

Abstract
Objective:
Apoptosis of chondrocytes in articular cartilage has been observed in rheumatoid arthritis patients. However, molecules involved in such chondrocyte apoptosis in arthritic joints have not been fully understood. We previously observed that apoptosis of chondrocytes is enhanced in a murine arthritis model induced by injection with anti–type II collagen antibodies and lipopolysaccharide (mAbs/LPS), and osteopontin (OPN) deficiency suppresses chondrocyte apoptosis in this arthritis model in vivo. To understand how OPN deficiency renders resistance against chondrocyte apoptosis, we examined the cellular basis for this protection.
Design:
Chondrocytes were prepared from wild-type and OPN-deficient mouse ribs, and tumor necrosis factor (TNF)–α–induced cell death was examined based on lactate dehydrogenase (LDH) release assay and TUNEL assay.
Results:
TNF-α treatment induced LDH release in wild-type chondrocytes, while OPN deficiency suppressed such LDH release in the cultures of these cells. TNF-α–induced increase in the number of TUNEL-positive cells was observed in wild-type chondrocytes, while OPN deficiency in chondrocytes suppressed the TNF-α induction of TUNEL-positive cells. OPN deficiency suppressed TNF-α–induced increase in caspase-3 activity in chondrocytes in culture. Furthermore, OPN overexpression in chondrocytes enhanced TNF-α–induced apoptosis.
Conclusion:
These results indicated that the presence of OPN in chondrocytes is involved in the susceptibility of these cells to TNF-α–induced apoptosis.
Keywords: biomechanics < general, animal models < general, chondrocytes < cells, chondrogenesis < cells
Introduction
Rheumatoid arthritis (RA) impairs quality of life due to joint destruction.1 Articular cartilage is lost in association with hyperproliferating synovia in which immune cells such as T cells and macrophages are infiltrated. Destruction of articular cartilage and bone loss progress and lead to pain and disability.2 In association with the destruction of joints in arthritis patients, chondrocyte apoptosis has been observed.3 However, the molecules involved in chondrocyte apoptosis in articular cartilage have not been fully understood.
Osteopontin (OPN) is a cytokine and a matrix protein and has been known to be involved in arthritis. In murine experimental arthritis models, apoptosis of chondrocytes was induced in wild-type, while OPN deficiency suppressed this apoptosis.1 Proinflammatory cytokines such as tumor necrosis factor (TNF)–α, interleukin (IL)–1, and nitric oxide (NO) are known to induce apoptosis in chondrocytes.4 In mouse models, lipopolysaccharide (LPS) injection induces high TNF-α protein expression levels in blood, while these levels were similar in OPN-deficient and wild-type mice.1 Therefore, it was not known whether OPN is involved in arthritic cartilage destruction directly or indirectly. The aim of this study was to investigate the effects of OPN deficiency on chondrocyte apoptosis induced by TNF-α. We found that OPN deficiency suppressed the levels of TNF-α–induced release of lactate dehydrogenase (LDH) in chondrocytes. Overexpression of OPN in chondrocytes enhanced TNF-α–induced increase in cytotoxicity. Thus, OPN is involved in TNF-α–induced apoptosis in chondrocytes.
Materials and Methods
Reagents
TNF-α was obtained from Chemicon International Inc. (Temecula, CA). LDH-Cytotoxic Test was obtained from Wako Pure Chemical Co. (Osaka, Japan).
Animals
Wild-type and OPN-deficient mice with a C57Bl/6x129/sv F2 background were produced as described previously.5 OPN-deficient mice were backcrossed to C57Bl/6 background 8 times. Wild-type and OPN-deficient mice were used at 2 to 3 weeks of age. All animal experiments were approved by the Animal Welfare Committee of Tokyo Medical and Dental University.
Cell Cultures
Chondrocytes were isolated from rib cartilage of 2- to 3-week-old mice as described previously.6 Briefly, cartilaginous rib cages were preincubated in the presence of 3 mg/mL collagenase (Sigma, St. Louis, MO) in DMEM for 45 minutes at 37 °C and rinsed with PBS. They were further incubated in 3 mg/mL of collagenase in DMEM at 37 °C for 5 hours, and the released cells were collected. Chondrocytes collected were rinsed with medium twice before they were seeded into tissue culture plates at 1.0 × 105 cells/cm2. The cells were grown to expand in DMEM supplemented with antibiotics (100 U/mL penicillin G sodium, 100 mg/mL streptomycin sulfate) and 10% FBS (GIBCO BRL, Carlsbad, CA) for 4 days. Chondrocytes were then collected and repeated into 96-well plates for cytotoxic and TUNEL assay and in 6-well plates for caspase assay. The cells were cultured in DMEM supplemented with 5% FBS. Some of the chondrocytes were transfected with lacZ and OPN containing adenovirus vectors.
RNA Extraction, cDNA Synthesis, and Polymerase Chain Reaction
Total RNA of chondrocytes was extracted according to the acid-guanium-phenol-chloroform (AGPC) method. First-strand cDNA was synthesized using 1 µg of the total RNA and Moloney murine leukemia virus reverse transcriptase (RT). Primers were synthesized based on the reported mouse cDNA sequences for GAPDH (GenBank accession: NM 008084) and OPN (GenBank accession: AF515708). Primers for polymerase chain reaction (PCR) were as follows: 5′-ACCACAGTCCATGCCATCAC-3′ (forward, GAPDH) and 5′-TCCACCACCCTGTTGCTGTA-3′ (reverse, GAPDH), 5′-CGACGATGATGACGATGATGAT-3′ (forward, OPN) and 5′-CTGGCTTTGGAACTTGCTTGAC-3′ (reverse, OPN). Amplification was carried out for 20 to 26 cycles each at 94 °C for 20 seconds, 60 °C for 30 seconds, and 72 °C for 40 seconds in a 25-µL reaction mixture containing 0.5 µL of each cDNA, 200 mM of each primer, 0.2 mM of dNTP, and 1 U of TaqDNA polymerase (Takara, Shiga, Japan). After amplification, 10 µL of each reaction mixture was analyzed by 1% agarose gel electrophoresis, and the bands were then visualized by ethidium bromide staining.
LDH Release
The levels of LDH activity released into medium were measured by using LDH-Cytotoxic Test (Wako Pure Chemical Co.) according to the manufacturer’s instructions. In brief, cells were plated at 2.0 × 104 cells/well in flat-bottomed 96-well culture plates and precultured in DMEM supplemented with 5% FBS. Chondrocytes were cultured in the presence of TNF-α. At the end of culture, the media were collected to measure LDH activity. To determine LDH activity, 20-µL aliquots of cell culture medium were added to the LDH assay mixture, and the increase in absorbance was measured at 560 nm. To detect LDH activity of the total cells, cells were treated with 0.5% Triton X-100, and the lysate was used for assay.
TUNEL Assay
TUNEL assay was conducted by using a TUNEL detection kit according to the manufacturer’s instructions (Takara Shuzo, Kyoto, Japan). Briefly, cells cultured in 96-well plates were fixed in 4% paraformaldehyde in PBS and then rinsed with PBS. Endogenous peroxidase was inactivated by the treatment with 0.3% H2O2 for 30 minutes at room temperature. Then, the cells were rinsed with PBS. The cells were incubated in permeabilization buffer on ice and then rinsed with PBS. The cells were further incubated in terminal deoxynucleotidyltransferase (TdT) buffer containing deoxynucleotidyl transferase and biotinylated dUTP in TdT buffer at 37 °C for 90 minutes and then rinsed with PBS. The cells were incubated at 37 °C for 30 minutes in the presence of anti-FITC horseradish peroxidase–conjugated antibody, and the signals were visualized using diaminobenzidine. For analysis of TUNEL-positive cells, over 50 cells were examined, and the fractions of positive cells were shown as a percentage.
Caspase-3 Activities
Caspase-3 activity was determined using a caspase-3/CPP32 colorimetric protease assay kit (Medical and Biological Laboratories, Nagoya, Japan) according to the manufacturer’s protocol. In brief, chondrocytes were seeded into 6-well plates at 105 cells/cm2. These cells were cultured in the presence of mouse recombinant TNF-α for the indicated periods of time. Cell lysates (100 µg of protein in 50 µL of lysis buffer) were incubated with DEVD-p-nitroanilide (pNA) as a substrate for 3 hours at 37 °C, and the amounts of pNA generated were determined based on spectrophotometry at 405 nm.
Statistical Analysis
The results were presented as means ± standard deviations (SDs), and statistical evaluation was performed based on the Student t test. All the experiments were conducted at least twice to verify reproducibility.
Results
We first examined the profile of the response of our chondrocyte preparation to TNF-α treatment based on LDH release. LDH is an enzyme present in the cytoplasm and is released from the cells upon cell death.7 We examined the levels of LDH release into the medium from the cultured chondrocytes 48 hours after the treatment with TNF-α. The levels of LDH release were low in chondrocytes, which were treated with vehicle alone (Fig. 1A, left column). When the chondrocytes were exposed to TNF-α, LDH release was increased in a dose-dependently manner (Fig. 1A). These data indicated that the cultures of chondrocytes prepared from mice underwent apoptosis upon the treatment with TNF-α.
Figure 1.
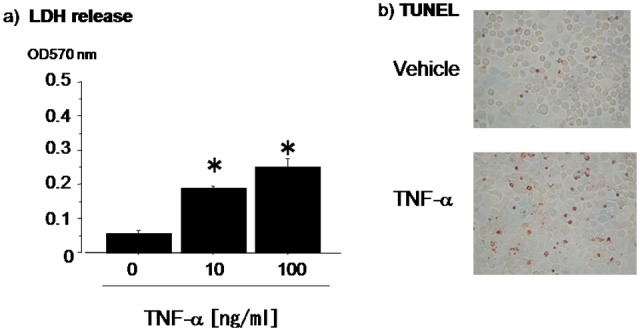
Tumor necrosis factor (TNF)–α induces cytotoxicity in chondrocytes in culture. TNF-α induced lactate dehydrogenase (LDH) release from chondrocytes in a dose-dependent manner (A). The number of TUNEL-positive cells was increased by the treatments of TNF-α in chondrocytes (B). Measurement of LDH release and TUNEL assay was performed 48 hours after TNF-α treatment. Data are expressed as means ± standard deviations. *P < 0.05, no treatments versus TNF-α treatments.
We further examined whether this TNF-α–induced apoptosis in the chondrocytes in culture can be detectable in individual cells. By using TUNEL assay, we examined morphologically the signals in chondrocytes in monolayer cultures. The baseline levels of the TUNEL signals were low in the chondrocyte cultures. However, the treatment with TNF-α significantly increased the appearance of chondrocytes exhibiting TUNEL signal (Fig. 1B). Thus, TNF-α treatment induces TUNEL-positive apoptosis in chondrocytes in vitro.
OPN Expression Is Enhanced upon TNF-α Treatment in the Chondrocytes in Culture
As we previously observed that OPN deficiency suppressed the levels of joint destruction in arthritis in association with the death of the chondrocytes in articular cartilage, we also tested these chondrocytes in monolayer cultures to see TNF-α effects on OPN expression. The chondrocytes were treated with TNF-α, and RNA levels were examined. RT-PCR analysis revealed that OPN mRNA levels were enhanced by TNF-α treatment. Therefore, OPN is the target of TNF-α signaling in the cultures of chondrocytes (Fig. 2).
Figure 2.
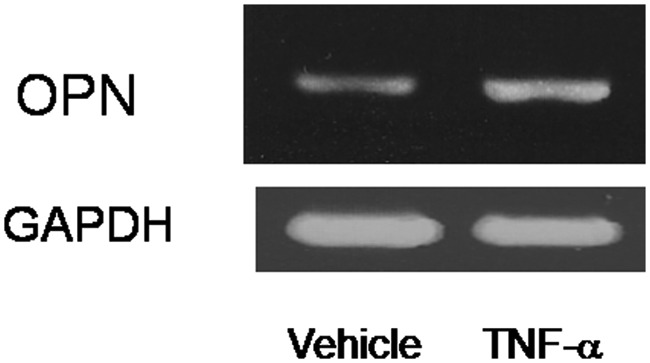
Osteopontin (OPN) expression is enhanced upon tumor necrosis factor (TNF)–α treatment in the chondrocytes in culture. RT-PCR analysis revealed that the expression of OPN mRNA in chondrocytes was increased by the treatment of TNF-α. Total RNA was exacted at 8 hours of TNF-α treatment.
OPN Deficiency Suppresses the Levels of TNF-α–Induced LDH Release into the Medium of Chondrocytes in Culture
To examine the effects of OPN deficiency on TNF-α–induced cytotoxicity, wild-type and OPN-deficient chondrocytes were cultured in the presence of TNF-α, and the levels of LDH release were examined. First, we determined the levels of the total LDH contents in the cells from wild-type and OPN-deficient chondrocytes. To obtain these data, we treated the chondrocytes in monolayer culture with Triton X-100 to release the total levels (lanes 1 and 2); total LDH levels in wild-type and OPN-deficient cells were similar.
In order to test the effects of OPN deficiency on the response to TNF-α, we cultured wild-type and OPN-deficient chondrocytes in the presence or absence of TNF-α for 48 hours. When OPN-deficient cells were cultured in the presence of vehicle alone, the levels of LDH release were similar to those in wild-type cells (Fig. 3A, lanes 3 and 4). TNF-α treatment increased LDH release into the medium in wild-type cells compared to the vehicle-treated cells. In contrast, OPN deficiency suppressed TNF-α–induced release of LDH into the medium down to the baseline levels of the vehicle-treated cells (Fig. 3A, lane 6 v. lanes 3 and 4). Therefore, OPN in the chondrocytes per se is involved in the TNF-α–induced LDH release.
Figure 3.

Osteopontin (OPN) deficiency suppresses the levels of tumor necrosis factor (TNF)–α–induced lactate dehydrogenase (LDH) release into the medium of chondrocytes in culture. The levels of LDH release induced by the treatment of Triton X-100 (TX-100) were similar in OPN-deficient chondrocytes compared with that in wild-type chondrocytes. On the other hand, the levels of LDH release induced by the treatment of TNF-α were significantly suppressed in OPN-deficient chondrocytes compared with that in wild-type chondrocytes (A). The fractions of TUNEL-positive cells were significantly increased by the treatment with TNF-α in wild-type chondrocytes. In contrast to wild-type, the increase of TUNEL-positive cells induced by TNF-α was suppressed in OPN-deficient chondrocytes (B, C). Data are expressed as means ± standard deviations. *P < 0.05.
OPN Deficiency Suppresses the Levels of Morphological Apoptosis Signals in Chondrocytes in Culture
To examine whether OPN deficiency suppresses TNF-α–induced morphological apoptosis signals in chondrocytes in monolayer culture, we subjected the cells to TUNEL assay. TNF-α treatment resulted in the appearance of TUNEL-positive cells in monolayer cultures of chondrocytes (Fig. 3B). In contrast, OPN deficiency suppressed such TNF-α–induced appearance of the TUNEL-positive chondrocytes in monolayer cultures to the baseline levels where the cells were treated with vehicle alone (Fig. 3B).
To quantify these observations, we counted the cells with TUNEL-positive (red color) signal and calculated the percentage of TUNEL-positive cells relative to the total number of the cells. In vehicle-treated cells, the baseline levels of TUNEL-positive cells were about 10% in either wild-type or OPN-deficient chondrocytes, and there was no statistically significant difference (Fig. 3C, lanes 1 and 2). Treatment with TNF-α increased TUNEL-positive cell number about 2-fold in the monolayer cultures of chondrocytes. In contrast, OPN deficiency suppressed such TNF-α–induced increase in the TUNEL-positive cell numbers down to baseline levels (Fig. 3C, lane 4 v. lane 3). These data indicate that OPN deficiency suppresses TNF-α–induced apoptosis of chondrocytes in culture based on at least 2 independent assays.
OPN Deficiency Suppresses TNF-α–Induced Increase in Caspase-3 Activities in Cultured Chondrocytes
The molecules involved in the effect of OPN deficiency on the TNF-α–induced apoptosis in chondrocytes were not clear. Therefore, we tested whether caspase-3 activities are modulated by OPN deficiency. In control cultures (vehicle alone), the chondrocytes from wild-type or OPN-deficient mice exhibited similar levels of baseline caspase-3 activities (Fig. 4, lanes 1 and 2). Upon treatment with TNF-α, wild-type chondrocytes exhibited about 2-fold increase in the levels of caspase-3 activity (Fig. 4, lane 3 v. lane 1). In contrast, OPN deficiency suppressed TNF-α–induced increase in caspase-3 activity down to the baseline levels (Fig. 4, lane 4 v. lane 2). These data indicated that OPN deficiency suppresses TNF-α–induced caspase-3 activity in chondrocytes.
Figure 4.
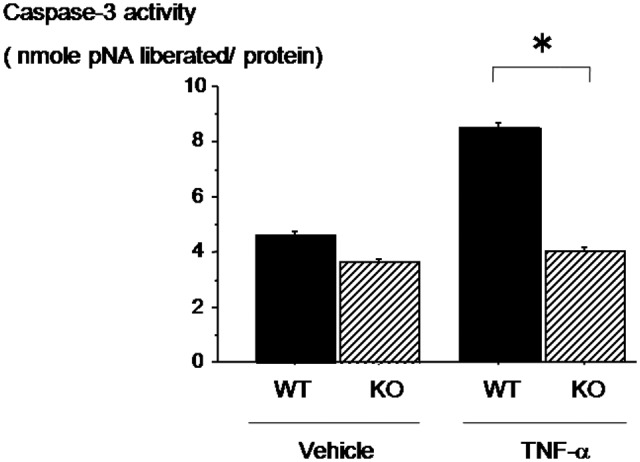
Osteopontin (OPN) deficiency suppresses tumor necrosis factor (TNF)–α–induced increase in caspase-3 activities in cultured chondrocytes. Caspase-3 activity was increased by the treatment of TNF-α in wild-type chondrocytes. However, OPN deficiency suppresses TNF-α–induced caspase-3 activity in chondrocytes. Data are expressed as means ± standard deviations. *P < 0.05.
Overexpression of OPN Increases the Levels of LDH Release from Chondrocytes in Monolayer Culture
Because OPN deficiency suppresses the levels of TNF-α–induced apoptosis as indicated by the release of LDH, we further examined whether overexpression of OPN enhances TNF-α–induced release of LDH. To do this, we constructed an adenovirus system. Chondrocytes were infected with either adenovirus carrying LacZ or OPN. LDH release was relatively low in the cells, which were infected with either adenovirus containing LacZ or OPN and were cultured in the presence of vehicle alone. Treatment with TNF-α increased LDH release from chondrocytes as seen before. Infection with adenovirus containing the LacZ gene as control did not alter TNF-α–induced increase in LDH release from the chondrocytes. In contrast, infection with adenovirus that expresses OPN further enhanced the levels of LDH release even in the cultures of chondrocytes, which were already releasing high levels of LDH after the treatment with TNF-α. (Fig. 5) These data further supported the idea that OPN is enhancing the chondrocyte apoptosis induced by TNF-α.
Figure 5.

Overexpression of osteopontin (OPN) increases the levels of lactate dehydrogenase (LDH) release from chondrocytes in monolayer culture. Chondrocytes were transfected with lacZ and OPN containing adenovirus vectors. Then, these cells were subjected to treatment with tumor necrosis factor (TNF)–α 24 hours later. LDH release was measured 48 hours after the treatment with TNF-α. Data are expressed as means ± standard deviations. *P < 0.05.
Discussion
In this study, we examined how OPN deficiency affects apoptosis of chondrocytes in culture. We showed biochemically that OPN deficiency suppressed TNF-α–induced release of LDH from the chondrocytes in culture. TNF-α–induced apoptosis of chondrocytes was also morphologically monitored based on TUNEL assay. OPN deficiency suppressed TNF-α–induced increase in TUNEL signals in chondrocytes. OPN deficiency suppresses TNF-α–induced increase in signaling. These suppressive effects on LDH release and TUNEL were associated with suppression of TNF-α–induced increase in caspase-3 activities in these chondrocytes in culture. Therefore, at least one of the target events affected by OPN deficiency is to suppress the levels of caspase-3 activities.
We previously observed that chondrocyte apoptosis in articular cartilage was reduced in OPN-deficient mice. In the joints of RA patients, apoptosis of chondrocytes has been observed.8 Cytotoxic mediators such as TNF-α, TRAIL, H2O2, and NO have been reported to induce apoptosis in chondrocytes.4,9,10 However, factors that modulate apoptosis in chondrocytes in arthritic joints have not been fully identified yet. Modulators for such apoptosis-inducing signals may alter the levels of severity and pathophysiology of joint destruction in RA. Variation in the levels of such modulators may lead to diversity in individual manifestations. In fact, the pathological features of RA are known to vary in individual patients. We have previously demonstrated that OPN deficiency suppressed chondrocyte apoptosis in an arthritic model induced by an injection with a mixture of the cocktail of anti–type II collagen antibodies followed by LPS treatment.1 Our current data indicated that OPN deficiency suppressed apoptosis of chondrocytes in culture subjected to treatment with TNF-α. Thus, OPN deficiency suppressed TNF-α–induced apoptosis of chondrocyte cells autonomously. This OPN deficiency suppression of apoptosis was via its suppression of the caspase-3 levels.
OPN is an extracellular matrix protein containing Arg-Gly-Asp (RGD).1,11,12,13 This motif has been shown to interact with integrins and to activate monocytes and other types of cells. The signaling through this RGD is important for the cell survival in endothelial cells and supports angiogenesis in tissue formation.1 In arthritis models, inflammatory cell infiltration and angiogenesis in synovium are observed upon an injection with monoclonal antibodies against type II collagen. OPN deficiency suppresses both manifestations.1 OPN is a cytokine and is involved in inflammatory events such as angiogenesis. It is also expressed in chondrocytes, while it is secreted and acts in the intracellular compartment. Involvement of OPN in several types of inflammatory models has been reported using the different tissues such as the brain. However, how OPN acts has not been fully known. We show here that OPN deficiency suppressed chondrocyte apoptosis induced by TNF-α in culture. Moreover, adenovirus infection experiments revealed that OPN overexpression exacerbates TNF-α–induced release of LDH as an indicator of chondrocyte apoptosis. These observations imply that in both isolated chondrocytes and articular cartilage in vivo, OPN is one of the factors to be in favor to exacerbate the destruction of the joint.
OPN mRNA expression was increased by TNF-α treatment. This may be also indicative of the action of this molecule as we have shown that OPN deficiency protects chondrocyte apoptosis induced by TNF-α. Therefore, one of the actions of OPN is to promote turnover of the cells upon the event of joint destruction or joint inflammation. OPN deficiency suppressed the number of apoptotic cells and caspase-3 activities induced by TNF-α. These data suggested that OPN is one of the targets for the possible measures to control the activity of RA that destroy the joint.
OPN overexpression with adenovirus vectors enhanced the cytotoxic release of LDH from chondrocytes upon the treatment with TNF-α. However, OPN overexpression per se did not alter the viability of normal chondrocytes, which were not treated with TNF-α. Therefore, it is suggested that OPN alone does not directly induce cytotoxity in chondrocytes. Rather, OPN would modulate (enhance) the TNF-α–mediated apoptosis signals. In this case, enhancement of OPN expression upon the treatment with TNF-α would be an endogeneous chondrocyte signal for the promotion of the inflammation. Furthermore, blocking of OPN activity or OPN signaling would provide opportunity for the suppression of the inflammatory signals in the joint that destroy cartilage and bone. It was shown that a neutralizing antibody against OPN, M5, is effective in arthritis. M5 antibody recognizes α9β1 integrin binding sites in OPN, which appears after the cleavage with thrombin as a cryptic site of the OPN to bind to a new repertoire of integrins. However, M5 antibody does not alter the cytotoxicity estimated based on the release of LDH induced by TNF-α (data not shown). Therefore, it is suggested that the function of OPN in TNF-α–induced apoptosis in our experiments would not be mediated by α9β1 integrin. Rather, it may be through the RGD sequence-dependent events. OPN was also reported to be located intracellularly. In the cytoplasmic compartment, OPN binds to CD44.14 Therefore, OPN may not only act as secreted molecule but it could also act through other activities inside the cells. This part of OPN functions is still to be indicated.
OPN was reported to be involved in inflammation through the function of chemotaxis14 and Th1 cytokine induction.12 OPN deficiency suppresses inflammatory animal models such as RA in joints and experimental autoimmune encephalomyelitis.1,15 On the other hand, the anti-inflammatory function of OPN was reported as well. OPN may inhibit IL-1–induced NO and prostaglandin production in human osteoarthritis (OA) chondrocytes.16 Thus, OPN may act in dual ways depending on the species or on the ages of humans or animals.
Our observation suggests that OPN is involved in the susceptibility to TNF-α–induced apoptosis in chondrocytes. It would be also intriguing if OPN promotes apoptosis in cultured related cells such as osteoblasts. Although we focus our efforts on chondrocytes this time, whether the overexpression of OPN affects the induction of apoptosis in the other cells is still an interesting question to be tested in the future.
In conclusion, we have shown that OPN deficiency suppresses cytotoxic cell death induced by TNF-α in chondrocytes in culture. Our observations on caspase-3 would at least in part explain the effects of OPN deficiency on articular cartilage destruction.
Footnotes
The authors declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This work was supported by grants-in-aid from the Japanese Ministry of Education (Global Center of Excellence [COE] Program, International Research Center for Molecular Science in Tooth and Bone Diseases: 18109011, 18659438, 18123456, 20013014) and grants from the Japan Space Forum, NASDA, and ABJS (Advanced Bone and Joint Science) Strategic Research Networks Projects (Japan Society for Promotion of Science Core to Core Program, Research for the Future Program, Genome Science).
References
- 1. Yumoto K, Rittling SR, Nifuji A, Uede T, Denhardt DT, Noda M, et al. Osteopontin deficiency protects joints against destruction in anti-type II collagen antibody-induced arthritis in mice. Proc Natl Acad Sci U S A. 2002;99(7):4556-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Seemayer CA, Distler O, Kuchen S, Muller-Ladner U, Michel BA, Neidhart M, et al. [Rheumatoid arthritis: new developments in the pathogenesis with special reference to synovial fibroblasts]. Z Rheumatol. 2001;60(5):309-18. [DOI] [PubMed] [Google Scholar]
- 3. Kim HA, Song YW. Apoptotic chondrocyte death in rheumatoid arthritis. Arthritis Rheum. 1999;42(7):1528-37. [DOI] [PubMed] [Google Scholar]
- 4. Schuerwegh AJ, Dombrecht EJ, Stevens WJ, Van Offel JF, Bridts CH, De Clerck LS. Influence of pro-inflammatory (IL-1 alpha, IL-6, TNF-alpha, IFN-gamma) and anti-inflammatory (IL-4) cytokines on chondrocyte function. Osteoarthritis Cartilage. 2003;11(9):681-7. [DOI] [PubMed] [Google Scholar]
- 5. Rittling SR, Matsumoto HN, McKee MD, Nanci A, Noda M, Denhardt DT, et al. Mice lacking osteopontin show normal development and bone structure but display altered osteoclast formation in vitro. J Bone Miner Res. 1998;13(7):1101-11. [DOI] [PubMed] [Google Scholar]
- 6. Yagi K, Nifuji A, Shinomiya K, Nakashima K, DeCrombrugghe B, Noda M, et al. Bone morphogenetic protein-2 enhances osterix gene expression in chondrocytes. J Cell Biochem. 2003;88(6):1077-83. [DOI] [PubMed] [Google Scholar]
- 7. Le Stunff H, Auger R, Kanellopoulos J, Raymond MN. The Pro-451 to Leu polymorphism within the C-terminal tail of P2X7 receptor impairs cell death but not phospholipase D activation in murine thymocytes. J Biol Chem. 2004;279(17):16918-26. [DOI] [PubMed] [Google Scholar]
- 8. Yatsugi N, Tsukazaki T, Osaki M, Koji T, Yamashita S, Shindo H. Apoptosis of articular chondrocytes in rheumatoid arthritis and osteoarthritis: correlation of apoptosis with degree of cartilage destruction and expression of apoptosis-related proteins of p53 and c-myc. J Orthop Sci. 2000;5(2):150-6. [DOI] [PubMed] [Google Scholar]
- 9. Kuhn K, Shikhman AR, Lotz M. Role of nitric oxide, reactive oxygen species, and p38 MAP kinase in the regulation of human chondrocyte apoptosis. J Cell Physiol. 2003;197(3):379-87. [DOI] [PubMed] [Google Scholar]
- 10. Yamamoto N, Sakai F, Kon S, Seki N, Fujii T, Uede T, et al. Essential role of the cryptic epitope SLAYGLR within osteopontin in a murine model of rheumatoid arthritis. J Clin Invest. 2003;112(2):181-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Asou Y, Rittling SR, Shinomiya K, Nifuji A, Denhardt DT, Noda M, et al. Osteopontin facilitates angiogenesis, accumulation of osteoclasts, and resorption in ectopic bone. Endocrinology. 2001;142(3):1325-32. [DOI] [PubMed] [Google Scholar]
- 12. Ashkar S, Weber GF, Rittling SR, Denhardt DT, Glimcher MJ, Cantor H, et al. Eta-1 (osteopontin): an early component of type-1 (cell-mediated) immunity. Science. 2000;287(5454):860-4. [DOI] [PubMed] [Google Scholar]
- 13. Gravallese EM. Osteopontin: a bridge between bone and the immune system. J Clin Invest. 2003;112(2):147-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhu B, Suzuki K, Goldberg HA, Rittling SR, Denhardt DT, Sodek J, et al. Osteopontin modulates CD44-dependent chemotaxis of peritoneal macrophages through G-protein-coupled receptors: evidence of a role for an intracellular form of osteopontin. J Cell Physiol. 2004;198(1):155-67. [DOI] [PubMed] [Google Scholar]
- 15. Chabas D, Baranzini SE, Rittling SR, Denhardt DT, Oksenberg JR, Steinman L, et al. The influence of the proinflammatory cytokine, osteopontin, on autoimmune demyelinating disease. Science. 2001;294(5547):1731-5. [DOI] [PubMed] [Google Scholar]
- 16. Potter MR, Rittling SR, Denhardt DT, Roper RJ, Teuscher C, Weis JJ, et al. Role of osteopontin in murine Lyme arthritis and host defense against Borrelia burgdorferi. Infect Immun. 2002;70(3):1372-81. [DOI] [PMC free article] [PubMed] [Google Scholar]