a control group, as cited in the FDA Guidance, specifically for larger defects where microfracture may not be indicated. However, the cost of ACI and the use of a competitor’s product may dissuade study sponsors from pursuing this option for a control group.
Clinical endpoints
The primary endpoint for cartilage repair therapies should include pain and/or function measurements using well-defined scales. Specifically, the measurements should a clinically significant improvement in the patient’s pain and function. An FDA Advisory Panel determined that both pain and function measurements should be included in the primary endpoint for cartilage repair therapies.15 A composite endpoint can be fashioned that includes pain, function, and lack of retreatment to create a robust endpoint. However, including too many individual measurements in a primary endpoint can be detrimental to the overall success rates of both treatment and control groups.
For physical function, validated measurements for inclusion in the primary endpoint are:
Knee Injury and Osteoarthritis Outcome Score (KOOS)
IKDC Subjective Knee Evaluation Form
Cincinnati Knee Rating System
Symptom Rating Form
Western Ontario and McMaster University Osteoarthritis Index (WOMAC)
Knee Society Score (KSS)
The specific functional measurement used in the primary endpoint depends on the repair procedure and the patient population studied. For example, the WOMAC was developed to assess function following total knee arthroplasty and validated for a patient population with advanced stages of osteoarthritis, whereas the KOOS was developed as an extension of the WOMAC to include more sports and activity-related outcomes, creating a score more sensitive to smaller improvements from early-stage treatments.16 Recent recommendations by ICRS detail the attributes of these rating systems and provide detailed assessments of the positive and negative aspects of each of these scoring systems for the study of cartilage repair.17
Pain is commonly measured via a visual analog scale, a validated measurement for severity of pain. However, visual analog scale only measures the severity of pain, and frequency of pain may be another important measurement for the success of a cartilage repair therapy. Other options are to use the WOMAC or KOOS pain sections to quantify the effects of treatment on patient pain.
Additional clinical measurements should be considered for a clinical trial, as general health data, patient economic measurements, and reimbursement outcomes can be useful in the postapproval marketing of the cartilage repair therapy. These additional measurements can also be useful in obtaining positive insurance coverage for the procedure, as the Center for Medicare and Medicaid Services and private payors require different data for positive coverage decisions than the FDA requires for marketing approval.
Structural measurements of the quality of cartilage repair following treatment, such as histology or magnetic resonance imaging (MRI), can give valuable insight to the success or failure of a device by giving a site-specific visualization of the repair process and demonstrating the mechanism of action for the treatments. However, significant challenges in quantification of data and ethical concerns limit the utility of these measurements in a primary outcome measurement, and, if used, should be used as secondary confirmatory endpoints. Histological evaluation of biopsied repair tissue gives a clear view of the repair process, but its destructive nature would require both an invasive action to take a biopsy, and creation of a secondary defect in a tissue environment where healing is difficult. The ICRS has issued recommendations related to the usage of histology in nonclinical and clinical studies of cartilage repair,18 although additional arthroscopic intervention during clinical study outside of typical standard of care may cause difficulties in patient enrollment and follow up, not to mention institutional review board concerns over patient safety.
Measuring structural changes during cartilage repair via noninvasive methods can potentially demonstrate the quality of repair tissue. The use of MRI to determine the quality of cartilage repair is widely reported in the literature; however, significant challenges are present in extending these imaging technologies to numerous clinical sites across the country rather than in one specific university-caliber location. Specifically, the quality of MRI equipment is variable from hospital to hospital and provides significant challenges in standardization of measurement procedures. A 2009 FDA Advisory Panel has noted that MRI measurements are suggested as secondary endpoints until the methods for use can be properly developed, validated, and be available for implementation at various clinical study sites.15
For drugs intended to delay cartilage degradation from osteoarthritis, FDA has approved IND studies with structural measurements by x-ray or MRI. These measurements are meant to show a delay in the structural progression of osteoarthritis in the joint19 and typically use radiographic joint space narrowing (via x-ray) or cartilage thickness or volume loss (via MRI) to demonstrate the product efficacy.20 Although these measurements provide structural information, the lack of sensitivity of these measurements (and the overall slow rate of cartilage degradation in osteoarthritis) has led to no disease-modifying efficacy demonstrated for any drugs tested to date.20
Clinical Study Considerations
While recent FDA guidance has focused on the requirements of an IND/IDE submission for a cartilage repair therapy, additional factors should be considered prior to the initiation of a clinical study. Sponsor companies should choose clinical investigators and sites carefully to allow quality enrollment, data collection, and patient follow-up compliance. The pace of patient enrollment can be affected by a number of factors, specifically the prevalence of the disease, ability of sites and investigators to draw a significant number of patients, and the availability of treatment alternatives to the control treatment. Significant patient enrollment may be difficult in a randomized study if the control treatment is unappealing to the patient compared with other options, such as nonsurgical controls or treatments with limited efficacy. Obtaining proper patient follow-up during the course of a clinical study is of great importance, as missing patient data points often require additional statistical consideration, and follow-up of less than 85% is looked at with concern by FDA reviewers who will consider those patients lost to follow-up as potential treatment failures.
Manufacturing
The manufacturing of a cartilage repair product can provide unique challenges and may adversely affect the regulatory approval process. Whereas FDA manufacturing requirements for drugs and devices are well established, cellular therapies have manufacturing processes that are constantly in development. As such, the approval process for the manufacturing of a cellular therapy may be just as challenging as the clinical study of these products. FDA has issued guidance on manufacturing techniques for human somatic cellular production with specific details on the process requirements for these types of products.21 Manufacturers are advised to work in coordination with the FDA early in the development process to craft the proper processes and controls in the clinical study phase that will ultimately lead to FDA approval.
For all cellular and biologic products, FDA requires potency testing as part of the release criteria.22 This assay can be biological (i.e., evaluating the product’s active ingredients within a living biological system) or nonbiological (biochemical, immunochemical, etc). For cellular products intended for cartilage repair, potency tests may include cell viability, the ability for cells to divide, cellular phenotyping, and/or the ability for cells to produce extracellular matrix (proteoglycan and/or collagen), depending on the exact application of the technology. The FDA will work with the cellular product manufacturer to determine the proper potency test for the product.
Regulatory Submissions
The regulatory submission process, whether in the form of an IND (drug), PMA (device), or BLA (biologic), requires the submission of information to prove safety and effectiveness of the product for the proposed use, and if the benefits of the product outweigh the risks. The preclinical data should be expanded upon as needed through the course of the clinical study and be submitted during annual reports to the FDA to clear up any preclinical issues the FDA may require. Clinical data (from phase III or pivotal studies) satisfying the primary endpoint and a thorough analysis of the clinical data, stratified by demographic data and comorbidities to confirm poolability will be required. Safety data and adverse events need to be reported to assist the FDA with making the proper risk–benefit determination.
Proper planning, analysis, and strategy are required in the clinical and regulatory submission phase to smoothly guide a product to approval. Specifically, having intimate knowledge of the study structure and inherent biases is necessary to plan analyses to minimize any and all flaws in the study structure. In addition, a thorough knowledge of the clinical dataset developed through rigorous data analysis is necessary to anticipate FDA concerns and address them as clearly as possible, from both a high level summary of the entire data set to subgroup analyses based on patient demographics and risk factors, to individual patient-level data. Furthermore, development of a rigorous statistical analysis plan and justification for that plan can provide the analytical framework to demonstrate a cartilage repair product’s safety and effectiveness.
Initial FDA review times vary widely based on the type of submission. For example, a PMA requires an initial decision within 180 days, whereas an NDA or a BLA requires an initial review time of 5 to 8 months. Several cycles of FDA review and deficiencies are likely, as is the need to present the product in front of an FDA Advisory Committee to help advise the FDA on the safety, effectiveness, and risk/benefit ratio of the product. The FDA will consider the views of the Advisory Committee in making a decision whether or not the product is approvable; however, the FDA makes the ultimate decision on the approvability of the product.
Discussion
The regulatory pathways for products intended for articular cartilage repair are often long and burdensome on study sponsors to obtain FDA approval, in both time and cost of clinical trials. In the United States, the only pathways to significantly shorten the path to market include the reclassification of cartilage repair devices from class III to class II or a change in medical device and biologics legislation. Because there are so many different technologies that use different mechanisms of actions, different materials, have different risks, and are indicated for different populations, the FDA would have a difficult task to develop special controls that would encompass all cartilage replacement technologies. In addition, as a paucity of products have obtained FDA approval to repair or regenerate articular cartilage to date, the FDA has little information from device clinical trials to downclassify these types of medical devices to class II. Even if the FDA were to downclassify cartilage repair devices to class II, the FDA would most likely still require clinical data for these technologies prior to obtaining premarket clearance, although likely a more limited dataset than is currently necessary for PMA approval.
Changing the regulatory pathway for cartilage repair products via legislation is also difficult. Many members of industry have lobbied for a European-style pathway for medical devices, demonstrating the efficacy of products in basic science and proof of principle animal studies with only a limited clinical proof of concept. However, recent news media and FDA attention to the safety issues with metal-on-metal hips23 and other products have provided pressure on legislators and the FDA to make the premarket approval process even more stringent for pre-market clearance or approval based on safety concerns. Industry and the scientific community will need to actively press for legislation to allow for a less burdensome approach to bring cartilage repair therapies to market, as well as work with the FDA early in the development process to allow the least burdensome approach to bring the product to market, while still addressing concerns about safety and effectiveness.
While the FDA strives for transparency in its expectations and the review process, details of clinical studies are only selectively revealed on clinicaltrials.gov, and results are only presented in public via FDA Advisory Committee meetings, industry sponsor publication, and professional labeling once approval is obtained. Although much of the lack of transparency is necessary to maintain confidentiality of details of a sponsor’s technology while undergoing review, this also makes the public and competitor companies unaware of the current thinking of the FDA on various products, particularly those that do not complete a clinical study or submit a marketing application to the FDA. Even though a recent US law requires all clinical trials for use in marketing applications to be listed on clinicaltrials.gov,24 industry can not only gain a greater degree of transparency about ongoing clinical trials but also see that there is no truly consistent clinical study design because of the differences in indications or technology mechanism of action.
Working with the FDA at early stages in development through presubmission pathways25 will allow industry to gain valuable insight during the initial development process and take necessary steps to address FDA’s concerns early in the process. Developing a rapport with FDA reviewers through the clinical study approval process and during the clinical study is necessary to inform the FDA about the product and allow the FDA to obtain a degree of comfort with the product prior to submission of a marketing application.
The path to regulatory approval for a cartilage repair therapy is challenging and time-consuming, involving often multiple phases of clinical study and data analysis. However, with the proper planning and attention to the details associated with the clinical trial, the process can be made easier, eventually saving the company time and money by bringing a product to the market in the most expeditious process possible.
Footnotes
Acknowledgments and Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval: The authors declare that no animals or human specimens were used in this study.
References
- 1. Falah M, Nierenberg G, Soudry M, Hayden M, Volpin G. Treatment of articular cartilage lesions of the knee. Int Orthop. 2010;34(5):621-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hunter W. Of the structure and disease of articulating cartilages: 1745. Clin Orthop Relat Res. 1995;317:3-6. [PubMed] [Google Scholar]
- 3. Browne JE, Branch TP. Surgical alternatives for treatment of articular cartilage lesions. J Am Acad Orthop Surg. 2000; 8(3):180-9. [DOI] [PubMed] [Google Scholar]
- 4. Shapiro F, Koide S, Glimcher MJ. Cell origin and differentiation in the repair of full-thickness defects of articular cartilage. J Bone Joint Surg Am. 1993;75(4):532-53. [DOI] [PubMed] [Google Scholar]
- 5. Sah RL, Klein TJ, Schmidt TA, Albrecht DR, Bae WC, Nugent GE, et al. Articular cartilage repair, regeneration, and replacement. In: Koopman WJ, Moreland LW, editors. Arthritis and allied conditions: a textbook of rheumatology. 15th ed Philadelphia: Lippincott Williams & Wilkins; 2005. P. 2277-301. [Google Scholar]
- 6. Levine DW, Mondano L, Halpin M. FDA regulatory pathways for knee cartilage repair products. Sports Med Arthrosc. 2008;16(4):202-7. [DOI] [PubMed] [Google Scholar]
- 7. US Food and Drug Administration. Guidance on application for products comprised of living autologous cells manipulated ex vivo and intended for structural repair or reconstruction. Washington, DC: US Food and Drug Administration; 1996. [Google Scholar]
- 8. Foy JR, Buch BD. Orthopaedic joint devices: the FDA’s short answers to your questions. J Am Acad Orthop Surg. 2008;16(suppl 1):S123-8. [DOI] [PubMed] [Google Scholar]
- 9. US Food and Drug Administration. 21 CFR parts 16, 1270, and 1271 current good tissue practice for human cell, tissue, and cellular and tissue-based product establishments; inspection and enforcement; final rule. [PubMed]
- 10. US Food and Drug Administration. FDA Cellular, Tissue, and Gene Therapies Advisory Committee Meeting, March 3, 2005. [Google Scholar]
- 11. Chu CR, Szczodry M, Bruno S. Animal models for cartilage regeneration and repair. Tissue Eng Part B Rev. 2010;16(1):105-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hurtig MB, Buschmann MD, Fortier LA, Hoemann CD, Hunziker EB, Jurvelin JS, et al. Preclinical studies for cartilage repair: recommendations from the International Cartilage Repair Society. Cartilage. 2011;2(2):137-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. US Food and Drug Administration. Guidance for industry: preparation of IDEs and INDs for products intended to repair or replace knee cartilage. Washington, DC: US Food and Drug Administration; 2011. [Google Scholar]
- 14. Mithoefer K, Saris DB, Farr J, Kon E, Zaslav K, Cole BJ, et al. Guidelines for the design and conduct of clinical studies in knee articular cartilage repair: International Cartilage Repair Society recommendations based on current scientific evidence and standards of clinical care. Cartilage. 2011;2(2):100-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. US Food and Drug Administration. FDA Cellular, Tissue, and Gene Therapies Advisory Committee Meeting, May 15, 2009. [Google Scholar]
- 16. Roos EM, Toksvig-Larsen S. Knee Injury and Osteoarthritis Outcome Score (KOOS)—validation and comparison to the WOMAC in total knee replacement. Health Qual Life Outcomes. 2003;1:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roos EM, Engelhart L, Ranstam J, Anderson AF, Irrgang JJ, Marx RG, et al. ICRS recommendation document: patient-reported outcome instruments for use in patients with articular cartilage defects. Cartilage. 2011;2(2):122-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hoemann C, Kandel R, Roberts S, Saris DB, Creemers L, Mainil-Varlet P, et al. International Cartilage Repair Society (ICRS) recommended guidelines for histological endpoints for cartilage repair studies in animal models and clinical trials. Cartilage. 2011;2(2):153-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. US Food and Drug Administration. Draft guidance for industry: clinical development programs for drugs, devices, and biological products intended for the treatment of osteoarthritis. Washington, DC: US Food and Drug Administration; 1999. [Google Scholar]
- 20. Le Graverand-Gastineau MP. Disease modifying osteoarthritis drugs: facing development challenges and choosing molecular targets. Curr Drug Targets. 2010;11(5):528-35. [DOI] [PubMed] [Google Scholar]
- 21. US Food and Drug Administration. Guidance for FDA reviewers and sponsors: content and review of Chemistry, Manufacturing, and Control (CMC) Information for Human Somatic Cell Therapy Investigational New Drug Applications (INDs). Washington, DC: US Food and Drug Administration; 2008. [Google Scholar]
- 22. US Food and Drug Administration. Draft guidance for industry: potency tests for cellular and gene therapy products. Washington, DC: US Food and Drug Administration; 2008. [Google Scholar]
- 23. US Food and Drug Administration. FDA Orthopaedic and Rehabilitative Devices Advisory Panel Meeting, June 27-28, 2012. [Google Scholar]
- 24. US Food and Drug Administration. Guidance for sponsors, investigators, and institutional review boards: questions and answers on informed consent elements, 21 CFR § 50.25(c). Washington, DC: US Food and Drug Administration; 2012. [Google Scholar]
- 25. US Food and Drug Administration. Draft guidance for industry and FDA staff medical devices: the pre-submission program and meetings with FDA staff. Washington, DC: US Food and Drug Administration; 2012. [Google Scholar]