

Abstract
Objective. To demonstrate posttraumatic chondrocyte apoptosis in the murine xiphoid after a crush-type injury and to ultimately determine the pathway (i.e., intrinsic or extrinsic) by which chondrocytes undergo apoptosis in response to mechanical injury. Design. The xiphoids of adult female wild-type mice were injured with the use of a modified Kelly clamp. Postinjury xiphoid cartilage was analyzed via 3 well-described independent means of assessing apoptosis in chondrocytes: hematoxylin and eosin staining, terminal deoxynucleotidyl transferase dUTP nick end labeling assay, and activated caspase-3 staining. Results. Injured specimens contained many chondrocytes with evidence of apoptosis, which is characterized by cell shrinkage, chromatin condensation, nuclear fragmentation, and the liberation of apoptotic bodies. There was a statistically significant increase in the number of chondrocytes undergoing apoptosis in the injured specimens as compared with the uninjured specimens. Conclusions. Chondrocytes can be stimulated to undergo apoptosis as a result of mechanical injury. These experiments involving predominantly cartilaginous murine xiphoid in vivo establish a baseline for future investigations that employ the genetic and therapeutic modulation of chondrocyte apoptosis in response to mechanical injury.
Keywords: injury, apoptosis, murine, chondrocytes, xiphoid
Introduction
Apoptosis is one of major causes of chondrocyte death in patients with septic arthritis,1 rheumatoid arthritis,2 primary osteoarthritis, and posttraumatic osteoarthritis.3-13 Considering the significance of maintaining the integrity of the extracellular matrix in cartilage, the prevention of chondrocyte apoptosis after traumatic injury has been pursued in an effort to reduce cartilage degeneration and posttraumatic osteoarthritis after injury.11,14 Efforts to prevent chondrocyte apoptosis after mechanical injury have thus far not been successful, in part, because of the fact that the mechanisms by which chondrocytes undergo apoptosis remain unclear.14-17
Apoptosis in eukaryotic cells proceeds commonly via one or two initially different pathways that ultimately converge and result in cell death.14-18 The extrinsic pathway is initiated by receptors at the cell membrane, and the intrinsic pathway is initiated within the cell’s mitochondria. Although the pathways differ with regard to their origins, they both lead to a final common pathway that terminates in cell death.19 But the molecular mechanisms responsible for chondrocyte apoptosis after traumatic or mechanical injury are less clear. Recent evidence from an in vitro study suggests that chondrocyte apoptosis that results from mechanical injury occurs via the intrinsic pathway in response to the calcium channel–mediated depolarization of the mitochondrial membrane.20 Further study of this molecular mechanism in vivo is necessary for the in-depth exploration of the different steps within each of the pathways by which apoptosis is known to occur.
Despite the existence of multiple animal models available for the study of cartilage injury, the molecular mechanisms responsible for chondrocyte apoptosis after mechanical injury are still not completely understood.5,9,21-26 Because genetically engineered murine strains already exist for the study of apoptotic pathways, a mouse injury model would thus be convenient and would enhance the current understanding of the molecular mechanisms by which injury-mediated chondrocyte apoptosis is induced. Two current models that are used to study the injury of cartilage are the destabilization of medial meniscus model and the intra-articular fracture model. Unfortunately, neither model is effective for the study of mechanically induced apoptosis in chondrocytes. The destabilization of medial meniscus model is commonly used to investigate chronic cartilage degradation via the introduction of meniscus instability, but it is not designed to induce acute tissue damage and apoptosis.24,27 The intra-articular fracture model involves the creation of an underlying bone fracture and the infliction of damage to the surrounding joint tissues.23 Although this model aims to study cartilaginous changes as a result of severe trauma, the cause of chondrocyte death is multifactorial, and it may be influenced by residual joint instability after fracture, leukocyte-induced inflammation, and residual joint incongruity after healing, all of which make the systematic study of chondrocyte apoptosis after traumatic injury very complicated.
In this study, the use of murine xiphoid as a system to study chondrocyte apoptosis after mechanical injury is proposed. The xiphoid is a hyaline cartilage–filled structure that is found at the end of the sternum in mice and rats. Several recent studies showed reproducible cartilage formation in the defect of rat xiphoid.28-30 They also showed that the formation of neocartilage in rat xiphoid was increased with fibroblast growth factor-2 in the absence of exogenous chondrocytes.30 Since xiphoid cartilage in mice is subcutaneous, it allows for easy access; only limited surgical exposure is required, and the placement of the tissue allows for the application of consistent and quantifiable mechanical injury. Xiphoid process also provides an environment with little or no presence of inflammatory cells to study the molecular mechanisms of apoptosis in response to mechanical injury, although the phenotypic difference between articular cartilage and xiphoid cartilage remains to be determined. Therefore, the objective of this investigation was to study whether xiphoid chondrocytes could be induced to undergo apoptosis in response to static mechanical injury. We hypothesized that static mechanical injury would induce apoptosis in murine xiphoid cartilage.
Materials and Methods
Experimental Cartilage Injury
All animal care and surgical procedures in this study were performed in accordance with University of Texas Southwestern institutional guidelines and approved by the Institutional Animal Care and Use Committee. Female wild-type C57/B6 mice were acquired from Jackson Laboratories (Jax Mice and Services, Bar Harbor, ME). The animals were anesthetized and placed in the supine position, with their chests and upper abdomens clipped and prepped. The xiphoid process was exposed via a 1-cm incision that was made at the base of the sternum.
To create the xiphoid injury, a modified Kelly clamp (BH443R Aesculap Inc., Center Valley, PA) was used. The tip of the Kelly clamp was milled down to a size of 9.2 mm long x 4.6 mm wide, and the grips of the Kelly clamp were crosshatched to allow for the application of mechanical stress to an area of 42 mm2. Several pilot tests performed with the use of pressure-sensitive film (Pressurex Low [350-1400 psi] and Pressurex Medium [1400-7100 psi]; Sensor Products, Madison, NJ) showed that this clamp delivered an average peak stress of 22.4 MPa across the xiphoid when it was pressed down to the first of 3 interlocking positions (Fig. 1A). This level of stress has previously been shown to induce acute apoptosis in cartilage.25,31,32 To ensure chondrocyte injury across the xiphoid, the clamp was applied 3 separate times with slight positional variation across the xiphoid for 2 minutes each time, with a 1-minute pause between clampings (Fig. 1A and B).
Figure 1.

A modified Kelly clamp (BH443R Aesculap Inc., Center Valley, PA) was used to induce injury. The grasping portion of the clamp has been shortened to minimize the risk of injury to adjacent tissue. (A) Peak pressure distribution was affected by the number of applications to the xiphoid as measured by Pressurex Low and Medium film (Sensor Products Inc., Madison, NJ). (B) The modified Kelly clamp was applied in 3 different orientations across the xiphoid while the mouse was anesthetized.
After injury, the xiphoid was irrigated. The soft tissues were closed in layers with 3-0 Vicryl suture (Ethicon, Inc, Somerville, NJ) for the subcutaneous tissues and with Dermabond (Ethicon, Inc.) for the skin. Postoperatively, the animals were allowed activity ad libitum. Control (sham) animals underwent an identical surgical approach and closure with exposure of the xiphoid for a predetermined period of time (10 minutes) but were not injured. Six animals were used in each group and euthanized at 6, 12, 24, 48, and 96 hours after injury. To limit the number of animals used in this study, a total of 15 animals undergoing sham surgery (n = 15) were euthanized at 6, 12, 24, 48, and 96 hours after treatment.
Hematoxylin and Eosin Staining and Safranin-O Staining
At the time of euthanasia, the xiphoid was removed with a portion of the sternum and the lower ribs to facilitate positioning of the specimens during the embedding process. After excision, the specimens were fixed in 10% neutral-buffered formalin (VWR International, West Chester, PA) for 20 hours and subsequently processed and embedded in paraffin (Thermo-Shandon Excelsior Tissue Processor, Pittsburgh, PA). The xiphoids were positioned to ensure uniform transverse sectioning from rostral to caudal and perpendicular to the axis of the sternum. At this point, 5-µm paraffin serial sections were made from each xiphoid process in groups of three, beginning at the tip of the remaining bony sternal core (i.e., approximately 1.5 mm block-face distance from the embedding plane). Two slides from each sample were stained with hematoxylin and eosin (H&E; Sigma, St. Louis, MO) and analyzed with light microscopy for hallmarks of cartilage apoptosis, including cell shrinkage, nuclear condensation and fragmentation, the liberation of apoptotic bodies, and inflammatory cell infiltration.
Adjacent or near-adjacent slides to those stained with H&E were subjected to Safranin-O staining. Briefly, this special stain involves the use of Weigert’s iron hematoxylin (Richard-Allan Scientific, Kalamazoo, MI) to counterstain nuclei, 0.01% Fast Green solution (Sigma) to stain cytoplasm and bone matrix, and 0.1% Safranin-O solution (Sigma) to stain proteoglycans. Slides were analyzed with light microscopy to confirm the presence of proteoglycans in the murine xiphoid matrix.
Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling of Tissue Sections
A third set of xiphoid slides was subjected to terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining to detect nicked DNA in dead and dying cells (DeadEnd Fluorometric TUNEL System, Promega Corporation, Madison, WI) in accordance with the vendor’s protocol. In brief, the sections were deparaffinized and prepared for TUNEL detection via permeabilization with proteinase-K. Subsequently, slides were incubated for 1 hour at 37°C with terminal deoxynucleotidyl transferase reaction mix. The reaction was halted with sodium chloride and sodium citrate buffer, and nuclear counterstaining was performed with propidium iodide (PI). Slides were cover slipped with the use of Vectashield mounting medium (Vector Labs, Burlingame, CA). Specimens were analyzed with the use of fluorescence microscopy at 470 nm (TUNEL) and 535 nm (PI) excitation wavelengths. Images (320 µm x 480 µm) that were taken from the central region close to the middle line (±1100 µm) of the xiphoid were used for morphometric analysis, because this area received a more uniform injury as a result of the elliptical shape of the xiphoid and the mechanical action of the injury-producing clamp. Image J software (National Institutes of Health, Bethesda, MD) was used to identify and enumerate total chondrocyte nuclei (i.e., PI-positive nuclei) and total TUNEL-positive chondrocyte nuclei. The image processor was blinded with regard to the treatment rendered to each processed specimen. After computer-aided image analysis, the manual verification of the TUNEL-positive nuclei number and the PI-positive nuclei number was performed to ensure that all cells were counted at least once by two independent observers who were blinded to the treatment groups.
Activated Caspase-3 Immunohistochemistry
In response to the extensive TUNEL positivity found at all postinjury time points and in the light of the body of literature that demonstrates that the expression of activated caspase-3 precedes DNA fragmentation,33,34 only a randomly selected subset (i.e., 2 from each group) of injury and control specimens from the 12-, 24-, 48-, and 96-hour postinjury time points were stained immunohistochemically for the presence of cleaved caspase-3. In brief, sections were deparaffinized before antigen retrieval with 0.05% citraconic anhydride, and they were then blocked with 3% normal goat serum. Samples were incubated with primary rabbit anti-cleaved caspase-3 antibody (Cat. #9661; Cell Signaling Technology, Danvers, MA) overnight at 4°C, and binding was subsequently detected with biotinylated goat anti-rabbit secondary antibody (BA-1000; Vector Laboratories). Streptavidin peroxidase (SA-5400; Vector Laboratories) and DAB chromogen (K3468; Dako North America, Carpinteria, CA) were used to reveal bound primary–secondary antibodies, and sections were counterstained with hematoxylin (Sigma). Cover slips were applied with permanent mounting media, and slides were analyzed with light microscopy.
Statistical Analysis
TUNEL data was subjected to statistical analysis that consisted of a 2-tailed Student’s t test to assess the significance between each postinjury group’s treatment samples and its control samples. A one-way analysis of variance with Tukey’s post hoc test was also used to compare the means among the postinjury groups. To assess interobserver variability for the TUNEL assay, Cohen’s kappa coefficient was calculated. All of the statistics were determined with the use of Systat 10.2 (Systat Software, Chicago, IL) and Excel (Microsoft, Redmond, WA). Statistical significance for all compared values was assumed at P < 0.05.
Results
Histological Findings: Hematoxylin and Eosin Staining and Safranin-O/Fast Green Staining
Low-power (4x) light microscopy was used to visualize the morphology of the xiphoid. The murine xiphoid was consistently found to have an elliptical shape that was widest in the center and that tapered toward its outermost tip (Fig. 2A). Under higher magnification (60x), the morphology of the chondrocytes and the surrounding matrix of the control specimens appeared to be normal and uninjured (Fig. 2B). Cells that appeared to be healthy were visible throughout each specimen; each chondrocyte was symmetrical, and these cells contained large oval nuclei with readily apparent nucleoli and ample cytoplasm with abundant organelles. These observations contrasted with those of the injured specimens, which contained many chondrocytes with the morphologic hallmarks of apoptosis: cell shrinkage, chromatin condensation, and numerous apoptotic bodies scattered about the lacunae (Fig. 2C). At the later time points (i.e., 96 hours after injury), polymorphonuclear cells in the periphery of the tissue were frequently evident.
Figure 2.

(A) Hematoxylin and eosin–stained specimen (4x) demonstrating the elliptical shape of the xiphoid cartilage. Muscle and loose connective tissue surround the xiphoid. (B) Hematoxylin and eosin–stained control specimen (20x) demonstrating consistent chondrocytes with large oval nuclei, readily apparent nucleoli, and abundant cytoplasm. (C) Hematoxylin and eosin–stained injured specimen (60x) 24 hours after injury. Several of the chondrocytes demonstrate shrinkage, nuclear blebbing (arrows), chromatin condensation, and fragmentation with the liberation of numerous apoptotic bodies (arrowheads). Bar = 100 µm.
Strong Safranin-O staining that was consistent with abundant proteoglycan matrix surrounding the chondrocyte lacunae was seen in both the control and injured specimens (Fig. 3A). Qualitatively, there was no difference in proteoglycan content between the control and injured specimens. No loss of proteoglycan or matrix damage was seen in the injured samples at 24 hours (Fig. 3B). Alternatively, at 96 hours, empty lacunae were seen in 2 of the 6 injured specimens.
Figure 3.

(A) Safranin-O–stained injured specimen 24 hours after injury demonstrating abundant proteoglycan (light red) within the xiphoid process. (B) The dashed box in A indicates the enlarged view of the stained sample shown here. Bar = 150 µm.
Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling Findings
TUNEL staining confirmed the presence of DNA fragmentation within the nucleus of the chondrocytes, and it allowed for the quantification of apoptosis frequency occurring within the injured and control specimens. Of most importance is that TUNEL-positive chondrocytes were located in the area (according to adjacent slides) where apoptotic cells with typical morphologic changes (as determined by light microscopy) were found. Results were calculated as the number of TUNEL-positive nuclei versus the number of PI-positive nuclei (Fig. 4A and B). A good agreement was found between 2 observers (κ = 0.635). Little or no chondrocyte apoptosis (3.3% ± 1.1%) was found within the control specimens. As compared with control specimens, a significant increase in TUNEL-positive cells were identified in each of the injured specimens at each time point that was studied (Fig. 5; P = 0.026, P = 0.001, P = 0.008, P < 0.001, and P < 0.001 for 6, 12, 24, 48, and 96 hours after injury, respectively). There was a steady increase in TUNEL-positive cells from 6 to 48 hours, but there was no further increase after 96 hours.
Figure 4.
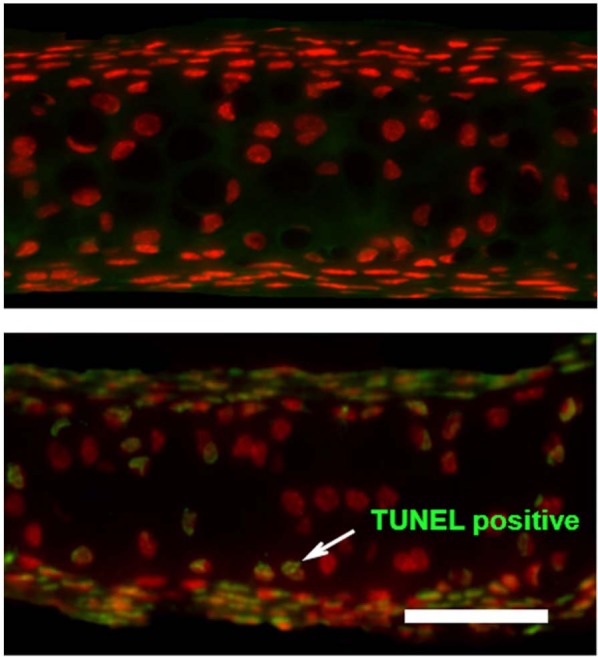
Terminal deoxynucleotidyl transferase dUTP nick end labeling assay and propidium iodide–labeled xiphoid specimens. (A) Control specimen. (B) Injured specimen 24 hours after injury. Chondrocytes that are stained red represent viable cells; those that are stained green represent chondrocytes undergoing apoptosis. Bar = 100 µm.
Figure 5.
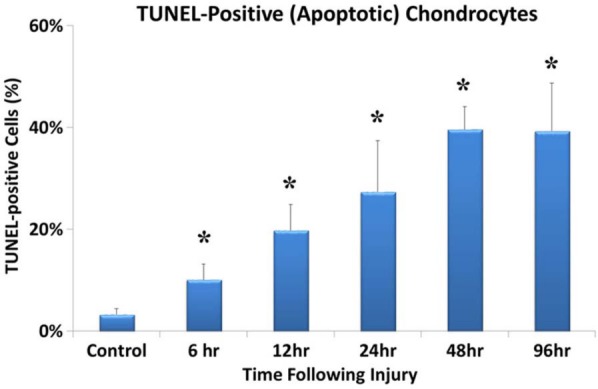
Significant increases in the percentage of terminal deoxynucleotidyl transferase dUTP nick end labeling assay positive chondrocytes after injury (n = 6 for all injury groups) as compared with sham controls (n = 15). The presence of an asterisk (*) indicates that the P value is less than 0.05.
Activated Caspase-3
Although the postinjury time points of xiphoid collection at 12, 24, 48, and 96 hours may not include the entire spectrum of injury progression as indicated by the presence of DNA fragmentation previously shown via light microscopy and TUNEL analysis, cleaved caspase-3 immunohistochemistry was performed on a subset of specimens to determine the presence of activated caspase. Cleaved caspase-3 immunostaining was noted in a marked number of chondrocytes in the injured specimens, whereas no cleaved caspase-3 immunostaining was seen in the control specimens (Fig. 6).
Figure 6.

Activated caspase-3–stained (A) injured and (B) uninjured specimens at 96 hours after injury. Brown cytoplasmic staining (arrows) in the injured specimen demonstrates the presence of activated caspase-3. The control specimen has a notable lack of cytoplasmic staining, thereby demonstrating the lack of activated caspase-3. Bar = 50 µm.
Discussion
As indicated by 3 well-established and independent means of assessing apoptosis in chondrocytes (i.e., histology, TUNEL staining, and activated caspase-3), chondrocyte apoptosis in the mouse xiphoid was stimulated by the application of mechanical injury. On the basis of the light microscopy analysis of H&E-stained and Safranin-O–stained specimens, the mouse xiphoid is a proteoglycan-rich tissue that contains an abundance of chondrocytes within the lacunae. These findings show that chondrocyte apoptosis and histological changes in murine xiphoid are similar to those in articular cartilage after mechanical injury.7,31
The hallmark of apoptosis is the enzymatic fragmentation of DNA as detected by the TUNEL assay. In this study of the mouse xiphoid, a statistically significant increase in the percentage of TUNEL-positive chondrocytes undergoing apoptosis was found in the injured specimens as compared with the uninjured control specimens. The maximum amount of TUNEL-positive apoptotic chondrocytes was evident 96 hours after injury. As compared with the studies of rabbit and bovine articular cartilage, the development of chondrocyte apoptosis appears much sooner in the mouse xiphoid after injury as evidenced by similar frequencies of TUNEL-positive chondrocytes being present between 12 and 96 hours after xiphoid injury.4,7,35 A further increase in apoptosis in the mouse xiphoid may occur more than 96 hours after injury, but this was not studied with the current experiments.35 However, it is also possible that this is due in part to differences among tissues, differences among species, and the ages of the animals studied. Younger and smaller animals are likely to have faster metabolic rates as compared with older and larger animals; this could explain the accelerated apoptotic rate.
Detractors of the use of TUNEL report the false-positive TUNEL staining of cells that are undergoing necrosis as well as apoptosis occurring as a result of endogenous endonucleases acting on cellular DNA before the processing of the tissue.7,36,37 Further confounding the issue is the fact that other investigators have pointed out that, after most types of injury, there is a mixture of different types of cell death (i.e., apoptosis, necrosis, and autophagy) and that these cell death pathways are affected by extensive crosstalk.38 The examination of tissue sections also showed evidence of scattered chondrocytes with necrotic morphology, although the percentage of necrotic cells was not estimated as a result of the limitation of the histological analysis that was used in the present study. These necrotic chondrocytes appeared to be more proximal to the point of compression injury, and they appeared at early time points after injury (data not presented). To distinguish between cells that are undergoing necrosis and those that are undergoing apoptosis, immunohistochemistry for activated caspase-3 was employed to confirm TUNEL-positive cells in consecutive sections in a subset of injured specimens. As the main executioner protein, activated caspase-3 binds with apoptotic protease-activating factor 1, cytochrome C, and adenosine triphosphate to form an apoptosome complex; this is common to both the intrinsic and the extrinsic apoptosis pathways.4,19,39 However, this complex interaction of essential proteins is absent during cell necrosis. In the current study, it was found that activated caspase-3–positive chondrocytes were only located in the TUNEL-positive regions of injured xiphoid specimens and not in the control specimens. This finding confirms that TUNEL-positive chondrocytes in the xiphoid injury model are apoptotic cells rather than necrotic cells.
Morphological changes seen with H&E staining provided further corroboration of TUNEL-positive apoptosis via the identification of cells exhibiting the hallmark morphological features of apoptosis, including cell shrinkage, nuclear condensation and fragmentation, and the liberation of apoptotic bodies (Fig. 2B). Apoptotic body formation distinguishes apoptotic cells from necrotic ones: necrotic degradation proceeds via a loss of membrane integrity and the release of cellular components, typically with an associated inflammatory response; this does not result in the liberation of apoptotic bodies. Cell destruction occurs within autophagic vacuoles, which then bleb into the lacunae; this leads to the complete self-destruction of the chondrocyte, which is visualized by the presence of empty lacunae.40,41 Only small portions of the visualized sections showed such changes, which limits the applicability of these changes to being a confirmatory tool. Still, the injured mouse xiphoid specimens clearly contained more cells that exhibited the aforementioned characteristics of apoptosis as compared with the control specimens. This further corroborates the TUNEL and activated caspase-3 findings, thereby confirming that chondrocytes can be induced to undergo apoptosis in response to mechanical injury.
Some limitations of this study should be addressed. First, xiphoid is not articular cartilage. To the authors’ knowledge, no study has compared the biomechanical, biophysical, biochemical, and structural differences between xiphoid cartilage and articular cartilage. However, in the present study, the light-microscopic analysis of H&E-stained and Safranin-O–stained xiphoid tissue reveals a cellular arrangement and a matrix structure and makeup that are similar to those of articular cartilage (e.g., round cells with oval nuclei, well-formed nucleoli, cytoplasmic organelles within well-formed lacunae). In addition, the extracellular matrix of xiphoid contains Safranin-O–staining material that is consistent with proteoglycans and collagen fibers. These findings suggest that xiphoid contains cartilaginous tissue and that it has a similar histological appearance to that of articular cartilage. Second, the current study was not intended to reproduce blunt trauma but rather to provide an overload injury model for the study of chondrocyte apoptosis agonists and mediators. In this study, the presence of chondrocytes that were undergoing apoptosis in greater numbers in mechanically injured xiphoid has been demonstrated by morphological changes, TUNEL staining, and activated caspase-3 staining. This suggests that consistent chondrocyte apoptosis can be produced in xiphoid tissue in response to overload injury. Another limitation of this study is that necrotic cells can be TUNEL positive 48 hours after traumatic injury, as previously described.7 Future studies with the use of the live/dead assay will be helpful for determining the contribution of necrosis to this injury model.
In summary, the present study found that the application of mechanical overload can produce consistent chondrocyte apoptosis in murine xiphoid; this is similar to what happens to chondrocytes in injured articular cartilage. This xiphoid injury model allows for the study of chondrocyte apoptosis with the use of established genetically modified mice and for the further investigation of the molecular mechanisms by which chondrocytes undergo apoptosis in response to mechanical injury.
Acknowledgments
The authors thank Ms. Kathy Stroud and Ms. Maia Frank for their technical and editorial assistance with this article. All authors have made substantial contributions to the conception and design of the study, the acquisition of data, the analysis and interpretation of that data, and the drafting and revising of the important intellectual content of this article.
Footnotes
This study was supported in part by a grant from the Orthopaedic Trauma Association. The association had no role in the study design; in the collection, analysis, or interpretation of data; in the writing of the report; or in the decision to submit the paper for publication.
Declaration of Conflicting Interests: The authors(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
Ethical Approval: This study was approved by the Institutional Animal Care and Use Committee at UT Southwestern Medical Center (APN# 2007-0160).
References
- 1. Lee MS, Yen CY, Ueng SW, Shih CH, Chao CC. Signal transduction pathways and apoptosis in bacteria infected chondrocytes. J Orthop Res. 2001;19:696-702. [DOI] [PubMed] [Google Scholar]
- 2. Chou CT, Yang JS, Lee MR. Apoptosis in rheumatoid arthritis—expression of Fas, Fas-L, p53, and Bcl-2 in rheumatoid synovial tissues. J Pathol. 2001;193:110-6. [DOI] [PubMed] [Google Scholar]
- 3. Blanco FJ, Guitian R, Vázquez-Martul E, de Toro FJ, Galdo F. Osteoarthritis chondrocytes die by apoptosis. A possible pathway for osteoarthritis pathology. Arthritis Rheum. 1998;41:284-9. [DOI] [PubMed] [Google Scholar]
- 4. Borrelli J., Jr Chondrocyte apoptosis and posttraumatic arthrosis. J Orthop Trauma. 2006;20:726-31. [DOI] [PubMed] [Google Scholar]
- 5. Borrelli J, Jr, Ricci WM. Acute effects of cartilage impact. Clin Orthop Relat Res. 2004;(423):33-9. [DOI] [PubMed] [Google Scholar]
- 6. Borrelli J, Jr, Tinsley K, Ricci WM, Burns M, Karl IE, Hotchkiss R. Induction of chondrocyte apoptosis following impact load. J Orthop Trauma. 2003;17:635-41. [DOI] [PubMed] [Google Scholar]
- 7. Chen CT, Burton-Wurster N, Borden C, Hueffer K, Bloom SE, Lust G. Chondrocyte necrosis and apoptosis in impact damaged articular cartilage. J Orthop Res. 2001;19:703-11. [DOI] [PubMed] [Google Scholar]
- 8. D’Lima DD, Hashimoto S, Chen PC, Lotz MK, Colwell CW., Jr Cartilage injury induces chondrocyte apoptosis. J Bone Joint Surg Am. 2001;83(Suppl 2):19-21. [DOI] [PubMed] [Google Scholar]
- 9. Jeffrey JE, Gregory DW, Aspden RM. Matrix damage and chondrocyte viability following a single impact load on articular cartilage. Arch Biochem Biophys. 1995;322:87-96. [DOI] [PubMed] [Google Scholar]
- 10. Loening AM, James IE, Levenston ME, Badger AM, Frank EH, Kurz B, et al. Injurious mechanical compression of bovine articular cartilage induces chondrocyte apoptosis. Arch Biochem Biophys. 2000;381:205-12. [DOI] [PubMed] [Google Scholar]
- 11. Ding L, Heying E, Nicholson N, Stroud NJ, Homandberg GA, Buckwalter JA, et al. Mechanical impact induces cartilage degradation via mitogen activated protein kinases. Osteoarthritis Cartilage. 2010;18:1509-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carames B, Taniguchi N, Seino D, Blanco FJ, D’Lima D, Lotz M. Mechanical injury suppresses autophagy regulators and pharmacologic activation of autophagy results in chondroprotection. Arthritis Rheum. 2012;64:1182-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rosenzweig DH, Djap MJ, Ou SJ, Quinn TM. Mechanical injury of bovine cartilage explants induces depth-dependent, transient changes in MAP kinase activity associated with apoptosis. Osteoarthritis Cartilage. 2012;20:1591-602. [DOI] [PubMed] [Google Scholar]
- 14. Huser CA, Peacock M, Davies ME. Inhibition of caspase-9 reduces chondrocyte apoptosis and proteoglycan loss following mechanical trauma. Osteoarthritis Cartilage. 2006;14: 1002-10. [DOI] [PubMed] [Google Scholar]
- 15. Nuttall ME, Nadeau DP, Fisher PW, Wang F, Keller PM, DeWolf WE, Jr, et al. Inhibition of caspase-3-like activity prevents apoptosis while retaining functionality of human chondrocytes in vitro. J Orthop Res. 2000;18:356-63. [DOI] [PubMed] [Google Scholar]
- 16. D’Lima DD, Hashimoto S, Chen PC, Colwell CW, Jr, Lotz MK. Impact of mechanical trauma on matrix and cells. Clin Orthop Relat Res. 2001;(391 Suppl):S90-9. [DOI] [PubMed] [Google Scholar]
- 17. Dang AC, Warren AP, Kim HT. Beneficial effects of intra-articular caspase inhibition therapy following osteochondral injury. Osteoarthritis Cartilage. 2006;14:526-32. [DOI] [PubMed] [Google Scholar]
- 18. Feng L, Balakir R, Precht P, Horton WE., Jr Bcl-2 regulates chondrocyte morphology and aggrecan gene expression independent of caspase activation and full apoptosis. J Cell Biochem. 1999;74:576-86. [PubMed] [Google Scholar]
- 19. Martin DA, Elkon KB. Mechanisms of apoptosis. Rheum Dis Clin North Am. 2004;30:441-54. [DOI] [PubMed] [Google Scholar]
- 20. Huser CA, Davies ME. Calcium signaling leads to mitochondrial depolarization in impact-induced chondrocyte death in equine articular cartilage explants. Arthritis Rheum. 2007;56:2322-34. [DOI] [PubMed] [Google Scholar]
- 21. Brandt KD, Myers SL, Burr D, Albrecht M. Osteoarthritic changes in canine articular cartilage, subchondral bone, and synovium fifty-four months after transection of the anterior cruciate ligament. Arthritis Rheum. 1991;34:1560-70. [DOI] [PubMed] [Google Scholar]
- 22. D’Lima DD, Hashimoto S, Chen PC, Lotz MK, Colwell CW., Jr In vitro and in vivo models of cartilage injury. J Bone Joint Surg Am. 2001;83(Suppl 2):22-4. [DOI] [PubMed] [Google Scholar]
- 23. Furman BD, Strand J, Hembree WC, Ward BD, Guilak F, Olson SA. Joint degeneration following closed intraarticular fracture in the mouse knee: a model of posttraumatic arthritis. J Orthop Res. 2007;25:578-92. [DOI] [PubMed] [Google Scholar]
- 24. Glasson SS, Askew R, Sheppard B, Carito B, Blanchet T, Ma HL, et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature. 2005;434:644-8. [DOI] [PubMed] [Google Scholar]
- 25. Milentijevic D, Rubel IF, Liew AS, Helfet DL, Torzilli PA. An in vivo rabbit model for cartilage trauma: a preliminary study of the influence of impact stress magnitude on chondrocyte death and matrix damage. J Orthop Trauma. 2005;19:466-73. [DOI] [PubMed] [Google Scholar]
- 26. Rundell SA, Baars DC, Phillips DM, Haut RC. The limitation of acute necrosis in retro-patellar cartilage after a severe blunt impact to the in vivo rabbit patello-femoral joint. J Orthop Res. 2005;23:1363-9. [DOI] [PubMed] [Google Scholar]
- 27. Majumdar MK, Chockalingam PS, Bhat RA, Sheldon R, Keohan C, Blanchet T, et al. Immortalized mouse articular cartilage cell lines retain chondrocyte phenotype and respond to both anabolic factor BMP-2 and pro-inflammatory factor IL-1. J Cell Physiol. 2008;215:68-76. [DOI] [PubMed] [Google Scholar]
- 28. Moyer HR, Wang Y, Farooque T, Wick T, Singh KA, Xie L, et al. A new animal model for assessing cartilage repair and regeneration at a nonarticular site. Tissue Eng Part A. 2010;16:2321-30. [DOI] [PubMed] [Google Scholar]
- 29. Singh K, Moyer H, Williams JK, Schwartz Z, Boyan BD. Fibrin glue: a scaffold for cellular-based therapy in a critical-sized defect. Ann Plast Surg. 2011;66:301-5. [DOI] [PubMed] [Google Scholar]
- 30. Wang Y, Huang YC, Gertzman AA, Xie L, Nizkorodov A, Hyzy SL, et al. Endogenous regeneration of critical-size chondral defects in immunocompromised rat xiphoid cartilage using decellularized human bone matrix scaffolds. Tissue Eng Part A. 2012;18:2332-42. [DOI] [PubMed] [Google Scholar]
- 31. Borrelli J, Jr, Torzilli PA, Grigiene R, Helfet DL. Effect of impact load on articular cartilage: development of an intra-articular fracture model. J Orthop Trauma. 1997;11:319-26. [DOI] [PubMed] [Google Scholar]
- 32. Torzilli PA, Grigiene R, Borrelli J, Jr, Helfet DL. Effect of impact load on articular cartilage: cell metabolism and viability, and matrix water content. J Biomech Eng. 1999;121:433-41. [DOI] [PubMed] [Google Scholar]
- 33. Matsuo M, Nishida K, Yoshida A, Murakami T, Inoue H. Expression of caspase-3 and -9 relevant to cartilage destruction and chondrocyte apoptosis in human osteoarthritic cartilage. Acta Med Okayama. 2001;55:333-40. [DOI] [PubMed] [Google Scholar]
- 34. Grogan SP, Aklin B, Frenz M, Brunner T, Schaffner T, Mainil-Varlet P. In vitro model for the study of necrosis and apoptosis in native cartilage. J Pathol. 2002;198:5-13. [DOI] [PubMed] [Google Scholar]
- 35. Levin A, Burton-Wurster N, Chen CT, Lust G. Intercellular signaling as a cause of cell death in cyclically impacted cartilage explants. Osteoarthritis Cartilage. 2001;9:702-11. [DOI] [PubMed] [Google Scholar]
- 36. Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Bursch W, Schulte-Hermann R. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology. 1995;21:1465-8. [DOI] [PubMed] [Google Scholar]
- 37. Stahelin BJ, Marti U, Solioz M, Zimmermann H, Reichen J. False positive staining in the TUNEL assay to detect apoptosis in liver and intestine is caused by endogenous nucleases and inhibited by diethyl pyrocarbonate. Mol Pathol. 1998;51:204-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Perl M, Chung CS, Ayala A. Apoptosis. Crit Care Med. 2005;33(12 Suppl):S526-9. [DOI] [PubMed] [Google Scholar]
- 40. Roach HI, Aigner T, Kouri JB. Chondroptosis: a variant of apoptotic cell death in chondrocytes? Apoptosis. 2004;9:265-77. [DOI] [PubMed] [Google Scholar]
- 41. Roach HI, Clarke NM. “Cell paralysis” as an intermediate stage in the programmed cell death of epiphyseal chondrocytes during development. J Bone Miner Res. 1999;14:1367-78. [DOI] [PubMed] [Google Scholar]