

Abstract
Background:
Our goal was to set up an ex vivo culture system to assess whether cartilage wounding (partial-thickness defects) can induce morphological changes in neighboring chondrocytes and whether these cells can translocate to the surface of the defect.
Methods:
Two-millimeter partial-depth defects were created in human osteochondral explants followed by culture for up to 4 weeks. Frozen sections of defects and defect-free regions were labeled using immunofluorescence for a plasma membrane protein, CD44, and actin with TRITC-phalloidin. Viable nuclei were detected with Hoechst 33342. Differential interference contrast (DIC), confocal, and transmission electron microscopy (TEM) were used to examine process extension.
Results:
Significant changes in cell morphology occurred in response to wounding in the superficial and deep cartilage zones. These included cell flattening, polarization of the actin cytoskeleton, extension of pseudopods projecting towards the edge of the defect, and interactions of these filopodia with collagen fibers. Cell density decreased progressively in the 300-µm zone adjacent to the defect to an average of approximately 25% to 35% after 3 weeks. Concomitant increases in cell density in the defect margin were observed. By contrast, minimal changes were seen in the middle cartilage zone.
Conclusions:
These novel observations strongly suggest active cartilage cell responses and movements in response to wounding. It is proposed that cartilage cells use contact guidance on fibrillated collagen to move into and populate defect areas in the superficial and deep zones.
Keywords: cartilage, pseudopod, motility, collagen, repair
Introduction
There are many causes of cartilage injury. Small defects can be the result of direct blunt trauma, unexpected and sudden joint loading, and torsional injuries.1-3 In one study, 20% of knees with acute hemarthrosis had a chondral injury on arthroscopy.4 Partial-thickness chondral injuries result in a poor healing response. Blood vessels, marrow mesenchymal cells, and macrophages are unable to gain access to chondral injuries through intact subchondral bone.1,2,5-7 In damaged articular cartilage, a transient increase in matrix production and clonal proliferation of chondrocytes has been described,8 but chondrocytes are not thought to play an effective role in the repair of partial-thickness articular cartilage injuries.1-3,7 Indeed, an absence of chondrocytes in the area surrounding defects has been noted.9 In an in vitro model, bovine cartilage explants were wounded with a trephine. Only approximately 20% of the chondrocytes within 200 µm of the wound were necrotic/apoptotic as determined by the TUNEL assay.10
When surrounded by the pericellular matrix, the chondrocyte exhibits a rounded morphology, appears stationary, and has little if any mitotic activity.8 However, Bush et al. examined osteochondral explants and observed chondrocyte cell processes in articular cartilage extending up to 30 µm in length.11 These processes tended to be aligned with collagen fibrils and were hypothesized to be either secondary to microdamage to the surrounding matrix or a result of pericellular matrix degradation. When chondrons, which are chondrocytes with their native pericellular matrix, are grown in monolayer, the cells extend pseudopodia that contain actin and shed their pericellular matrix (Lee, unpublished observations). Also, when chondrocytes grown in monolayer are exposed to type II collagen fibrils, adhesions via β1 integrins occur between the collagen fibrils and the chondrocytes’ pseudopodia. The contractile force exerted through this contact is sufficient to bend type II collagen fibrils.12 Further, translocation of cultured chondrocytes into devitalized cartilage has been observed,13 and chondrocyte migration has also been reported in monolayer culture.14,15 Chondrocytes migrate on 2-dimensional fibronectin substrata without dedifferentiating to produce collagen I and maintain expression of collagen II.15
In the superficial zone of articular cartilage, collagen fibrils are oriented largely parallel to the joint surface. In the middle and deep zones, collagen fibrils are arranged perpendicular to the joint surface.16,17 Therefore, in a partial-thickness defect, collagen fibrils are predicted to be mostly perpendicular to the defect surface in the superficial zone, parallel in the middle zone, and perpendicular again in the deep zone (Fig. 1). We used osteochondral explants with partial-thickness defects to better characterize the response of chondrocytes in articular cartilage to acute matrix injury. We predicted that injury would create fibrillated areas in cartilage containing exposed large type II collagen fibrils and that this might stimulate more process formation and adhesion of cell processes to the free collagen. We proposed that chondrocytes would respond to local matrix damage by forming processes and eventually migrating. A secondary hypothesis was that the direction of cell migration would be dependent on the orientation of the collagen fibrils.
Figure 1.
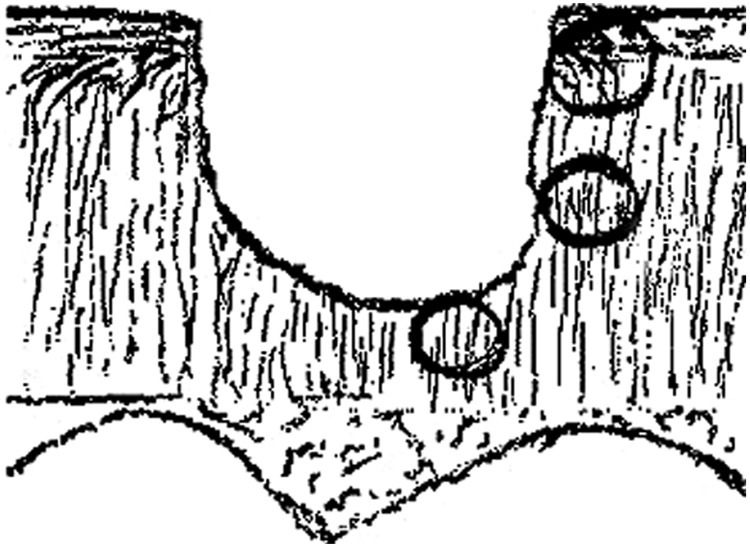
Drawing depicting a partial-thickness cartilage injury and relative predicted average collagen orientation in 3 regions adjacent to the defect. In the superficial zone (top circle) and deep zone (bottom circle), the majority of collagen fibrils are perpendicular to the defect edge. In the middle zone, most fibrils are predicted to run parallel to the defect edge.
Methods
Using differential interference contrast (DIC), fluorescence (confocal and conventional), and transmission electron microscopy (TEM), we studied the formation of actin-containing cell processes from chondrocytes in normal articular cartilage adjacent to defects and the time-related increase in length of these cell processes. We followed these injuries for 4 weeks.
Specimens and Culture
Osteochondral explants 2 to 3 cm in diameter were obtained from 10 patients at the time of total knee arthroplasty, total hip arthroplasty, or hip hemiarthroplasty. Mean patient age was 62 years (range = 18-92 years). There were 7 knees (5 osteoarthritic and 2 normal cartilage from tumor amputation) and 3 hips (2 from hip fractures and 1 from spastic hip dislocation with cerebral palsy). Explants were taken from grossly normal-appearing (nonarthritic) areas of the femoral condyles and femoral heads. There was no synovial tissue with the explants. Explants were rinsed with phosphate-buffered saline (PBS) and twice in DMEM with gentamycin and kanamycin. Cultures were maintained in 15 mL Opti-MEM with GlutaMax1 (cat. # 51985-034, Life Technologies, Carlsbad, CA) medium with penicillin/streptomycin, 2% FBS, 2.7 mM CaCl2, and 25 µg/mL L-ascorbic acid-2-monophosphate (Sigma, St. Louis, MO). Explant cultures were incubated at 37 °C, 5% CO2, in 50 mL sterile, conical polypropylene tubes. The media were changed every 2 to 3 days.
Creation and Harvest of Defects
Defects were created within 3 days of explant harvest using a Dremel Minimite drill (Racine, WI) and a 2.8-mm drill bit. Four to 6 defects were made in each specimen. Only defects that were determined to be partial thickness by gross examination were cultured. At 0 days, 3 days, and 1, 2, 3, and 4 weeks, the explants were harvested. For immunofluorescence, the explants were fixed and stored in 4:1 methanol:DMSO at −20 °C.18 Prior to embedding in O.C.T. medium (Tissue-Tek, Sakura Finetek U.S.A.), explants were rehydrated in PBS for 30 minutes. For phalloidin labeling, explants were fixed in 3.7% formaldehyde in PBS for 30 minutes, rinsed in PBS, and then embedded in O.C.T. Twenty-micrometer sections were cut using a cryostat (Frigocut 2800, Reichert-Jung, Leica Microsystems, Wetzlar, Germany). The sections were not allowed to dry.
Immunofluorescence
The sections were rinsed in PBS after mounting and blocked with 1% BSA in PBS. The primary antibodies used were H4C4 to CD44 (prepared by J.E.K. Hildreth and J.T. August, Developmental Studies Hybridoma Bank) and monoclonal antibody C4F6 to type II collagen (provided by Clinton Chichester). Both antibodies were specific.18-21 TRITC-labeled phalloidin (Sigma) was used to stain sections for actin at each time point. Controls included omitting the primary antibody or TRITC-phalloidin from duplicate sections. The control sections were incubated in buffer + 1% BSA, while experimental sections were in solution with the primary antibody or phalloidin. For detection of viable chondrocytes, all sections were stained with Hoechst 33342 (10 µg/mL, Molecular Probes, Eugene, OR). Sections were mounted with polyvinyl alcohol mounting medium22 containing 1% n-propyl gallate and examined with a Leica confocal microscope (Wetzlar, Germany) using a 40x oil immersion objective or an Olympus IX70 fluorescence microscope equipped with a chilled CCD camera (Orca, Olympus, Tokyo, Japan). Confocal micrographs were produced by combining multiple focal levels from a single section.
Determination of Cartilage Cellularity
After Hoechst staining, viable nuclei were detected with fluorescence microscopy.18 Using a stage micrometer, the 40x dry objective and the 1.5x magnifier with an Olympus IX70 microscope, it was determined that one high power field was approximately 300 µm in diameter. High power fields touching the edge of the defect were defined as adjacent fields, and high power fields greater than 300 µm away from the defect edge were defined as distal. Using a hand-held counter, intact nuclei were counted per high power field in superficial, middle, and deep zones adjacent and distal to the defect edge. Sections from 3 separate explants from the same patient were used to compile the data. Sections were examined from osteochondral explants 3 days and 1, 2, 3, and 4 weeks after defect creation.
TEM
TEM was used to evaluate cell processes near the fibrillated border. Explants containing defects 4 weeks old were fixed overnight in 2% paraformaldehyde/2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4, at 4 °C. Following several buffer washes, the samples were decalcified in 0.1 M sodium EDTA in 0.1 M sodium cacodylate, pH 7.4, until the underlying bone softened. After decalcification, the cartilage pieces were postfixed in a mixture of 1% osmium tetroxide and 1.25% potassium ferrocyanide buffered with 0.1 M sodium cacodylate, pH 7.4, for 2 hours. The specimens were dehydrated with increasing concentrations of ethanol, transferred to propylene oxide, and gradually infiltrated with Spurr’s resin (Ted Pella Inc., Redding, CA). Following polymerization, the blocks were ultrathin sectioned at 70 nm perpendicular to the articular surface. Grid-mounted sections were double stained with 4% aqueous acetate and 4% lead citrate and observed on a LEO EM-910 transmission electron microscope (LEO Electron Microscopy Inc., Thornwood, NY) at an accelerating voltage of 80 kV.
Results
Overall Observations
In human articular cartilage, adjacent to the experimentally produced partial-thickness defects, we were able to demonstrate the formation of actin-containing cell processes from chondrocytes. We also found a time-related increase in length of these cell processes. Figure 1 is a sketch of a defect that illustrates the direction of the collagen fibers in relation to the defect. Over 4 weeks, fibrillation developed and appeared to increase in the deep and superficial zones of cartilage. We found this fibrillation difficult to quantify. In those areas where fibrillation was seen, cell polarization and process formation were greatest. There was also a time-related hypocellularity that developed in the matrix adjacent to the defects, and after 4 weeks, many cells could be clearly seen lining the surface of the defect. Frequently, vacated lacunae could be seen below these lining cells.Figures 2 to 9 strongly support these observations, as detailed below.
Figure 2.

Chondrocytes extend cell processes toward the edge of the defect. The red fluorescence is CD44 labeling of plasma membranes, and the green is Hoechst staining of nuclei (images are pseudocolor). (A) Confocal microscope image of a defect edge in the middle zone on the day of defect creation. Chondrocytes were rounded and found in abundance adjacent to the defect edge. Bar = 10 µm. (B) Confocal image of a defect edge in the deep zone at 3 days. A cell process extends from the chondrocyte to the fibrillated edge of the defect. Bar = 10 µm. (C) Confocal image at 4 weeks demonstrating multiple cells with extensions toward the defect and cells on the edge of the defect. Bar = 10 µm. (D) DIC image of defect edge in the deep zone at 3 days (corresponding to Fig. 2B). A small cleft extends from the chondrocyte to the fibrillated edge of the defect (arrows). Bar = 10 µm.
Figure 9.
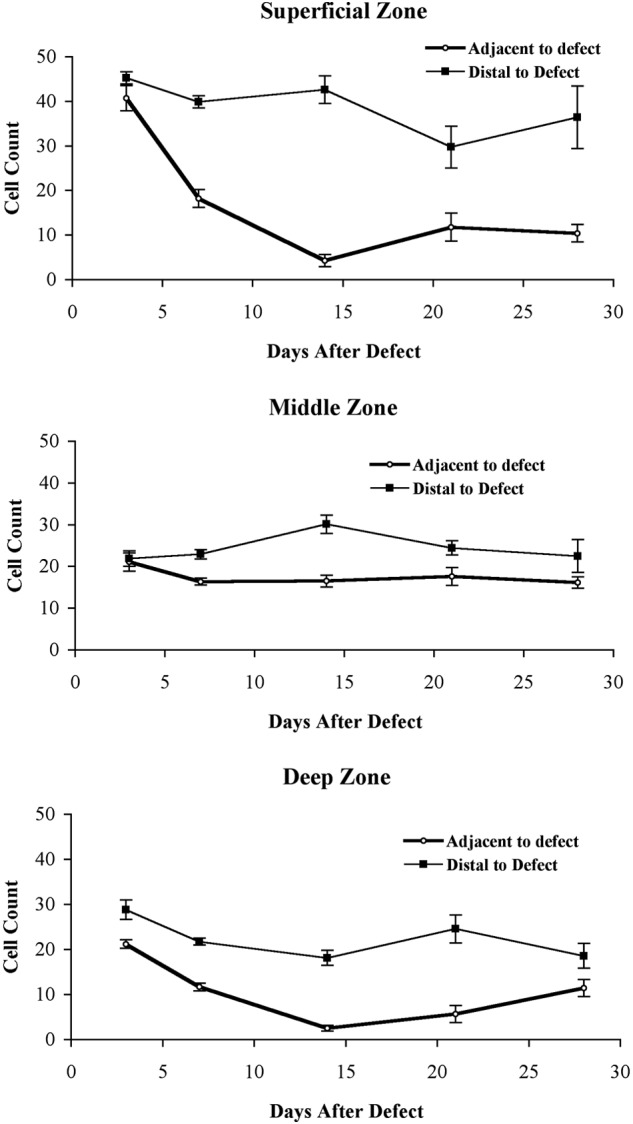
Graphs comparing the change of cartilage cellularity adjacent and distal to the defect edges in 3 different zones of the explants. Cells were counted in fields of 300 µm diameter, and the graph shows the total number of cells counted per field for each of the 3 cartilage zones at different times. To provide a semiquantitative estimate of cells per unit area, we calculated the initial cell numbers for the superficial, middle, and deep zones of the area adjacent to the defect, and these were 5.6, 2.8, and 2.8 cells per 100 µm2 (not shown). (A) The changes in cellularity of the superficial zone adjacent to the defect (diamond) and distal to the defect (square) are compared. Over 300 µm from the defect, the cellularity of the cartilage did not change significantly, while within 300 µm, the cellularity dramatically decreased during the first 2 weeks after the defect was created. (B) Small changes in the middle zone are seen. Again, greater than 300 µm from the defect, the cellularity was not affected dramatically by culture conditions. The cellularity adjacent to the defects was also unchanged. (C) Changes in the deep zone are demonstrated. These included a gradual decrease in the cellularity in the deep zone over the first 2 weeks. The cellularity of the deep zone distal to the defects, like that of the middle and superficial zones, remained relatively stable throughout the 4 weeks. In the area adjacent to the defect, the cellularity increased during the last 2 weeks (n = 3 for all points; mean ± SD is shown).
Cells Extend Processes
The analysis of sections from sequential time points revealed a time-related progression of cell process formation. At day 0 after defect creation, cells adjacent to the defect edge remained rounded, as exemplified in Figure 2A. At day 3 after defect creation, early cell process formation was detected in some of the cells by CD44 immunolabeling (Fig. 2B and 2D), particularly along the superficial and deep aspects of the defect. The length of the processes varied and appeared to increase over the first 3 weeks. Figure 2C shows cells close to the edge of the defect at 4 weeks and clearly demonstrates that the cells have elongated and that their long axes are directed towards the wound site. At this time, the number of cells directly adjacent to and lining the defect edge had also increased.
Cell Processes Contain Actin
Labeling with TRITC-phalloidin was used to detect actin in cellular processes near the defect. The presence of actin provided further support that the processes observed with CD44 immunolabeling were cell extensions. The timing and direction of process formation detected by phalloidin (Fig. 3) appeared quite similar to those detected by CD44 immunofluorescence (Fig. 2). Immediately following the formation of the defect, cells near the edge of the defect appeared round (Fig. 3B). After several days, these cells began to extend actin-containing processes toward the edge of the defect, and over time, these extensions increased in length (Fig. 3B, 3D, 3F, and 3H). At lower magnification, the increase over time in cells with extensions is evident (Fig. 4). At 2 weeks, there was significant variability in the number of cells with extensions per section. In some sections, multiple cells with extensions were observed (Fig. 4E and 4F), but other sections showed chondrocytes with minimal changes in cell morphology. This may reflect a natural variability in the response of cartilage from different donors during a time frame when critical changes in cell translocation start to take place. After 4 weeks, each section showed consistent changes in cell morphology, and by this time, empty lacunae could be clearly seen in some sections (Fig. 4G and 4H).
Figure 3.

Cell processes contain actin as demonstrated by phalloidin labeling. Images show the increasing length over time of actin-containing cellular extensions toward the defect in the superficial zone. Images were obtained on day 0 (A, B), 3 (C, D), 7 (E, F), 14 (G, H), and 28 (I, J). Note in D that the cell process, as seen in this section, is approximately equal in length to the cell body after 3 days of culture, while the pseudopods shown in F and H are 2 to 4 times the observed body length at 7 to 14 days. Arrows indicate edges of defects in the DIC images. Bar = 10 µm.
Figure 4.

Low magnification images of the superficial zone of defect edges demonstrating the orientation and number of cellular extensions with time. Phalloidin labeling for actin at day 0 (A, B), 7 (C, D), 14 (E, F), and 28 (G, H) shows the adjacent chondrocytes extending cell processes toward the edge of the defect over time. By 4 weeks, resurfacing of the defect edge has occurred. Bar = 30 µm.
Direction of Process Extension along the Direction of Collagen Fibers
Process extension was roughly parallel to predicted collagen bundle orientation. Areas around defects that exhibited the most fibrillation were the deep and superficial zones, and these findings are consistent with the observed process formation. The predicted collagen fibril orientation is perpendicular to the defect surface (Fig. 1) in these zones. The predicted collagen fibril orientation for the middle zone is parallel to the cut edge, and less fibrillation was observed in these areas. Immediately following the formation of the defect and during the first 2 weeks in culture, a fibrillated border was not evident in the superficial and deep zones. After 4 weeks, the fibrillation was observed in these zones. Using confocal microscopy, fibrillated defect edges demonstrated exposed type II collagen fibrils in sheets (Fig. 5). Further from the defect, the matrix appeared denser with proteoglycans and other matrix proteins interspersed throughout the collagen infrastructure.
Figure 5.
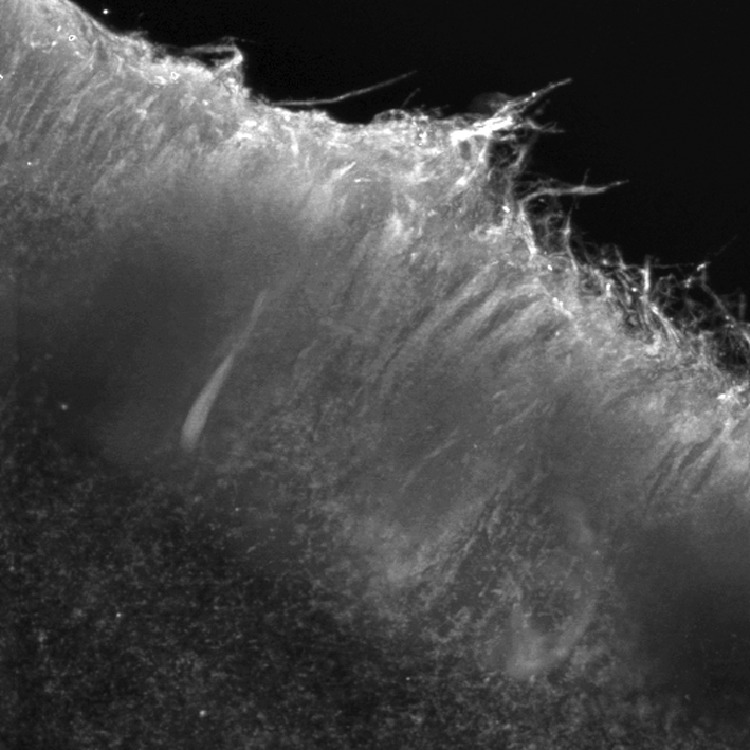
Orientation of collagen fibrils adjacent to defect edges at 4 weeks. The image was centered on the deep zone of the defect, with the defect edge located superiorly. Confocal image of a defect edge labeled with C4F6 for type II collagen.
The association of cell processes with collagen fibrils was evident in transmission electron micrographs at 4 weeks after wounding. These pseudopodia were not visualized in continuity because TEM sections were only 70 nm thick. In Figure 6A, a cell process has extended through the pericellular matrix and is attached to a collagen fibril. In a different cell, several smaller pseudopodia are seen adhering to collagen fibrils in the interterritorial region of the matrix (Fig. 6B). Smaller pseudopodia adhering to collagen fibrils may direct larger cell extensions along collagen bundles. Processes contain large amounts of rough endoplasmic reticulum and Golgi apparatus and are consistent with an increase in matrix production after matrix injury (Fig. 6B).
Figure 6.

Transmission electron micrographs showing chondrocyte processes adjacent to defects at 4 weeks after wounding. (A) A process has extended through the pericellular matrix and attached to collagen fibrils. (B) A different cell shows a larger process with smaller filipodia adhering directly to a collagen fibril.
Process Formation Precedes Cell Translocations
By day 3, process formations were identified; however, an increase in the number of chondrocyte nuclei adjacent to matrix defects was not observed until 3 weeks following defect formation. Because each image represents a point in time, actual migration could not be observed directly. Chondrocyte migration was inferred based on cell morphology (polarization towards the surface edge, process formation), position of nuclei outside of lacunae, and the hypocellularity (and empty lacunae) of matrix adjacent to defects. Importantly, the topography of the cell decrease in areas adjacent to the defect coincided with increases in cellularity along the defect edge (see below).
Resurfacing of Matrix Defects
In the majority of samples, apparent cellular migration resulted in a lining of the defect edge with chondrocytes. This was usually evident by 3 to 4 weeks (Fig. 7A). Many of these cells seemed to retain their processes, which were seen within the cut edge of the defect (Fig. 7B and 7C). Zones of hypocellularity were seen deep to all defect regions lined by chondrocytes, suggesting a pattern of migration. This was observed in both superficial and deep zones, but not usually in the middle zone, where areas devoid of lining cells and underlying hypocellularity were noted (Fig. 8).
Figure 7.
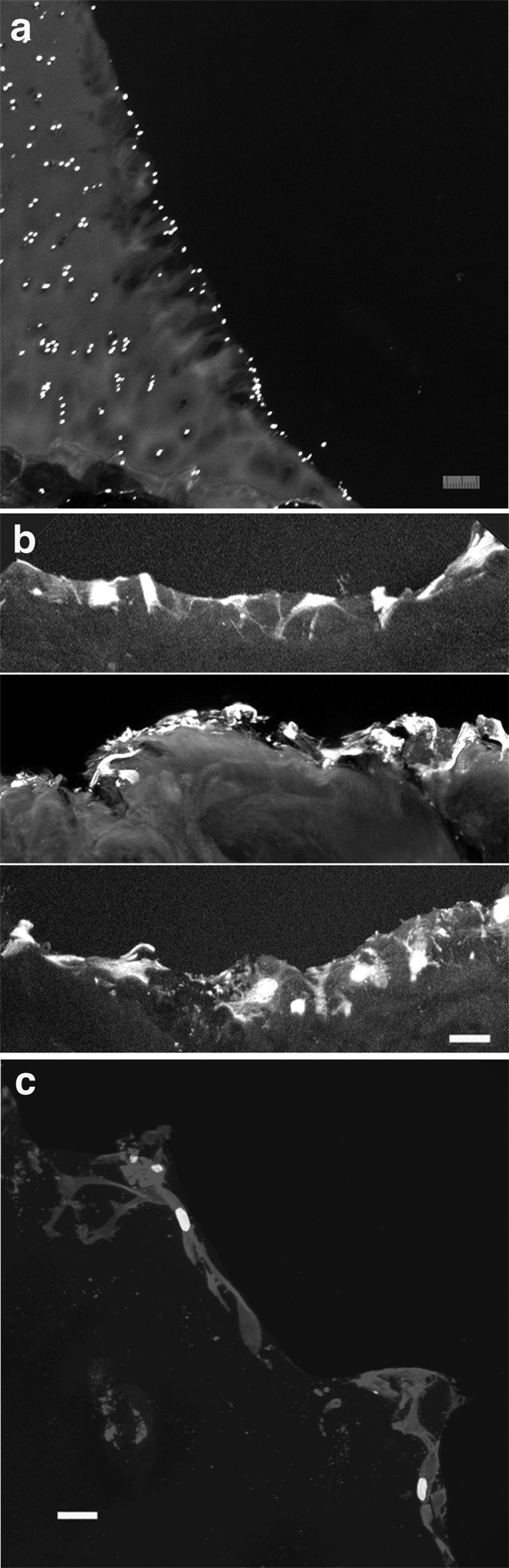
Resurfacing of defect edges after 4 weeks. (A) Sections were stained with Hoechst to identify viable nuclei. Viable chondrocytes have lined a fibrillated edge along the deep zone of a defect. Note the relatively hypocellular matrix adjacent to the defect. Bar = 30 µm. (B) Collection of confocal images of different sections after CD44 immunolabeling for plasma membranes showing the chondrocytes extending across the surface of the defect. Bar = 30 µm. (C) Higher magnification of another section shows CD44-labeled chondrocytes spread over the defect edge. Bright spots in the center of the chondrocytes are nuclei (Hoechst stain). Bar = 10 µm.
Figure 8.

Nuclei are shown in the middle cartilage zone in a region corresponding to the circle pointed by the arrow in the diagram.
Changes in the Cellularity of the Superficial, Middle, and Deep Zones
Regions of hypocellularity with empty lacunae containing remnants of cell processes were seen in many sections adjacent to the superficial and deep zones of cartilage defects but not in the middle zone of the defects (Fig. 4G and 4H show examples of empty lacunae after 4 weeks close to the edge of the defect in the superficial zone). In order to quantify the amount of chondrocyte attrition near defects, chondrocyte counts were performed in explants from 3 different donor sites. The cell counting area extended 300 µm from the defect edge and included viable cells within the cartilage but not cells located on the defect edge. In this area adjacent to the defects, superficial zone cellularity decreased during the first 2 weeks and seemed to stabilize approximately 10% to 25% of initial cell density between 2 and 4 weeks (Fig. 9A). By marked contrast, the cellularity of the cells more distal from the defect remained relatively stable (decreased to ~85% of initial density by 4 weeks). In the middle zone, there was an initial very small decrease (~20%) in the cellularity adjacent to the defect. The cellularity then did not appreciably change over the following 4 weeks (Fig. 9B). In the deep zone (Fig. 9C), there was a rapid decrease in the number of viable cells similar to that seen in the superficial zone during the first 2 weeks to approximately 20% of initial cell density, and then an apparent partial recovery was observed in the last 2 weeks.
It is important to note that in all zones, the cellularity of the area extending from 300 to 600 µm from the defect edge (distal) remained constant over the entire 4-week period, showing that neither migration nor cell death was caused by explant harvesting or culture conditions.
Discussion
The natural history of partial-thickness cartilage injuries in humans is a topic of debate. While it is clear that large partial-thickness lesions have no ability to heal,1,2,5,23-25 many authors feel that small partial-thickness injuries may resolve without symptomatic sequelae.2,5,6 Proposed mechanisms for the resolution of small partial-thickness injuries have included the contribution of synovial cells,23 short-term clonal proliferation and increased matrix production by chondrocytes,1,2,5 and “fluid flow” of the matrix.6 Our study suggests the novel concept that chondrocyte migration contributes to the resurfacing of cartilage lesions in vitro.
These results suggest a previously undescribed response of chondrocytes to partial-thickness cartilage injuries, including formation of actin-rich processes and orientation of chondrocytes with their long axes projecting towards the defect site as well as the appearance of cells on the edge of the defect coincident with loss of cells in the adjacent border. These responses occurred largely in the deep and superficial zones and were not evident in the middle zones. The cell processes are adherent to collagen fibrils, and the association between the orientation of the collagen fibrils and the direction of process extension suggests that the collagen infrastructure directs process formation and/or alignment. Our observations are consistent with the possibility that following injury and fibrillation of the collagen network, chondrocytes align towards and slowly migrate to the edge of the defect using contact guidance on the exposed collagen fibers. The phenomenon of process formation by chondrocytes has been described previously,12 but their elaboration following injury, their association with collagen fibers, and their projection into wounded sites are all novel observations. Process extension in cartilage is consistent with the behavior of chondrocytes in monolayer as well as chondrocytes cultured in the presence of type II collagen (Lee, unpublished data).12
The loss of cells adjacent to defect edges has been described5,24 and attributed to apoptosis and necrosis.10 However, it is noteworthy that in an in vitro model using adult bovine cartilage, only 20% of the chondrocytes adjacent to the defect were positive with the TUNEL assay, indicating that while cell death occurred, the majority of the chondrocytes survived.10 We counted viable cells to quantify decreases in cellularity over time. Areas that showed cells lining the defect edge were always associated with an adjacent region of hypocellularity.
Cell migration is a dynamic process that was not fully captured in our study. Nonetheless, the changes in chondrocyte morphology (polarization, process extension, orientation towards the defect) and the loss of cells in areas immediately below those populated by lining cells are most consistent with chondrocyte migration. This concept is strengthened by previous observations that chondrocytes are able to migrate slowly on 2-dimensional surfaces.14,15 In the present 3-dimensional model, the injury and fibrillation of the matrix expose planar surfaces on which the chondrocyte may attach and locomote. Further, the translocation of cells across 300 µm of the cartilage surface within the 4-week period is consistent with the measured low speed range of chondrocyte movement (assuming directed migration over the full period, the average speed would be 0.52 µm/h).
Osteochondral explants are an excellent system for studying chondrocyte behavior. Explants contain no synovium, and therefore, the possibility of cells coming from vascular or synovial elements is eliminated. The subchondral bone is intact, providing the full thickness of the articular surface and serving as a buttress to facilitate the creation of partial-thickness defects. Chondrocytes in the cartilage remained viable and did not change in density in fields greater than 300 µm away from the created defect. However, there are several limitations to generalizing from osteochondral explants to the in vivo situation. The explants did not receive any mechanical stimulation. In addition, the culture medium differs significantly from synovial fluid. In monolayer culture, it has been shown that chondrocytes can gradually dedifferentiate in serum-containing medium.26 Future research directions will include investigation of the differentiation state of the polarized cells within fibrillated matrix and on the defect edges. Koeling et al.27 recently showed that in advanced osteoarthritic cartilage, the tidemark is breached, and a cell population with progenitor characteristics penetrates the cartilage. While we avoided areas of gross degeneration in our study, it is not possible to rule out that progenitor cells resident in cartilage28,29 contribute to the migration patterns. Progenitor cell populations are an integral part of the superficial zone of normal cartilages,28,29 and less well-characterized cells expressing markers associated with progenitor cells are found in deeper zones of normal and osteoarthritic cartilage.30 The issue of potential progenitor cell migrations in cartilage requires a detailed evaluation and will be addressed in separate investigations.
In addition, exposed cells in the bone portion of the explant may release factors affecting the behavior of chondrocytes. Conceivably, a few osteocytes may have migrated and contributed to the observations, and/or a few cells released into the medium could have adhered to fibrillated sites on the explants. Thus, ectopic cell contamination cannot be fully ruled out, but the potential contributions of these cells do not adequately explain all of our observations. The following findings are most compatible with outward migration of cartilage cells: 1) cartilage cells near the defects polarized in the direction of the wound and attached to collagen within the cartilage matrix, and further, they did so selectively within the superficial and deep zones but not the vast middle zone; 2) the cells in these areas were lost, while cells appeared on the outside cut edges, adjacent only to the select cartilage areas where cells had previously polarized.
We propose a model for cartilage cell response to partial-thickness cartilage injury. Matrix injury promotes a gradual process of collagen fibrillation in the superficial and deep zones. This may serve to expose more collagen fibrils and stimulate migration. Processes extend along collagen fibrils, chondrocytes elongate, and migration is initiated. As migrating chondrocytes reach the surface of a defect, they flatten out along the edge, resurface the defect, and may help to prevent further fibrillation and matrix injury. It is clear that not every partial-thickness injury is resurfaced successfully in vivo, but it is possible that many injuries resolve uneventfully without further symptoms. Further studies in this area are needed to determine the significance of the migration of cartilage cells.
Acknowledgments
The authors thank Marianne Tioran and Vicki Madden for technical assistance and Dr. C. William Davis and the UNC Cystic Fibrosis Center for the use of the Leica confocal microscope. Dr. Teresa Morales (Harvard Medical School) contributed by helping to write the article.
Footnotes
This work was funded by NIAMS (AR43883).
The authors declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- 1. Buckwalter JA. Articular cartilage: injuries and potential for healing. J Orthop Sports Phys Therap. 1998;28:192-202. [DOI] [PubMed] [Google Scholar]
- 2. Campbell CJ. The healing of cartilage defects. Clin Orthop. 1969;64:45-63. [PubMed] [Google Scholar]
- 3. Newman AP. Articular cartilage repair. Am J Sports Med. 1998;26:309-24. [DOI] [PubMed] [Google Scholar]
- 4. Noyes FR, Bassett RW, Grood ES, Butler DL. Arthroscopy in acute traumatic hemarthrosis of the knee. J Bone Joint Surg. 1980;62-A:687-95. [PubMed] [Google Scholar]
- 5. Buckwalter JA, Mankin HJ. Articular cartilage: degeneration and osteoarthrosis, repair, regeneration, and transplantation. J Bone Joint Surg. 1998;79-A:612-32. [PubMed] [Google Scholar]
- 6. Ghadially FN, Thomas I, Oryschak AF, Lalonde J-M. Long-term results of superficial defects in articular cartilage: a scanning electron microscope study. J Path. 1977;121:213-7. [DOI] [PubMed] [Google Scholar]
- 7. Hunziker EB. Articular cartilage repair: basic science and clinical progress. A review of current status and prospects. Osteoarthritis Cartilage. 2002;10(6):432-63. [DOI] [PubMed] [Google Scholar]
- 8. Mankin HJ. The response of articular cartilage to mechanical injury. J Bone Joint Surg. 1982;64-A:460-6. [PubMed] [Google Scholar]
- 9. Mankin HJ. Localization of tritiated thymidine in articular cartilage of rabbits, II: repair in immature cartilage. J Bone Joint Surg Am. 1962;44A:688-98. [Google Scholar]
- 10. Tew SR, Kwan APL, Hann A, Thomson BM, Archer CW. The reactions of articular cartilage to experimental wounding: role of apoptosis. Arthritis Rheum. 2000;43:215-25. [DOI] [PubMed] [Google Scholar]
- 11. Bush PG, Hall AC. The volume and morphology of chondrocytes within non-degenerate and degenerate human articular cartilage. Osteoarthritis Cartilage. 2003;11(4):242-51. [DOI] [PubMed] [Google Scholar]
- 12. Lee GM, Loeser RF. Cell surface receptors transmit sufficient force to bend collagen fibrils. Exp Cell Res. 1999;248:294-305. [DOI] [PubMed] [Google Scholar]
- 13. Peretti GM, Randolph MA, Caruso EM, Rossetti F, Zaleske DJ. Bonding of cartilage matrices with cultured chondrocytes: an experimental model. J Orthop Res. 1998;16:89-95. [DOI] [PubMed] [Google Scholar]
- 14. Frenkel SR, Clancy RM, Ricci JL, Di Cesare PE, Rediske JJ, Abramson SB. Effects of nitric oxide on chondrocyte migration, adhesion, and cytoskeletal assembly. Arthritis Rheum. 1996;39:1905-12. [DOI] [PubMed] [Google Scholar]
- 15. Chang C, Lauffenburger DL, Morales TI. Motile chondrocytes from newborn calf: migration properties and synthesis of collagen II. Osteoarthritis Cartilage. 2003;11:603-12. [DOI] [PubMed] [Google Scholar]
- 16. Speer DP, Dahners LE. The collagenous architecture of articular cartilage: correlation of scanning electron microscopy and polarized light microscopy observations. Clin Orthop. 1979;139:267-75. [PubMed] [Google Scholar]
- 17. Jeffery AK, Blunn GW, Archer CW, Bentley G. Three-dimensional collagen architecture in bovine articular cartilage. J Bone Joint Surg. 1991;73-B:795-801. [DOI] [PubMed] [Google Scholar]
- 18. Lee GM, Poole CA, Kelley SS, Chang J, Caterson B. Isolated chondrons: a viable alternative for studies of chondrocytes metabolism in vitro. Osteoarthritis Cartilage. 1997;5:261-74. [DOI] [PubMed] [Google Scholar]
- 19. Belitsos PC, Hildreth JE, August JT. Homotypic cell aggregation induced by anti-CD44 (Pgp-1) monoclonal antibodies and related to CD44 (Pgp-1) expression. J Immunol. 1990;144(5):1661-70. [PubMed] [Google Scholar]
- 20. Srinivas GR, Barrach HJ, Chichester CO. Quantitative immunoassays for type II collagen and its cyanogens bromide peptides. J Immunol Methods. 1993;159(1-2):53-62. [DOI] [PubMed] [Google Scholar]
- 21. Srinivas GR, Chichester CO, Barrach HJ, Pillai V, Matoney AL. Production of type II collagen specific monoclonal antibodies. Immunol Invest. 1994;23(2):85-98. [DOI] [PubMed] [Google Scholar]
- 22. Osborne M, Weber K. Immunofluorescence and immunocytochemical procedures with affinity purified antibodies: tubulin-containing structures. Meth Cell Biol. 1982;24:97-132. [DOI] [PubMed] [Google Scholar]
- 23. Hunziker EB, Rosenberg LC. Repair of partial-thickness defects in articular cartilage: cell recruitment from the synovial membrane. J Bone Joint Surg. 1996;78-A:721-33. [DOI] [PubMed] [Google Scholar]
- 24. Hunziker EB, Quinn TM. Surgical removal of articular cartilage leads to loss of chondrocytes from the wound edges. Trans Orthop Res Soc. 2000;46:185. [DOI] [PubMed] [Google Scholar]
- 25. Walker JM. Pathomechanics and classification of cartilage lesions, facilitation of repair. J Orthop Sports Phys Therap. 1998;28:216-31. [DOI] [PubMed] [Google Scholar]
- 26. Stewart MC, Saunders KM, Burton-Wurster N, Macleod JN. Phenotypic stability of articular chondrocytes in vitro: the effects of culture models, bone morphogenic protein 2, and serum supplementation. J Bone Min Res. 2000;15:166-74. [DOI] [PubMed] [Google Scholar]
- 27. Koelling S, Kruegel J, Irmer M, Path JR, Sadowski B, Miro X, et al. Migratory chondrogenic progenitor cells from repair tissue during the later stages of human osteoarthritis. Cell Stem Cell. 2009;4:324-35. [DOI] [PubMed] [Google Scholar]
- 28. Dowthwaite GP, Bishop JC, Redman SN, Khan IM, Rooney P, Evans DJR, et al. The surface of articular cartilage contains a progenitor cell population. J Cell Sci. 2004;117:889-97. [DOI] [PubMed] [Google Scholar]
- 29. Alsalameh S, Amin R, Gemba T, Lotz M. Identification of mesenchymal progenitor cells in normal and osteoarthritic human articular cartilage. Arthritis Rheum. 2004;50:1522-32. [DOI] [PubMed] [Google Scholar]
- 30. Grogan SP, Miyaki S, Asahara H, D’Lima DD, Lotz MK. Mesenchymal progenitor cell markers in human articular cartilage: normal distribution and changes in osteoarthritis. Arthritis Res Ther. 2009;11(3):R85. [DOI] [PMC free article] [PubMed] [Google Scholar]